

Abstract
Increasing experimental and clinical evidence has revealed a critical role for myeloid cells in the development and progression of cancer. The ability of monocytes and macrophages to regulate inflammation allows them to manipulate the tumor microenvironment to support the growth and development of malignant cells. Recent studies have shown that macrophages can exist in several functional states depending on the microenvironment they encounter in the tissue. These functional phenotypes not only influence the genesis and propagation of tumors, but also the efficacy of cancer therapies particularly radiation. Early classification of the macrophage phenotypes, or “polarization states”, identified two major states, M1 and M2, that have cytotoxic and wound repair capacity respectively. In the context of tumors, classically activated or M1 macrophages driven by IFN-gamma support anti-tumor immunity while alternatively activated or M2 macrophages generated in part from interleukin-4 exposure hinder anti-tumor immunity by suppressing cytotoxic responses against a tumor. In this review, we discuss the role that the functional phenotype of a macrophage population plays in tumor development. We will then focus more specifically on how macrophages and myeloid cells regulate the tumor response to radiation therapy.
Keywords: macrophage, macrophage polarization, radiation therapy, tumorassociated macrophages
Introduction
Macrophages arise from the myeloid cell lineage in the bone marrow. They begin life by entering the blood stream as monocytes and in response to a variety of inflammatory stimuli migrate into the tissue and become mature macrophages. Once in the tissue they can differentiate into several different types of mature macrophages. These macrophages play pivotal roles in the initiation, propagation and resolution of inflammation.1 In the tissue, macrophages are highly responsive to environmental cues including cytokines and other inflammatory stimuli. Macrophages undergo phenotypic changes upon encountering these triggers to acquire functions that can support or inhibit an inflammatory response.2 Tumors actively recruit myeloid cells and express various cytokines and cell surface molecules that push recruited myeloid cells to differentiate into macrophages that can support tumor growth and inhibit the tumor response to therapies such chemotherapy and radiation.3 Tumor-associated macrophages inhibit the response to therapies through multiple mechanisms including inhibition of the anti-tumor immune response stimulated by therapy-induced cell death and production of vascular and epithelial growth factors. In this review, we will discuss the current understanding of macrophage phenotypes, the role of macrophages in cancer and finally how macrophages and their functional phenotypes regulate the response to radiation therapy.
Macrophage polarization: functional diversity of macrophages
Recent studies have revealed tremendous functional diversity among macrophages ranging from their cytotoxic killing abilities to their central role in tissue repair. This diversity likely arises from the multitude of situations requiring phagocytic cells throughout the body. Found in virtually every tissue of the body, tissue and bone marrow derived macrophages encounter a tremendous number of agents they have to deal with in order to maintain tissue homeostasis. Thus, their diversity arises from the need to prepare macrophages for the things they will encounter anywhere from infected cells to damaged tissue. Early classification schemas attempted to categorize macrophage functional states into either the classically activated macrophages (M1) phenotype or the alternatively activated macrophages (M2) phenotype, mirroring the Th1/Th2 polarization states found in T cells.4 With respect to tumors, M1 macrophages due to their cytotoxic capacity is often considered the “anti-tumor” phenotype whereas the M2 macrophages due to their immunosuppressive and angiogenic capacioty are thought to be the “pro-tumor” phenotype. More recent studies examining macrophage populations in vivo have shown that the M1/M2 classification grossly oversimplifies the wide spectrum of functional macrophage phenotypes found in the body.5,6 Nevertheless, for the purposes of this discussion it remains helpful to broadly classify macrophages into their earlier M1/M2 nomenclature recognizing that this subdivision likely does not capture a complete understanding of the macrophage phenotypes involved.
M1 macrophages: the “anti-tumor” phenotype
Macrophages have long been recognized as the first line of defense against foreign pathogens in innate immunity. 7 Th1-related cytokines like interferon-γ (IFN-γ) and microbicidal stimuli such as lipopolysaccharide (LPS) prime macrophages to produce a cytotoxic activation state that characterizes the M1 phenotype.8 In these M1 macrophages, downstream signaling of IFNs and toll-like receptors (TLRs), through activation of signal transducer and activator of transcription 1 (STAT1) and nuclear factor-kB (NF-kB) drives the expression of a transcriptional program including the chemokines C-C motif ligand 15 (CCL15), C-X-C motif ligand 10 (CXCL10), chemokine receptors such as C-C chemokine receptor type 7 (CCR7) and reactive oxygen species (particularly inducible nitric oxide synthase, iNOS).9 M1 macrophages have been identified by both surface markers and expression of several key genes such as interleukin (IL)-12, IL-1 and tumor necrosis factor (TNF).10–12 Studies examining the interaction between macrophages and cancer cells suggest that M1 macrophages both directly kill cancer cells and support the cytotoxic activity of other immune cells including T cell and NK cells thus the M1 phenotype is often considered the “anti-tumor” phenotype.13
M2 macrophages: “pro-tumor” phenotype
Originally identified in response to metazoan parasite infections and allergens, M2 macrophages form when macrophages encounter Th2-associated cytokines including IL-4 and IL-13 which activate STAT6 leading to expression of targets that were found to be key not only in mediating anti-helminthic immunity but also tissue repair.14 M2-polarized macrophages possess higher levels of arginase (Arg-1) activity, allowing them to convert arginine to ornithine, a precursor of polyamines and collagen, contributing to the production of extracellular matrix.15 M2 macrophages are also known to secrete other factors associated with wound healing such as vascular endothelial growth factor (VEGF), colony stimulating factor 1 (CSF1) and IL-8 which promote angiogenesis, lymphangiogenesis and fibrosis.2,16 They are characterized by expression of immunosuppressive cytokines, chemokines and surface markers such as IL-10, CCL17 and CD206.9 Tumor-associated macrophages (TAMs) share many of the same expression patterns as M2 macrophages. TAMs play a crucial role in the initiation, promotion and metastasis of cancer cells by encouraging angiogenesis and remodeling of the stromal matrix to help establish the premalignant niche. Thus, M2-polarized macrophages are often considered a “pro-tumor” phenotype.17,18
Myeloid-macrophage cells in cancer
Myeloid cells and macrophages have been associated with both the development and progression of cancer.16 Several larger retrospective clinical studies found that increasing numbers of TAMs correlate with higher grade tumors in multiple tumor types including breast, lung and prostate.19 Tumors actively recruit macrophages as they grow in order to establish a favorable microenvironment through macrophage-derived growth signals, tissue remodeling and immunosuppression.20
Recruitment of TAMs to tumors
Multiple cytokines and chemokines coordinate the recruitment and differentiation of macrophages in sites of tumor formation. Tumors produce macrophage colony-stimulating factor (M-CSF) and CCL-2 both of which regulate the influx and survival of TAMs.21 CSF1 regulates the production of myeloid cells in the bone marrow and is also responsible for attracting macrophages to the tumor from the circulation. CCL2 and ligation of its receptor CCR2 on macrophages induce their chemotaxis and retention in tumors. Other inflammatory mediators released by tumors such as TNF-α, IL-6 and VEGF also play a role in macrophage recruitment to the tumors.22
TAMs promote tumor growth
Once in the tumors, TAMs secrete a series of cytokines, chemokines and growth factors to promote tumor growth by setting up an immunosuppressive microenvironment, supporting de novo angiogenesis and enhancing the metastatic potential of malignant cells (Table 1).
Table 1.
A simplified summary of the functions of the cytokines, chemokines and growth factors that produced by TAMs in tumor development.
| Factors | Functions | Reference |
|---|---|---|
| PD-L1, PD-L2 | immunosuppression by inhibiting T cell functions | 26 |
| TGF-β | immunosuppression by inducing Tregs, metastasis | 166 |
| IL-10 | immunosuppression by suppressing antigen presentation | 167 |
| Prostaglandin E2 | immunosuppression by suppressing DC activation | 168 |
| Arginase-1 | immunosuppression by inhibiting T cell functions | 27 |
| CCL18 | Metastasis | 169 |
| MMPs | metastasis, angiogenesis, matrix remodeling | 170 |
| TNF-α | metastasis, angiogenesis | 171 |
| VEGF | metastasis, angiogenesis | 172 |
| Fibroblast growth factor 2 | Angiogenesis | 173 |
In accordance with their ability to set up a favorable immune environment for tumor growth, TAMs possess the ability to strongly suppress anti-tumor immunity. One mechanism employed by macrophages to suppress anti-tumor immunity is expression of the inhibitory programmed death-1 (PD-1) and its ligands, PD-L1 and PD-L2.23,24 Engagement of PD-1 on T cells and macrophages themselves leads to suppression of CTLs and inhibition of phagocytosis respectively.23,25,26 In addition to the expression of inhibitory signals such as PD-L1 and 2, TAMs also produce Arg-1 which depletes arginine from the tumor microenvironment leading to further inhibition of effector T cell responses as T cells require arginine for activation and because the catabolic byproducts of arginine themselves are immunosuppressive.27–29
In addition to establishing an immunosuppressive microenvironment in tumors, TAMs also enhance angiogenesis in tumors. Often found in association with tumor vasculature, macrophages are drawn to hypoxic areas within tissue.30 Once there, lactic acid-mediated hypoxia-inducible factor-1α (HIF-1α) activation in macrophages leads to transcription of VEGF which induces angiogenesis in tumors.31,32 Macrophages also alter the tumor matrix making it more favorable for tumor growth through the release of tissue remodeling factors such as matrix metalloproteinase (MMP-9).33 MMP-mediated remodeling of the extracellular matrix liberates “damage” signals which promotes ongoing chronic inflammation as well as altering the architecture of the tissue both of which further stimulate tumor cell invasion and subsequent metastases.34,35
As a result of the crucial role that macrophages play in the development and progression of cancer, many preclinical and clinical studies have directed their efforts at targeting macrophages and their activity.
Targeting macrophages in cancer
The presence of TAMs has been associated with chemotherapy resistance and poor clinical prognosis in multiple cancer types including pancreatic, prostate and breast.36–38 Hence, multiple agents that deplete and/or prevent the infiltration of TAMs are currently under investigation in several disease sites (Table 2).
Table 2.
