

ABSTRACT
Helicobacter pylori is an organism known to colonize the normal human stomach. Previous studies have shown that the bacterium does this by elevating its periplasmic pH via the hydrolysis of urea. However, the value of the periplasmic pH was calculated indirectly from the proton motive force equation. To measure the periplasmic pH directly in H. pylori, we fused enhanced green fluorescent protein (EGFP) to the predicted twin-arginine signal peptides of HydA and KapA from H. pylori and TorA from Escherichia coli. The fusion proteins were expressed in the H. pylori genome under the control of the cagA promoter. Confocal microscopic and cell fractionation/immunoblotting analyses detected TorA-EGFP in the periplasm and KapA-EGFP in both the periplasm and cytoplasm, while the mature form of HydA-EGFP was seen at low levels in the periplasm, with major cytoplasmic retention of the precursor form. With H. pylori expressing TorA-EGFP, we established a system to directly measure periplasmic pH based on the pH-sensitive fluorimetry of EGFP. These measurements demonstrated that the addition of 5 mM urea has little effect on the periplasmic pH at a medium pH higher than pH 6.5 but rapidly increases the periplasmic pH to pH 6.1 at an acidic medium pH (pH 5.0), corresponding to the opening of the proton-gated channel, UreI, and confirming the basis of gastric colonization. Measurements of the periplasmic pH in an HP0244 (FlgS)-deficient mutant of H. pylori expressing TorA-EGFP revealed a significant loss of the urea-dependent increase in the periplasmic pH at an acidic medium pH, providing additional evidence that FlgS is responsible for recruitment of urease to the inner membrane in association with UreI.
IMPORTANCE Helicobacter pylori has been identified as the major cause of chronic superficial gastritis and peptic ulcer disease. In addition, persistent infection with H. pylori, which, if untreated, lasts for the lifetime of an infected individual, predisposes one to gastric malignancies, such as adenocarcinoma and mucosa-associated lymphoid tissue (MALT) lymphoma. A unique feature of the neutralophilic bacterium H. pylori is its ability to survive in the extremely acidic environment of the stomach through its acid acclimation mechanism. The presented results on measurements of periplasmic pH in H. pylori based on fluorimetry of fully active green fluorescent protein fusion proteins exported with the twin-arginine translocase system provide a reliable and rapid tool for the investigation of acid acclimation in H. pylori.
KEYWORDS: EGFP fluorimetry, Helicobacter pylori, pH measurement, periplasmic pH
INTRODUCTION
Helicobacter pylori, the major causative agent of peptic ulcer disease, mucosa-associated lymphoid tissue (MALT) lymphoma, and gastric cancer, is a bacterium known to colonize the normal acid-secreting human stomach (1). Being a neutralophile, H. pylori has evolved a unique strategy to combat gastric acidity, known as acid acclimation, which is the ability to buffer its periplasm to pH ∼6.1 in strongly acidic media (2). By buffering the periplasm to near neutrality, the organism is able to effectively maintain its cytoplasmic pH within a range compatible with not only survival but also bacterial replication. The mechanism of acid acclimation relies on the highly expressed neutral-pH-optimum cytoplasmic urease (3), a proton-gated urea channel (UreI) (4–6), and a pair of carbonic anhydrases, one expressed in the cytoplasm and one expressed in the periplasm (7, 8). With acidification, UreI is activated, allowing urea to move into the cytoplasm, where it is hydrolyzed by UreI-associated urease at the membrane. Urea hydrolysis produces 2NH3 and H2CO3, and subsequently, H2CO3 is converted to CO2 by cytoplasmic β-carbonic anhydrase (β-CA). Since it is a gas, CO2 exits rapidly into the periplasm. Protons entering the cytoplasm are neutralized by NH3, forming NH4+. In addition, NH3 effluxes into the periplasm, neutralizing entering protons, and CO2 is converted to HCO3− by a periplasmic membrane-anchored α-carbonic anhydrase. Hence, NH3 in the periplasm consumes both the entering protons and the protons produced by the action of α-carbonic anhydrase on CO2 and HCO3− and buffers the periplasmic space to near neutrality.
H. pylori has at least 2 two-component system histidine kinases that sense the pH changes in the periplasm and cytoplasm and regulate the acid-induced transcription of genes responsible for acid acclimation. The two-component system HP0165-HP0166 (ArsSR) provides one of the signaling pathways to regulate the pH-responsive transcriptional control of the urease gene cluster and of other genes involved in acid acclimation (9–13). The sensor histidine kinase HP0165 (ArsS) has two transmembrane domains, putting its histidine-rich input domain in the periplasm, where the pH changes can be sensed. The other histidine kinase that is required for pH homoeostasis, HP0244 (FlgS), appears to be the only sensor protein that has no transmembrane domain in H. pylori, indicating its location in cytoplasm and a possible role in responding to changes in the cytoplasmic pH. The response regulator for the acid response is yet to be identified, although FlgS belongs to the FlgRS two-component system, which regulates flagellar gene expression via HP0703 (FlgR), which is not implicated in the response to acidity. FlgS regulates several pH acclimation genes overlapping ArsS at extreme acidity (14, 15).
At acidic pH, membrane assembly of active urease takes place for the recruitment of urease to the inner membrane along with the nickel insertion proteins, which allows the activation of urease at the membrane in the vicinity of UreI (16, 17). The membrane association of urease activity provides localized increases in levels of NH3, NH4+, and H2CO3. Our previous studies demonstrated that the cytoplasmic histidine kinase FlgS is required for the pH-regulated membrane assembly of urease with UreI and its metabolites CO2, NH3, and NH4+ (14). The assembly of a pH-regulatory complex of active urease with UreI provides an advantage for periplasmic buffering because of the immediate access of urea to the urease.
