

ABSTRACT
Aggregatibacter species are commensal bacteria of human mucosal surfaces that are sometimes involved in serious invasive infections. During the investigation of strains cultured from various clinical specimens, we encountered a coherent group of 10 isolates that could not be allocated to any validly named species by phenotype, mass spectrometry, or partial 16S rRNA gene sequencing. Whole-genome sequencing revealed a phylogenetic cluster related to but separate from Aggregatibacter aphrophilus. The mean in silico DNA hybridization value for strains of the new cluster versus A. aphrophilus was 56% (range, 53.7 to 58.0%), whereas the average nucleotide identity was 94.4% (range, 93.9 to 94.8%). The new cluster exhibited aggregative properties typical of the genus Aggregatibacter. Key phenotypic tests for discrimination of the new cluster from validly named Aggregatibacter species are alanine-phenylalanine-proline arylamidase, N-acetylglucosamine, and β-galactosidase. The name Aggregatibacter kilianii is proposed, with PN_528 (CCUG 70536T or DSM 105094T) as the type strain.
KEYWORDS: average nucleotide identity, HACEK, Haemophilus, phylogeny, abscesses
INTRODUCTION
The bacterial genus Aggregatibacter accommodates species previously classified in the genera Actinobacillus and Haemophilus. The genus comprises three validly named species, namely, Aggregatibacter actinomycetemcomitans, Aggregatibacter aphrophilus, and Aggregatibacter segnis. The species are part of the human oral microbiota but can cause severe infections. The name of the genus designates a rod-shaped bacterium that aggregates with others (1).
Aggregatibacter species are included in the so-called HACEK (Haemophilus, Aggregatibacter, Cardiobacterium, Eikenella, and Kingella) group, a recognized but unusual cause of infective endocarditis. Although Aggregatibacter species share a propensity for aggregation that may include adherence to cardiac valve tissue, noticeable differences exist with respect to association with particular diseases. A. actinomycetemcomitans was originally coisolated with Actinomyces from actinomycotic lesions, and case reports confirmed this association (2, 3). However, A. actinomycetemcomitans has attracted attention because of its association with periodontitis (4, 5). In particular, a single serotype b clonal lineage with enhanced production of leukotoxin is linked with the aggressive form of periodontitis in adolescents from a limited geographical and ethnic host range (6). A. aphrophilus was originally cultured from a case of infective endocarditis, but later observations have linked the bacterium with infections of the central nervous system. In 1962, King and Tatum characterized invasive strains referred to the Centers for Disease Control and Prevention (CDC) (Atlanta, GA) during a 10-year period, and they noticed that 10 of 34 A. aphrophilus strains originated from brain abscesses, in marked contrast to none of 33 A. actinomycetemcomitans strains (7). A. aphrophilus generally accounts for 2 to 7% of cultivable bacteria from intracranial abscesses (8, 9), while high-throughput sequencing identifies the species more frequently (10).
A. segnis may be difficult or impossible to distinguish from Haemophilus parainfluenzae biotype V by phenotypic means (11). Although it is a well-known but rare cause of infective endocarditis (12), the true prevalence of this bacterium in human infections is probably underreported (13). During investigation of Aggregatibacter strains cultured from bloodstream infections and other human infections, we encountered a coherent and distinct group of strains that could not be allocated to any previously described species.
MATERIALS AND METHODS
Bacterial strains and culture.
Aggregatibacter sp. and other HACEK group bacteria cultured from blood samples and other human specimens were collected from all Danish departments of clinical microbiology (14); additional strains were obtained from the Institute of Medical Microbiology (Zurich, Switzerland) (see below). Type and reference strains were obtained from culture collections (American Type Culture Collection [ATCC], Culture Collection, University of Gothenburg [CCUG], and National Collection of Type Cultures [NCTC]). Table S1 in the supplemental material lists all clinical, reference, and type strains investigated in the study, their origin, the experiments for which they were used, and their GenBank accession numbers. Strains were routinely cultured on chocolate agar plates at 35°C, in a humidified atmosphere supplemented with 5% CO2.
MALDI-TOF MS and phenotypic analyses.
For matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS), thin layers of bacterial smears were deposited on target plates and overlaid with 1 μl of matrix solution (a saturated solution of α-cyano-4-hydroxycinnamic acid in 50% acetonitrile with 2.5% trifluoroacetic acid). Pretreatment with acetonitrile overlay was used in some experiments. Samples were air dried at room temperature to allow cocrystallization of the matrix and sample. Measurements were performed with a Microflex mass spectrometer (Bruker Daltonics, Bremen, Germany), using FlexControl software (version 3.0) and default settings recommended by the manufacturer, and spectra were compared with the Biotyper database (version 3.1). MALDI-TOF MS was also performed with a Vitek mass spectrometer using the SARAMIS database, according to the manufacturer's instructions (bioMérieux, Marcy l'Etoile, France). For generation of the distance matrix and dendrogram, Biotyper software was used (15).
Key phenotypic tests (including dependence on NAD, synthesis of porphyrin, production of ortho-nitrophenyl-β-galactoside [ONPG], and tryptophanase [indole production], urease, and ornithine decarboxylase activity) were performed according to accepted guidelines (16) and supplemented with biochemical results from the Vitek 2 NH identification card (bioMérieux). The latter card includes fermentation of N-acetylglucosamine and enzymatic testing of alanine-phenylalanine-proline arylamidase.
DNA sequencing, genome assembly, and analysis.