Single-reagents that target TAMs for cancer treatments.
| Treatment | Type of study | References | |
|---|---|---|---|
| Targeting macrophage infiltration | |||
| Anti-CSF1/CSF1R antibody | In vivo; mice; breast, osteosarcoma, pancreatic, glioma cancer | 42–44,46,64 | |
| PLX3397 (CSF1R inhibitor) | Phase 1 clinical trial; solid, pancreatic cancers | 47,64 | |
| Emactuzumab (anti-CSF1R antibody) | Phase 1 clinical trial; soft tissue tumors | 48 | |
| Siltuximab (anti-IL-6 antibody) | Phase 1, 2 clinical trials; ovarian, prostate, renal cell, advanced solid cancer | 53–56 | |
| Carlumab (anti-CCL2 antibody) | Phase 1, 2 clinical trials; solid, prostate cancer | 51,52 | |
| Anti-CCL2 antibody | In vivo; mice; breast, glioma, prostate cancer | 45,126,127 | |
| CCL2 siRNA | In vivo; mice; breast cancer | 125 | |
| Treatment | Type of study | Functions | References |
| Targeting macrophage polarization/activation | |||
| P. aeruginosa mannose-sensitive hemagglutinin | Ex vivo | M1 promotion | 58 |
| Iron oxide nanoparticles | In vivo; mice; adenocarcinoma | M1 promotion | 59 |
| miRNAs | In vivo; mice; lung, liver cancer | M2 inhibition | 61,62 |
| STAT3 inhibition | Ex vivo; in vivo; mice; sarcoma | M2 inhibition | 174,175 |
| CD40 agonist | Phase 1 clinical trials; solid tumors and diffuse large B-cell lymphoma | M1 promotion | 66,176 |
| Dacetuzumab (anti-CD40 antibody) | Phase 1 clinical trial; non-Hodgkin’s lymphoma | M1 promotion | 67 |
| CD870,873 (anti-CD40 antibody) | Phase 1 clinical trials; pancreatic ductal adenocarcinoma; solid tumors | M1 promotion | 68–70 |
| β-glucan | Phase 1 clinical trials; pancreatic ductal adenocarcinoma; solid tumors | M1 promotion | 177 |
| Trabectedin (anti-tumor reagent) | In vivo; mice; sarcoma, ovarian cancer Phase 3 clinical trial; soft tissue sacorma |
M2 inhibition | 73–75 |
Experimental studies
Myeloid recruitment and expansion from progenitors into tumor-associated macrophages and other myeloid subpopulations, is dependent on three growth factors: M-CSF/CSF1, GM-CSF/CSF2 and G-CSF/CSF3. Targeting these growth factors by either blocking agents or genetic ablation reduced TAM accumulation leading to delayed tumor progression in models of breast and pancreatic neuroendocrine cancer.39–43 Not surprisingly, blocking GM-CSF or G-CSF led to a preferential decrease in CD11b+Gr1+ and Ly6G+ whereas CSF-1 blockade appears to have broader depletion of both CD11b+Gr-1+ and CD11b+Gr-1−.38,40 In addition to preventing the recruitment of monocytes, inhibition of the CSF1/CSF1R also blocks the polarization of TAMs into the pro-tumoral phenotype.44 Qian et al further similarly showed that targeting CCL2 in a murine model of breast cancer mirrors CSF-1 ablation with significantly reduced metastatic disease.45
Clinical studies
Based on the preclinical data, clinical trials using agents that block CSF1/CSF1R and CCL2 target myeloid cells have been examined. While these therapies have largely been deemed safe, their anti-tumor efficacy has been mixed. Clinical trials of CSF1/CSF1R inhibitors including monoclonal antibodies and tyrosine kinase inhibitors are ongoing in multiple different malignancies with variable results.46–48 In a phase 1 clinical trial, application of the monoclonal antibody emactuzumab (RG7155) inhibiting CSF1R activation achieved an objective response for 86% of patients with diffuse-type tenosynovial giant cell tumor with modest toxicity. One caveat, however, is that CSF-1R is overexpressed in this disease and thus the response may be due to direct activity on the receptor and not its effects on macrophages.48 Other studies with CSF-1/CSF-1R directed agents in other disease histologies have been less successful with most showing limited responses.49 Interestingly, in a study which sought to identify additional factors that mediate resistance to CSF-1R antibody, IL-4 treatment restored viability of emactuzumab-treated macrophages in vitro with this population of macrophages showing increased expression of CD206.50 This in vitro data was mirrored by data from melanomas with high levels of IL-4 expression which show more CD206+ macrophages infiltration upon emactuzumab treatment suggesting that the IL-4 pathway may be an important target in conjunction with CSF-1R directed agents.50 The phase 1 study with the CCL2-blocking agent (carlumab) in patients with advanced solid malignancies, showed evidence of transient free CCL2 suppression and preliminary anti-tumor activity with minimal toxicity.51 However, the follow-up phase 2 clinical trial in patients with metastatic prostate cancer, failed to show anti-tumor activity in part because cessation of carlumab resulted in rebound elevation of serum CCL2 levels likely due to compensatory increases in CCL2 production with antibody administration.52 Another potential explanation for the lack of anti-tumor activity with carlumab is the weak binding affinity of of the antibody which may have allowed for continued CCL2 signaling.52
Another strategy to target macrophages has been to target inflammatory cytokines that attract myeloid cells to sites of inflammation. One example of this is the anti-IL-6 antibody (siltuximab), which was shown to decrease circulating CCL-2 and CXCL-12 leading to reduced TAM infiltration in ovarian xenografts.53 Consistent with this preclinical data, siltuximab has demonstrated modest anti-tumor efficacy in patients with prostate54 and renal cancer55 though it has been less successful in other solid tumors.56 Thus, drugs targeting macrophage infiltration into tumors as single agents have had excellent safety and some modest responses. Given the limited clinical responses to agents directed at the entire macrophage population, other groups have pursued promising strategies for tumors that target macrophage functional phenotypes instead.
Targeting macrophage polarization in cancer
Experimental studies
As TAMs often express M2-like characteristics, several groups have pursued a therapeutic strategy of reprograming TAMs towards more cytotoxic, anti-tumor M1 phenotypes.57 To that end, two primary strategies to re-educate macrophages have been employed successfully: enhancing M1 polarization directly and preventing M2-polarization (Table 2). Therapies designed to increase M1 polarization are typically directed at activating the pathways used to clear damaged or infected cells. For example, bacterial products such as Pseudomonas aeruginosa mannose-sensitive hemagglutinin which activates toll-like receptors58 and nanoparticles like ferumoxytol, a bioconjugated manganese dioxide which stimulates production of reactive oxygen species have been used in murine models to target macrophages. Both treatments demonstrated reduced tumor growth and progression to metastases in models of lung and breast cancer.58–60 Alternatively, preventing M2 polarization has also demonstrated anti-tumor efficacy in several different models. Two of the most promising approaches to target the M2 phenotype in macrophages include elimination the DICER protein in macrophages which leads to overexpression of microRNAs miR-511-3p or miR-26a both of which inhibit signaling required for M2 polarization and CSF-1/CSF-1R blockade.61–63 Interestingly, CSF-1/CSF-1R blockade in murine models of pancreatic cancer and glioma led to selective killing of the M2 macrophages64 and repolarization of the remaining TAMs into the M1 phenotype.44 Overall, re-educating macrophages to an anti-tumor phenotype in murine models of cancer has been very effective and thus many of these strategies are starting to be explored in the clinical setting.
Clinical studies
The strategy of enhancing M1 macrophage activation has shown some clinical success. Two examples, CD40 agonist and β-glucan administration, whose primary mechanisms of action involve macrophages, have demonstrated early activity in both hematologic and solid malignancies. Anti-CD40 antibody triggers an anti-tumor immune response by signaling through CD40, a receptor of the TNF-α family widely expressed by antigen-presenting cells particularly macrophages. Trials with humanized anti-CD40 antibodies have demonstrated the ability to trigger T cell specific anti-tumor immune responses against diffuse large B cell lymphomas, melanoma and pancreatic cancer.65–70 β-glucan, a yeast-derived polysaccharide, that can differentiate TAMs into an M1 phenotype has also been shown to have modest activity in a phase II multi-cancer study.71
One agent with potent anti-tumor activity that involves macrophages currently in clinical use for the treatment of sarcomas is Trabectedin (ET743, Yondelis), a natural product derived from the marine tunicate Ecteinascidia turbinate.72–75 Though primarily thought of as a DNA-damaging agent,76 recent data has revealed that administration also leads to specific apoptosis of macrophages by activating the caspase-8 signaling pathway in macrophages77 and further that it inhibits in vitro differentiation of macrophages and the production of IL-6 and CCL2.77 Thus, many effective therapies, like trabectin, may have unappreciated effects on macrophages as part of their mechanism of action. The experimental and clinical studies highlighted here revel some of the complex role that macrophages play in tumor biology. Increasingly it has also been recognized that macrophages have the capacity to regulate not only the development and progression of tumors, but also the response to therapies particularly radiation.
Radiation and the Immune System
Radiation (RT) has been used for the treatment of cancer for over a century following its discovery by Wilhelm Roentgen in 1895.78 The term radiation can refer to multiple types of energy along the electromagnetic spectrum, however therapeutic radiation typically refers to ionizing radiation with energies from the kilovoltage to megavoltage range. In this range of energies, radiation creates free radicals which can damage DNA which is one of the main cell intrinsic mechanisms by which RT is thought to kill cancer cells.79 Advances in the delivery of RT over the last decade has made RT one of the mainstays of treatment for virtually every cancer type with approximately 60% of all cancer patients receiving RT at some point during their course of treatment.80 While the direct effects of RT on tumors cells has been well studied, the consequences of the cell damage induced by RT on the tumor stroma, particularly the tumor-associated immune cells, remains largely unexplored.
Radiation-induced inflammatory response and macrophages
Mechanistic studies about RT have long focused on the tumor cell intrinsic mechanisms and only recently has it been recognized that tumor cell extrinsic factors including the immune microenvironment play an equally important role in determining the overall response of a tumor to RT.81 Recent experimental evidence has demonstrated that RT can trigger an anti-tumor immune response and that an optimal response depends on the ability of RT to generate a productive anti-tumor immune response. Several groups including our own have shown that there is a characteristic inflammatory-response induced by RT (Fig 1). Akin to other immune reactions, the RT-induced inflammatory response consists of five phases: innate recognition, initiation of inflammation, antigen presentation, effector response and resolution. Macrophages play an important role in all phases of the RT-induced inflammatory response.
Fig. 1. Macrophages and the radiation-induced immune response.

Ionizing radiation (RT) induces an anti-tumor immune response within the tumor through the generation of inflammatory mediators including cytosolic dsDNA, HMGB1, ATP, calreticulin (CRT) and Hsp70 within the tumor cells. These molecules activate the resident immune cells such as macrophages to secrete a series of cytokines/chemokines including IL-1 and TNF-α, which further recruits more macrophages to the tumor site. Activated macrophages and DCs migrate to the lymphoid tissues bearing tumor antigens, where they present them to T cells. Activated T cells then re-enter the circulation and return to the tumor where they target malignant cells. However, the outcome of the response is in part determined by the ability of the T cell response to the microenvironment. If the malignant cells are completely eradicated, macrophages help restore normal tissue homeostasis by supporting angiogenesis and matrix remodeling. If there is an insufficient immune response, macrophages still attempt to restore tissue to its normal state but in so doing inadvertently support tumor regrowth.