The measurement of cytoplasmic and periplasmic pH is an important parameter for further studies of H. pylori acid acclimation. There are various techniques available to measure intracellular pH in bacteria, including microelectrodes (18), radiolabeled membrane-permeant probes (19), nuclear magnetic resonance (NMR) spectroscopy (20), and pH-sensitive fluorescent probes (21, 22). The measurement of cytoplasmic pH in H. pylori has been facilitated by the use of the trapped fluorescent indicator method with 2′,7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein (BCECF) (23–25). Measurement of the cytoplasmic pH and membrane potential using fluorescent probes under conditions of fixed medium acidity (24) between pH 3.0 and 6.0 in the presence of urea has provided critical information in deriving the mechanism of acid acclimation. Using the proton motive force (PMF) equation (PMF = −61ΔpH + Δψ, where ΔpH and Δψ are the differences in pH and membrane potential across the inner membrane separating the cytoplasm from the periplasm, respectively) (26), the periplasmic pH was calculated to be ∼6.1 (24). Since the periplasmic pH was calculated rather than directly measured, we sought to develop a method to directly measure the periplasmic pH.
Green fluorescent proteins (GFPs), as targeted pH indicators, have unique advantages over other pH-sensitive probes because they provide the ability to measure pH at specific intracellular sites with little background signal and no indicator leakage and without the toxicities associated with chemical indicators and invasive loading procedures (27). As a heterologous protein, the expression of GFP in the cytoplasm of Escherichia coli was first demonstrated in 1994 (28). However, the periplasmic expression of GFP fusion proteins was unsuccessful with Sec-targeting signal peptides, as functional GFP could not be detected in the periplasm (29). The inability to export functional GFP into the periplasm with the Sec system is due to the misfolding and improper chromophore formation of GFP in the periplasm (30), since the Sec system can transport only unfolded, nascent proteins that fold after they cross the membrane (31). This problem was solved by using the signal sequence of the twin-arginine translocation (Tat) pathway (TorA-GFP), which can export folded proteins into periplasm (32).
Recent studies have characterized the Tat system in H. pylori (33). Analysis of H. pylori genome sequences revealed the presence of single copies of tatA, tatB, and tatC, which are required for a fully functional Tat system (34, 35), sharing relatively strong similarity with the well-characterized E. coli Tat system. However, H. pylori has a different genomic organization and is missing the tatE ortholog. While the Tat system appears to be essential in H. pylori (33), searching the substrate proteins of the Tat system in H. pylori based on the presence of the twin-arginine (RR) conserved (S/T)RRXFLK motif in their signal sequence with Tat prediction programs (36–38) revealed that only four H. pylori proteins appear to be exported by the Tat system: the hydrogenase small-subunit protein (HydA), the catalase-associated protein (KapA), the biotin sulfoxide reductase (BisC), and the ubiquinol cytochrome oxidoreductase Rieske protein (FbcF) (33).
The aim of this study was to establish a system that can rapidly and reliably measure the periplasmic pH in H. pylori based on pH-sensitive fluorimetry of enhanced GFP (EGFP) expressed in the periplasmic compartment. Here, we report that with the TorA-RR signal peptide, EGFP retains proper folding and pH sensitivity and is targeted to the periplasm of H. pylori. Our initial experiments with this system have confirmed the effects of urea on the acid response in live H. pylori bacteria by directly measuring periplasmic pH. By integrating the PcagA-TorA-EGFP construct into an HP0244 (FlgS)-deficient mutant, we were able to measure the periplasmic pH after the addition of 5 mM urea to the medium at pH 5.0 and found a significant loss of urea-dependent periplasmic pH restoration in the FlgS-deficient mutant. The pH increased from 4.8 to 5.1, in contrast to the elevation to pH 6.1 found in the wild-type strain, providing additional evidence that FlgS is responsible for the recruitment of urease to the inner membrane in association with the urea channel, UreI. The ability to directly measure both cytoplasmic and periplasmic pH expands the experimental approach to pH regulation in H. pylori.
RESULTS
EGFP expression in H. pylori with different twin-arginine signal peptides.
We constructed H. pylori strains for the expression of EGFP to measure the periplasmic pH (Fig. 1). For this purpose, we used the RR signal peptides of the hydrogenase small subunit (HydA) and the catalase-associated protein (KapA) from H. pylori (33) as well as the trimethylamine N-oxide (TMAO) reductase (TorA) from E. coli (32), which are known to be exported by the twin-arginine translocation (Tat) pathway in E. coli (39). DNA fragments coding for these signal peptides were respectively fused in frame with the coding region of the EGFP gene and integrated into the H. pylori genome such that the expression of the Tat signal peptide tagged with EGFP is driven by the cagA promoter (PcagA) (Fig. 1). The Tat signal peptide-EGFP-coding sequence was followed by a chloramphenicol resistance cassette.
FIG 1.

Construction of H. pylori G27 strains expressing Tat-EGFP. The coding sequences for Tat signal peptide (SP)-EGFP fusion proteins (HydA-Tat-EGFP, KapA-Tat-EGFP, KapA-ΔTat-EGFP, and TorA-Tat-EGFP) followed by a chloramphenicol resistance cassette were integrated into the H. pylori G27 genome downstream of the cagA promoter (PcagA) via allelic-exchange recombination. The signal peptide sequences with the twin-arginine motif [the two arginines are shown in boldface type, and the consensus sequence (S/T)RRXFLK is underlined] for HydA and KapA from H. pylori were identified by the PRED-TAT, TATFIND, and TatP prediction programs (33), and the signal sequence for TorA from E. coli was used in a study reported previously (32). In the KapA-ΔTat-EGFP signal peptide, the twin-arginine motif was deleted.
