

Abstract
Resistance to aminoglycoside antibiotics has had a profound impact on clinical practice. Despite their powerful bactericidal activity, aminoglycosides were one of the first groups of antibiotics to meet the challenge of resistance. The most prevalent source of clinically relevant resistance against these therapeutics is conferred by the enzymatic modification of the antibiotic. Therefore, a deeper knowledge of the aminoglycoside-modifying enzymes and their interactions with the antibiotics and solvent is of paramount importance in order to facilitate the design of more effective and potent inhibitors and/or novel semisynthetic aminoglycosides that are not susceptible to modifying enzymes.
Keywords: antibiotic resistance, combination therapy, bi-substrate inhibitors, decoy acceptors
1. Introduction
The continuous appearance of bacterial strains resistant to most antibiotics already known, along with the scarce perspective of new bactericidal agents coming out in the near future, have placed bacterial multi-drug resistance (MDR) among the most pressing issues for worldwide health [1,2,3]. Furthermore, the increasing number of hospital MDR infections has boosted the interest for new aminoglycoside antibiotics for clinical use, and consequently research on this topic has drawn renewed attention. Aminoglycosides are a group of natural antibiotics from Streptomyces that have been used in clinical practice for more than 50 years [4]. Their potent bactericidal activity relies upon binding specifically to the 16S rRNA of the 30S ribosomal subunit, thus interfering with protein synthesis [5,6]. However, the first resistant bacterial strains began to appear in the 1960s due to a high rate of dissemination via R-plasmids, transposons, and integrons [7,8]. In an attempt to overcome this emerging resistance to natural antibiotics, the first semi-synthetic aminoglycosides, such as amikacin, dibekacin, isepamicin, and netilmicin, were introduced during the 1970s [9,10]. The discovery of gentamicins, a newer family of aminoglycosides isolated from Micromonospora, contributed to a come-back of this class of bactericidal agents in clinical practice [11]. These antibiotics were very active against Pseudomonas aeruginosa, a tougher micro-organism less susceptible to the original aminoglycosides, which unfortunately soon after developed resistance through the ANT(2″) enzyme [12,13]. Butirosine, another aminoglycoside discovered later, was able to avoid inactivation by APH(3′) and ANT(2″) enzymes [14]. There is a large number of aminoglycoside antibiotics, but the resistance mechanisms developed by micro-organisms increase with the frequency of their use.
2. Understanding the Modifying Enzymes
Tolerance towards antibiotics can be achieved through different mechanisms. However, the most prevalent in the clinic is that due to enzymatic modification that renders aminoglycosides of decreased affinity for their natural primary target, 16S rRNA [15,16,17]. There are three families of aminoglycoside-modifying enzymes (AMEs): N-acetyltransferases (AACs) that acetylate an amino group using acetyl-Coenzyme A; O-nucleotidyltransferases (ANTs) that transfer an adenyl group from ATP to a hydroxyl group of the antibiotic; and O-phosphotransferases (APHs), which phosphorylate a hydroxyl group also employing ATP (Figure 1). The emergence of bifunctional enzymes (i.e., APH(2″)-AAC(6′)) that are able to modify almost all aminoglycoside antibiotics presents a huge challenge that new aminoglycosides will have to overcome [18] (Figure 1).
Figure 1.