Identification by 16S rRNA gene sequencing of a 526-bp amplicon spanning variable regions V1 to V3 was performed as described previously (17, 18). For whole-genome sequencing (WGS), genomic DNA was extracted using the DNeasy blood and tissue kit (Qiagen, Hilden, Germany) and diluted to 0.30 ng/μl. DNA libraries were prepared from 1 ng of genomic DNA with a Janus NGS Express robot (PerkinElmer), using the Nextera XT DNA sample preparation kit in combination with the Nextera XT index kit v2, set D (Illumina, San Diego, CA, USA), according to the manufacturer's protocol. Quality control of the libraries was conducted by on-chip electrophoresis (high-sensitivity LabChip GX; Caliper, PerkinElmer) and by quantitative PCR (qPCR) (KAPA library quantification kit; Kapa Biosystems). Dual-indexed paired-end sequencing (2 by 150 bp), aiming at 200× coverage, was performed with an Illumina NextSeq 500 system using v2 chemistry with a medium flow cell (Illumina). Paired demultiplexed FASTQ files were generated using CASAVA software (Illumina), and initial quality control was performed using FastQC. Reads were assembled using the SPAdes genome assembler (version 3.9). Draft assemblies were used for quantitation of single-nucleotide polymorphisms (SNPs) using Parsnp analysis with recommended settings (19). In silico DNA hybridization was performed with the Genome-to-Genome Distance Calculator (version 2.1), using standard settings and the recommended identity/high-scoring segment pair length calculation (20). Average nucleotide identity (ANI) values were calculated using online tools (http://www.ezbiocloud.net/sw/oat) (21). Roary (22), a rapid, large-scale, prokaryote pan-genome analysis tool, was used with default settings for identification of core genes of the A. kilianii-A. aphrophilus cluster, as well as A. kilianii- and A. aphrophilus-specific genes. Roary creates clusters of genes that share amino acid sequence similarity and coverage above a given threshold and orders strains by the presence or absence of orthologs. To maximize the comparability of data, FASTA files for reference strains downloaded from GenBank (Table S1) were reannotated with Prokka (23), in parallel with study strains.
Autoaggregation assay.
Overnight cultures on chocolate agar were suspended in tubes containing brain heart infusion broth, to an optical density at 600 nm (OD600) of 0.8. Tubes were allowed to stand at room temperature, and OD600 values were measured after 0.5, 1, 2, 3, and 4 h.
Accession number(s).
The full-length 16S rRNA gene sequence of the type strain was deposited in GenBank under accession number MH160776. The GenBank accession number for the whole-genome sequence of the type strain is NRCQ00000000. This whole-genome shotgun sequencing project has been deposited in DDBJ/ENA/GenBank under accession numbers NRCJ00000000 to NRDG00000000. The accession numbers for individual study strains and type and reference strains used for comparison are listed in Table S1 in the supplemental material.
RESULTS
Delineation of study strains.
During characterization of Aggregatibacter strains cultured from various human infections, 7 isolates could not be identified to the species level by MALDI-TOF MS, using either the Bruker Microflex or the Vitek MS plus SARAMIS. The greatest similarities were obtained with deposited reference spectra for A. aphrophilus strains, but identification was unreliable, with log scores in the range of 1.46 to 1.75; single measurements could exceed the 1.7-log-score breakpoint and attain C genus-level identification (Bruker), but such results were not reproducible and acetonitrile overlay did not improve the scores. Three strains were subjected to identification by partial 16S rRNA gene sequencing of the V1 to V3 region; similarities in the range of 96 to 97% were obtained with the A. aphrophilus type strain and similarities of 94 to 95% were obtained with A. actinomycetemcomitans and A. segnis type strains, while similarities with other Pasteurellaceae type strains were below 94%. However, 99 to 100% similarity was observed with deposited sequences of Aggregatibacter strains cultured from various human infections that had been identified to the genus level by integrating conventional phenotypic methods and 16S rRNA gene sequence analysis (18). Three strains from Switzerland with deposited 16S rRNA sequences were included in the present study, and 10 strains with almost identical 16S rRNA gene sequences were subjected to further analysis. Four strains were cultured from sterile sites (blood, 2 strains; abdominal abscesses, 2 strains), and 6 strains were cultured from nonsterile sites (conjunctivitis, dacryocystitis, and paranasal sinuses, 5 strains; wound, 1 strain). Table S1 lists the geographic origin, year of sampling, and associated infection for the strains included in the study.
Comparison of study strains by MALDI-TOF MS and WGS (Parsnp calling).
The relationships of 26 Aggregatibacter study strains (including the A. actinomycetemcomitans, A. aphrophilus, and A. segnis type strains) plus 7 Haemophilus and 2 Pasteurella type strains, as revealed by MALDI-TOF MS, are shown in Fig. 1. Four, 4, and 4 clinical strains clustered with the A. actinomycetemcomitans, A. aphrophilus, and A. segnis type strains, respectively, while strain PN_491 (cultured from an animal bite) took a unique position. A group of 10 study strains, including the designated Aggregatibacter kilianii type strain (PN_528T), constituted a coherent and distinct cluster related to but separate from A. aphrophilus (Fig. 1, bottom). We generated reference spectra from independent measurements of strain PN_528T for inclusion in the local database, and all other isolates of the new species exhibited log scores of >2.0 with the designated type strain (data not shown).
FIG 1.