To identify the location of EGFP and to test whether this protein is active, H. pylori strains containing the Tat signal peptide tagged with EGFP (G27/PcagA-HydA-EGFP, G27/PcagA-KapA-EGFP, and G27/PcagA-TorA-EGFP) were examined by using confocal microscopy (Fig. 2). A very weak fluorescence signal was detected in the bacteria with HydA-EGFP (Fig. 2A); however, it appeared to localize in the periplasm. H. pylori strains expressing KapA-EGFP exhibit diffuse fluorescence extending throughout most of the interior (Fig. 2B), showing that the functional form of EGFP is located mainly in the cytoplasm (or located evenly in both the periplasm and cytoplasm). To confirm the location of KapA-EGFP, bacteria expressing KapA-ΔTat-EGFP (with the twin-arginine motif deleted in the KapA signal sequence) were used as a control, and the fluorescence location appeared to be like that of KapA-EGFP (Fig. 2C). The bacteria expressing TorA-EGFP displayed a prominent peripheral fluorescence signal virtually entirely as halos in the outer regions of the bacteria, which corresponds to the periplasm, with very little fluorescence being found in the interior region (Fig. 2D). These data strongly indicate that H. pylori with TorA-EGFP properly transported fully folded and active EGFP to the periplasm almost exclusively.
FIG 2.

Confocal microscopy of H. pylori expressing HydA-EGFP (A), KapA-EGFP (B), KapA-ΔTat-EGFP (C), and TorA-EGFP (D). H. pylori strains were analyzed by confocal microscopy with excitation at 488 nm and emission at 500 to 545 nm.
To further investigate the export of EGFP in H. pylori by the Tat pathway with different signal peptides, the localization and relative quantities of EGFP were analyzed by cell fractionation and immunoblotting (Fig. 3). In the bacteria expressing HydA-EGFP, two EGFP forms, of ∼35 kDa (HydA-EGFP precursor) and ∼26 kDa (mature-size EGFP), were detected. While significant amounts of HydA-EGFP are located in the cytoplasmic fraction (C) in its precursor form, some HydA-EGFP was transported across membranes to the periplasmic compartment (P), and only a portion was processed to the mature size, which became fully folded and active EGFP. This is consistent with its weak fluorescence location in the periplasm found by confocal microscopy (Fig. 2A). In bacteria expressing KapA-EGFP, only mature-sized (∼27-kDa) proteins were detected, and they were located almost equally in the periplasm and cytoplasm fractions. However, when the twin-arginine motif was deleted from the KapA-Tat signal peptide (with the bacteria expressing KapA-ΔTat-EGFP), the clear majority of the EGFP was found in the cytoplasmic fraction. These results suggest that although confocal images will not be able to clearly identify its periplasm location (Fig. 2B and C), H. pylori strains with KapA-EGFP export EGFP into the periplasm, although this transport is inefficient under these experimental conditions. In bacteria expressing TorA-EGFP, only mature-sized (∼27-kDa) EGFP was detected, with a much greater abundance being found in the periplasmic fraction, which confirmed the findings with confocal microscopy (Fig. 2D).
FIG 3.
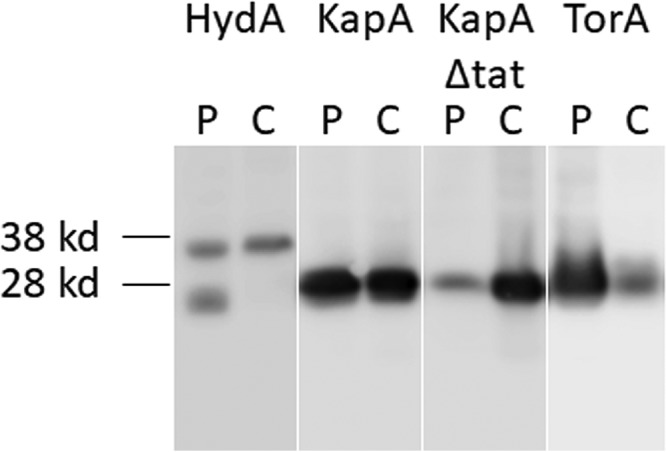
Export of EGFP by the Tat pathway with different RR signal peptides in H. pylori. The expression and localization of EGFP fused with different RR signal peptides were analyzed for H. pylori strains (G27/PcagA-HydA-EGFP, G27/PcagA-KapA-EGFP, G27/PcagA-KapA-Δtat-EGFP, and G27/PcagA-TorA-EGFP), indicated as HydA, KapA, KapA-Δtat, and TorA. Cells were fractionated to yield cytoplasmic (C) and periplasmic (P) samples and immunoblotted by using an antibody to GFP. Mobilities of molecular mass markers for 38 and 28 kDa are indicated.
Standard curves for the measurement of pH with KapA-ΔTat-EGFP and TorA-EGFP in live H. pylori bacteria.