Susceptible positions in Kan B to AME modification.
AACs are found both in Gram-positive and Gram-negative bacteria and are able to reduce the affinity of the antibiotic for the receptor 16S rRNA by 4 orders of magnitude (Table 1). This family of enzymes is the largest within the AMEs, with 48 sequences identified so far, and they all weigh around 20–25 kDa and comprise a wide range of positions susceptible to modification (6′, 2′, N-1, and N-3). Mutagenesis studies have shown that just the mutation of a single amino acid of the AAC can modulate the specificity for the antibiotic. For example, AAC(6′)-I and AAC(6′)-II share the capacity to modify kanamycin but they differ in their propensity to acetylate amikacin or gentamicin C. The structural gene of the AAC(3) and AAC(6′) is generally found on transposable elements [19]. AAC(3) enzymes were the first to be described to confer resistance to gentamicin (G), kanamycin (K), fortimicin (F), and tobramycin (T) [20] and can acetylate either N/O groups of the aminoglycoside. Five AAC(2′) enzymes are encoded in Mycobacteria and one has been crystalized from Mycobacterium tuberculosis. AAC(2′) from M. tuberculosis catalytically and enthalpically favors those aminoglycosides with amine/hydroxyl groups at the 2′ position and shows an increase in affinity for all antibiotics when CoA is present. This AME exhibits a high tolerance towards the antibiotic molecule (4,5- and 4,6-substitution), and it is thought to be modulated by water molecules that mediate between aminoglycoside and side chain carboxylate groups of the receptor. Moreover, side chain reorientation relative to the apo-enzyme upon binding of the substrates also contributes to the plasticity of this enzyme [21]. AAC(6′) enzymes are the most prevalent in clinical strains, conferring resistance to amikacin (A), gentamicin (G), kanamycin (K), neomycin (N), dibekacin (D), sisomicin (S), isepamicin (I), and tobramycin (T) and has an ordered kinetic mechanism. Three-dimensional (3D) structures of AAC(6′), AAC(3), and AAC(2′) have been described with a remarkable conservation of the overall structure, albeit with low amino acid sequence identity [22,23].
Table 1.
N-acetyltransferase (AAC)-modifying enzymes.
| Enzyme | Resistance Profile | Bacterial Source | Pdb Number | |
|---|---|---|---|---|
| AAC(6′) | I (a–d,e,f–z) | T, A, N, D, S, K, I | Salmonella enterica | 1S60, 2VBQ, 1S3Z, 1S5K, 2QIR |
| II | T, G, N, D, S, K | Enterococcus faecium | 2A4N, 5E96 | |
| Acinetobacter haemolyticus | 4F0Y, 4EVY, 4F0Y | |||
| Acinetobacter baumannii | 4E80 | |||
| Escherichia coli | 6BFF, 6BFH, 1V0C, 2BUE, 2VQY | |||
| Staphylococcus warneri | 4QC6 | |||
| AAC(3) | I (a–b) | G, S, F | Serratia marcesans | 1B04 |
| II (a–c) | T, G, N, D, S | Pseudomonas aeruginosa | 4YFJ | |
| III (a–c) | T, G, D, S, K, N, P, L | Klebsiella pneumoniae, | ||
| IV | T, S, N, D, S, A | Campylobacter jejuni | ||
| VII | G | Actinomycetes | ||
| AAC(2′) | I (a–c) | T, S, N, D, Ne | Providencia stuartii | 5US1 |
| Mycobacterium tuberculosis | 1M44, 1M4D, 1M4G, 1M41 | |||
| AAC(1) | Ia | P, L, R, AP | E. coli | |
| Campylobacter spp. |
Abbreviations: A, amikacin; AP, apramycin; D, dibekacin; F, fortimicn; H, hygromycin; I, isepamicin; G, gentamicin; K, kanamycin; L, lividomycin; N, netilmicin; Ne, neomycin; P, paromomycin; R, ribostamycin; S, sisomicin; T, tobramycin.
APHs are the second-most abundant family of AMEs and confer resistance to aminoglycosides in Enterococcus and Staphylococcus strains (Table 2). There are seven types of APHs with 30 kDa mass and high sequence homology (20–40%) in their C-terminal end [24]. APH(3′)-IIIa is usually used as a resistance marker and its 3D structure has been described several times [25,26]. APH(3′)-IIIa promiscuity seems to be governed by disordered elements that adopt well-defined conformations when the aminoglycoside is bound [27]. APH(3′)-IIa from Enterococcus faecalis is able to phosphorylate the 3′ and 5′-OH groups, giving rise to di-phosphorylated aminoglycosides [28] through an ordered sequential mechanism in which the binding of ATP is followed by the antibiotic [29]. An interesting property of this enzyme is that it is competitively inhibited by tobramycin, which one would expect not to be a substrate because it lacks a free 3′-hydroxylgroup. APH(2″) is a very promiscuous enzyme based on the range of susceptible positions in the antibiotic skeleton (2″, 3′, 3″, and 5″); it represents an important resistance element in Gram-positive bacteria, and this enzyme uses Guanosine Triphosphate (GTP) as the most efficient donor substrate over ATP. Oddly enough, many other APHs have been identified, such as APH(6), APH(3″), APH(9), APH(4), and APH(7), but their occurrence is strikingly uncommon in the clinic. APH(9)-Ia exhibits a similar folding to that of the APH(3′) and APH(2″) enzymes, but it differs significantly in its substrate binding area and in the fact that it undergoes a conformational change upon ligand binding.
Table 2.
O-phosphotransferase (APH)-modifying enzymes.
| Enzyme | Resistance Profile | Bacterial Source | Pdb | |
|---|---|---|---|---|
| APH(3′) | I (a–d) | K, Ne, R, L, P | Acinetobacter baumannii | 4FEV |
| II | K, Ne, B, P, R | Stenotrophomonas maltophilia | ||
| III (a–b) | K, Ne, P, B, L, R, B, A, I | |||
| IV | K, Ne, B, P, R | |||
| V | Ne, P, R | |||
| VI | K, Ne, P, R, B, A, I | Bacillus circulans | ||
| APH(2″) | I-a | K, G, T, S, D | ||
| I-(b,d) | K, G, T, N, D | Escherichia coli | 4DCA | |
| II-(a–b) | K, G, T | Enterococcus faecium | 3HAM, 3HAV | |
| IVa | G, K, S | Enterococcus cassaliflavus | 5C4K, 5C4L, 4N57, 4DT8, 4DT9, 4DTA, 4DTB, 3SG8, 3SG9 | |
| APH(3″) | I (a–b) | St | Acinetobacter baumannii | 4EJ7, 4FEU, 4FEV, 4FEX, 4FEW |
| III a | St | Enterococcus faecalis | 2BKK | |
| APH(7) | I a | H | Streptomyces hygroscopicus | |
| APH(4) | I-(a–b) | H | Escherichia coli | 3W0O, 3TYK, 3W0M, 3W0N |
| APH(6) | I-(a–d) | St | Streptomyces griseus | |
| APH(9) | I-(a–b) | Sp | Legionella pneumophila | 3I0O, 3I0Q, 3I1A, 3Q2M |
Abbreviations: A, amikacin; D, dibekacin; H, hygromycin; I, isepamicin; G, gentamicin; K, kanamycin; L, lividomycin; N, netilmicin; Ne, neomycin; P, paromomycin; R, ribostamycin; S, sisomicin; T, tobramycin; Sp, spectinomycin; St, streptomycin.
ANTs are the smallest family of AMEs, with four crystalized structures, ANT(2″), ANT(3″), ANT(4′), and ANT(6′), as can be seen in the following table (Table 3) [30,31,32,33,34,35].
Table 3.
O-nucleotidyltransferase (ANT)-modifying enzymes.
| Enzyme | Resistance Profile | Bacterial Host | Pdb Number |
|---|---|---|---|
| ANT(2″) | K, T, G, D, S | Pseudomonas aeruginosa | 4XJE, 5CFT, 5CFS, 5CFU |
| Klebsiella pneumoniae | 4WQK, 4WQL, 5KQJ | ||
| ANT(3″) | St, Sp | Salmonella enterica | 4CS6, 5G4A |
| ANT(4′) | K, Ne, T, A, D, I | Pseudomonas aeruginosa | 4EBJ, 4EBK |
| Staphylococcus aureus | 1KNY | ||
| ANT(6) | St | Bacillus subtilis | 2PBE, 1B87 |
| ANT(9) | Sp | Enterococcus avium |
Abbreviations: A, amikacin; D, dibekacin; I, isepamicin; G, gentamicin; K, kanamycin; Ne, neomycin; S, sisomicin; T, tobramycin; Sp, spectinomycin; St, streptomycin.
Genes coding for ANT enzymes are found in plasmids, transposons, and chromosomes. ANT(2″)-Ia is able to modify gentamicin, tobramycin, dibekacin, sisomicin, and kanamycin [30], while ANT(4′)-Ia is active against almost all aminoglycosides and other aminoglycosides with 4′/4″-OH groups [33]; ANT(3″) inactivates streptomicyn and spectinomycin [32], while ANT(6) and ANT(9) can modify only streptomycin and spectomycin, respectively [35,36]. ANT(2″)-Ia and ANT(4′)-Ia are clinically relevant proteins and only share 27% amino acid homology. ANT(4′)-Ia is a dimeric enzyme with two active sites, and recognizes all nucleotides triphosphate and almost all 4,5 or 4,6-aminoglycosides except those from the streptomycin family [37,38]. The 3D structure of ANT(4′)-Ia was the first one to be described, and its complex with kanamycin revealed that several active site residues interact via hydrogen bonds with the glucosamine and 2-deoxystreptamine moiety, but relatively fewer residues interact with the 3′-aminoglucose sugar, this being important knowledge for the design of antibiotics or inhibitors. The antibiotic binding pocket is covered by negatively charged residues, where Glutamic 145 acts as a general base for the activation of the 4′-OH group of kanamycin, thus preparing the antibiotic for the attack of the Mg-ATP stabilized by lysine 149, which facilitates the nucleophilic attack. Overall, the dominant role of electrostatics in aminoglycoside recognition, in combination with the enzyme anionic regions, confers to the protein/antibiotic complex a highly dynamic character. The kinetic mechanism is an ordered process, where the antibiotic binds first to the active site and later on to the Mg-ATP. Determination of the order of product release revealed that PPi is discharged first, followed by the AMP-aminoglycoside, all in agreement with an ordered Bi-Bi mechanism [39]. ANT(2″)-Ia from Klebsiella pneumoniae has an ordered sequential mechanism, where the presence of the two substrates at the same time is essential (Mg-ATP binds before the aminoglycoside) [40,41,42,43]. The observed promiscuity of ANT(2″)-Ia from Pseudomonas aeruginosa is not only due to binding cleft size, but also it can be controlled by ligand modulation on dynamic, disordered, and thermodynamic properties of ANT under cellular conditions [44]. ANT(6′), a 37 kDa protein, transfers an adenyl group from ATP to the 6′-hydroxy function on the aminoglycoside, thus leading to a sharp decrease in the drug affinity for its target RNA. The conformational behavior of streptomycin, both in the free and the protein-bound states, was studied by NMR experiments given that the 3D structure is in a free form [45,46]. The streptomycin is characterized by a high degree of flexibility in solution, but this equilibrium is clearly altered upon binding to the enzyme. ANT(6′) showed a clear specificity for nucleotides that incorporate a purine ring, and regarding the aminoglycoside counterpart, it is very specific: it only recognizes streptomycin. In contrast, the 3D structure and the kinetic mechanism of ANT(9) has not been resolved so far.
It was in 1986 when the first bifunctional enzyme was described, AAC(6′)-Ie-APH(2″)-Ia from Enterococcus faecalis [47]; a structure-function analysis with various aminoglycosidic substrates revealed an enzyme with a broad specificity in both enzymatic activities catalyzing N- and O-acetylation. The AAC(6′)-APH(2″) from Staphylococcus aureus enzyme confers resistance to gentamicin, kanamycin, tobramycin, and, when overexpressed, to amikacin, presenting a dramatic negative impact on clinical therapy [48]. Later on, other bifunctional enzymes were also described: ANT(3″)-Ii-AAC(6′)-IId from Serratia marcescens [49] and AAC(3)-Ib-AAC(6′)-Ib from Pseudomonas aeruginosa [50] (Table 4).
Table 4.
Bifunctional modifying enzymes.
| Enzyme | Resistance Profile | Bacterial Source | Pdb Number |
|---|---|---|---|
| Enterococcus faecalis | |||
| AAC(6′)-Ie-APH(2″)-IVa | G, K, T, A | Staphylococcus aureus | 4ORQ |
| APH(2″)-Id-APH(2″)-IVa | K, G, T, S, D | Enterococcus casseliflavus | 4DBX, 4DE4, 4DFB |
| APH(2″)-Ia-APH(6′)-Ie | K, G, T, S, D, St | Staphylococcus aureus | 5IQF |
| ANT(3)-Ib-AAC(6′)-IId | T, A, N, D, S, K, St, Sp | Serratia marcescens | |
| AAC(3)-Ib-AAC(6′)-Ib | G, S, F, T, A, N, D, K, I | Pseudomonas aeruginosa |
Abbreviations: A, amikacin; D, dibekacin; F, fortimicn; I, isepamicin; G, gentamicin; K, kanamycin; N, netilmicin; S, sisomicin; T, tobramycin; Sp, spectinomycin; St, streptomycin.
The adenyltransferase domain of ANT(3″)-Ii-AAC(6′)-IId appears to be highly specific for the aminoglycoside, while the acetyltransferase domain shows a broad substrate tolerance [51]. Kinetic analysis of the mechanism of ANT(3″)-Ii points towards a Theorell–Chance type of reaction, with ATP binding to the active site before the aminoglycoside, and once the reaction has occurred, the product is the last to be released. However, the AAC(6′)-IId domain follows an ordered Bi-Bi mechanism in which the antibiotic is the first to bind in the active site, and CoA is released prior to the modified aminoglycoside. Also, structural studies have revealed that this enzyme contains dynamic segments that modulate before and after aminoglycoside binding. In the case of AAC(3)-Ib-AAC(6′)-Ib, both domains follow a sequential ordered kinetic mechanism in which the acetyl-CoA binds first to the active site, followed by the aminoglycoside, and the CoA is the last product to be released [52]. Despite this interesting behavior, to date only the crystal structure of some APH(2″) domains have been described.
3. Semi-Synthetic Aminoglycoside Derivatives
The emergence of resistance towards the first generation of aminoglycosides led to increased efforts to identify similar antibiotics that were not susceptible to resistance [53,54,55,56,57,58]. From a conceptual point of view, one of the most simple and straightforward strategies to generate new derivatives that are not susceptible to enzymatic inactivation relies on the modification or elimination of those functional groups (OH/NH2) that can be altered by the enzyme. However, this should only be taken into consideration when such functional groups are not involved in key contacts with the receptor (16S rRNA) so that their biological activity is maintained. Tobramycin (3′-deoxy-kanamycin B) showed high activity against strains expressing APH(3′), and it is a good competitive inhibitor of this enzyme [59]. Dibekacin (3′,4′-dideoxy-kanamycin B) was the first rationally designed semi-synthetic aminoglycoside based on the 3′ phosphorylation of kanamycin B, being effective against Staphylococcus and Pseudomonas, but unfortunately still susceptible to ANT(2″) [60] and the bifunctional enzyme AAC(6′)-APH(2″) [61]. In contrast, fewer examples of deoxygenation on neomycin-related antibiotics have been reported [62]. The number and positions of amino groups play a significant role in the aminoglycoside activity. Methylation of the N-6′ and N-3″ positions in kanamycin B yielded a compound (6′,3″-di-N-methyl-kanamycin B) that displayed activity against resistant strains, but showed a weaker bactericidal activity than its natural antibiotic [63] (Figure 2).
Figure 2.