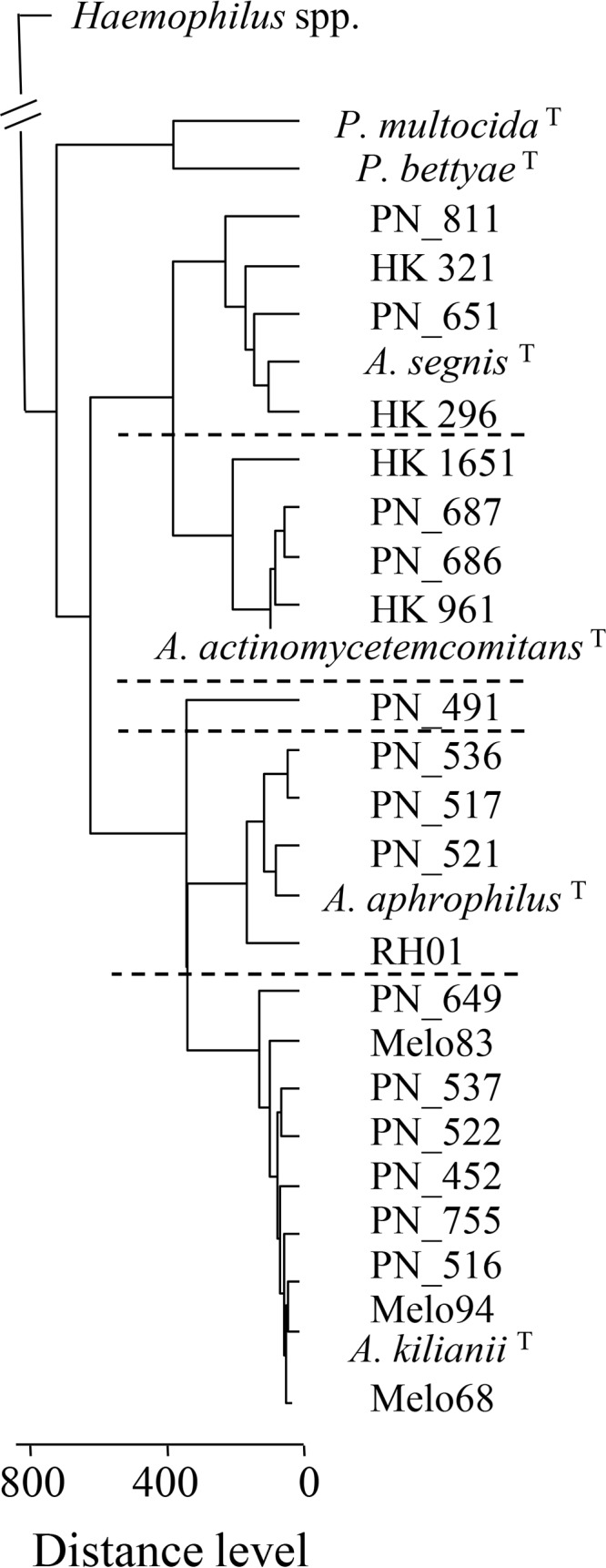
Relationships of 23 study strains plus selected Pasteurella and Haemophilus type strains (see Table S1 in the supplemental material), as revealed by MALDI-TOF MS. Dotted lines separate individual species of the genus Aggregatibacter (PN_491 is unclustered). Superscript T denotes type strains. The distance matrix and dendrogram were calculated with Biotyper software (Bruker); distance levels above 500 are uninformative (15).
The 10 strains of the new cluster plus 14 other study strains were subjected to WGS, and draft assemblies (23 to 121 contigs) were prepared. Figure 2 shows a phylogenetic reconstruction based on SNPs of 24 study strains plus 4 previously deposited type and reference strain sequences. Strain PN_491 remained unclustered, although it was more related to A. actinomycetemcomitans by SNP calling than by MALDI-TOF MS analysis. The existence of a coherent and distinct cluster related to but separate from A. aphrophilus was confirmed and was supported by a bootstrap value of 100%.
FIG 2.
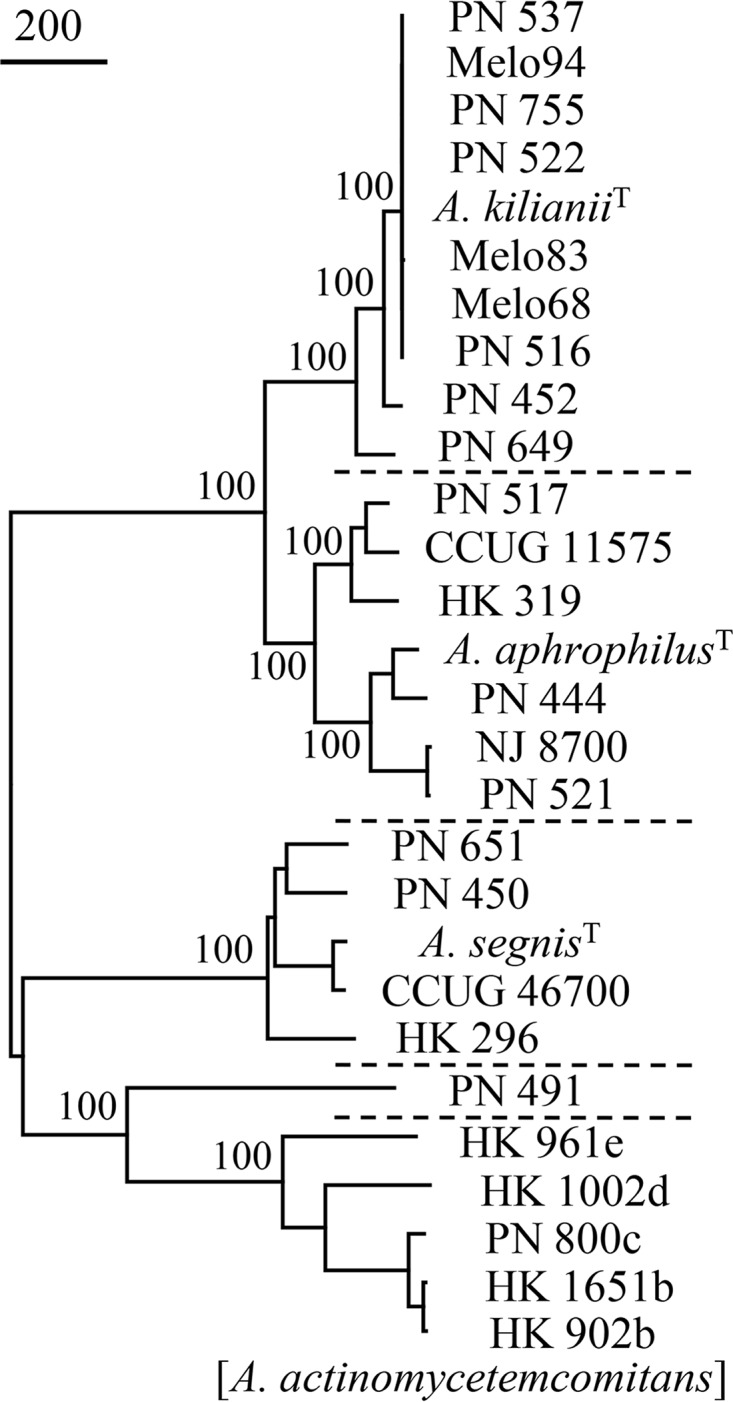
Relationships of 24 Aggregatibacter study strains plus 4 reference strains, as revealed by WGS (Parsnp analysis, neighbor-joining tree). Dotted lines separate individual species of the genus Aggregatibacter (PN_491 is unclustered). Superscript T denotes type strains; the sequence of the type strain of A. actinomycetemcomitans has not been deposited. A total of 3,155 positions with SNPs were included in the data set. Values to the left of the nodes represent percent bootstrap support for the nodes (500 replicates). The bar indicates 200 SNP substitutions.
Genome-to-genome distance and average nucleotide identity.
We performed in silico DNA hybridization by use of the Genome-to-Genome Distance Calculator 2.1, which estimates the DNA-DNA hybridization (DDH) values that would have resulted from classic hybridization experiments (20). Nine strains of the new cluster were highly similar and belonged to the same subspecies (DDH values of >80%), while the 10th strain (PN_649) was within the 70% species boundary. Interspecies in silico DDH values clearly separated the three validly named Aggregatibacter species, which demonstrated DNA-DNA binding values of less than 30% (Table 1). In silico DDH values for strains of the new cluster versus A. aphrophilus were 56.4 ± 1.5%, confirming that these phylogenetic groups belong to different species (24). Among indices of overall genome relatedness developed to