To correlate the fluorescence intensity with intracellular pH and generate standard curves, the fluorescence signals from H. pylori expressing KapA-ΔTat-EGFP (Fig. 4A), KapA-EGFP (Fig. 4B), and TorA-EGFP (Fig. 4C) at an excitation wavelength 488 nm were recorded in the presence of 10 μM TCS (3,3′,4′,5-tetrachloro-salicylamide), which collapses the transmembrane proton gradient and equalizes the cytoplasmic pH to the external pH (40). The regression equations generated from these standard curves (pH 5.0 to pH 8.0) were used to convert signal intensities to pH units to determine the intracellular pH values during all the pH-measuring experiments. For H. pylori expressing KapA-ΔTat-EGFP, the curve of the fluorescence signal as a function of external pH without TCS showed a smaller slope (52,705) than the one in the presence of TCS (164,214) (Fig. 5A). This is consistent with the detection of KapA-ΔTat-EGFP in the cytoplasm by confocal microscopy combined with immunoblotting (Fig. 2C and 3) and again suggests that the active form of KapA-ΔTat-EGFP is located mainly in the cytoplasm of H. pylori. On the other hand, for H. pylori expressing TorA-EGFP, both curves (with and without TCS) are a pair of very close parallel lines with very little difference (within 1 standard deviation) (Fig. 5B), consistent with the detection of TorA-EGFP in the periplasm by confocal microscopy and immunoblotting (Fig. 2D and 3) and with previous data demonstrating that the periplasmic pH is equal or similar to the medium pH (41). For the subsequent pH measurement studies, standard curves were prepared for each individual strain preparation, and the regression equations were generated with the standard curves in the presence of TCS for cytoplasmic pH and in the absence of TCS for periplasmic pH. The cytoplasmic fluorescence profiles of the strain expressing KapA-ΔTat-EGFP allowed us to use it as a control when we measured periplasmic pH with TorA-EGFP.
FIG 4.

Excitation spectra for KapA-ΔTat-EGFP, KapA-EGFP, and TorA-EGFP as a function of medium pH. H. pylori strains G27/PcagA-KapA-Δtat-EGFP (A), G27/PcagA-KapA-EGFP (B), and G27/PcagA-TorA-EGFP (C) were suspended in HP medium with 20 mM buffer at different pHs (pH 4.5 to 8.5) without 3,3′,4′,5-tetrachloro-salicylanilide (TCS). The emission wavelength was 507 nm. At least 3 independent experiments with separated cell preparations were performed for each strain, and similar results were obtained for each strain. Data from a representative experiment for each strain are shown.
FIG 5.

Standard curves of intracellular pH as a function of the fluorescence signal and pH homeostasis. (A and B) The fluorescence signals from H. pylori expressing KapA-ΔTat-EGFP (A) and TorA-EGFP (B) were measured at an excitation wavelength 488 nm without and with 10 μM TCS (to collapse ΔpH). The standard curves were derived by using the regression equation from the data (pH 5.0 to pH 8.0) in the presence of TCS. (C and D) Cytoplasmic pH (C) and periplasmic pH (D) values were determined based on the standard curves. Error bars represent standard deviations of means (n = 5), and they are within the data point if not visible.
We then used the standard curves from KapA-ΔTat-EGFP and TorA-EGFP to measure the cytoplasmic and periplasmic pHs of H. pylori maintaining pH homeostasis. Fluorescence signals of bacteria in the medium with different pHs in the absence of TCS (Fig. 5A and B, blue lines for KapA-ΔTat-EGFP and TorA-EGFP, respectively) were converted to pH values (Fig. 5C and D) by using the regression equations for the standard curves (Fig. 5A and B, dotted lines). In H. pylori (HP) medium buffered at pH 5.0 to 8.0, H. pylori maintained a cytoplasmic pH (measured from bacteria expressing KapA-ΔTat-EGFP) within a range of approximately pH 6.6 to 7.6, similar to the measurements using BCECF-acetoxymethyl ester (AM), a membrane-permeant fluorescent pH indicator (25), while the periplasmic pH (measured with bacteria expressing TorA-EGFP) was between pH 4.8 and 7.7, close to the medium pHs.
Effect of external pH shift on intracellular pH in H. pylori strains expressing KapA-EGFP and TorA-EGFP.
To investigate the ability of H. pylori to maintain intracellular pH, time course studies were performed to compare the perturbation and recovery of cytoplasmic and periplasmic pHs following rapid external acidification. Cultures of H. pylori expressing KapA-ΔTat-EGFP and TorA-EGFP grown overnight were harvested and suspended in HP medium buffered with 5 mM N-tris(hydroxymethyl)methyl-3-aminopropanesulfonic acid (TAPS) adjusted to pH 7.5. The initial intracellular pHs were pH ∼7.65 to 7.75 for H. pylori expressing KapA-ΔTat-EGFP and about pH 7.5 for the bacteria expressing TorA-EGFP, as determined by the standard curves with bacteria from the same cultures. For KapA-ΔTat-EGFP (reflecting cytoplasmic pH) (Fig. 6A), after the addition of HCl (8.5 mM) at 60 s (here, the external pH was decreased to pH 2.27), the cytoplasmic pH dropped to pH ∼4.17 to 4.46 within 10 s, with an average drop of 3.32 pH units compared to the initial pH. A rapid recovery that started within 5 s after the lowest point and increased the cytoplasmic pH to about pH 6.38 to 6.63 within 60 s was followed by a slower recovery during the next several minutes, bringing the cytoplasmic pH back to pH ∼7.05 to 7.33 at 360 s. At the end of the time course, the external pH of the cultures was between pH 2.27 and 2.31. For TorA-EGFP (Fig. 6B), the periplasmic pH fell to pH ∼3.9 to 4.0 (an average drop of 3.53 pH units compared to the initial pH) within 5 s after the addition of HCl, with a small recovery that increased the periplasmic pH to pH ∼4.3 to 4.4 at the end of the time course (360 s). Since the measurements are based on standard curves that are limited to a pH range of between pH 5.0 and 8.0, the extrapolated pH values for both cytoplasmic and periplasmic pH after the addition of HCl are not accurate, but they are clearly below pH 5.0.