Structures of Kanamycin B and 6’,3’’-di-N-Methyl Kanamycin B.
The observation that butirosin, a N-1 derivative of the 2-deoxystreptamine moiety, is poorly modified by APH(3’), prompted the synthesis of several kanamycin and neomycin derivatives bearing an N-1 modification. Kanamycin derivatization at this position with a (S)-4-amino-2-hydroxybutyryl (AHB) group gave rise to amikacin, which has proven to be a very effective aminoglycoside antibiotic in clinic [64] (Figure 3). This compound was able to arrest the cell growth of strains expressing the AAC(1), APH(3′)-Ia, and ANT(2′) enzymes [65]. The N-1 modification of dibekacin with an AHB group provided arbekacin, which was used in clinic against Pseudomonas and Staphylococcus [66] (Figure 3). Arbekacin is a substrate of the bifunctional enzyme AAC(6′)-APH(2″), but cannot be modified by the APH(3′) or ANT(4′) present in some Methicillin-resistant Staphylococcus aureus (MRSA) strains [67,68]. The trend of antibacterial activity of these aminoglycoside derivatives with an N-1 AHB group is similar to a 3′-deoxygenation in the antibiotic skeleton. Some other functionalities at the N-1 position were also studied, but were much less active against P. aeruginosa.
Figure 3.

Structure of kanamycin class of aminoglycosides.
Further elaboration upon these N-1 derivatives yielded two new derivatives, JLN027 and etimicin, that are more effective than gentamicin, amikacin, and tobramycicn [69,70,71], but still have encountered resistance to some MRSA strains [72,73,74,75] (Figure 4).
Figure 4.

Aminoglycosides with N-1 kanamycin derivatives.
Modification of the N-6′, N-2′, N-3, and N-1 positions through the synthesis of deaminated aminoglycosides (neamine or kanamycin B) has shown a dramatic loss of enzymatic susceptibility towards APH(3′)-Ia/IIa while still retaining the antibacterial activity, probably due to a decreased overall positive charge on the antibiotic. AMEs do not show activity against pyranmycins, which are a group of semisynthetic derivatives of aminoglycosides that differ from regular aminoglycosides in that they contain a pyranose in place of a furanose at the O-5 position of neamine, giving rise to a derivative with low cytotoxicity (TC005 derivative) [76] (Figure 5). In order to improve the derivatives, Chang’s group prepared some 3′,4′-dideoxygenated pyranmycin and kanamycin derivatives giving resistance to AME (RR501) [77,78] (Figure 5). These simplified skeletons proved highly effective against several pathogenic bacterial strains, such as P. aeruginosa and S. aureus.
Figure 5.

Structure of kanamycin B analogs (Pyranmycin).
Selective modification of the N-3″ amino of kanamycin A into a guanidine group gave N-3″-guanidino kanamycin A (Figure 6), which presented protection against inactivation performed by ANT(4′), APH(3′), and AAC(6′), while maintaining its antibiotic activity [79]. The protecting effect of the guanidine group at the 3″-position was rationalized in terms of a binding hindrance with the respective inactivating enzymes, which presumably does not take place within the A-site.
Figure 6.

Structure of the N-3’’-Guanidino Kanamycin A and Plazomicin.
Plazomicin (PLZ, formerly ACHN-490) represents another example of amino modification, where the N-6’, N-1, and N-3″ positions have been substituted with different ramifications (Figure 6). This aminoglycoside is not affected by any known AME except for AAC(2′), making it one of the few examples that is currently in clinical use, retaining activity against most of the clinical isolates (Minimum Inhibitory Concentration, MIC 4 μg/mL) [80,81,82]. These new aminoglycosides constitute our most promising defense alternative for the treatment of resistant MRSA strains. PLZ is currently in Phase 3 clinical trials for patients with bloodstream infections or nosocomial pneumonia and it is an important new weapon in the pipeline to fight antibiotic resistance.
The aminosugar ring in aminoglycosides provides these molecules with high affinity for the prokaryotic A-site, since it penetrates deeply into the major groove, thus displacing A1492 of 16S rRNA, giving the ribosome a continuous “on” state during the translation process. Structural studies of the interactions between aminoglycosides and the RNA or AMEs involved in their enzymatic inactivation have been performed to identify the molecular nature of these recognition processes. The 4,5 or 4,6-disubstituted aminoglycosides specifically bind to the A-site with several conserved contacts, the majority of them corresponding to the pseudo-disaccharide neamine [83,84,85]. The synthesis of hybrid (4,5/4,6-) aminoglycosides, based on the superimposition of crystallographic structures, originated a new family of 4,5,6-aminoglycoside derivatives with a better prognosis against AMEs than the corresponding natural antibiotics [86] (Figure 7). Another approach based on structural data showed that the antibiotic scaffold presents different conformations when bound to the A-site or to the AME. This realization translated into the design of a conformationally locked aminoglycoside that retained the antibiotic activity but was not susceptible to enzymatic modification [87,88] (Figure 7).
Figure 7.

Structure of the 4,5,6-Neomycin derivative and constrained neomycin.
In order to increase the receptor binding affinity, aminoglycoside dimers were conceived and synthesized. Crystallographic studies indicated that neither the 6″-OH group in kanamycin nor the 5-OH group in neamine were essential for RNA binding; thus, they were selected as anchoring points for the synthesis of various dimers equipped with different linkers [89] (Figure 8). Kanamycin dimers (Figure 8) exhibited the same activity than the parent compound, but were also inactivated by the same AMEs (ANT(4′), APH(3′), and AAC(6′)). In the case of the neamine dimers [90] (Figure 8), these presented the same biological activity (lower than neomycin or kanamycin), but as well were inactivated by the AMEs. However, these compounds are interesting probes for strains not expressing AMEs, since they exhibit higher affinity for the receptor RNA.
Figure 8.

Structure of the Neamine and kanamycin dimers.
The molecular recognition of aminoglycosides with their receptors, in most cases, is stabilized by a significant number of salt-bridges and polar contacts, but it seems to be promoted by CH/π stacking interactions involving aromatic residues of the protein/RNA with the antibiotic. So, the role played by CH/π stacking interactions in the molecular recognition of aminoglycosides by its receptors has been evaluated and proven to be significant [91]. The modification of natural aminoglycosides is a promising direction to search for novel aminoglycosides with potency against resistant strains.
Recently, Crich et al. prepared a semisynthetic paromomycin derivative that displays similar antibacterial activity to the parent compound against clinical strains of E. coli and MRSA (ANT(4′,4″), APH-(3′,5″), and AAC(6′)). The enhanced activity on the ribosome has been demonstrated to depend on the equatorial hydroxyl group at the 6′-position, thereby providing support for the crystallographically derived models of aminoglycoside–ribosome interactions [92] (Figure 9).
Figure 9.

Structure of the Paromomycin derivative4. Inhibitors of Aminoglycoside Modifying Enzymes.
Another way to evade aminoglycoside resistance by AMEs is through the use of specific inhibitors of these enzymes. Some attempts have been made to produce inhibitors of one or more of the AMEs [93,94,95]. Bi-substrate analogs have been synthesized as inhibitors of AAC and ANT/APH enzymes based on the proposed kinetic mechanism. For instance, the crystallographic structure of AAC(6′)-Ii in complex with kanamycin B-CoA provided a new insight into chemical optimization [96] (Figure 10). This adduct is a good inhibitor of the AAC family, but unfortunately does not have any bacterial activity due to a poor permeability of the cell wall [97].
Figure 10.