replace the problematic DDH methods, ANI has been most widely used. ANI is calculated for two genome sequences by breaking the genome sequence of the query strain into 1,020-bp-long fragments. Then, nucleotide identity values for individual fragments of the query strain and the genome of the subject strain are calculated using the NCBI BLASTn program. The ANI is the mean of these nucleotide identity values; usually, only matches with at least 30% overall sequence identity are considered (25). ANI analysis confirmed that the 10 strains of the putative new species are highly related, with a mean ANI value of 99.06% (Table 1). ANI was also calculated between species; again, the new taxon was most closely related to A. aphrophilus, but the distance between strains of the two phylogenetic groups (mean ANI of 70 comparisons, 94.38% [range, 93.90 to 94.76%]) is below the 95% ANI threshold value that is considered the species boundary (26, 27).
TABLE 1.
DNA distances within and between Aggregatibacter species, based on whole-genome sequences of 23 study strains and 4 reference strains
| Speciesa | No. of comparisons | DDH (%)b |
ANI (%)c |
||||
|---|---|---|---|---|---|---|---|
| Mean | SD | Range | Mean | SD | Range | ||
| A. actinomycetemcomitans | 10 | 72.48 | 16.43 | 54.0–94.4 | 96.49 | 2.34 | 93.76–99.35 |
| A. aphrophilus | 21 | 71.16 | 7.12 | 65.2–94.8 | 96.71 | 0.84 | 96.01–99.37 |
| A. segnis | 10 | 63.31 | 7.93 | 59.0–85.4 | 95.41 | 1.10 | 94.53–98.34 |
| A. kilianii | 45 | 95.41 | 1.10 | 94.5–98.3 | 99.06 | 1.21 | 96.85–99.97 |
| A. kilianii vs A. actinomycetemcomitans | 50 | 24.07 | 0.11 | 23.9–24.3 | 81.51 | 0.06 | 81.23–81.98 |
| A. kilianii vs A. aphrophilus | 70 | 55.98 | 1.57 | 53.7–58.0 | 94.38 | 0.26 | 93.90–94.76 |
| A. kilianii vs A. segnis | 50 | 28.66 | 0.42 | 28.0–29.7 | 84.83 | 0.28 | 84.33–85.20 |
| A. segnis vs A. aphrophilus | 35 | 28.17 | 0.31 | 27.8–28.8 | 84.52 | 0.19 | 84.12–84.93 |
| A. segnis vs A. actinomycetemcomitans | 25 | 24.24 | 0.12 | 24.1–24.5 | 81.62 | 0.10 | 81.48–81.86 |
| A. aphrophilus vs A. actinomycetemcomitans | 35 | 24.31 | 0.33 | 23.8–24.8 | 81.55 | 0.26 | 81.12–81.98 |
Sequences of A. actinomycetemcomitans, A. aphrophilus, and A. segnis from GenBank plus locally generated genome sequences were selected to represent the diversity of individual species (strains and sequence accession numbers are listed in Table S1 in the supplemental material).
DNA-DNA hybridization values were calculated using the NCBI BLASTn program (20).