The initial response to RT involves the release of innate danger signals known as damage-associated molecular patterns (DAMPs) from irradiated cells in response to the damage induced by RT.82 This immunogenic cell death process is characterized in part by release of high mobility group protein box 1 (HMGB1),83 the expression of calreticulin (CRT) on the surface of the tumor cells, 84 release of ATP into the extracellular space,85 production of heat-shock proteins (HSPs)86 and leakage of double strand DNA into the cytosol.87 Macrophages can sense many of these innate inflammatory molecules through their expression of TLR4 for HMGB1, NOD-like protein receptor 3 (NLRP3) for ATP, low-density lipoprotein-receptor-related protein (LRP) for calreticulin and cyclic GMP-AMP (cGAMP) synthase (cGAS) and its downstream adaptor stimulator of interferon genes (STING) for cytosolic DNA.88 Downstream signaling from these receptors leads to release of pro-inflammatory cytokines that initiate the inflammatory cascade. Unlike the other damage associated molecules, early CRT expression allows tumor cells to be efficiently engulfed by macrophages and dendritic cells (DCs), thereby setting the stage for efficient presentation of tumor specific antigen to CTLs.84
Following innate recognition of the DAMPs, downstream signaling from the sensing of DAMPs converges upon activation of the NF-kB pathway leading to the production of pro-inflammatory cytokines to initiate inflammation.89 Much of this signaling occurs in innate cells such as macrophages, dendritic cells and, to a lesser extent, in the tumor cells themselves. Analysis of the tumor stroma following RT revealed an increased number of macrophages due to their resistance to RT-mediated death and enhanced recruitment.38,90,91 In addition to release of cytokines from innate immune cells following RT, several different cancer cell lines have demonstrated increased production of the cytokines IL-1α, IL-6, GM-CSF and IL-8 following exposure to RT compared to unirradiated controls.92,93 Tumor cells also release other pro-inflammatory molecules including chemokines following RT. For example, in a murine mammary carcinoma model, the RT-induced chemokine CXCL16 recruits tumor infiltrating lymphocytes, amplifying the immune response.94 The combination of these cytokines and chemokines acts as a positive feedback loop recruiting more immune cells chief among them are myeloid-macrophage cells which then release more pro-inflammatory molecules.
Once a response has been initiated, macrophages and DCs migrate to the lymphoid tissues carrying tumor antigens for presentation to T cells to generate an anti-tumor immune response. RT strongly enhances antigen presentation by inducing GM-CSF and expression of costimulatory molecules. Increased GM-CSF secretion following RT results in increased differentiation of DCs which augments tumor recognition by the host immune system.95 Furthermore, T cell costimulatory molecules including ICAM-1 and B7.1 and 2 necessary for T cell activation can be induced within in the tumor following RT, which enhances the development of an anti-tumor immune response.96,97 Once the T cell response is underway, release of IFN-γ from T cells as a result of RT further upregulates the expression of MHC I molecules on both tumor and stromal cells, leading to better antigen presentation.98,99 Myeloid cells including macrophages play a critical role in the presentation of antigen and costimulation to T cells within the tumor microenvironment allowing for the development of an anti-tumor immune response.
As T cells become activated, they migrate back into tumors in response to factors such as CXCL16 released by macrophages, other immune cells and tumor cells as described above. These effector T cells produce a potent anti-tumor immune response that is one of the key mechanisms by which RT works therapeutically. Mediated primarily by IFN-γ producing CD8+ T cells,100 several recent studies have revealed that macrophages also play a role in supporting the initial anti-tumor immune response through both direct tumor cytotoxicity and the production of inflammatory cytokines.101 However, even though RT produces a potent anti-tumor immune response in most patients, the magnitude and durability of the response to RT is highly variable. This occurs in part because simultaneously with the initial RT-induced anti-tumor response, RT also triggers potent immunosuppressive and healing mechanisms that support tumor regrowth and/or resistance (Fig 1). Consistent with this hypothesis, experimental data from several murine tumor models have shown that RT recruits both M1 and M2 macrophages from bone marrow derived myeloid cells.102–104 For example, in a murine prostate cancer model, both single dose (25 Grays/Gys) and fractionated irradiation (15 X 4 Gys) resulted in intratumoral macrophages with both higher expression of both M1 markers such as COX-2 and iNOS as well as M2 markers including Arg-1.105 Interestingly, the balance of M1 versus M2 macrophages produced following RT may depend on the radiation dose. In a model of pancreatic adenocarcinoma, low-dose γ irradiation led to the differentiation of iNOS+ M1 macrophages which promoted efficient recruitment of tumor-specific T cells by helping normalize the tumor vasculature.106 Due to their plasticity and potent regulatory potential, existing macrophages and those subsequently recruited following RT play an important role in both the initial anti-tumor immune response and the later creation of an immunosuppressive pro-tumoral microenvironment.
Similar to other immune responses, the RT-induced effector phase is then followed by a resolution phase in which tissue homeostasis is restored by suppressing any ongoing immune responses and repairing the tissue by restoring the matrix integrity and blood supply through angiogenesis. Myeloid cells and tissue macrophages dominate this phase of immune responses having both immunosuppressive and tissue repair activity. RT-mediated inflammation also induces the pathways used for the resolution of immune responses in which macrophages play the major role. For example, RT induces the transcription of HIF-1α which leads to increased expression of CXCL-12, CCL-2, CSF1 and VEGF which support angiogenesis, recruit macrophages and promote their immunosuppressive function.107–110 HIF-1α and IFN-γ signaling also induces the expression of PD-L1 in TAMs and tumor cells which suppresses the anti-tumor immune response.111 Additionally, RT causes cancer cell death partially via apoptosis which is known to induce immunosuppressive and anti-inflammatory phenotypes in macrophages as they clear dying cells and antigens.112 Apoptotic cells drive differentiation of macrophages into the M2 phenotype with enhanced secretion of anti-inflammatory cytokines such as TGF-β and IL-10 and upregulation of Arg-1.113,114 In fact, in non-tumor settings, systemic administration of apoptotic cells is an efficient means to generate antigen-specific tolerance.115 Thus, RT induces a complex immune response that includes strong immune stimulatory effects but also many immune suppressive pathways in the tumor microenvironment both of which depend on RT’s effects on myeloid cells including macrophages.
Combining macrophage targeting with radiation therapy
As we outlined above, radiation recruits large numbers of myeloid cells to tumors in response to both immunogenic cell death and the ensuing hypoxia from microvessel apoptosis. Given the increased recruitment of myeloid cells post-RT and the limited efficacy of macrophage targeting alone,44,116 the myeloid-macrophage compartment makes an ideal target for combining with RT to enhance its anti-tumor efficacy.
Targeting macrophage infiltration in combination with RT
Experimental studies
While macrophages play an important part in the initiation of the anti-tumor immune response following RT, they have a much more diverse and extensive role in suppressing post-RT anti-tumor immunity and supporting tumor regrowth. Both in vitro and in vivo TAMs isolated from irradiated tumors support tumor growth105 and resistance to RT.117 This resistance may be attributed in part to the “pro-tumor” M2-like phenotype of macrophages following RT with increased expression of PD-L1 and Arginase-1 as well as secretion of VEGF118 and MMP-9.119 Thus, strategies eliminating or inhibiting macrophages in combination with RT have demonstrated enhanced anti-tumor efficacy.120,121 Two methods for targeting macrophages in conjunction with RT have shown efficacy experimentally, depleting macrophages and preventing their migration to tumors (Table 3).
Table 3.
Studies that combine radiation therapy with macrophage manipulation for cancer treatment.
| Treatment | Type of study | Reference | |
|---|---|---|---|
| Targeting macrophage infiltration | |||
| Anti-CD11b antibody | In vivo; mice; squamous cell carcinoma | 90 | |
| PLX3397 (CSF1 inhibitor) | In vivo; mice; prostate, glioblastoma, breast cancer | 38,49,101,107 | |
| HIF-1 inhibitor | In vivo; mice; glioblastoma | 102 | |
| Ola-peg (CXCL12 inhibitor) | In vivo; rat; brain tumor | 131 | |
| AMD3100 (CXCL12/CCR4 inhibition) | In vivo; mice; prostate, cervical, lung cancer | 122,133–135 | |
| CCX771 (CXCR antagonist) | In vivo; mice, rat; brain tumor | 136 | |
| CXCL12 siRNA | In vivo; mice; astrocytoma | 178 | |
| Anti-CCL2 antibody | In vivo; mice; pancreatic ductal adenocarcinoma | 128 | |
| Treatment | Type of study | Functions | Reference |
| Targeting macrophage polarization/activation | |||
| Anti-CSF1 antibody | In vivo; mice; pancreatic ductal adenocarcinoma | M2 inhibition | 124 |
| Anti-VEGF/VEGFR | In vivo; mice; melanoma, ovarian, breast, | M2 inhibition | 118,179–181 |
| Anti-TNFR antibody | In vivo; mice; melanoma | M2 inhibition | 118 |
| Paclitaxel (cell cycle inhibitor) | In vivo; mice; breast cancer | M1 promotion | 140 |
| CTLA-4 blockade | In vivo; mice; breast, melanoma cancer; Case reports, phase 1 clinical trial; melanoma, non-small cell lung carcinoma |
M2 inhibition |
148,182,183 183–185 |
| TLR7 agonist imiquimod | In vivo; mice; breast cancer | M1 promotion | 138 |
| TLR9 agonist CpG ODN | Phase 1/2 clinical trial; mycosis fungoides | M1 promotion | 156 |
| IDO inhibitor | Phase 1b/2 clinical trial; melanoma | M2 inhibition | 154 |
| IL-2 | In vivo; mice; sarcoma, melanoma Phase 1 clinical trial; renal cell carcinoma |
M2 inhibition | 186,187 |
| Sunitinib (Tyrosine kinase inhibitor) | In vivo; mice; pancreatic adenocarcinoma; Phase 2 clinical trial; prostate cancer |
M2 inhibition | 159,160 |
| Sorafenib (Tyrosine kinase inhibitor) | Patient case report; renal cell carcinoma; Phase 2 clinical trial; hepatocellular carcinoma |
M2 inhibition | 161,162 |
Early evidence that macrophages impact the efficacy of RT came from depletion of bone marrow-derived cells from tumors (consisting largely of macrophages) by whole body radiation which delayed lung tumor regrowth after local irradiation.122 Subsequently, another group reported that elimination of TAMs by the macrophage-depleting agent liposomal clodronate increased the response to RT in a murine melanoma model.118 These early studies suggested that macrophages in general limit the tumor response to RT and thus other groups have developed other strategies to target macrophages as whole-body RT and liposomal clodronate have limited clinical utility.
Elimination of macrophages in tumors remains challenging as there are limited agents with adequate depleting capabilities that can be used outside the experimental setting, thus most of the current agents targeting macrophages with RT have focused on inhibiting macrophage migration into tumors. Several pathways that have been effectively targeted in combination with RT include the adhesion molecule CD11b, the macrophage cytokine and its receptor CSF-1/CSF-1R, and the chemokines CCL2 and CXCL12 and their respective receptors CCR2 and CXCR4.