Structure of Kanamycin –CoA inhibitor to AME.
Using the same approach, a nucleotide–aminoglycoside complex for the inhibition of APHs/ANTs has been described (Figure 11). Such a tethered bi-substrate design contains a neamine core with the 3′-OH linked to adenosine via a non-hydrolyzable linker in place of the triphosphate group [98].
Figure 11.

Structure of nucleotide-neamine complex as inhibitor of APHs and ANTs.
Regarding the modification of discrete functional groups targeted by enzymatic resistance, different aminoglycoside analogs have been synthesized, which turn into suicide inactivators upon enzymatic phosphorylation. Such is the case of 2-nitro-2’-deaminokanamycin, a good inhibitor of APH(3′)-Ia and APH(3′)-IIa [99] (Figure 12), which generates in situ a nitro-alkene intermediate susceptible to Michael addition by close nucleophilic residues, thus rendering a covalent intermediate that blocks the active site and abolishes the activity (Figure 12). Another interesting example is the synthesis of 3′-ketokanamycin, where the keto group is known to exist in equilibrium with its ketal form, so that the phosphorylated ketal can be transformed back into a keto form by eliminating a dibasic phosphate, and then it can further re-regenerate the ketal. Interestingly, both 3′-ketal- and 2′-nitro-kanamycin derivatives can inactivate the APH(3′) in an irreversible manner. Most probably, these compounds are inhibitors of other kinases too.
Figure 12.

Structure and mode of action of the 2’-nitro-2’deaminokanamycin B.
Allen et al. reported that α-hydroxytropolone plus the appropriate aminoglycoside substrates were active against resistant bacteria possessing the adenylyltransferase phenotype [100]. Recently, α-hydroxytropolone derivatives have also been described as good competitive inhibitors of ATP in the binding site of ANT(2″)-Ia [101] (Figure 13).
Figure 13.

Structure of hydroxytropolone derivatives.
Garneau-Tsodikova et al. have reported a sulfonamide scaffold that served as a pharmacophore to generate inhibitors of AAC(2″) from Mycobacterium tuberculosis, whose upregulation causes resistance to the aminoglycosides [102] (Figure 14).
Figure 14.
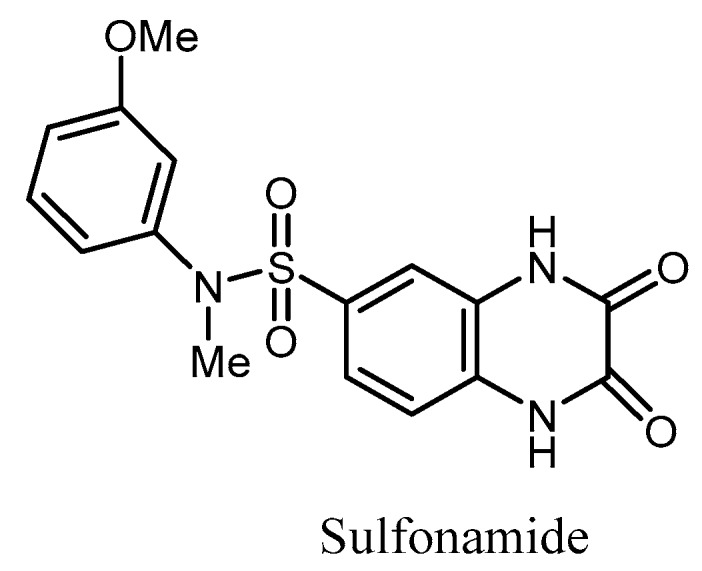
Structure of a sulfonamide as inhibitor of the AAC(2’’).
Given that AMEs share common binding features and that many of them also bind peptides and proteins, cationic peptides could serve as lead molecules in the development of new inhibitors of these enzymes. Therefore, cationic peptides have been tested as inhibitors of APH(3′)-IIIa, AAC(6′)-Ii, and AAC(6′)-APH(2″), and the results showed that the indolicidin moiety and its analogs have an inhibitory effect against both ACC and APH enzymes, albeit by different mechanisms. These peptides constitute the first example of broad-spectrum inhibitors of AMEs, but unfortunately none of them showed any inhibitory effect in vivo. Known inhibitors of eukaryotic protein kinases have been studied too to determine whether they were active against APH(3′)-IIIa and AAC(6′)-APH(2″) because of the structural relation found between these enzymes [95].
Some aminoglycoside dimers have also been used as inhibitors of the AMEs. Neamine dimers were investigated for their antibacterial activity and their capability to inhibit the action of bifunctional AAC(6′)-APH(2″), and were proven to be active against clinically isolated strains of P. aeruginosa. However, the synthesis of one universal inhibitor for all AMEs seems still unreachable, since good inhibition relies on many mechanistic and kinetic factors that can vary between families (AAC, ANT, and APH) and between types too (i.e., APH(3′)-I, II).
4. Combination Therapy
The use of a combination therapy can help in solving the problem of resistance by AMEs. This approach relies on the rescue of original aminoglycosides (gentamicin, amikacin, or etimicin) through the co-administration of the corresponding inhibitors for each enzyme. Ideally, the adjuvant compound (inhibitor) would be targeted preferentially by the resistance enzymes, thus freeing the antibiotic to bind the target A-site. An all-purpose inhibitor would be a compound that mimics the charge and shape of an aminoglycoside (common for the three families of AMEs), having in mind that all AMEs have a highly negatively charged surface in their binding sites such that a must-have feature should be an overall positive charge. A proof-of-concept for this strategy is that the use of streptidine along with streptomycin in the cell culture restores the activity of the streptomycin against ANT(6), because streptidine competes for the binding site with the streptomycin, acting as a “decoy acceptor” of the enzyme [103] (Figure 15). Thus, streptidine could be a good starting compound for the design of more efficient “decoy acceptors” of AMEs targeting streptomycin, or 2-deoxystreptamine for those targeting 4,5- or 4,6-aminoglycosides (Figure 15).
Figure 15.

Structure of the streptomycin and streptidine as “decoy acceptor”.
Hybrid antibiotics have also been developed to battle bacterial resistance [104,105,106,107]. The ability to slow down the emergence of resistance is probably one of the most important advantages of hybrid drugs [108,109]. This strategy connects two antibiotics that have different modes of action into a single molecule. The main difficulty so far has been to find the correct linker that connects the two drugs to provide better inhibition on both targets. The synthesis of the Cipro-NeoB/Cipro-KanA, which was active against a wide range of strains, is one of the most successful examples of this approach (Figure 16). The MIC values of these hybrids were the same against resistant and non-resistant strains (AAC(6′), APH(3′), and AAC(6′)/APH(2″) [110,111]. This kind of aminoglycoside derivative, based on a hybrid structure, provides a promising drug with an unusual dual mechanism of action, a potent profile against AMEs, and reduced potential for generating bacterial resistance.
Figure 16.