ANI values were calculated with the OrthoANI algorithm (21).
Comparative genomic analysis of A. aphrophilus and the new cluster.
To clarify differences between these closely related species, complete protein-coding gene sets of 10 strains of A. aphrophilus and 10 strains of the new cluster (designated Aggregatibacter kilianii) were generated by using Prokka (23) and compared with Roary (22). A total of 4,463 gene clusters were identified, and the results of the clustering procedure were visualized as a presence/absence matrix (Fig. 3). The core genome of the A. aphrophilus/A. kilianii cluster consisted of 1,187 genes present in all 20 strains, while 1,417 “soft core” genes were present in 19 of 20 strains. A total of 153 clusters with homologous genes that were present in all 10 of the A. aphrophilus genomes and absent in all 10 of the A. kilianii genomes were found; 94 of these clusters were annotated, while 59 coded for hypothetical proteins. A total of 188 clusters with homologous genes that were present in all 10 of the A. kilianii genomes and absent in all 10 of the A. aphrophilus genomes were found; 125 of these clusters were annotated, while 63 coded for hypothetical proteins. We performed a similar comparison of 20 strains of Haemophilus influenzae (including phylogenetic group II strains) and 10 strains of Haemophilus haemolyticus (including cryptic genospecies biotype IV strains) and identified 6,996 gene clusters (data not shown). The core genome consisted of 512 genes present in all 30 strains, while 654 soft core genes were present in 29 strains; 80 genes were present in all H. influenzae genomes and absent in all H. haemolyticus genomes, while 130 genes were present in all H. haemolyticus genomes and absent in all H. influenzae genomes. The data indicate greater diversity within and between species of the H. influenzae/H. haemolyticus cluster than that observed for the A. aphrophilus/A. kilianii cluster.
FIG 3.

Homologous gene cluster (core and accessory) presence/absence matrix. Fifteen study strains of A. kilianii and A. aphrophilus, plus 5 reference strains of A. aphrophilus, were included (Table S1). The neighbor-joining dendrogram shows the phylogenetic relationships of strains as evaluated by Parsnp analysis. In the matrix, genomes are shown as rows and homologous gene clusters are shown as columns. The presence of a gene cluster in a genome is indicated by blue. Core clusters found in all genomes are shown on the left of the matrix; arrows indicate bundles of gene clusters exclusive to A. kilianii (top) or A. aphrophilus (bottom).
Phenotypic characterization.
Table 2 shows phenotypic characteristics of importance for characterization of genus and differentiation of species. All species of Aggregatibacter are capable of synthesizing porphyrin (X factor not required), while synthesis of NadV was variable; A. segnis requires exogenously supplied NAD for growth, while this factor (designated V for vitamin-like) is not required for A. actinomycetemcomitans and A. kilianii sp. nov. A. aphrophilus encompasses both phenotypes, previously designated Haemophilus aphrophilus and Haemophilus paraphrophilus (1). A single strain of A. kilianii sp. nov. (PN_452) exhibited NAD dependence; the NAD-dependent phenotype could be ascribed to a single-nucleotide deletion in the nadV gene at positions 282 to 286, where a 6-nucleotide homopolymer was terminated after 5 deoxythymidine residues, resulting in a reading frameshift and a pseudogene of 1,388 nucleotides (GenBank accession number NRCQ00000000). Transformation to NAD-independent growth failed, using previously applied methods (1); the strain appeared to be incompetent for transformation (transformation efficiency below 10−8, the detection limit of the experimental setup). According to accepted criteria, ≥90% positive strains corresponds to a positive result for this characteristic, as specified for NadV synthesis by A. kilianii sp. nov. in Table 2; however, it is possible that examination of a larger number of strains may change the result for this characteristic to variable.
TABLE 2.
Differential phenotypic characteristics of Aggregatibacter species
| Characteristic | Findinga |
|||
|---|---|---|---|---|
| A. actinomycetemcomitans | A. aphrophilus | A. segnis | A kilianii sp. nov. | |
| Porphyrin synthesis (X factor not required) | + | + | + | + |
| NadV synthesis (V factor not required) | + | v | 0 | + |
| Catalase | + | 0 | v | 0 |
| Hemolysis | 0b | 0 | 0 | 0 |
| β-Galactosidase (ONPG) | 0 | + | v | + |
| Tryptophanase (indole) | 0 | 0 | 0 | 0 |
| Urease | 0 | 0 | 0 | 0 |
| Ornithine decarboxylase | 0 | 0 | 0 | 0 |
| Proline arylamidase | 0 | v | 0 | + |
| Alanine-phenylalanine-proline arylamidase | 0 | 0 | 0 | + |
| Phenylphosphonate | v | + | v | + |
| Acid from | ||||
| Maltotriose | v | v | v | 0 |
| N-Acetylglucosamine | v | 0 | 0 | + |
+, ≥90% of strains positive; v, 11 to 89% of strains positive; 0, ≤10% of strains positive.
Isolates with overexpression of leukotoxin may exhibit a zone of hemolysis (35).
All species of Aggregatibacter were negative in the three tests used for biotyping of H. influenzae and H. parainfluenzae (indole, urease, and ornithine decarboxylase), a combination that corresponds to H. parainfluenzae biotype V. A decisive phenotypic test for identification of A. kilianii sp. nov. is alanine-phenylalanine-proline arylamidase as tested by the Vitek NH card, which is positive for A. kilianii and negative for the three other Aggregatibacter species; A. kilianii can be separated from A. aphrophilus and A. segnis by testing for N-acetylglucosamine and from A. actinomycetemcomitans by testing for catalase and β-galactosidase (ONPG) (Table 2).