CD11b, the α-subunit of the CD18 integrin, is expressed primarily on monocytes and macrophages123 and administration of a CD11b-neutralizing antibody resulted in an improved response to RT in a murine squamous cell carcinoma xenografts model.90 Similarly, in CD18-deficient mice, implanted tumors are more sensitive to irradiation compared to their wild type littermates,90 though interestingly mice genetically-deficient in CD11b have a similar level of radiosensitivity compared to wild type mice.
In irradiated tumors, the production of the macrophage cytokine CSF1 increases by around two-fold compared to non-irradiated tumors, likely due to the recruitment of the DNA damage-induced kinase ABL1 into the cell nucleus where it binds with the promoter of CSF1 to enhance its expression.49 PLX3397 (Plexxicon) is a small molecule that selectively inhibits the tyrosine kinase activity of CSF1R. In murine models of prostate cancer, breast cancer and glioblastoma, application of PLX3397 with RT suppress tumor growth more effectively than RT alone.49,101,107 In each of these models, blockade of CSF1R signaling with either a small-molecule inhibitor or a CSF-1 neutralizing antibody dramatically decreased the mobilization of TAMs and improved the therapeutic efficacy in a CD8+ T-cell dependent manner.101 Interestingly, CSF-1 inhibition may not only affect the presence of TAMs, but also their suppressive activity. A neutralizing antibody against CSF1 prevented RT from altering the phenotype of macrophages in tumors to M2 macrophages and increased the efficacy of RT in a murine model of pancreatic cancer.124
Similar to blocking CSF-1/CSF-1R, several agents have been developed to target the CCL2-CCR2 axis to inhibit the migration of monocytes into tumors. In several different mouse models of cancers, inhibition of CCL2 either by siRNA or monoclonal antibodies markedly reduced infiltration of macrophages leading to increased survival in tumor-bearing animals.45,125–127 For example, using a syngeneic murine model of pancreatic ductal adenocarcinoma, Kalbasi et al found that RT induced a significant increase in CCL2 production in tumors with subsequent recruitment of CCR2+ inflammatory monocytes.128 Administration of an anti-CCL2 antibody inhibited RT-induced monocyte recruitment and delayed tumor growth but only when given in combination with RT.128 Surprisingly, another study with a similar antibody found that interruption of CCL2 blockade led to enhanced metastases and reduced survival. This effect was attributed in part to enhanced angiogenesis from excess VEGF-A production and increased proliferation of metastatic cells from elevated IL-6 signaling both of which occurred as a result of rebound macrophage infiltration following cessation of CCL2 inhibition.129 This highlights a need for caution when utilizing anti-CCL2 agents and perhaps all agents targeting macrophages as the monotherapy in metastatic cancer.
In a glioblastoma multiforme (GBM) xenograft model, pharmacologic inhibition of the CXCL12/CXCR4 interaction blocked the recruitment of CD11b+ monocytes to the tumor and significantly slowed tumor regrowth.102,130 In a autochthonous rat brain tumor model, treatment with the CXCL12 inhibitor Ola-peg with RT significantly prolonged survival compared to irradiation alone.131 Treatment with AMD3100, a small bicyclam molecule that inhibits the binding between CXCL12/CXCR4, in combination with RT also significantly delayed tumor regrowth in xenograft and syngeneic models of breast, prostate and lung carcinoma.122,132–134 Interestingly, the enhanced efficacy observed with AMD3100 is lost when it was administered 5 days after radiation, suggesting that the CXCR4 signal is responsible for macrophage recruitment only in the period shortly following RT.122 In cervical cancer, analysis of human surgical specimens found that CXCR4 expression correlates with cancer severity. In orthotopic cervical cancer xenografts, addition of AMD3100 with standard radiation therapy and chemotherapy improved anti-tumor immune responses and reduced metastases without increased toxicity.135 The importance of CXCL12 following RT is further underscored by studies targeting CXCR7, the high-affinity receptor for CXCL12. In murine and human GBM xenograft models, inhibition of CXCR7 by a specific antagonist (CCX771) post-RT prevented tumor recurrence and significantly prolonged survival.136
In sum, multiple agents targeting macrophages have shown synergy when administered with RT. While many of these agents have limited efficacy alone, more clinical trials to further explore the interaction between RT and macrophages are warranted.
Clinical studies
No clinical outcomes using macrophage targeting agents with RT have been reported to date, however several trials are underway in glioblastoma and other histologies testing the efficacy of adding agents that target macrophages to standard courses of RT.
Targeting macrophage polarization in combination with RT
Experimental studies
As targeting macrophages in combination with RT has shown early promise, other strategies, particularly those targeting macrophage polarization, have been explored to understand whether it might be possible to enhance the synergy with RT even further by promoting the anti-tumor activity of macrophages while limiting their tumor promoting functions. Similar to the studies targeting macrophage polarization alone described previously, the two main strategies of enhancing the anti-tumor (M1) macrophages or reducing pro-tumor (M2) macrophages have been employed in combination with RT (Table 3).
TLR agonists represent a novel approach to stimulate an anti-tumor immune response by activating cytotoxic activity in macrophages and thereby enhancing the subsequent innate and adaptive anti-tumor immune response.137 In a murine model of skin-involving breast cancer, topical treatment with the TLR7 agonist imiquimod significantly slowed tumor growth when administered with RT versus imiquimod alone with responding tumors showing increased tumor infiltration by CD11c+ dendritic cells, CD4+, and CD8+ T cells.138 Moreover, low-dose chemotherapy cyclophosphamide which is thought to deplete myeloid-derived suppressor cells (MDSCs) given before start of treatment with imiquimod and RT further improved tumor inhibition.138 Other chemotherapies have also been found to enhance the efficacy of RT in part through their effects on macrophages. For example, in a murine mammary carcinoma model, paclitaxel enhanced the effect of RT and was found to promote IL-12 production from TAMs to inhibit their suppression of T cell cytotoxic activity.139,140
Innate immune cells can also detect the presence of cancer cells and trigger an adaptive anti-tumor response. The STING pathway which senses cytosolic tumor-derived DNA promotes type I IFNs production and boost the anti-tumor immune response. Recent studies revealed that the therapeutic effect of RT depends on the STING pathway, through the production of type I IFNs and the downstream T cell response.87 Importantly, treatment with a STING agonist and RT synergistically amplify the anti-tumor immune response in several tumor-bearing murine models.87,88
In addition to augmenting the anti-tumor activity, it has become increasingly recognized that targeting the immunosuppressive microenvironment in tumors is crucial to producing a successful anti-tumor immune response. Macrophages have a large role in establishing the immunosuppressive microenvironment in tumors. They acquire this immunosuppressive capability through a number of pathways including IL-4, TGF-β and Axl/MerTK. Multiple recent studies have demonstrated improved responses to RT when combined with agents that target these pathways.
In a syngeneic orthotopic murine model of breast cancer IL-4 blockade in combination with RT significantly delayed tumor regrowth compared to RT alone. This enhanced response to RT was associated with an increased number of CD8+ T cells and reduced number of CD4+ T cells.101 Indeed, neutralization of IL-4 led to a significant enhancement of anti-tumor immunity while limiting the development of immunosuppressive macrophage phenotypes.101 This study suggests that therapeutic targeting of Th2 cytokines enhances the efficacy of RT by altering the macrophage phenotype to favor anti-tumor activity following RT.
Overexpression of TGF-β in tumors is associated with early metastatic recurrences and poor patient outcome.141 In the MMTV-PyMT (expression of the polyoma middle T antigen under the control of the murine mammary tumor virus) transgenic model of metastatic breast cancer, both RT and chemotherapy increase the levels of circulating TGF-β which promotes lung metastasis during tumor regrowth.142 Focal tumor radiation also sharply increases intratumoral TGF-β in a dose dependent manner.143 Application of TGF-β neutralizing antibody enhances the local response to RT and abrogates the RT-induced increase in lung metastases in part through its effects on macrophages and the formation of regulatory T cells.142,143 Macrophages express the full complement of TGF-β receptors and when exposed to TGF-β, macrophages adopt a strongly immunosuppressive, regulatory phenotype.144 TGF-β has also long been known to be critical for the formation of regulatory T cells.145 Thus, given the upregulation of TGF-β following RT, targeting TGF-β in combination with RT may have synergy because of the combination’s potential to target multiple suppressive pathways in tumors.
The Tyro3/Axl/Mer family of tyrosine kinase receptors are strongly expressed on myeloid cells and associated with tumor invasion and metastasis.146 These tyrosine kinases regulate the function of mature macrophages through their control of macrophage mediated apoptosis.147 The receptor Axl is highly expressed in the radioresistant tumors but not in radiosensitive tumors. Genetic ablation of Axl enhanced antigen presentation, altered cytokine secretion and restored radiosensitivity.148 Another receptor, Mertk, in the Tyro3/Axl/Mer family also mediates the development of a suppressive macrophage phenotype following RT. Ligation of Mertk on macrophages resulted in suppressive cytokine release via NF-κB p50 upregulation, which in turn limited tumor control following RT.149 Elimination of this pathway led to enhanced anti-tumor immune responses following RT.
Metabolic reprogramming of the tumor has also been shown to alter the immune composition of the tumor microenvironment primarily through its effects on macrophages. Administration of a glycolysis inhibitor, 2-Deoxy-D-Glucose (2-DG), improved the radiosensitivity by increasing the RT-induced anti-tumor immunity in a murine model of Ehrlich ascites tumor.150 A follow up study showed that the combined treatment of RT and 2-DG not only enhanced the functional activation of macrophages but also skews the macrophages towards an M1 phenotype. 151
Thus, multiple lines of experimental evidence indicate that targeting macrophage phenotypes in combination with RT can dramatically enhance the response of tumors to RT. Strategies that augment the cytotoxic, classically activated M1 phenotype or reduce the immunosuppressive, alternatively activated M2 phenotype have all shown success in preclinical models. Thus, these strategies which directly or indirectly target myeloid cells and their functional activity are currently being explored in the clinical setting.
Clinical studies
Though no agents specifically targeting macrophage phenotype have been tested in combination with RT, several agents that strongly impact myeloid functional phenotypes have been successfully employed in clinical trials with RT. These agents include indoleamine-2,2-dioxygenase (IDO) inhibitors, TLR9 agonists and the tyrosine kinase inhibitor sunitinib.