FIG 6.

Effect of external acid shift on intracellular pH of H. pylori. Cultures of strains expressing KapA-ΔTat-EGFP (A) and TorA-EGFP (B) grown overnight were harvested and suspended in HP medium (5 mM TAPS, pH 7.5). At 60 s, the external pH was shifted from pH 7.5 to acidic pH by the addition of 8.5 mM HCl. Fluorescence was converted to pH units by using the standard curves for KapA-ΔTat-EGFP and TorA-EFGP, respectively. For each strain, three independent cultures were tested. Error bars at 60, 67, 120, and 300 s represent standard deviations of means (n = 3), and they are within the data point if not visible.
Effect of urea on the intracellular pH in live H. pylori bacteria expressing KapA-ΔTat-EGFP and TorA-EGFP.
The effect of urea on the intracellular pH is of particular interest because H. pylori uses a unique pH homeostasis strategy that relies on its high constitutive levels of urease (3, 42). Urea in acidic media enters the cytoplasm via the inner membrane-localized, proton-gated urea channel, UreI, and is converted into carbon dioxide and ammonia in the cytoplasm by the action of urease, which eventually buffers the periplasmic pH to close to 6.0. Periplasmic buffering is facilitated by the conversion of carbon dioxide to bicarbonate with the aid of the cytoplasmic β-carbonic anhydrase (β-CA) and periplasmic α-carbonic anhydrase (2, 26). Cultures from H. pylori bacteria expressing KapA-EGFP and TorA-EGFP grown overnight were harvested and suspended in HP medium with 5 mM buffer adjusted to pH 7.5, 6.5, or 5.0. For KapA-ΔTat-EGFP (Fig. 7A), the initial cytoplasmic pHs in the corresponding media were pH ∼7.6, ∼7.3, and ∼6.6. After the addition of 5 mM urea at 60 s, very little change was detected in the medium that was buffered at pH 7.5 and 6.5, while in the medium buffered at pH 5.0, the cytoplasmic pH increased rapidly from pH 6.6 to pH 7.08 and reached pH 7.12 at the end of the time course (240 s). For TorA-EGFP (Fig. 7B), the initial periplasmic pHs in the corresponding media were ∼7.5, ∼6.8, and ∼4.8. After the addition of 5 mM urea at 60 s, no periplasmic pH change was seen in media buffered at pH 7.5. In media buffered at pH 6.5, a small increase in the periplasmic pH was detected, from pH 6.8 to pH 7.1, by the end of the time course. In media buffered at pH 5.0, the periplasmic pH increased from pH 4.8 to pH 6.0 within 10 s and continued to increase at a slower pace to reach pH 6.1 at the 240-s time point. At the end of the time course, the external pHs of the cultures were measured and found to be ∼8.2, ∼6.7, and ∼6.4, corresponding to initial medium pHs of 7.5, 6.5, and 5.0, respectively. These results suggest that urea has little effect on the cytoplasmic or periplasmic pH at neutral or basic medium pH but has a significant impact at acidic medium pH, which is consistent with previous findings showing that the UreI inner membrane urea channel is proton gated (43).
FIG 7.

Urea increases the intracellular pH in acidic medium. Cultures of H. pylori strains G27/PcagA-KapA-Δtat-EGFP (A) and G27/PcagA-TorA-EGFP (B) were harvested and suspended in 5 mM HP medium buffers, adjusted to pH 7.5, 6.5, and 5.0. At 60 s, 5 mM urea was added, and the fluorescence signal was converted to pH units, as described in Materials and Methods. Signals were averaged for three independent cultures under each condition. Error bars at 60, 70, and 210 s represent standard deviations of means (n = 3), and they are within the data point if not visible. pHex indicates external pH.
Urea-dependent periplasmic pH restoration is lost in the FlgS-deficient mutant.
As a first step to expand this periplasmic pH-measuring system to acid acclimation investigation in H. pylori, the PcagA-TorA-EGFP construct was integrated into the H. pylori genome in which the flgS gene had been deleted to generate strain G27/ΔFlgS-PcagA-TorA-EGFP. Time course experiments were performed to compare the urea effects on the periplasmic pH between G27/ΔFlgS-PcagA-TorA-EGFP and wild-type strain G27/PcagA-TorA-EGFP. As shown in Fig. 8, in media buffered at pH 5.0, after the addition of 5 mM urea, the periplasmic pH for G27/ΔFlgS-PcagA-TorA-EGFP increased from pH 4.8 to pH 5.1 (Fig. 8B), only a fraction of the periplasmic pH increase found in wild-type strain G27/PcagA-TorA-EGFP, which increased from pH 4.8 to pH 6.1 (Fig. 8A). The difference in mean values of the measured periplasmic pHs between wild-type and ΔFlgS mutant strains (3 independent experiments for each group) after the addition of 5 mM urea at pH 5.0 (time course from 70 to 300 s) was analyzed by using unpaired Student's t test and was highly significant (P < 0.001). These results suggest a significant loss of urea-dependent periplasmic pH restoration in the deficient mutant. This provides additional evidence that FlgS is responsible for the recruitment of urease to the inner membrane in association with UreI, the urea channel, thereby accelerating periplasmic buffering.