Structure of the kanamycin B-Cipro complex as hybrid antibiotic.
5. Conclusions
In this review, we have aimed to cover the most relevant semi-synthetic aminoglycosides, inhibitors, and decoy acceptors of the AMEs. By far, the most successful chemical approach to modify the natural aminoglycosides has been the modification of the N-1/N-3″ amino groups with the AHB group or a guanidino substituent that has retained the parent antibiotic activity. Alkylation at the N-6′ position of the antibiotic resulted in a decrease of affinity for resistant enzymes, but a concomitant decrease in bacterial activity has been observed as well. Chemical deoxygenation of the 3′/3′,4′-OH groups of aminoglycosides resulted in compounds with high affinity for the RNA and resistance against MRSA. Comparatively, fewer inhibitors or decoy acceptors of the AMEs have been described.
On the other hand, ANT(4′)-Ia, ANT(2″)-Ia, ANT(3″)(9), AAC(3)-IIIb, and APH(3′)-IIIa have been described as highly dynamic enzymes and display properties of intrinsically disordered proteins in the absence of the aminoglycosides [112]. The degree of promiscuity by AMEs is governed by the dynamics of the protein, which is strongly influenced by the ligand and the cofactor, as well as its interaction with the solvent, and it should be taken into consideration in the design of new drugs.
Structural knowledge of both the RNA- and AME-aminoglycoside complexes has helped in the design of new antibiotics that can escape the action of the enzymatic modification [113]. In order to avoid or inhibit the activity of AMEs, obtaining structural and mechanistic information is of paramount importance.
Acknowledgments
The authors thank the Spanish Ministerio de Economía y Competitividad (Grant MAT2015-65184-C2-2-R and CTQ2016-79255-P) and the CSIC project iCOOPB20237 for their support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Davies J., Davies D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010;74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Livermore D.M. Fourteen years in resistance. Int. J. Antimicrob. Agents. 2012;39:283–294. doi: 10.1016/j.ijantimicag.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 3.Arias C.A., Murray B.E. Antibiotic-resistant bugs in the 21st century—A clinical super-challenge. N. Engl. J. Med. 2009;360:439–443. doi: 10.1056/NEJMp0804651. [DOI] [PubMed] [Google Scholar]
- 4.Greenwood D., Finch R., Davey P., Wilcox M. Antimicrobial Chemotherapy. 4th ed. Oxford University Press; Oxford, UK: 2000. pp. 32–48. [Google Scholar]
- 5.Chen G.-H., Pan P., Yao Y., Cheng Y., Meng X.-B., Li Z.-J. Regioselective modification of amino groups in aminoglycosides based on cyclic carbamate formation. Tetrahedron. 2008;64:9078–9087. doi: 10.1016/j.tet.2008.07.022. [DOI] [Google Scholar]
- 6.Chen G.-H., Pan P., Yao Y., Chen Y., Meng X.-B., Li Z.-J. Selective deprotection of the Cbz amine protecting group for the facile synthesis of kanamycin A dimers linked at N-3″ position Original. Tetrahedron. 2009;65:5922–5927. doi: 10.1016/j.tet.2009.06.002. [DOI] [Google Scholar]
- 7.Umezawa H., Okanishi M., Kondo S., Hamana K., Utahara R., Maeda K., Mitsuhashi S. Phosphorylative inactivation of aminoglycosidic antibiotics by Escherichia coli carrying R factor. Science. 1967;157:1559–1561. doi: 10.1126/science.157.3796.1559. [DOI] [PubMed] [Google Scholar]
- 8.Doi O., Miyamoto M., Tanaka N., Umezawa H. Inactivation and phosphorylation of kanamycin by drug-resistant Staphylococcus aureus. Appl. Microbiol. 1968;16:1282–1284. doi: 10.1128/am.16.9.1282-1284.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller G.H., Sabatelli F.J., Naples L., Hare R.S., Shaw K.J. The changing nature of aminoglycoside resistance mechanisms and the role of isepamicin—A new broad-spectrum aminoglycoside. The Aminoglycoside Resistance Study Groups. J. Chemother. 1995;7:31–44. [PubMed] [Google Scholar]
- 10.Mitscher L.A. Antibiotics and Antimicrobial Agents. Lippincott Williams & Wilkins; Baltimore, PA, USA: 2002. pp. 788–791. [Google Scholar]
- 11.Weinstein M.J., Luedemann G.M., Oden E.M., Wagman G.H. Gentamicin, a new broad-spectrum antibiotic complex. Antimicrob. Agents Chemother. 1963;161:1–7. [PubMed] [Google Scholar]
- 12.Martin C.M., Ikari N.S., Zimmerman J., Waitz J.A. A virulent nosocomial Klebsiella with a transferable R factor for gentamicin: Emergence and suppression. J. Infect. Dis. 1971;124:24–29. doi: 10.1093/infdis/124.Supplement_1.S24. [DOI] [PubMed] [Google Scholar]
- 13.Benveniste R., Davies J. R-factor mediated gentamicin resistance: A new enzyme which modifies aminoglycoside antibiotics. FEBS Lett. 1971;14:293–296. doi: 10.1016/0014-5793(71)80282-X. [DOI] [PubMed] [Google Scholar]
- 14.Woo P., Dion H., Bartz Q. Butirosins A and B, aminoglycoside antibiotics. III. structures. Tetrahedron Lett. 1971;12:2625–2628. doi: 10.1016/S0040-4039(01)96935-7. [DOI] [Google Scholar]
- 15.Hayashi S.F., Norcia L.J., Seibel S.B., Silvia A.M. Structure activity relationships of hygromycin A and its analogs: Protein synthesis inhibition activity in a cell free system. J. Antibiot. 1997;50:514–521. doi: 10.7164/antibiotics.50.514. [DOI] [PubMed] [Google Scholar]
- 16.Shaw K.J., Rather P.N., Hare R.S., Miller G.H. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 1993;57:138–163. doi: 10.1128/mr.57.1.138-163.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hotta K., Zhu C.B., Ogata T., Sunada A., Ishikawa J., Mizuno S., Kondo S. Enzymatic 2′-N-acetylation of arbekacin and antibiotic activity of its product. J. Antibiot. 1996;49:458–464. doi: 10.7164/antibiotics.49.458. [DOI] [PubMed] [Google Scholar]
- 18.Labby K.J., Garneau-Tsodikova S. Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Future Med. Chem. 2013;5 doi: 10.4155/fmc.13.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gillings M.R., Paulsen I.T., Tetu S.G. Genomics and the evolution of antibiotic resistance. Ann. N. Y. Acad. Sci. 2017;1388:92–107. doi: 10.1111/nyas.13268. [DOI] [PubMed] [Google Scholar]
- 20.Wolf E., Vassilev A., Makino Y., Sali A., Nakatani Y., Burley S.K. Crystal structure of a GCN5-related N-acetyltransferase: Serratia marcescens aminoglycoside 3-N-acetyltransferase. Cell. 1998;94:439–449. doi: 10.1016/S0092-8674(00)81585-8. [DOI] [PubMed] [Google Scholar]
- 21.Vetting M.W., Hegde S., Javid-Majd F., Blanchard J.S., Roderick S.L. Aminoglycoside 2′-N-acetyltrasferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat. Struct. Biol. 2002;9:653–658. doi: 10.1038/nsb830. [DOI] [PubMed] [Google Scholar]
- 22.Lovering A.M., White L.O., Reeves D.S. AAC-(1): A new aminoglycoside-acetylating enzyme modifying the Cl aminogroup of apramycin. J. Antimicrob. Chemother. 1987;20:803–813. doi: 10.1093/jac/20.6.803. [DOI] [PubMed] [Google Scholar]
- 23.Sunada A., Nakajima M., Ikeda Y., Kondo S., Hotta K. Enzymatic 1-N-acetylation of paromomycin by an actinomycete strain 8 with multiple aminoglycoside resistance and paromomycin sensitivity. J. Antibiot. 1999;52:809–814. doi: 10.7164/antibiotics.52.809. [DOI] [PubMed] [Google Scholar]
- 24.Brenner S. Phosphotransferase sequence homology. Nature. 1987;329:6134. doi: 10.1038/329021a0. [DOI] [PubMed] [Google Scholar]
- 25.Burk D.L., Hon W.C., Leung A.K., Berghuis A.M. Structural analysis of nucleotide binding to an aminoglycoside phosphotransferase. Biochemistry. 2001;40:8756–8764. doi: 10.1021/bi010504p. [DOI] [PubMed] [Google Scholar]
- 26.Fong D.H., Berghuis A.M. Substrate promiscuity of an aminoglycoside antibiotic resistance enzyme via target mimicry. EMBO J. 2002;21:2323–2331. doi: 10.1093/emboj/21.10.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Norris A.L., Serpersu E.H. Ligand promiscuity through the eyes of the aminoglycoside N3 acetyltransferase IIa. Protein Sci. 2013;22:916–928. doi: 10.1002/pro.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson P.R., Hughes D.W., Wright G.D. Regiospecificity of aminoglycoside phosphotransferase from Enterococci and Staphylococci (APH(3’)-IIIa) Biochemistry. 1996;35:8686–8695. doi: 10.1021/bi960389w. [DOI] [PubMed] [Google Scholar]