TAMs and MDSCs highly express the tryptophan catabolic enzyme IDO, which induces T-cell dysfunction by depletion of tryptophan and accumulation of toxic catabolites. As IDO is in part responsible for tumor resistance to anti-CTLA-4 and anti-PD-1 therapy,152 administration of an IDO inhibitor in combination with anti-CTLA-4 or anti-PD-1 has been tested in combination with dual checkpoint blockade. Indeed, similar to the preclinical data153, the combination of an IDO inhibitor and an anti-PD-1 antibody demonstrated excellent clinical responses with the objective response rates reaching to 53% in late-stage melanoma patients.154
Several preclinical and clinical studies have also shown that TLR9 agonists such as CpG reduce the number and suppressive activity of tumor-infiltrating MDSCs and induces their maturation into M1 macrophages.155 In patients with mycosis fungoides, a subtype of cutaneous T-cell lymphoma, combining RT and in situ vaccination with CpG showed a 33% response rate in a small phase I trial.156 Preclinical data in mice and dogs combining CpG, RT and IDO inhibition suggest that optimal responses may require combinations of immunotherapies that both augment cytotoxic responses and inhibit immunosuppressive activity with RT.153,157
Several tyrosine kinase inhibitors are currently approved for the treatment of multiple cancer types. Mechanistically, they inhibit cellular signaling by targeting multiple receptor tyrosine kinases such as platelet-derived growth factor (PDGF), VEGFRs and c-Kit. 158 With respect to macrophages, sunitinib has been shown to limit macrophage M2 polarization by inhibiting STAT3 signaling which allows more macrophages to retain their M1 phenotype despite suppressive signaling. In a murine model of pancreatic cancer, sunitinib sensitized pancreatic tumors making them more susceptible to RT.159 In a phase 2 trial of patients with localized high-risk prostate cancer, prolonged disease control was observed with the combined application of sunitinib, chemotherapy and RT.160 Pathologic analysis of the tissue suggested that the combination of RT, chemotherapy and a similar tyrosine kinase inhibitor, sorafenib, can reverse TAMs to an anti-tumor phenotype.161 Similar enhanced efficacy was seen in metastatic renal cell carcinoma161 and unresectable hepatocellular carcinoma.162
In summary, while these agents all undoubtedly have effects outside of their impact on myeloid-macrophage cells, some of their bioactivity is likely attributable to their ability to influence the functional phenotype of macrophages. As macrophages appear to have an outsized role in regulating the tumor response to RT, therapies directed at pathways that increase the number of cytotoxic macrophages and reduce the formation of immunosuppressive, pro-tumor macrophages may prove to have potent synergy with RT.
Future macrophage-directed drug targets in combination with RT
Macrophages play a critical role in regulating the RT-induced inflammatory response. Thus, immunotherapies targeting macrophages and/or their functional polarization represent logical targets to be given in conjunction with RT and multiple preclinical studies and early clinical data supports this approach (Fig 2). Again, strategies to augment the anti-tumor immune response and prevent the late tumor and RT mediated M2 polarization of the myeloid-macrophage cells are the most promising targets to combine with RT. One of the most promising agent that augments the immune response to RT in preclinical models is STING agonists. As macrophages produce type I IFNs in a STING-dependent manner in response to cytosolic DNA produced by RT,88 application of a STING agonist with RT may augment the RT-induced inflammation leading to enhanced anti-tumor immunity.
Fig. 2. Potential immune targets to combine with radiation therapy.
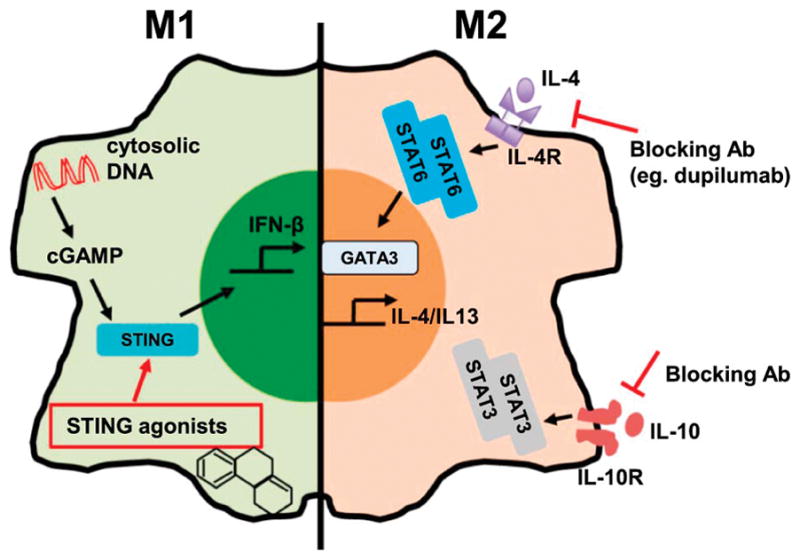
The cGAS-STING cytosolic DNA sensing pathway is essential for production of type I IFNs such as IFN-β. An RT-induced DNA exonuclease Trex1 degrades cytosolic DNA to dampen the production of type I IFNs in response to RT. However, STING agonists can bypass this exonuclease and thus may directly activate macrophages to augment the response to RT. Ligation of IL-10 receptor activates STAT3, which is critical for the expression of its own cytokine IL-10. Blockade of the IL-10 pathway may disrupt this feedback and combined with RT to boost anti-tumor immunity. Upon IL-4 stimulation, the transcriptional regulator GATA3 induces a program that polarizes macrophages into an M2 phenotype. Agents that target the IL-4 pathway may improve the efficacy of RT by preventing formation of the M2 phenotype in macrophages in response to RT-induced IL-4 production.
With the success of PD-1/PD-L1 directed therapies, it has become increasingly evident that reducing the immunosuppressive tumor microenvironment is necessary for the development of anti-tumor immunity. Macrophages play a key role in establishing this immunosuppression and thus agents that target the immunosuppressive function of macrophages will likely prove to be important targets in future trials. Two such pathways critical for establishing the immunosuppressive macrophage phenotype are the IL-10 and IL-4 pathways. IL-10 is a key immunosuppressive cytokine that promotes the regulatory functions of macrophages. Blockade of IL-10 in multiple animal models has led to delayed tumor growth and improved overall survival,163,164 therefore targeting IL-10 in combination with RT may lead to better control of tumor progression via modulation of TAM-mediated immunosuppression. IL-4 is best known for its role in mediating Th2 immunity, however it has also long been associated with M2 polarization of macrophages as well. Binding of IL-4 to the IL-4 receptor results in the phosphorylation and dimerization of STAT6. STAT6 dimers translocate into the nucleus where they promote expression of the Th2 master regulator GATA-binding protein 3 (GATA3) which leads to transcription of the M2 program in macrophages.165 Targeting this pathway through IL-4 or IL-4 receptor blockade and/or inhibition of downstream signaling has been shown in preclinical data to lead to significant enhancement of the efficacy of RT. Given the critical role of the IL-4 pathway in mediating the M2 macrophage phenotype, this remains an excellent target for future combinatorial clinical studies.
Conclusions
Macrophages participate and regulate nearly every aspect of tumor development from the initial establishment of the premalignant niche to the angiogenesis and immunosuppression needed for tumor progression and the development of metastases. They exhibit tremendous plasticity and can be polarized into phenotypes that range from the pro-inflammatory, anti-tumor M1 phenotype to the immunosuppressive, pro-tumor M2 phenotype in response to the different environmental stimuli encountered in tumors. Radiation therapy can affect the development of both the M1 and M2 activation pathways. The initial cell damage created by RT initiates a cytotoxic program characterized by Th1 cytokine secretion and CD8+ T cells that favors the development of anti-tumor macrophage phenotypes. However, simultaneously RT activates immunosuppressive pathways including IL-4, IL-10 and TGF-β that lead to the formation of immunosuppressive, pro-tumor M2 macrophages. Targeting macrophage infiltration and polarization in combination with RT has demonstrated tremendous synergy in preclinical models and early phase clinical trials. Further clinical trials are currently underway exploring combinations of RT with therapeutics directed at macrophages to produce enhanced cytotoxic activity while reducing or reversing the development of immunosuppressive macrophage phenotypes.
Acknowledgments
We thank all the lab members in Dr. Stephen L. Shiao and Dr. David M. Underhill laboratories for their support.
Conflicts of Interest: All authors have read the journal’s policy on disclosure of potential conflicts of interest and have none to declare.
All authors have read the journal’s authorship agreement and that the manuscript has been reviewed by and approved by all named authors.