Aggregative properties were profiled by subjecting 10 strains of each of the four species to tests for autoaggregation, in which suspended cells at room temperature may form clumps and settle at the bottom of the tube, leaving a clear broth. Effective autoaggregation was apparent with all species of Aggregatibacter, although the property varied between strains (Fig. 4). Nonefficient aggregation and settling were not restricted to reference strains propagated for years but could be observed among recently cultured, invasive isolates. High levels of autoaggregation appeared more prevalent among strains of A. aphrophilus.
FIG 4.

Autoaggregation quantitated as sedimentation of bacterial cell suspensions at room temperature. Results are presented as the mean of three separate experiments; standard deviations for representative strains are shown. Ten strains of A. actinomycetemcomitans (A), A. aphrophilus (B), A. segnis (C), and A. kilianii (D) were assessed. Solid lines represent strains cultured from bloodstream infections and abdominal abscesses; dotted lines represent strains cultured from subgingival plaques, sputum, and superficial wounds (see Table S1 for the origins of strains).
DISCUSSION
There is no widely accepted concept of species for prokaryotes, and assignment of isolates to species is based on measures of phenotypic or genome similarity. Over the past 50 years, DNA-DNA hybridization has been the gold standard for bacterial species demarcation, providing a constant numerical threshold for the species boundary (“The phylogenetic definition of a species generally would include strains with approximately 70% or greater DNA-DNA relatedness” [28]). To circumvent the labor-intensive and error-prone nature of DDH experiments, there has been a continuous demand for an alternative genotypic standard. We used two different algorithms based on WGS, namely, Genome-to-Genome Distance Calculator 2.1 and OrthoANI, for evaluation of the putative new species. Both algorithms confirmed the existence of a new species, closely related to but separate from A. aphrophilus. However, analysis of reference strains from the other species of the genus Aggregatibacter underscored miscellaneous taxonomic challenges. A. actinomycetemcomitans strain HK_961 of serotype e failed to cluster sufficiently with the other strains of this species, both by in silico DDH (54.0 to 54.6%) and by ANI (93.76 to 93.98%). The strain belongs to the so-called clade e (29), which perhaps should be excluded from the species (30). An unexpectedly high level of divergence was observed among five strains of A. segnis. Only strain CCUG_46700 was related to the type strain of A. segnis in accordance with accepted species boundaries; the 3 other clinical strains exhibited in silico DDH values of 59.0 to 60.8% and ANI values of 94.53 to 95.0% versus the type strain. The species has received little attention and, prior to this study, only two genome sequences had been deposited in GenBank. Additional investigations are required to clarify whether this species is in need of a division into separate species.
Comparative genomic analysis is a powerful tool for examination of closely related species that may provide new testing schemes for their discrimination. Nonhemolytic strains of H. haemolyticus are exceedingly difficult to discriminate from the more pathogenic H. influenzae by phenotype (11, 17), but recent comparisons based on WGS have suggested a dual-target, PCR-based testing scheme that can be used for efficient discrimination (31). The ANI values for five deposited genomes of H. haemolyticus versus selected reference strains of H. influenzae were in the range 91.24 to 92.70% (data not shown). Thus, the distance between H. haemolyticus and H. influenzae exceeds the distance between A. kilianii sp. nov. and A. aphrophilus. Genes unique to A. kilianii or unique to A. aphrophilus (Fig. 3) can be tested by reverse transcription-PCR assays and evaluated for discriminative potential.
The newly discovered phylogenetic group belongs to genus Aggregatibacter by genomic sequence comparison and by phenotype. It cannot be assigned to any named species by phenotype, mass spectrometry, 16S rRNA gene sequence comparison, or WGS. Strains with almost identical 16S rRNA gene sequences have occasionally been cultured from various clinical samples (GenBank accession numbers AF227858, AY538693, and KC866198) (18, 32). Microbiota sequence investigations have documented uncultured bacteria of the putative new species in throat and skin samples (33, 34). The phylogenetic group has aggregative properties in common with other species of Aggregatibacter. We propose to name the new species Aggregatibacter kilianii. Validation as a species with specific entries in culture collections, public sequence databases, and automated identification systems will facilitate the recognition, reporting, and knowledge of A. kilianii in human infections.
Description of Aggregatibacter kilianii.
Aggregatibacter kilianii (kil.i.an′i.i. N.L. gen. masc. n. kilianii of Kilian, named in honor of Mogens Kilian for his substantial contributions to Haemophilus and Aggregatibacter research). Gram-negative, nonmotile, facultatively anaerobic, short, regular rods (0.5 by 1.5 to 1.7 mm), with occasional filamentous forms. Colonies on chocolate agar incubated in air supplemented with 5 to 10% extra CO2 are highly convex, granular, yellowish, and opaque and reach a diameter of 1.0 to 1.5 mm within 24 h. When plates are incubated without extra CO2, the growth characteristically shows very small colonies interspersed with a few larger colonies. Growth in broth may be granular, with heavy sediment on the bottom of the tube. Porphyrins are synthesized from δ-aminolaevulinic acid (X factor is not required), and nicotinamide mononucleotide is synthesized from nicotinamide and 5-phosphoribosyl pyrophosphate (V factor is not required). Results are negative in the three tests used for biotyping of H. influenzae and H. parainfluenzae (indole, urease, and ornithine decarboxylase).
Acid is produced from glucose, while mannose, galactose, maltotriose, ribose, and xylose are not fermented. Variable fermentation is observed with maltose and sucrose. H2O2 is not decomposed; ONPG is hydrolyzed. Key tests for discrimination between Aggregatibacter kilianii and the other species of genus Aggregatibacter are alanine-phenylalanine-proline arylamidase, N-acetylglucosamine, catalase, and β-galactosidase.