- 29.McKay G.A., Wright G.D. Kinetic mechanism of aminoglycoside phosphotransferase type IIIa. Evidence for a Theorell-Chance mechanism. J. Biol. Chem. 1995;270:24686–24692. doi: 10.1074/jbc.270.42.24686. [DOI] [PubMed] [Google Scholar]
- 30.Cox G., Stogios P.J., Savchenko A., Wright G.D. Structural and Molecular Basis for Resistance to Aminoglycoside Antibiotics by the Adenylyltransferase ANT(2’’)-Ia. Mbio. 2015;6 doi: 10.1128/mBio.02180-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bassenden A.V., Rodionov D., Shi K., Berghuis A.M. Structural Analysis of the Tobramycin and Gentamicin Clinical Resistome Reveals Limitations for Next-generation Aminoglycoside Design. ACS Chem. Biol. 2016;11:1339–1346. doi: 10.1021/acschembio.5b01070. [DOI] [PubMed] [Google Scholar]
- 32.Chen Y., Nasvall J., Wu S., Andersson D.I., Selmer M. Crystal structure of AadA an aminoglycoside adenyltransferase. Acta Crystallogr. D Biol. Crystallogr. 2015;71:2267. doi: 10.1107/S1399004715016429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pedersen L.C., Benning M.M., Holden H.M. Structural investigation of the antibiotic and ATP-binding sites in kanamycin nucleotidyltransferase. Biochemistry. 1995;34:13305–13311. doi: 10.1021/bi00041a005. [DOI] [PubMed] [Google Scholar]
- 34.Stogios P.J., Wawrzak Z., Minasov G.A., Evdokimova E., Egorova O., Yim V., Kudritska M., Courvalin P., Savchenko A., Anderson W.F. Crystal structure of aminoglycoside 4′-O-adenylyltransferase ANT(4’)-IIb, apo. Cent. Struct. Genom. Infect. Dis. (CSGID) 2012 doi: 10.2210/pdb4ebj/pdb. [DOI] [Google Scholar]
- 35.Tyagi R., Eswaramoorthy S., Burley S.K., Swaminathan S. New York SGX Research Center for Structural Genomics. Crystal structure of an aminoglycoside 6-adenyltransferase from Bacillus subtilis. 2007. Unpublished work.
- 36.Murphy E. Nucleotide sequence of a spectinomycin adenyltransferase AAD(9) determinant from Staphylococcus aureus and its relationship to AAD(3’)(9) Mol. Gen. Genet. 1985;200:33–39. doi: 10.1007/BF00383309. [DOI] [PubMed] [Google Scholar]
- 37.Matesanz R., Díaz J.F., Corzana F., Santana A.G., Bastida A., Asensio J.L. Multiple keys for a single lock: The unusual structural plasticity of the nucleotidyltransferase (4’)/kanamycin complex. Chemistry. 2012;18:2875–2889. doi: 10.1002/chem.201101888. [DOI] [PubMed] [Google Scholar]
- 38.Revuelta J., Corzana F., Bastida A., Asensio J.L. The unusual nucleotide recognition properties of the resistance enzyme ANT(4’): Inorganic tri/polyphosphate as a substrate for aminoglycoside inactivation. Chem. A Eur. J. 2010;16:8635–8640. doi: 10.1002/chem.201000641. [DOI] [PubMed] [Google Scholar]
- 39.Chen-Goodspeed M., Vanhooke J.L., Holden H.M., Raushel F.M. Kinetic mechanism of kanamycin nucleotidyltransferase from Staphylococcus aureus. Bioorg. Chem. 1999;27:395–408. doi: 10.1006/bioo.1999.1144. [DOI] [Google Scholar]
- 40.Magnet S., Blanchard J.S. Molecular Insights into Aminoglycoside Action and Resistance. Chem. Rev. 2005;105:477–497. doi: 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]
- 41.Van Pelt J.E., Northrop D.B. Purification and properties of gentamicin nucleotidyltransferase from Escherichia coli: Nucleotide specificity, pH optimum, and the separation of two electrophoretic variants. Arch. Biochem. Biophys. 1984;230:250–263. doi: 10.1016/0003-9861(84)90106-1. [DOI] [PubMed] [Google Scholar]
- 42.Ramirez M.S., Tolmasky M.E. Aminoglycoside Modifying Enzymes. Drug Resist. Update. 2010;13:151–171. doi: 10.1016/j.drup.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gates C.A., Northrop D.B. Alternative substrate and inhibition kinetics of aminoglycoside nucleotidyltransferase 2”-I in support of a Theorell-Chance kinetic mechanism. Biochemistry. 1988;27:3826–3833. doi: 10.1021/bi00410a046. [DOI] [PubMed] [Google Scholar]
- 44.Bacot-D V.R., Bassenden A.V., Sprules T., Berghuis A.M. Effect of solvent and protein dynamics in ligand recognition and inhibition of aminoglycoside adenyltransferase 2″-Ia. Protein Sci. 2017;26:1852–1863. doi: 10.1002/pro.3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Corzana F., Cuesta I., Bastida A., Hidalgo A., Latorre M., González C., García-Junceda E., Jiménez-Barbero J., Asensio J.L. Molecular recognition of aminoglycoside antibiotics by bacterial defense proteins: NMR analysis of Streptomycin inactivation by Bacillus subtilis Aminoglycoside-6-adenyl Transferas. Chem. Eur. J. 2005;11:5102–5113. doi: 10.1002/chem.200400941. [DOI] [PubMed] [Google Scholar]
- 46.Latorre M., Revuelta J., García-Junceda E., Bastida A. 6-O-Nucleotidyltransferase: An aminoglycoside-modifying enzyme specific for streptomycin/streptidine. MedChemComm. 2016;7:177–183. doi: 10.1039/C5MD00496A. [DOI] [Google Scholar]
- 47.Ferretti J.J., Gilmore K.S., Courvalin P. Nucleotide-sequence analysis of the gene specifying the bifunctional 6’-aminoglycoside acetyltransferase 2’’-aminoglycoside phosphotransferase enzyme in Streptococcus faecalis and identification and cloning of gene regions specifying the two activities. J. Bacteriol. 1986;167:631–638. doi: 10.1128/jb.167.2.631-638.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yıldız Ö., Çoban A.Y., Şener A.G., Coşkuner S.A., Bayramoğlu G., Güdücüoğlu H., Özyurt M., Tatman-Otkun M., Karabiber N., Özkütük N., et al. Antimicrobial susceptibility and resistance mechanisms of methicillin resistant Staphylococcus aureus isolated from 12 Hospitals in Turkey. Ann. Clin. Microbiol. Antimicrob. 2014;16:44–49. doi: 10.1186/s12941-014-0044-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Centron D., Roy P.H. Presence of a group II intron in a multiresistant Serratia marcescens strain that harbors three integrons and a novel gene fusion. Antimicrob. Agents Chemother. 2002;46:1402–1409. doi: 10.1128/AAC.46.5.1402-1409.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dubois W., Poirel L., Marie C., Arpin C., Nordmann P., Quentin C. Molecular characterization of a novel class 1 integron containing blaGES-1 and a fused product of aac(3)-Ib/aac(6’)-Ib’ gene cassettes in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2002;46:638–645. doi: 10.1128/AAC.46.3.638-645.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim C., Hesek D., Zajícek J., Vakulenko S.B., Mobashery S. Characterization of the bifunctional aminoglycoside-modifying enzyme ANT(3’’)-Ii/AAC(6’)-IId from Serratia marcescens. Biochemistry. 2006;45:8368–8377. doi: 10.1021/bi060723g. [DOI] [PubMed] [Google Scholar]
- 52.Kim C., Villegas-Estrada A., Hesek D., Mobashery S. Mechanistic characterization of the bifunctional aminoglycoside-modifying enzyme AAC(3)-Ib/AAC(6’)-Ib’ from Pseudomonas aeruginosa. Biochemistry. 2007;46:5270–5282. doi: 10.1021/bi700111z. [DOI] [PubMed] [Google Scholar]
- 53.Shaul P., Green K.D., Rutenberg R., Kramer M., Berkov-Zrihen Y., Breiner-Goldstein E., Garneau-Tsodikova S., Fridman M. Assessment of 6’- and 6””-N-acylation of aminoglycosides as a strategy to overcome bacterial resistance. Org. Biomol. Chem. 2011;9:4057–4063. doi: 10.1039/c0ob01133a. [DOI] [PubMed] [Google Scholar]
- 54.Bastida A., Hidalgo A., Chiara J.L., Torrado M., Corzana F., Pérez-Cañadillas J.M., Groves P., García-Junceda E., González C., Jiménez-Barbero J., et al. Exploring the use of conformationally locked aminoglycosides as a new strategy to o.vercome bacterial resistance. J. Am. Chem. Soc. 2006;128:100–116. doi: 10.1021/ja0543144. [DOI] [PubMed] [Google Scholar]