FIG 8.

Urea-dependent periplasmic pH restoration in acidic medium is lost in FlgS-deficient mutant. Cultures of H. pylori wild-type strain G27/PcagA-TorA-EGFP (A) and strain G27/ΔFlgS-PcagA-TorA-EGFP (B) were harvested and suspended in HP medium with 5 mM HOMOPIPES buffer (pH 5.0). At 60 s, 5 mM urea was added, and the fluorescence signal was converted to pH units, as described in Materials and Methods. For each strain, three independent cultures were tested. Error bars at 60, 70, and 240 s represent standard deviations of means (n = 3), and they are within the data point if not visible.
DISCUSSION
In this study, we have shown that EGFP can be exported to the H. pylori periplasm using the HydA RR signal peptide (Fig. 2A). HydA is a small-subunit protein of the H. pylori hydrogenase. This enzyme, shown previously to be membrane bound (44), was later found to contain a twin-arginine consensus region in the sequence of the small-subunit protein HydA (33), suggesting that this protein can be a target of the Tat system. Our results for the first time provide direct experimental evidence that the hydrogenase small-subunit protein HydA is exported to the periplasm via a Tat system in H. pylori. However, cell fractionation with immunoblotting studies (Fig. 3) revealed that there are significant amounts of HydA-EGFP retained in the cytoplasm in the precursor form that will not generate any fluorescence signal, and only about 50% of the HydA-EGFP that was transported across membranes to the periplasm was processed to the mature form, which became the fully folded and active EGFP. Therefore, the fluorescence signal is not strong enough to use HydA-EGFP as a tool to measure periplasmic pH.
The catalase-associated protein (KapA; HP0874) possessing a predicted signal sequence with a Tat motif (33, 44) is believed to be responsible for the translocation of catalase (KatA; HP0875) to the periplasmic side of the membrane (45). Although the confocal images showed that the fluorescence signal for KapA-EGFP is distributed evenly throughout the bacteria, which is very similar to that of KapA-ΔTat-EGFP (with the twin-arginine motif deleted in the KapA signal sequence, supposedly located in the cytoplasm) (Fig. 2B and C), the data with fractionation and immunoblotting (Fig. 3) demonstrated that the mature form of KapA-EGFP is located almost equally in the periplasm and cytoplasm, whereas with the bacteria expressing KapA-ΔTat-EGFP, most of the mature form of the proteins was found in the cytoplasmic fraction. Therefore, the data presented here indicate that the H. pylori KapA signal peptide enables EGFP transport to periplasm, although it is not fully efficient under these experimental conditions.
With the TorA-RR signal peptide from trimethylamine N-oxide (TMAO) reductases of E. coli, which has been shown to be translocated by the Tat system (34), H. pylori is capable of exporting EGFP to the periplasm in a fully folded and active form (Fig. 2C and 3). Our initial attempts to export a functional form of EGFP into the periplasm with a signal peptide from H. pylori α-carbonic anhydrase, which is believed to be a Sec system for translocation, were unsuccessful (our unpublished results). This confirms that the Sec pathway is incapable of exporting folded functional proteins such as GFP (which is known to be stably folded) (29). Previous studies (32, 46, 47) have shown that GFP, a heterologous protein, can be translocated in an active form across the cytoplasmic membrane by the Tat system in E. coli with the TorA-RR signal peptide by using the pBAD24 vector containing the arabinose-inducible PBAD promoter (22). In the presence of arabinose to induce TorA-GFP expression, the export system was ineffective, retaining GFP in the cytoplasm. The change in growth conditions by the removal of arabinose after a 2-h induction promoted a massive increase in the export efficiency, resulting in the majority of the GFP being transferred into the periplasm (46). We integrated the TorA-EGFP fusion open reading frame (ORF) into the cag locus of the H. pylori genome. The expression of TorA-EGFP under the control of the cagA promoter PcagA appears to be compatible with the Tat translocation system in H. pylori, and this experimental system eliminated the arabinose-adding/removing process. To our knowledge, this study shows for the first time that EGFP was successfully exported in an active form by the Tat system in H. pylori.
Previous studies used fluorimetry of TorA-GFPmut3* (22) to measure the intracellular pH of E. coli, which allowed the assessment of the cellular rate of pH change in response to a rapid shift in the pH of the external medium. After the addition of HCl (8.5 mM) to medium buffered at pH 7.5, both cytoplasmic and periplasmic pHs fell to below pH 6.0 within 10 to 20 s. While the cytoplasmic pH began to recover within 4 s, the periplasmic pH showed minimal recovery. In these studies with H. pylori, similar results (Fig. 6A and B) were observed for both cytoplasmic and periplasmic pHs in the absence of urea, except that the addition of HCl caused much larger drops in both cytoplasmic and periplasmic pHs (to around pH 4.0). These differences may be due to the different buffers used in these studies. They may also reflect different pH homeostasis mechanisms in E. coli and H. pylori.