Abbreviations
- RT
radiation therapy
- IFN-γ
interferon-γ
- LPS
lipopolysaccharide
- TLR
toll-like receptor
- STAT
signal transducer and activator of transcription
- NF-kB
nuclear factor-kB
- CXCLs
C-X-C motif ligands
- CCLs
C-C motif ligands
- INOS
inducible nitric oxide synthase
- IL
interleukin
- TNF
tumor necrosis factor
- Arg-1
arginase-1
- VEGF
vascular endothelial growth factor
- CSF1
colony stimulating factor 1
- TAMs
tumor-associated macrophages
- M-CSF
macrophage colony-stimulating factor
- PD-1
programed death-1
- HIF-1α
hypoxia-inducible factor-1α
- MMP
matrix metalloproteinase
- DAMP
damage-associated molecular pattern
- HMGB1
high mobility group protein box 1
- CRT
calreticulin
- HSPs
heat-shock proteins
- NLRP3
NOD-like protein receptor 3
- LRP
low-density lipoprotein-receptor-related protein
- CGAS
cyclic GMP-AMP synthase
- STING
stimulator of interferon genes
- DCs
dendritic cells
- Gy
Gray
- MiRNA
microRNA
- GBM
glioblastoma multiforme
- MDSCs
myeloid-derived suppressor cells
- IDO
indoleamine-2,2-dioxygenase
- PDGF
platelet-derived growth factor
- GATA3
GATA-binding protein 3
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cassetta L, Cassol E, Poli G. Macrophage Polarization in Health and Disease. Sci World J. 2011;11:2391–2402. doi: 10.1100/2011/213962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nature immunology. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 3.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends in immunology. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 4.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. Journal of immunology. 2000;164:6166–6173. doi: 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- 5.Murray Peter J, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xue J, et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity. 2014;40:274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nature reviews. Immunology. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dale DC, Boxer L, Liles WC. The phagocytes: neutrophils and monocytes. Blood. 2008;112:935–945. doi: 10.1182/blood-2007-12-077917. [DOI] [PubMed] [Google Scholar]
- 9.Gordon S. Alternative activation of macrophages. Nature reviews. Immunology. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 10.Fleetwood AJ, Dinh H, Cook AD, Hertzog PJ, Hamilton JA. GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on Type I interferon signaling. Journal of Leukocyte Biology. 2009;86:411–421. doi: 10.1189/jlb.1108702. [DOI] [PubMed] [Google Scholar]
- 11.Krausgruber T, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011;12:231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 12.Bonizzi G, Karin M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends in immunology. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 13.Romieu-Mourez R, et al. Distinct roles for IFN regulatory factor (IRF)-3 and IRF-7 in the activation of antitumor properties of human macrophages. Cancer research. 2006;66:10576–10585. doi: 10.1158/0008-5472.CAN-06-1279. [DOI] [PubMed] [Google Scholar]
- 14.Biswas SK, Chittezhath M, Shalova IN, Lim JY. Macrophage polarization and plasticity in health and disease. Immunologic research. 2012;53:11–24. doi: 10.1007/s12026-012-8291-9. [DOI] [PubMed] [Google Scholar]
- 15.Kreider T, Anthony RM, Urban JF, Gause WC. Alternatively activated macrophages in helminth infections. Current opinion in immunology. 2007;19:448–453. doi: 10.1016/j.coi.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ji RC. Macrophages are important mediators of either tumor-or inflammation-induced lymphangiogenesis. Cellular and Molecular Life Sciences. 2012;69:897–914. doi: 10.1007/s00018-011-0848-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer research. 2006;66:605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 18.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 19.de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- 20.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Kempen LCL, de Visser KE, Coussens LM. Inflammation, proteases and cancer. European journal of cancer. 2006;42:728–734. doi: 10.1016/j.ejca.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 22.Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Critical Reviews in Oncology/Hematology. 2008;66:1–9. doi: 10.1016/j.critrevonc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Gordon SR, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495–499. doi: 10.1038/nature22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spary LK, et al. Tumor stroma-derived factors skew monocyte to dendritic cell differentiation toward a suppressive CD14+ PD-L1+ phenotype in prostate cancer. Oncoimmunology. 2014;3:e955331. doi: 10.4161/21624011.2014.955331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ascierto PA, Marincola FM. 2015: the year of anti-PD-1/PD-L1s against melanoma and beyond. EBioMedicine. 2015;2:92–93. doi: 10.1016/j.ebiom.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishimura H, Honjo T. PD-1: an inhibitory immunoreceptor involved in peripheral tolerance. Trends in immunology. 2001;22:265–268. doi: 10.1016/s1471-4906(01)01888-9. [DOI] [PubMed] [Google Scholar]
- 27.Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends in immunology. 2003;24:301–305. doi: 10.1016/s1471-4906(03)00132-7. [DOI] [PubMed] [Google Scholar]
- 28.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nature reviews. Immunology. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 29.Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunological reviews. 2008;222:180–191. doi: 10.1111/j.1600-065X.2008.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 31.Semenza GL. HIF-1 and tumor progression: pathophysiology and therapeutics. Trends in molecular medicine. 2002;8:S62–S67. doi: 10.1016/s1471-4914(02)02317-1. [DOI] [PubMed] [Google Scholar]
- 32.Colegio OR, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finkernagel F, et al. The transcriptional signature of human ovarian carcinoma macrophages is associated with extracellular matrix reorganization. Oncotarget. 2016;7:75339–75352. doi: 10.18632/oncotarget.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugimura K, et al. High infiltration of tumor-associated macrophages is associated with a poor response to chemotherapy and poor prognosis of patients undergoing neoadjuvant chemotherapy for esophageal cancer. Journal of surgical oncology. 2015;111:752–759. doi: 10.1002/jso.23881. [DOI] [PubMed] [Google Scholar]
- 37.Kurahara H, et al. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. Journal of Surgical Research. 2011;167:e211–e219. doi: 10.1016/j.jss.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 38.DeNardo DG, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer discovery. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. The Journal of experimental medicine. 2001;193:727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bronte V, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. The Journal of Immunology. 1999;162:5728–5737. [PMC free article] [PubMed] [Google Scholar]
- 41.Kowanetz M, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+ Ly6C+ granulocytes. Proc Natl Acad Sci. 2010;107:21248–21255. doi: 10.1073/pnas.1015855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hollmén M, et al. G-CSF regulates macrophage phenotype and associates with poor overall survival in human triple-negative breast cancer. Oncoimmunology. 2016;5:e1115177. doi: 10.1080/2162402X.2015.1115177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kubota Y, et al. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. The Journal of experimental medicine. 2009;206:1089–1102. doi: 10.1084/jem.20081605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pyonteck SM, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nature medicine. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qian BZ, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ries CH, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer cell. 2014;25:846–859. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 47.Anthony S, et al. Pharmacodynamic activity demonstrated in phase I for PLX3397, a selective inhibitor of FMS and Kit. Journal of Clinical Oncology. 2011;29:3093–3093. [Google Scholar]
- 48.Cassier PA, et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. The Lancet Oncology. 2015;16:949–956. doi: 10.1016/S1470-2045(15)00132-1. [DOI] [PubMed] [Google Scholar]
- 49.Xu J, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73:2782–2794. doi: 10.1158/0008-5472.CAN-12-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pradel LP, et al. Macrophage susceptibility to emactuzumab (RG7155) treatment. Molecular cancer therapeutics. 2016;15:3077–3086. doi: 10.1158/1535-7163.MCT-16-0157. [DOI] [PubMed] [Google Scholar]
- 51.Sandhu SK, et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer chemotherapy and pharmacology. 2013;71:1041–1050. doi: 10.1007/s00280-013-2099-8. [DOI] [PubMed] [Google Scholar]
- 52.Pienta KJ, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investigational new drugs. 2013;31:760–768. doi: 10.1007/s10637-012-9869-8. [DOI] [PubMed] [Google Scholar]
- 53.Coward J, et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clinical cancer research. 2011;17:6083–6096. doi: 10.1158/1078-0432.CCR-11-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karkera J, et al. The anti-interleukin-6 antibody siltuximab down-regulates genes implicated in tumorigenesis in prostate cancer patients from a phase I study. The Prostate. 2011;71:1455–1465. doi: 10.1002/pros.21362. [DOI] [PubMed] [Google Scholar]
- 55.Rossi J, et al. A phase I/II study of siltuximab (CNTO 328), an anti-interleukin-6 monoclonal antibody, in metastatic renal cell cancer. British journal of cancer. 2010;103:1154. doi: 10.1038/sj.bjc.6605872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Angevin E, et al. A phase 1/2, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clinical Cancer Research, clincanres. 2014;2200:2013. doi: 10.1158/1078-0432.CCR-13-2200. [DOI] [PubMed] [Google Scholar]
- 57.Xu M, et al. Intratumoral delivery of IL-21 overcomes anti-Her2/Neu resistance through shifting tumor-associated macrophages from M2 to M1 phenotype. Journal of immunology. 2015;194:4997–5006. doi: 10.4049/jimmunol.1402603. [DOI] [PubMed] [Google Scholar]
- 58.Yang L, et al. CD163+ tumor-associated macrophage is a prognostic biomarker and is associated with therapeutic effect on malignant pleural effusion of lung cancer patients. Oncotarget. 2015;6:10592. doi: 10.18632/oncotarget.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zanganeh S, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotech. 2016;11:986–994. doi: 10.1038/nnano.2016.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ortega RA, et al. Biocompatible mannosylated endosomal-escape nanoparticles enhance selective delivery of short nucleotide sequences to tumor associated macrophages. Nanoscale. 2015;7:500–510. doi: 10.1039/c4nr03962a. [DOI] [PubMed] [Google Scholar]
- 61.Squadrito ML, et al. miR-511–3p modulates genetic programs of tumor-associated macrophages. Cell Rep. 2012;1:141–154. doi: 10.1016/j.celrep.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 62.Chai ZT, et al. microRNA-26a suppresses recruitment of macrophages by down-regulating macrophage colony-stimulating factor expression through the PI3K/Akt pathway in hepatocellular carcinoma. Journal of hematology & oncology. 2015;8:56. doi: 10.1186/s13045-015-0150-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baer C, et al. Suppression of microRNA activity amplifies IFN-[gamma]-induced macrophage activation and promotes anti-tumour immunity. Nature cell biology. 2016 doi: 10.1038/ncb3371. [DOI] [PubMed] [Google Scholar]
- 64.Zhu Y, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer research. 2014;74:5057–5069. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson P, et al. Clinical and biological effects of an agonist anti-CD40 antibody: a Cancer Research UK phase I study. Clinical Cancer Research. 2015;21:1321–1328. doi: 10.1158/1078-0432.CCR-14-2355. [DOI] [PubMed] [Google Scholar]
- 66.Chowdhury F, Johnson PW, Glennie MJ, Williams AP. Ex vivo assays of dendritic cell activation and cytokine profiles as predictors of in vivo effects in an anti-human CD40 monoclonal antibody ChiLob 7/4 phase I trial. Cancer immunology research. 2014;2:229–240. doi: 10.1158/2326-6066.CIR-13-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Advani R, et al. Phase I study of the humanized anti-CD40 monoclonal antibody dacetuzumab in refractory or recurrent non-Hodgkin’s lymphoma. Journal of Clinical Oncology. 2009;27:4371–4377. doi: 10.1200/JCO.2008.21.3017. [DOI] [PubMed] [Google Scholar]