- 55.Fair R.J., McCoy L.S., Hensler M.E., Aguilar B., Nizet V., Tor Y. Singly modified amikacin and tobramycin derivatives show increased rRNA A-site binding and higher potency against resistant bacteria. ChemMedChem. 2014;9:2164–2171. doi: 10.1002/cmdc.201402175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fair R.J., Hensler M.E., Thienphrapa W., Dam Q.N., Nizet V., Tor Y. Selectively guanidinylated aminoglycosides as antibiotics. ChemMedChem. 2012;7:1237–1244. doi: 10.1002/cmdc.201200150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang L., Ye X-S. Development of Aminoglycoside Antibiotics Effective Against Resistant Bacterial Strains. Curr. Top. Med. Chem. 2010;10:1898–1926. doi: 10.2174/156802610793176684. [DOI] [PubMed] [Google Scholar]
- 58.Yan R.-B., Yuan M., Wu Y., You X., Ye X.-S. Rational design and synthesis of potent aminoglycoside antibiotics against resistant bacterial strains. Bioorg. Med. Chem. 2011;19:30–40. doi: 10.1016/j.bmc.2010.11.065. [DOI] [PubMed] [Google Scholar]
- 59.Umezawa H., Umezawa S., Tsuchiya T., Okazaki I. 3’,4’-Dideoxykanamycin B active against kanamycin-resistant Escherichia coli and Pseudomonas aeruginosa. J. Antibiot. 1971;24:485–487. doi: 10.7164/antibiotics.24.485. [DOI] [PubMed] [Google Scholar]
- 60.Schmidt F.R., Nucken E.J., Henschke R.B. Nucleotide sequence analysis of 2’’-aminoglycoside nucleotidyl-transferase ANT(2’’) from Tn4000: Its relationship with AAD(3’) and impact on Tn21 evolution. Mol. Microbiol. 1988;2:709–717. doi: 10.1111/j.1365-2958.1988.tb00081.x. [DOI] [PubMed] [Google Scholar]
- 61.Daigle D.M., Hughes D.W., Wright G.D. Prodigious substrate specificity of AAC(6’)-APH(2”), an aminoglycoside antibiotic resistance determinant in enterococci and staphylococci. Chem. Biol. 1999;6:99–110. doi: 10.1016/S1074-5521(99)80006-4. [DOI] [PubMed] [Google Scholar]
- 62.Woo P.W.K., Haskell T.H. Deoxy derivatives of butirosin A and 5”-amino-5”-deoxybutirosin A, aminoglycoside antibiotics resistant to bacterial 3’-phosphorylative enzymatic inactivation. Synthesis and NMR studies. J. Antibiot. 1982;35:692–702. doi: 10.7164/antibiotics.35.692. [DOI] [PubMed] [Google Scholar]
- 63.Maeda K., Fujii F., Kondo S., Umezawa H. Chemical derivation of antibiotics active against resistant bacteria. Jpn. J. Med. Sci. Biol. 1967;21:224–227. [PubMed] [Google Scholar]
- 64.Kagawuchi H., Naito T., Nakagawa S., Fujisawa K.I. B-K8, a new semisynthetic aminoglycoside antibiotic. J. Antibiot. 1972;25:695–708. doi: 10.7164/antibiotics.25.695. [DOI] [PubMed] [Google Scholar]
- 65.Kim Y.A., Park Y.S., Youk T., Lee H., Lee K. Correlation of Aminoglycoside Consumption and Amikacin- or Gentamicin-Resistant Pseudomonas aeruginosa in Long-Term Nationwide Analysis: Is Antibiotic Cycling an Effective Policy for Reducing Antimicrobial Resistance? Ann. Lab. Med. 2018;38:176–178. doi: 10.3343/alm.2018.38.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kondo S., Iinuma K., Yamamoto H., Maeda K., Umezawa H. Letter: Synthesis of 1-N-{(S)-4-amino-2-hydroxybutyryl}-kanamycin B and -3’,4’-dideoxykanamycin B active against kanamycin resistant bacteria. J. Antibiot. 1973;26:412–415. doi: 10.7164/antibiotics.26.412. [DOI] [PubMed] [Google Scholar]
- 67.Ubutaka K., Yamashita N., Gotoh A., Konno M. Purification and characterization of aminoglycoside-modifying enzymes from Staphylococcus aureus and Staphylococcus epidermidis. Antimicrob. Agents Chemother. 1984;25:754–759. doi: 10.1128/AAC.25.6.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kondo S., Hotta K. Semisynthetic aminoglycoside antibiotics: Development and enzymatic modifications. J. Infect. Chemother. 1999;5:1–9. doi: 10.1007/s101560050001. [DOI] [PubMed] [Google Scholar]
- 69.Bennett J.E., Dolin R., Blaser M. Principles and Practice of Infectious Diseases. 8th ed. Douglas & Bennett’s, Churchill, Livingston; New York, NY, USA: 1995. pp. 279–301. [Google Scholar]
- 70.Montie T., Patamasucon P. Aminoglycosides: The complex problem of antibiotic mechanisms and clinical applications. Eur. J. Clin. Microbiol. Infect. Dis. 1995;14:85–87. doi: 10.1007/BF02111863. [DOI] [PubMed] [Google Scholar]
- 71.Edson R.S., Terrell C.L. The aminoglycosides. Mayo Clin. Proc. 1991;66:1158–1164. doi: 10.1016/S0025-6196(12)65798-X. [DOI] [PubMed] [Google Scholar]
- 72.Zhao C., Li J., Hou J., Guo M., Zhang Y., Chen Y. A randomized controlled clinical trial, on etimicin, a new aminoglycoside antibiotic, versus netilmicin in the treatment of bacterial infections. Clin. Med. J. 2000;113:1026–1030. [PubMed] [Google Scholar]
- 73.Lortholary O., Tod M., Cohen Y., Petitjean O. Aminoglycosides. Med. Clin. N. Am. 1995;79:761–787. doi: 10.1016/S0025-7125(16)30038-4. [DOI] [PubMed] [Google Scholar]
- 74.Kaelowsky J.A., Zelenitsky S.A., Zhanel G.G. Aminoglycoside adaptive resistance. Pharmacotherapy. 1997;17:549–555. doi: 10.1002/j.1875-9114.1997.tb03063.x. [DOI] [PubMed] [Google Scholar]
- 75.Neu H.C. The crisis in antibiotic resistance. Science. 1992;257:1064–1073. doi: 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- 76.Chang C.-W.T., Hui Y., Elchert B., Wang J., Li J., Rai R. Pyranmycins, a novel class of aminoglycosides with improved acid stability: The SAR of D-pyranoses on ring III of pyranmycin. Org. Lett. 2002;4:4603–4606. doi: 10.1021/ol0269042. [DOI] [PubMed] [Google Scholar]
- 77.Rai R., Chang H., Chang C.-W.T. Novel Method for the Synthesis of 3’,4’-Dideoxygenated Pyranmycin and Kanamycin Compounds, and Studies of Their Antibacterial Activity against Aminoglycoside Resistant Bacteria. J. Carbohydr. Chem. 2005;24:131–143. doi: 10.1081/CAR-200059968. [DOI] [Google Scholar]
- 78.Rai R., Chen H., Czyryca P.G., Li J., Chang C.-W.T. Design and Synthesis of Pyrankacin: A Pyranmycin Class Broad Spectrum Aminoglycoside Antibiotic. Org. Lett. 2006;8:887–889. doi: 10.1021/ol0529750. [DOI] [PubMed] [Google Scholar]
- 79.Santana A.G., Zárate S.G., Asensio J.L., Revuelta J., Bastida A. Selective modification of the 3′′-amino group of kanamycin prevents significant loss of activity in resistant bacterial strains. Org. Biomol. Chem. 2016;14:516–525. doi: 10.1039/C5OB01599E. [DOI] [PubMed] [Google Scholar]
- 80.Galani I., Souli M., Daikos G.L., Chrysouli Z., Poulakou G., Psichogiou M., Panagea T., Argyropoulou, Stefanou I., Plakias G., Giamarellou H., et al. Activity of Plazomicin (ACHN-490) against MDR clinical isolates of Klebsiella pneumoniae, Escherichia coli, and Enterobacter spp. J. Chemother. 2012;24:191–194. doi: 10.1179/1973947812Y.0000000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mega W.M., Doyle-Eisele M., Cass R.T., Kostrub C.F., Sherwood R.L., Metz M.A., Cirz R.T. Plazomicin is effective in a non-human primate pneumonic plague model. Bioorg. Med. Chem. 2016;24:6429–6439. doi: 10.1016/j.bmc.2016.08.049. [DOI] [PubMed] [Google Scholar]
- 82.Livermore D.M., Mushtaq S., Warner M., Zhang J.C., Maharjan S., Doumith M. Activity of aminoglycosides, including ACHN-490, against carbapenem-resistant Enterobacteriaceae isolates. J. Antimicrob. Chemother. 2011;66:48–53. doi: 10.1093/jac/dkq408. [DOI] [PubMed] [Google Scholar]
- 83.Francois B., Russell R.J.M., Murray J.B., Aboul-ela F., Masquida B., Vicens Q., Westhof E. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: Role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res. 2005;33:5677–5690. doi: 10.1093/nar/gki862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nuthanakanti A., Boerneke M.A., Hermann T., Srivatsan S.G. Structure of the Ribosomal RNA Decoding Site Containing a Selenium-Modified Responsive Fluorescent Ribonucleoside Probe. Angew. Chem. Int. Ed. Engl. 2017;56:2640–2644. doi: 10.1002/anie.201611700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Murray J.B., Meroueh S.O., Russell R.J., Lentzen G., Haddad J., Mobashery S. Interactions of designer antibiotics and the bacterial ribosomal aminoacyl-tRNA site. Chem. Biol. 2006;13:129–138. doi: 10.1016/j.chembiol.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 86.Revuelta J., Vacas T., Corzana F., González C., Bastida A., Asensio J.L. Structure-Based Design of Highly Crowded Ribostamycin/Kanamycin Hybrids as a new Family of Antibiotics. Chem. A Eur. J. 2010;16:2986–2991. doi: 10.1002/chem.200903003. [DOI] [PubMed] [Google Scholar]