H. pylori survives gastric acidity by expressing urease at a level higher than that of any other known microbe (3, 23). Previous studies measuring H. pylori transmembrane potential and internal pH with BCECF-AM at medium pHs of 3.0, 4.0, and 6.0 in the presence of urea demonstrated that cytoplasmic urease can compensate for medium acidity by the hydrolysis of urea when the external pH is decreased to <6.0 (24). When 5 mM urea was added, while the membrane potential was elevated to about −105 mV, there was a rapid rise of the measured internal pH that then declined to a steady state at all acidic medium pH levels. This steady state provided a calculated internal pH value of 6.2, although this was never directly measured. We assumed that this was the periplasmic pH, since we could calculate the proton motive force across the inner membrane (24). The use of free BCECF showed that the periplasmic pH was elevated by the addition of urea at various medium pH values, but it was not possible to directly measure the pH in the periplasm with this method (48). In this study, we are now able to directly measure periplasmic pH with TorA-EGFP expressed in H. pylori and prove that 5 mM urea has little effect on periplasmic pH at a medium pH of >6.5 but increases the periplasmic pH rapidly to pH 6.2 at acidic medium pH, corresponding to the opening of UreI (Fig. 7). The ability to maintain the periplasmic pH at about pH 6.2 is likely to be essential for gastric acid survival and colonization. These results are consistent with the deduced periplasmic pH based on the measurement of membrane potential and calculation of the proton motive force equation and suggest that we have developed a reliable tool for the direct and rapid investigation of acid acclimation in H. pylori.
To colonize at the highly variable pH of the stomach, H. pylori expresses two pH sensor histidine kinases. ArsS responds to a moderate fall in the periplasmic pH, and FlgS responds to cytoplasmic acidification (15). Our previous studies have shown that the acid activation of FlgS leads to the recruitment of the urease structural subunits UreA and UreB and the Ni2+ insertion protein UreE to UreI at the inner membrane, resulting in the activation of urease activity (14). The deletion of FlgS abolishes urease activation and membrane assembly (14), leading to impaired cytoplasmic and periplasmic pH homeostasis and a loss of acid survival (15). In the present study, to further determine the role of FlgS in periplasmic pH homeostasis, we used a PcagA-TorA-EGFP periplasmic pH-measuring system to investigate the changes in periplasmic pH in an FlgS-deficient mutant. At pH 5.0, unlike the wild-type strain, the FlgS-deficient mutant was unable to restore periplasmic pH to 6.1 with the addition of 5 mM urea (with only a fraction of the periplasmic pH increase found for the wild-type strain) (Fig. 8), providing additional evidence that HP0244 is required for the recruitment of urease to the inner membrane in association with UreI, the urea channel. The association of the urease complex with UreI results in the transport of NH3, NH4+, and CO2 through UreI into the periplasm to maintain periplasmic pH homeostasis. Therefore, the FlgS-dependent assembly of a pH-regulatory complex of active urease with UreI offers an advantage for periplasmic buffering, conferring acid resistance and facilitating gastric colonization by H. pylori.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
H. pylori strain G27 (49) was the parental strain. The strains expressing EGFP under the control of the cagA promoter (G27/PcagA-HydA-EGFP, G27/PcagA-KapA-EGFP, G27/PcagA-KapA-Δtat-EGFP, and G27/PcagA-TorA-EGFP) were constructed by allelic exchange using a chloramphenicol resistance cassette, as described below. Bacteria were grown under microaerobic conditions (5% O2, 10% CO2, 85% N2) on either tryptic soy agar (TSA) plates supplemented with 5% sheep blood (Becton Dickinson) or brain heart infusion (BHI) agar plates supplemented with 7% horse serum (Gibco BRL-Life Technologies Inc.) and 0.25% yeast extract (Difco Laboratories). All bacteria were grown in media in the presence of Dent selective supplement (Oxoid Limited), and the strains expressing EGFP were always grown in the presence of 10 μg/ml chloramphenicol (Sigma Chemical Co.).
For intracellular pH measurements, cultures grown overnight were harvested from BHI agar plates and resuspended to optical density at 600 nm (OD600) values of 0.4 for strain G27/PcagA-KapA-EGFP, 0.1 for strain G27/PcagA-KapA-Δtat-EGFP, and 0.25 for strains G27/PcagA-TorA-EGFP and G27/ΔFlgS-PcagA-TorA-EGFP in HP medium (140 mM Na2Cl, 5 mM KCl, 1.3 mM CaCl2, 0.5 mM MgSO4, 10 mM glucose, 1 mM glutamine) buffered with a 5 to 20 mM concentration (depending on the experiment) of the appropriate buffer [pH 4.5 to 5.0, homopiperazine-N,N′-bis-2-(ethanesulfonic acid) (HOMOPIPES); pH 5.5 to 6.0, 2-(N-morpholino)ethanesulfonic acid (MES); pH 6.5 to 7.0, 3-(N-morpholino)propanesulfonic acid (MOPS); pH 7.5 to 8.5, N-tris(hydroxymethyl)methyl-3-aminopropanesulfonic acid (TAPS)]. With these cell densities, each strain provides a fluorescence signal intensity that is high enough to be detectable and avoids the scattering of the excitation or emission beam from the concentrated sample, which reduces the apparent signal intensity.
Construction of H. pylori strains expressing Tat-EGFP under the control of the cagA promoter PcagA.