A. kilianii is a commensal of the upper respiratory tract of humans. It is occasionally involved in human infections and has been isolated from conjunctivitis, wounds, abdominal abscesses, and blood. The type strain PN_528 (CCUG 70536T or DSM 105094T) was isolated from an abdominal abscess in Denmark in 2009. The G+C content of the DNA of the type strain is 42.9 mol%.
Supplementary Material
ACKNOWLEDGMENTS
The members of the Danish HACEK Study Group (Ming Chen, Sønderborg Hospital; Jens J. Christensen, Slagelse Hospital; Esad Dzajic, Esbjerg Hospital; Kurt Fuursted and Marianne Voldstedlund, State Serum Institute, Copenhagen; Gitte Hartmeyer, Odense University Hospital; Jenny D. Knudsen, Hvidovre Hospital; Claus Moser, Rigshospitalet; Bente Olesen, Herlev and Gentofte Hospital; Flemming Rosenvinge, Vejle Hospital; Henrik C. Schønheyder, Aalborg University Hospital) are gratefully acknowledged for the culture, storage, and donation of strains. Anders Jensen, Department of Biomedicine, Aarhus University (Aarhus, Denmark), is thanked for probing of 16S rRNA gene sequences at the Sequence Read Archive.
This research received no specific grants from any funding agency in the public, commercial, or not-for-profit sectors.
M.M., L.L., and N.N.-L. conceived and planned the study, R.Z. and M.L. identified cryptic strains by 16S rRNA gene sequencing, M.M. and R.Z. performed the phenotypic characterization, P.V. and N.N.-L. performed WGS analysis, and N.N.-L. analyzed the data and wrote the first draft of the manuscript, which was revised critically for important intellectual content by M.M. and L.L. All authors approved the final version of the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JCM.00053-18.
REFERENCES
- 1.Nørskov-Lauritsen N, Kilian M. 2006. Reclassification of Actinobacillus actinomycetemcomitans, Haemophilus aphrophilus, Haemophilus paraphrophilus and Haemophilus segnis as Aggregatibacter actinomycetemcomitans gen. nov., comb. nov., Aggregatibacter aphrophilus comb. nov. and Aggregatibacter segnis comb. nov., and emended description of Aggregatibacter aphrophilus to include V factor-dependent and V factor-independent isolates. Int J Syst Evol Microbiol 56:2135–2146. doi: 10.1099/ijs.0.64207-0. [DOI] [PubMed] [Google Scholar]
- 2.Clarridge JE, Zhang Q. 2002. Genotypic diversity of clinical Actinomyces species: phenotype, source, and disease correlation among genospecies. J Clin Microbiol 40:3442–3448. doi: 10.1128/JCM.40.9.3442-3448.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moskowitz SM, Shailam R, Mark EJ. 2015. Case records of the Massachusetts General Hospital: case 25-2015: an 8-year-old girl with a chest-wall mass and a pleural effusion. N Engl J Med 373:657–667. doi: 10.1056/NEJMcpc1400836. [DOI] [PubMed] [Google Scholar]
- 4.Fine DH, Markowitz K, Furgang D, Fairlie K, Ferrandiz J, Nasri C, McKiernan M, Gunsolley J. 2007. Aggregatibacter actinomycetemcomitans and its relationship to initiation of localized aggressive periodontitis: longitudinal cohort study of initially healthy adolescents. J Clin Microbiol 45:3859–3869. doi: 10.1128/JCM.00653-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henderson B, Ward JM, Ready D. 2010. Aggregatibacter (Actinobacillus) actinomycetemcomitans: a triple A* periodontopathogen? Periodontol 2000 54:78–105. doi: 10.1111/j.1600-0757.2009.00331.x. [DOI] [PubMed] [Google Scholar]
- 6.Haubek D, Ennibi OK, Poulsen K, Vaeth M, Poulsen S, Kilian M. 2008. Risk of aggressive periodontitis in adolescent carriers of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans in Morocco: a prospective longitudinal cohort study. Lancet 371:237–242. doi: 10.1016/S0140-6736(08)60135-X. [DOI] [PubMed] [Google Scholar]
- 7.King EO, Tatum HW. 1962. Actinobacillus actinomycetemcomitans and Haemophilus aphrophilus. J Infect Dis 111:85–94. doi: 10.1093/infdis/111.2.85. [DOI] [PubMed] [Google Scholar]
- 8.Roche M, Humphreys H, Smyth E, Phillips J, Cunney R, McNamara E, O'Brien D, McArdle O. 2003. A twelve-year review of central nervous system bacterial abscesses; presentation and aetiology. Clin Microbiol Infect 9:803–809. doi: 10.1046/j.1469-0691.2003.00651.x. [DOI] [PubMed] [Google Scholar]
- 9.Al Masalma M, Lonjon M, Richet H, Dufour H, Roche PH, Drancourt M, Raoult D, Fournier PE. 2012. Metagenomic analysis of brain abscesses identifies specific bacterial associations. Clin Infect Dis 54:202–210. doi: 10.1093/cid/cir797. [DOI] [PubMed] [Google Scholar]