- 87.Asensio J.L., Hidalgo A., Bastida A., Torrado M., Corzana F., García-Junceda E., Cañada J., Chiara J.L., Jimenez-Barbero J. A simple structural-based approach to prevent aminoglycoside inactivation by bacterial defense proteins. Conformational restriction provides effective protection against neomycin-B nucleotidylation by ANT4”. J. Am. Chem. Soc. 2005;127:8278–8279. doi: 10.1021/ja051722z. [DOI] [PubMed] [Google Scholar]
- 88.Blount K.F., Zhao F., Hermann T., Tor Y. Conformational constraint as a means for understanding RNA-aminoglycoside specificity. J. Am. Chem. Soc. 2005;127:9818–9829. doi: 10.1021/ja050918w. [DOI] [PubMed] [Google Scholar]
- 89.Santana A.G., Bastida A., Martínez del Campo T., Asensio J.L., Revuelta J. An efficient and general route to the synthesis of novel aminoglycosides for RNA binding. Synlett. 2011:219–222. doi: 10.1055/s-0030-1259305. [DOI] [Google Scholar]
- 90.Sucheck S.J., Wong A.L., Koeller K.M., Boehr D.D., Draker K., Sears P., Wright G.D. Design of bifunctional antibiotics that target bacterial rRNA and inhibit resistance-causing enzymes. J. Am. Chem. Soc. 2000;122:5230–5231. doi: 10.1021/ja000575w. [DOI] [Google Scholar]
- 91.Vacas T., Corzana F., Jiménez-Osés G., González C., Gómez A.M., Bastida A., Revuelta J., Asensio J.L. Role of aromatic rings in the molecular recognition of aminoglycoside antibiotics: Implications for drug design. J. Am. Chem. Soc. 2010;132:12074–12090. doi: 10.1021/ja1046439. [DOI] [PubMed] [Google Scholar]
- 92.Mandhapati A.R., Yang G., Kato T., Shcherbakov D., Hobbie S.N., Vasella A., Boöttger E.C., Crich D. Structure-Based Design and Synthesis of Apramycin−Paromomycin Analogues: Importance of the Configuration at the 6′-Position and Differences between the 6′-Amino and Hydroxy Series. J. Am. Chem. Soc. 2017;139:14611–14619. doi: 10.1021/jacs.7b07754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Leban N., Kaplan E., Chaloin L., Godreuil S., Lionne C. Kinetic characterization and molecular docking of novel allosteric inhibitors of aminoglycoside phosphotransferases. Biochim. Biophys. Acta. 2017;1861:3464–3473. doi: 10.1016/j.bbagen.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 94.Kotra L.P., Haddad J., Mobashery S. Aminoglycosides: Perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob. Agents Chemother. 2000;44:3249–3256. doi: 10.1128/AAC.44.12.3249-3256.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boehr D.D., Draker K.A., Koteva K., Bains M., Hancock R.E., Wright G.D. Broad-spectrum peptide inhibitors of aminoglycoside antibiotic resistance enzymes. Chem. Biol. 2003;10:189–196. doi: 10.1016/S1074-5521(03)00026-7. [DOI] [PubMed] [Google Scholar]
- 96.Gao F., Yan X., Baetting O.M., Berghuis A.M., Auclair K. Regio- and chemoselective 6’-N-derivatization of aminoglycosides: Bisubstrate inhibitors as probes to study aminoglycoside 6’-N-acetyltransferases. Angew. Chem. Int. Ed. 2005;44:6859–6862. doi: 10.1002/anie.200501399. [DOI] [PubMed] [Google Scholar]
- 97.Williams J.W., Northrop D.B. Synthesis of a tight-binding, multisubstrate analog inhibitor of gentamicin acetyltransferase I. J. Antibiot. (Tokyo) 1979;32:1147–1154. doi: 10.7164/antibiotics.32.1147. [DOI] [PubMed] [Google Scholar]
- 98.Liu M., Haddad J., Azucena E., Kotra L.P., Kirzhner M., Mobashery S. Tethered bisubstrate derivatives as probes for mechanism and as inhibitors of aminoglycoside 3’-phosphotransferases. J. Org. Chem. 2000;65:7422–7431. doi: 10.1021/jo000589k. [DOI] [PubMed] [Google Scholar]
- 99.Roestamadji J., Grapsas I., Mobashery S. Mechanism-based inactivation of bacterial aminoglycoside 3’-phosphotransferases. J. Am. Chem. Soc. 1995;117:80–84. doi: 10.1021/ja00106a009. [DOI] [Google Scholar]
- 100.Allen N.E., Alborn W.E., Jr., Hobbs J.N., Jr., Kirst H.A. 7´hydroxytropolone: An inhibitor of aminoglycoside-2’’-O-adenyltrasfersae. Antimicrob. Agents Chemother. 1982;22:824–831. doi: 10.1128/AAC.22.5.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hirsch D.R., Cox G., D’Erasmo M.P., Shakya T., Meck C., Mohda N., Wright G.D., Murelli R.P. Inhibition of the ANT(2’’)-Ia resistance enzyme and rescue of aminoglycoside antibiotic activity by synthetic α-hydroxytropolones. Bioorg. Med. Chem. Lett. 2014;24:4943–4947. doi: 10.1016/j.bmcl.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Garzan A., Willby M.J., Green K.D., Gajadeera C.S., Hou C., Tsodikov O.V., Posey J.E., Garneau-Tsodikova S. Sulfonamide-Based Inhibitors of Aminoglycoside Acetyltransferase Eis Abolish Resistance to Kanamycin in Mycobacterium tuberculosis. J. Med. Chem. 2016;59:10619–10628. doi: 10.1021/acs.jmedchem.6b01161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Latorre M., Peñalver P., Revuelta J., Luis Asensio J.L., García-Junceda E., Bastida A. Rescue of the streptomycin antibiotic activity by using streptidine as a “decoy acceptor” for the aminoglycoside-inactivating enzyme adenyl transferase. Chem. Commun. 2007:2829–2831. doi: 10.1039/B704785A. [DOI] [PubMed] [Google Scholar]
- 104.Pokrovskaya V., Baasov T. Dual-acting hybrid antibiotics: A promising strategy to combat bacterial resistance. Expert Opin. Drug Discov. 2010;5:883–902. doi: 10.1517/17460441.2010.508069. [DOI] [PubMed] [Google Scholar]
- 105.Berkov-Zrihen Y., Green K.D., Labby K.J., Feldman M., Garneau-Tsodikova S., Fridman M. Synthesis and evaluation of hetero- and homodimers of ribosome-targeting antibiotics: Antimicrobial activity, in vitro inhibition of translation, and drug resistance. J. Med. Chem. 2013;56:5613–5625. doi: 10.1021/jm400707f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xiao Z.P., Wang X.D., Wang P.F. Design, synthesis, and evaluation of novel fluoroquinolone-flavonoid hybrids as potent antibiotics against drug-resistant microorganisms. Eur. J. Med. Chem. 2014;80:92–100. doi: 10.1016/j.ejmech.2014.04.037. [DOI] [PubMed] [Google Scholar]
- 107.Wong W.T., Chan K.C., So P.K. Increased structural flexibility at the active site of a fluorophore-conjugated beta-lactamase distinctively impacts its binding toward diverse cephalosporin antibiotics. J. Biol. Chem. 2011;286:31771–31780. doi: 10.1074/jbc.M110.198895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bremner J.B., Ambrus J.I., Samosorn S. Dual action-based approaches to antibacterial agents. Curr. Med. Chem. 2007;14:1459–1477. doi: 10.2174/092986707780831168. [DOI] [PubMed] [Google Scholar]
- 109.Hainrichson M., Pokrovskaya V., Shallom-Shezifi D. Branched aminoglycosides: Biochemical studies and antibacterial activity of neomycin B derivatives. Bioorg. Med. Chem. 2005;13:5797–5807. doi: 10.1016/j.bmc.2005.05.058. [DOI] [PubMed] [Google Scholar]
- 110.Shavit M., Pokrovskaya V., Belakhov V., Baasov T. Covalently linked kanamycin–Ciprofloxacin hybrid antibiotics as a toolto fight bacterial resistance. Bioorg. Med. Chem. 2017;25:2917–2925. doi: 10.1016/j.bmc.2017.02.068. [DOI] [PubMed] [Google Scholar]
- 111.Pokrovskaya V., Nudelman I., Kandasamy J., Baasov T. Aminoglycosides: Redesign Strategies for Improved Antibiotics and Compounds for Treatment of Human Genetic Diseases. In: Fukuda M., editor. Methods in Enzymology. 1st ed. Volume 478. Elsevier Inc.; Amsterdam, The Netherlands: 2010. pp. 437–462. Charper 21 Glycomics. [DOI] [PubMed] [Google Scholar]
- 112.Serpersu E.H., Norris A.L. Advances in Carbohydrate Chemistry and Biochemistry. Volume 67. Elsevier Inc.; Amsterdam, The Netherlands: 2012. Effect of protein dynamics and solvent in ligand recognition by promiscuous aminoglycoside-modifying enzymes; pp. 222–243. [DOI] [PubMed] [Google Scholar]
- 113.Herzog I.M., Zada S.L., Fridman M. Effects of 5-O-Ribosylation of Aminoglycosides on Antimicrobial Activity and Selective Perturbation of Bacterial Translation. J. Med. Chem. 2016;59:8008–8018. doi: 10.1021/acs.jmedchem.6b00793. [DOI] [PubMed] [Google Scholar]