- 68.Beatty GL, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clinical cancer research, clincanres. 2013 doi: 10.1158/1078-0432.CCR-13-1320. 1320.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vonderheide RH, et al. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology. 2013;2:e23033. doi: 10.4161/onci.23033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vonderheide RH, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. Journal of Clinical Oncology. 2007;25:876–883. doi: 10.1200/JCO.2006.08.3311. [DOI] [PubMed] [Google Scholar]
- 71.Segal NH, et al. A phase II efficacy and safety, open-label, multicenter study of imprime PGG injection in combination with cetuximab in patients with stage IV KRAS-mutant colorectal cancer. Clinical colorectal cancer. 2016;15:222–227. doi: 10.1016/j.clcc.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guan Y, Sakai R, Rinehart K, Wang A. Molecular and crystal structures of ecteinascidins: potent antitumor compounds from the Caribbean tunicate Ecteinascidia turbinata. Journal of biomolecular structure & dynamics. 1993;10:793–818. [PubMed] [Google Scholar]
- 73.Germano G, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer cell. 2013;23:249–262. doi: 10.1016/j.ccr.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 74.Germano G, et al. Antitumor and anti-inflammatory effects of trabectedin on human myxoid liposarcoma cells. Cancer research. 2010;70:2235–2244. doi: 10.1158/0008-5472.CAN-09-2335. [DOI] [PubMed] [Google Scholar]
- 75.Demetri GD, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. Journal of Clinical Oncology. 2015;34:786–793. doi: 10.1200/JCO.2015.62.4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gordon EM, Sankhala KK, Chawla N, Chawla SP. Trabectedin for soft tissue sarcoma: current status and future perspectives. Advances in therapy. 2016;33:1055–1071. doi: 10.1007/s12325-016-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Allavena P, et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer research. 2005;65:2964–2971. doi: 10.1158/0008-5472.CAN-04-4037. [DOI] [PubMed] [Google Scholar]
- 78.Roentgen W. On a new kind of ray (first report) nchener medizinische Wochenschrift (1950) 1959;101:1237–1239. [PubMed] [Google Scholar]
- 79.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Canc. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 80.Thariat J, Hannoun-Levi JM, Myint AS, Vuong T, Gérard JP. Past, present, and future of radiotherapy for the benefit of patients. Nat Rev Clini Oncol. 2013;10:52–60. doi: 10.1038/nrclinonc.2012.203. [DOI] [PubMed] [Google Scholar]
- 81.Demaria S, Golden EB, Formenti SC. Role of Local Radiation Therapy in Cancer Immunotherapy. JAMA oncology. 2015;1:1325–1332. doi: 10.1001/jamaoncol.2015.2756. [DOI] [PubMed] [Google Scholar]
- 82.Ahmed MM, Guha C, Hodge JW, Jaffee E. Immunobiology of radiotherapy: new paradigms. Radiation research. 2014;182:123–125. doi: 10.1667/RR13849.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Apetoh L, et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nature medicine. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 84.Obeid M, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nature medicine. 2007;13:54. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 85.Ghiringhelli F, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β–dependent adaptive immunity against tumors. Nature medicine. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 86.Lumniczky K, Sáfrány G. The impact of radiation therapy on the antitumor immunity: local effects and systemic consequences. Cancer letters. 2015;356:114–125. doi: 10.1016/j.canlet.2013.08.024. [DOI] [PubMed] [Google Scholar]
- 87.Deng L, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41:843–852. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vanpouille-Box C, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8 doi: 10.1038/ncomms15618. ncomms15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Richmond A. NF-κB, chemokine gene transcription and tumour growth. Nature reviews. Immunology. 2002;2:664–674. doi: 10.1038/nri887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ahn GO, et al. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci. 2010;107:8363–8368. doi: 10.1073/pnas.0911378107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Crittenden MR, et al. Expression of NF-κB p50 in tumor stroma limits the control of tumors by radiation therapy. PloS one. 2012;7:e39295. doi: 10.1371/journal.pone.0039295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang J, Nakatsugawa S, Niwa O, Ju G, Liu S. Ionizing radiation-induced IL-1 alpha, IL-6 and GM-CSF production by human lung cancer cells. Chinese medical journal. 1994;107:653–657. [PubMed] [Google Scholar]
- 93.Yamanaka R, Tanaka R, Yoshida S. Effects of irradiation on cytokine production in glioma cell lines. Neurologia medico-chirurgica. 1993;33:744–748. doi: 10.2176/nmc.33.744. [DOI] [PubMed] [Google Scholar]
- 94.Matsumura S, et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. Journal of immunology. 2008;181:3099–3107. doi: 10.4049/jimmunol.181.5.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gallo PM, Gallucci S. Implications for autoimmunity. 2013. The dendritic cell response to classic, emerging, and homeostatic danger signals. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Seo A, et al. Enhancement of B7–1 (CD80) expression on B-lymphoma cells by irradiation. Immunology. 1999;96:642–648. doi: 10.1046/j.1365-2567.1999.00720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vereecque R, et al. γ-Ray irradiation induces B7. 1 expression in myeloid leukaemic cells. British journal of haematology. 2000;108:825–831. doi: 10.1046/j.1365-2141.2000.01967.x. [DOI] [PubMed] [Google Scholar]
- 98.Reits EA, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. Journal of Experimental Medicine. 2006;203:1259–1271. doi: 10.1084/jem.20052494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lugade AA, et al. Radiation-induced IFN-γ production within the tumor microenvironment influences antitumor immunity. Journal of immunology. 2008;180:3132–3139. doi: 10.4049/jimmunol.180.5.3132. [DOI] [PubMed] [Google Scholar]
- 100.Reeves E, James E. Antigen processing and immune regulation in the response to tumours. Immunology. 2016 doi: 10.1111/imm.12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shiao SL, et al. TH2-Polarized CD4(+) T Cells and Macrophages Limit Efficacy of Radiotherapy. Cancer immunology research. 2015;3:518–525. doi: 10.1158/2326-6066.CIR-14-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kioi M, et al. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120:694–705. doi: 10.1172/JCI40283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen FH, et al. Radiotherapy decreases vascular density and causes hypoxia with macrophage aggregation in TRAMP-C1 prostate tumors. Clinical Cancer Research. 2009;15:1721–1729. doi: 10.1158/1078-0432.CCR-08-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Russell J, Brown J. The irradiated tumor microenvironment: role of tumor-associated macrophages in vascular recovery. Frontiers in Physiology. 2013;4 doi: 10.3389/fphys.2013.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsai CS, et al. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. International Journal of Radiation Oncology* Biology* Physics. 2007;68:499–507. doi: 10.1016/j.ijrobp.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 106.Klug F, et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer cell. 2013;24:589–602. doi: 10.1016/j.ccr.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 107.Stafford JH, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro-oncology. 2015:nov272. doi: 10.1093/neuonc/nov272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Du R, et al. HIF1α induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ceradini DJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nature medicine. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 110.Mojsilovic-Petrovic J, et al. Hypoxia-inducible factor-1 (HIF-1) is involved in the regulation of hypoxia-stimulated expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) and MCP-5 (Ccl12) in astrocytes. Journal of neuroinflammation. 2007;4:12. doi: 10.1186/1742-2094-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Noman MZ, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. Journal of Experimental Medicine. 2014 doi: 10.1084/jem.20131916. jem. 20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Melcher A, Gough M, Todryk S, Vile R. Apoptosis or necrosis for tumor immunotherapy: what’s in a name? Journal of molecular medicine. 1999;77:824–833. doi: 10.1007/s001099900066. [DOI] [PubMed] [Google Scholar]
- 113.Freire-de-Lima CG, et al. Apoptotic cells, through transforming growth factor-β, coordinately induce anti-inflammatory and suppress pro-inflammatory eicosanoid and NO synthesis in murine macrophages. Journal of Biological Chemistry. 2006;281:38376–38384. doi: 10.1074/jbc.M605146200. [DOI] [PubMed] [Google Scholar]
- 114.Fadok V, McDonald P, Bratton D, Henson P. Regulation of macrophage cytokine production by phagocytosis of apoptotic and post-apoptotic cells. Biochemical Society Transactions. 1998;26:653–656. doi: 10.1042/bst0260653. [DOI] [PubMed] [Google Scholar]
- 115.Noelia A, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–258. doi: 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Manthey CL, et al. JNJ-28312141, a novel orally active colony-stimulating factor-1 receptor/FMS-related receptor tyrosine kinase-3 receptor tyrosine kinase inhibitor with potential utility in solid tumors, bone metastases, and acute myeloid leukemia. Molecular cancer therapeutics. 2009;8:3151–3161. doi: 10.1158/1535-7163.MCT-09-0255. [DOI] [PubMed] [Google Scholar]
- 117.Milas L. Tumor bed effect in murine tumors: relationship to tumor take and tumor macrophage content. Radiation research. 1990;123:232–236. [PubMed] [Google Scholar]
- 118.Meng Y, et al. Blockade of tumor necrosis factor α signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer research. 2010;70:1534–1543. doi: 10.1158/0008-5472.CAN-09-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ahn GO, Brown JM. Matrix Metalloproteinase-9 Is Required for Tumor Vasculogenesis but Not for Angiogenesis: Role of Bone Marrow-Derived Myelomonocytic Cells. Cancer cell. 2008;13:193–205. doi: 10.1016/j.ccr.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer cell. 2013;23:277–286. doi: 10.1016/j.ccr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 121.Hughes R, et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer research. 2015;75:3479–3491. doi: 10.1158/0008-5472.CAN-14-3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kozin SV, et al. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer research. 2010;70:5679–5685. doi: 10.1158/0008-5472.CAN-09-4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Arnaout MA. Structure and function of the leukocyte adhesion molecules CD11/CD18. Blood. 1990;75:1037–1050. [PubMed] [Google Scholar]
- 124.Seifert L, et al. Radiation Therapy Induces Macrophages to Suppress T-Cell Responses Against Pancreatic Tumors in Mice. Gastroenterology. 2016;150:1659–1672.e1655. doi: 10.1053/j.gastro.2016.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fang WB, et al. Targeted gene silencing of CCL2 inhibits triple negative breast cancer progression by blocking cancer stem cell renewal and M2 macrophage recruitment. Oncotarget. 2016;7:49349. doi: 10.18632/oncotarget.9885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Loberg RD, et al. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer research. 2007;67:9417–9424. doi: 10.1158/0008-5472.CAN-07-1286. [DOI] [PubMed] [Google Scholar]
- 127.Zhu X, Fujita M, Snyder LA, Okada H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. Journal of neuro-oncology. 2011;104:83–92. doi: 10.1007/s11060-010-0473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kalbasi A, et al. Tumor-derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clinical Cancer Research. 2017;23:137–148. doi: 10.1158/1078-0432.CCR-16-0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bonapace L, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515:130–133. doi: 10.1038/nature13862. [DOI] [PubMed] [Google Scholar]
- 130.Chau NM, et al. Identification of novel small molecule inhibitors of hypoxia-inducible factor-1 that differentially block hypoxia-inducible factor-1 activity and hypoxia-inducible factor-1α induction in response to hypoxic stress and growth factors. Cancer Research. 2005;65:4918–4928. doi: 10.1158/0008-5472.CAN-04-4453. [DOI] [PubMed] [Google Scholar]