- 10.Kommedal Ø, Wilhelmsen MT, Skrede S, Meisal R, Jakovljev A, Gautad P, Hermansen NO, Vik-Mo E, Solheim O, Ambur OH, Sæbø Ø, Høstmælingen CT, Helland C. 2014. Massive parallel sequencing provides new perspectives on bacterial brain abscesses. J Clin Microbiol 52:1990–1997. doi: 10.1128/JCM.00346-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nørskov-Lauritsen N. 2014. Classification, identification, and clinical significance of Haemophilus and Aggregatibacter species with host specificity for humans. Clin Microbiol Rev 27:214–240. doi: 10.1128/CMR.00103-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chambers ST, Murdoch D, Morris A, Holland D, Pappas P, Almela M, Fernandez-Hidalgo N, Almirante B, Bouza E, Forno D, del Rio A, Hannan MM, Harkness J, Kanafani ZA, Lalani T, Lang S, Raymond N, Read K, Vinogradova T, Woods CW, Wray D, Corey GR, Chu VH. 2013. HACEK infective endocarditis: characteristics and outcomes from a large, multi-national cohort. PLoS One 8:e63181. doi: 10.1371/journal.pone.0063181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lau SK, Woo PC, Mok MY, Teng JL, Tam VK, Chan KK, Yuen KY. 2004. Characterization of Haemophilus segnis, an important cause of bacteremia, by 16S rRNA gene sequencing. J Clin Microbiol 42:877–880. doi: 10.1128/JCM.42.2.877-880.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lützen L, Olesen B, Voldstedlund M, Christensen JJ, Moser C, Knudsen JD, Fuursted K, Hartmeyer GN, Chen M, Søndergaard TS, Rosenvinge FS, Dzajic E, Schønheyder HC, Nørskov-Lauritsen N. 2018. Incidence of HACEK bacteraemia in Denmark: a 6-year population-based study. Int J Infect Dis 68:83–87. doi: 10.1016/j.ijid.2018.01.025. [DOI] [PubMed] [Google Scholar]
- 15.Sauer S, Freiwald A, Maier T, Kube M, Reinhardt R, Kostrzewa M, Geider K. 2008. Classification and identification of bacteria by mass spectrometry and computational analysis. PLoS One 3:e2843. doi: 10.1371/journal.pone.0002843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Versalovic J, Carroll KC, Funke G, Jorgensen JH, Landry ML, Warnock DW (ed). 2011. Manual of clinical microbiology, 10th ed ASM Press, Washington, DC. [Google Scholar]
- 17.Nørskov-Lauritsen N, Bruun B, Andersen C, Kilian M. 2012. Identification of haemolytic Haemophilus species isolated from human clinical specimens and description of Haemophilus sputorum sp. nov. Int J Med Microbiol 302:78–83. doi: 10.1016/j.ijmm.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 18.de Melo Oliveira MG, Abels S, Zbinden R, Bloemberg GV, Zbinden A. 2013. Accurate identification of fastidious Gram-negative rods: integration of both conventional phenotypic methods and 16S rRNA gene analysis. BMC Microbiol 13:162. doi: 10.1186/1471-2180-13-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Treangen TJ, Ondov BD, Koren S, Phillippy AM. 2014. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol 15:524. doi: 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. 2013. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee I, Kim YO, Park SC, Chun J. 2016. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol 66:1100–1103. doi: 10.1099/ijsem.0.000760. [DOI] [PubMed] [Google Scholar]
- 22.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, Fookes M, Falush D, Keane JA, Parkhill J. 2015. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 24.Gevers D, Cohan FM, Lawrence JG, Spratt BG, Coenye T, Feil EJ, Stackebrandt E, Van de Peer Y, Vandamme P, Thompson FL, Swings J. 2005. Opinion: re-evaluating prokaryotic species. Nat Rev Microbiol 3:733–739. doi: 10.1038/nrmicro1236. [DOI] [PubMed] [Google Scholar]
- 25.Chun J, Rainey FA. 2014. Integrating genomics into the taxonomy and systematics of the Bacteria and Archaea. Int J Syst Evol Microbiol 64:316–324. doi: 10.1099/ijs.0.054171-0. [DOI] [PubMed] [Google Scholar]
- 26.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. 2007. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 27.Richter M, Rosselló-Móra R. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A 106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wayne L, Brenner DJ, Colwell RR, Grimont PAD, Kandler O, Krichevsky MI, Moore LH, Moore WEC, Murray RGE, Stackebrandt E, Starr MP, Truper HG. 1987. Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Int J Syst Bacteriol 37:463–464. doi: 10.1099/00207713-37-4-463. [DOI] [Google Scholar]
- 29.Kittichotirat W, Bumgarner RE, Chen C. 2016. Evolutionary divergence of Aggregatibacter actinomycetemcomitans. J Dent Res 95:94–101. doi: 10.1177/0022034515608163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jorth P, Whiteley M. 2012. An evolutionary link between natural transformation and CRISPR adaptive immunity. mBio 3:e00309-12. doi: 10.1128/mBio.00309-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu F, Rishishwar L, Sivadas A, Mitchell GJ, Jordan IK, Murphy TF, Gilsdorf JR, Mayer LW, Wang X. 2016. Comparative genomic analysis of Haemophilus haemolyticus and nontypeable Haemophilus influenzae and a new testing scheme for their discrimination. J Clin Microbiol 54:3010–3017. doi: 10.1128/JCM.01511-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drancourt M, Bollet C, Carlioz A, Martelin R, Gayral JP, Raoult D. 2000. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol 38:3623–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, NISC Comparative Sequence Program, Murray PR, Turner ML, Segre JA. 2012. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 22:850–859. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, Gamonal J, Diaz PI. 2013. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J 7:1016–1025. doi: 10.1038/ismej.2012.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haubek D, Dirienzo JM, Tinoco EM, Westergaard J, Lopez NJ, Chung CP, Poulsen K, Kilian M. 1997. Racial tropism of a highly toxic clone of Actinobacillus actinomycetemcomitans associated with juvenile periodontitis. J Clin Microbiol 35:3037–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.