- 131.Liu S-C, et al. Blockade of SDF-1 after irradiation inhibits tumor recurrences of autochthonous brain tumors in rats. Neuro-oncology. 2013:not149. doi: 10.1093/neuonc/not149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jo DY. CXCR4 antagonists in hematologic malignancies: more than just mobilizers? Korean J Hematol. 2011;46:209–210. doi: 10.5045/kjh.2011.46.4.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen FH, et al. Combination of Vessel-Targeting Agents and Fractionated Radiation Therapy: The Role of the SDF-1/CXCR4 Pathway. Int J Rad Oncol Biol Phy. 2013;86:777–784. doi: 10.1016/j.ijrobp.2013.02.036. [DOI] [PubMed] [Google Scholar]
- 134.Domanska UM, et al. CXCR4 inhibition enhances radiosensitivity, while inducing cancer cell mobilization in a prostate cancer mouse model. Clinical & experimental metastasis. 2014;31:829–839. doi: 10.1007/s10585-014-9673-2. [DOI] [PubMed] [Google Scholar]
- 135.Chaudary N, et al. Plerixafor Improves Primary Tumor Response and Reduces Metastases in Cervical Cancer Treated with Radio-Chemotherapy. Clinical Cancer Research. 2017;23:1242–1249. doi: 10.1158/1078-0432.CCR-16-1730. [DOI] [PubMed] [Google Scholar]
- 136.Walters M, et al. Inhibition of CXCR7 extends survival following irradiation of brain tumours in mice and rats. British journal of cancer. 2014;110:1179–1188. doi: 10.1038/bjc.2013.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Krieg AM. Development of TLR9 agonists for cancer therapy. Journal of Clinical Investigation. 2007;117:1184. doi: 10.1172/JCI31414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dewan MZ, et al. Synergy of topical toll-like receptor 7 agonist with radiation and low-dose cyclophosphamide in a mouse model of cutaneous breast cancer. Clinical Cancer Research. 2012;18:6668–6678. doi: 10.1158/1078-0432.CCR-12-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mullins DW, Burger CJ, Elgert KD. Paclitaxel enhances macrophage IL-12 production in tumor-bearing hosts through nitric oxide. Journal of immunology. 1999;162:6811–6818. [PubMed] [Google Scholar]
- 140.Milas L, Hunter NR, Mason KA, Kurdoglu B, Peters LJ. Enhancement of tumor radioresponse of a murine mammary carcinoma by paclitaxel. Cancer research. 1994;54:3506–3510. [PubMed] [Google Scholar]
- 141.Wojtowicz-Praga S. Reversal of tumor-induced immunosuppression by TGF-β inhibitors. Investigational new drugs. 2003;21:21–32. doi: 10.1023/a:1022951824806. [DOI] [PubMed] [Google Scholar]
- 142.Biswas S, et al. Inhibition of TGF-β with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. Journal of Clinical Investigation. 2007;117:1305. doi: 10.1172/JCI30740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Vanpouille-Box C, et al. TGFbeta Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015;75:2232–2242. doi: 10.1158/0008-5472.CAN-14-3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kaminska B, Wesolowska A, Danilkiewicz M. TGF beta signalling and its role in tumour pathogenesis. Acta Biochim Pol. 2005;52:329. [PubMed] [Google Scholar]
- 145.Chen W, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. The Journal of experimental medicine. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Canc. 2014;14:769–785. doi: 10.1038/nrc3847. [DOI] [PubMed] [Google Scholar]
- 147.Zhang Y, et al. Breakdown of immune homeostasis in the testis of mice lacking Tyro3, Axl and Mer receptor tyrosine kinases. Immunology and cell biology. 2013;91:416–426. doi: 10.1038/icb.2013.22. [DOI] [PubMed] [Google Scholar]
- 148.Aguilera TA, et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat Commun. 2016:7. doi: 10.1038/ncomms13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Crittenden MR, et al. Mertk on tumor macrophages is a therapeutic target to prevent tumor recurrence following radiation therapy. Oncotarget. 2016;7:78653. doi: 10.18632/oncotarget.11823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Farooque A, Singh N, Adhikari JS, Afrin F, Dwarakanath BSR. Enhanced antitumor immunity contributes to the radio-sensitization of Ehrlich Ascites Tumor by the glycolytic inhibitor 2-deoxy-D-glucose in mice. PloS one. 2014;9:e108131. doi: 10.1371/journal.pone.0108131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Farooque A, Afrin F, Adhikari JS, Dwarakanath BSR. Polarization of macrophages towards M1 phenotype by a combination of 2-deoxy-d-glucose and radiation: implications for tumor therapy. Immunobiology. 2016;221:269–281. doi: 10.1016/j.imbio.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 152.Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2, 3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. Journal of Experimental Medicine. 2013 doi: 10.1084/jem.20130066. jem. 20130066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Monjazeb AM, et al. Blocking indolamine-2, 3-dioxygenase rebound immune suppression boosts antitumor effects of radio-immunotherapy in murine models and spontaneous canine malignancies. Clinical Cancer Research. 2016;22:4328–4340. doi: 10.1158/1078-0432.CCR-15-3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zakharia Y, et al. Updates on phase1b/2 trial of the indoleamine 2, 3-dioxygenase pathway (IDO) inhibitor indoximod plus checkpoint inhibitors for the treatment of unresectable stage 3 or 4 melanoma. J Clin Oncol. 2016 [Google Scholar]
- 155.Shirota Y, Shirota H, Klinman DM. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. Journal of immunology. 2012;188:1592–1599. doi: 10.4049/jimmunol.1101304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kim YH, et al. In situ vaccination against mycosis fungoides by intratumoral injection of a TLR9 agonist combined with radiation: a phase 1/2 study. Blood. 2012;119:355–363. doi: 10.1182/blood-2011-05-355222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Monjazeb AM, et al. Combined radiotherapy and immunotherapy using CPG oligodeoxynucleotides and indolamine 2, 3 dioxygenase (IDO) blockade. J Immunother Cancer. 2013;1:P256. [Google Scholar]
- 158.Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. Journal of Pharmacology and Experimental Therapeutics. 2005;315:971–979. doi: 10.1124/jpet.105.084145. [DOI] [PubMed] [Google Scholar]
- 159.Cuneo KC, et al. SU11248 (sunitinib) sensitizes pancreatic cancer to the cytotoxic effects of ionizing radiation. Int J Rad Oncol Biol Phy. 2008;71:873–879. doi: 10.1016/j.ijrobp.2008.02.062. [DOI] [PubMed] [Google Scholar]
- 160.Armstrong A, et al. A phase 2 multimodality trial of docetaxel/prednisone with sunitinib followed by salvage radiation therapy in men with PSA recurrent prostate cancer after radical prostatectomy. Prostate cancer and prostatic diseases. 2016;19:100. doi: 10.1038/pcan.2015.59. [DOI] [PubMed] [Google Scholar]
- 161.Kasibhatla M, Steinberg P, Meyer J, Ernstoff MS, George DJ. Radiation therapy and sorafenib: clinical data and rationale for the combination in metastatic renal cell carcinoma. Clinical genitourinary cancer. 2007;5:291–294. doi: 10.3816/CGC.2007.n.007. [DOI] [PubMed] [Google Scholar]
- 162.Chen SW, et al. Phase 2 study of combined sorafenib and radiation therapy in patients with advanced hepatocellular carcinoma. Int J Rad Oncol Biol Phy. 2014;88:1041–1047. doi: 10.1016/j.ijrobp.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 163.Matar P, Rozados VR, Gervasoni SI, Scharovsky OG. Down regulation of T-cell-derived IL-10 production by low-dose cyclophosphamide treatment in tumor-bearing rats restores in vitro normal lymphoproliferative response. International immunopharmacology. 2001;1:307–319. doi: 10.1016/s1567-5769(00)00028-x. [DOI] [PubMed] [Google Scholar]
- 164.Kim BG, et al. Inhibition of interleukin-10 (IL-10) production from MOPC 315 tumor cells by IL-10 antisense oligodeoxynucleotides enhances cell-mediated immune responses. Cancer immunology, immunotherapy. 2000;49:433–440. doi: 10.1007/s002620000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Maier E, Duschl A, Horejs-Hoeck J. STAT6-dependent and -independent mechanisms in Th2 polarization. European journal of immunology. 2012;42:2827–2833. doi: 10.1002/eji.201242433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sheng J, Chen W, Zhu HJ. The immune suppressive function of transforming growth factor-β (TGF-β) in human diseases. Growth Factors. 2015;33:92–101. doi: 10.3109/08977194.2015.1010645. [DOI] [PubMed] [Google Scholar]
- 167.Grütz G. New insights into the molecular mechanism of interleukin-10-mediated immunosuppression. Journal of leukocyte biology. 2005;77:3–15. doi: 10.1189/jlb.0904484. [DOI] [PubMed] [Google Scholar]
- 168.O’Callaghan G, Houston A. Prostaglandin E2 and the EP receptors in malignancy: possible therapeutic targets? British Journal of Pharmacology. 2015;172:5239–5250. doi: 10.1111/bph.13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chen J, et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer cell. 2011;19:541–555. doi: 10.1016/j.ccr.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 171.Naylor MS, Stamp GW, Foulkes WD, Eccles D, Balkwill FR. Tumor necrosis factor and its receptors in human ovarian cancer. Potential role in disease progression. J Clin Invest. 1993;91:2194–2206. doi: 10.1172/JCI116446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Duyndam MCA, et al. Vascular Endothelial Growth Factor-165 Overexpression Stimulates Angiogenesis and Induces Cyst Formation and Macrophage Infiltration in Human Ovarian Cancer Xenografts. Am J Path. 2002;160:537–548. doi: 10.1016/s0002-9440(10)64873-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Presta M, Chiodelli P, Giacomini A, Rusnati M, Ronca R. Fibroblast growth factors (FGFs) in cancer: FGF traps as a new therapeutic approach. Pharmacology & Therapeutics. doi: 10.1016/j.pharmthera.2017.05.013. [DOI] [PubMed] [Google Scholar]
- 174.Hussain SF, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer research. 2007;67:9630–9636. doi: 10.1158/0008-5472.CAN-07-1243. [DOI] [PubMed] [Google Scholar]
- 175.Fujiwara Y, Takeya M, Komohara Y. A novel strategy for inducing the antitumor effects of triterpenoid compounds: blocking the protumoral functions of tumor-associated macrophages via STAT3 inhibition. BioMed research international. 2014;2014 doi: 10.1155/2014/348539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hugo W, et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell. 2015;162:1271–1285. doi: 10.1016/j.cell.2015.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kushner BH, et al. Phase I trial of a bivalent gangliosides vaccine in combination with β-glucan for high-risk neuroblastoma in second or later remission. Clinical cancer research. 2014;20:1375–1382. doi: 10.1158/1078-0432.CCR-13-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wang SC, Yu CF, Hong JH, Tsai CS, Chiang CS. Radiation therapy-induced tumor invasiveness is associated with SDF-1-regulated macrophage mobilization and vasculogenesis. PLoS One. 2013;8:e69182. doi: 10.1371/journal.pone.0069182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Winkler F, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer cell. 2004;6:553–563. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 180.Myers AL, Williams RF, Ng CY, Hartwich JE, Davidoff AM. Bevacizumab-induced tumor vessel remodeling in rhabdomyosarcoma xenografts increases the effectiveness of adjuvant ionizing radiation. Journal of pediatric surgery. 2010;45:1080–1085. doi: 10.1016/j.jpedsurg.2010.02.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dings RP, et al. Scheduling of radiation with angiogenesis inhibitors anginex and Avastin improves therapeutic outcome via vessel normalization. Clinical Cancer Research. 2007;13:3395–3402. doi: 10.1158/1078-0432.CCR-06-2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Demaria S, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:728–734. [PubMed] [Google Scholar]
- 183.Twyman-Saint Victor C, et al. Radiation and dual checkpoint blockade activates non-redundant immune mechanisms in cancer. Nature. 2015;520:373. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Golden EB, Demaria S, Schiff PB, Chachoua A, Formenti SC. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer immunology research. 2013;1:365–372. doi: 10.1158/2326-6066.CIR-13-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Postow MA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. New England Journal of Medicine. 2012;366:925–931. doi: 10.1056/NEJMoa1112824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Rosenberg SA, Mule J, Spiess PJ, Reichert CM, Schwarz SL. Regression of established pulmonary metastases and subcutaneous tumor mediated by the systemic administration of high-dose recombinant interleukin 2. Journal of Experimental Medicine. 1985;161:1169–1188. doi: 10.1084/jem.161.5.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Seung SK, et al. Phase 1 study of stereotactic body radiotherapy and interleukin-2—tumor and immunological responses. Science translational medicine. 2012;4:137ra174–137ra174. doi: 10.1126/scitranslmed.3003649. [DOI] [PubMed] [Google Scholar]