Plasmids for the integration of EGFP attached with one of the three Tat signal peptides (HydA and KapA from H. pylori and TorA from E. coli) into the cag locus of H. pylori G27 were constructed by using a Genesmart seamless cloning and assembly kit (Invitrogen by Life Technologies), as follows (Fig. 1). A 1,421-bp DNA fragment containing a 216-bp sequence encoding amino acids 1 to 72 of CagC (HP0545), a 348-bp sequence encoding the complete CagB (HP0546) protein, and an 833-bp intergenic sequence between the cagB and cagA genes were amplified from chromosomal DNA of H. pylori 26695 with primer pair HP0545-5P(400-430)-KpnI/HP0547-3P(1785-1860). PCR performed with primer pair HP0547-5P(1808-1828)/HP0547-3P(1542-1569)-SacI generated a 731-bp fragment encoding amino acids 1 to 243 of CagA (HP0547). A 920-bp chloramphenicol resistance cassette was amplified from pBC SK+ with primers Ch-5 and Ch-3. A 717-bp fragment encoding EGFP, a GFPmut1 variant (50) which contains the double-amino-acid replacement of Phe-64 to Leu and Ser-65 to Thr, was amplified from pEGFP-C1 (BD Biosciences-Clontech) with primer pair EGFP-5/EGFP-3. For the fragments encoding Tat signal peptides, PCR performed with primer pairs HydA-F/HydA-R, KapA-F/KapA-R, and KapA-Δtat-F/KapA-R, using chromosomal DNA of H. pylori G27 as a template, yielded a 256-bp fragment for the HydA signal peptide, a 121-bp fragment for the KapA signal peptide, and a 100-bp fragment for the KapA signal peptide with its twin-arginine motif deleted, and a 176-bp fragment for the TorA signal peptide was amplified with primer pair TorA-F/TorA-R (32) from chromosomal DNA of E. coli Top10. The primers used for generating inserts were designed to have each DNA fragment share 15-bp-end-terminal homology with the adjacent fragment (including the cloning vector pBluescript). The seamless cloning and assembly reactions with 5 inserts and a KpnI/SacI-linearized pBluescript vector resulted in four constructs containing a Tat signal peptide (HydA, KapA, KapA-ΔTat, or TorA) fused with an EGFP-chloramphenicol resistance cassette flanked by cagC-cagB-PcagA upstream and cagA downstream, which was cloned into the KpnI/SacI sites of the pBluescript vector. All the primer sequences are listed in Table 1. The plasmid constructs carrying a Tat signal peptide tagged with EGFP and a chloramphenicol resistance cassette were introduced into H. pylori strain G27 by natural transformation. For the strain that targets EGFP to the periplasm of the FlgS-deficient mutant, the construct containing TorA-EGFP was transformed into G27/ΔFlgS::Km (15). The resulting strains (G27/PcagA-HydA-EGFP, G27/PcagA-KapA-EGFP, G27/PcagA-KapA-Δtat-EGFP, G27/PcagA-TorA-EGFP, and G27/ΔFlgS-PcagA-TorA-EGFP) had the Tat-EGFP fusion ORF integrated into the cag locus of H. pylori G27 or G27/ΔFlgS and directly under the control of the cagA promoter PcagA. The chloramphenicol-resistant transformants were confirmed by PCR analysis for the correct integration of the Tat-EGFP ORF and the chloramphenicol resistance cassette in the genomic cag locus of H. pylori.
TABLE 1.
Oligonucleotide primers used in this study
| Primer | Sequence (5′–3′)a | Siteb | Templatec |
|---|---|---|---|
| HP0545-5P(400-430)-KpnI | gggcgaattgggtaccTTTCTTGTCTTTCAAATTTTTG | KpnI | H. pylori |
| HP0547-3P(1785-1860) | TGTTTCTCCTTACTATACCTAG | H. pylori | |
| HP0547-5P(1808-1828) | ataataagcggatgaATGACTAACGAAACTATTGATC | H. pylori | |
| HP0547-3P(1542-1569)-SacI | caaaagctggagctcTTGGACATGGGGAACTGG | SacI | H. pylori |
| Ch-5 | gaattcctgcagAAATCCTGGTGTCCCTGTTG | pBC SK+ | |
| Ch-3 | TCATCCGCTTATTATCACT | pBC SK+ | |
| EGFP-5 | GTGAGCAAGGGCGAGGAGCTGTTCACCGG | pEGFP-C1 | |
| EGFP-3 | ctgcaggaattcTTACTTGTACAGCTCGTCCAT | pEGFP-C1 | |
| HydA-F | ctaggtatagtaaggagaaacaATGTTCTACGATGAAAAAAAGACC | H. pylori | |
| HydA-R | cagctcctcgcccttgctcacCGCCTTCAAAGTCAAGGGAGCAAAAC | H. pylori | |
| KapA-F | ctaggtatagtaaggagaaacaATGAAACGAAGGGATTTTATTAAAAC | H. pylori | |
| KapA-Δtat-F | ctaggtatagtaaggagaaacaATGACGACTACTTTAGGCGCTACAGGTGC | H. pylori | |
| KapA-R | cagctcctcgcccttgctcacTGCCTGCAAAATCTGTGCTCC | H. pylori | |
| TorA-F | ctaggtatagtaaggagaaacaATGAACAATAACGATCTCTTTCAGG | E. coli | |
| TorA-R | ctaggtatagtaaggagaaacaCGCCGCTTGCGCCGCAGTCGCAC | E. coli |
Sequences in uppercase type are derived from the genome sequences of H. pylori 26695 (51) and E. coli (32) and vector sequences. Sequences introduced for cloning purposes (end-terminal homology with the adjacent fragment) are in lowercase type, and restriction recognition sites are underlined.
Restriction recognition sites.
Genomic DNA from H. pylori strain G27 and E. coli Top10 and plasmid DNA from pBS SK+ and pEGFP-C1 served as templates for PCR.
Cell fractionations and immunoblotting.
Periplasm and cytoplasm fractions were prepared by using methods described previously by Thomas et al. (32). Cultures grown overnight were harvested from 2 to 3 BHI agar plates by centrifugation at 4,000 × g for 10 min at 4°C. The pellets were resuspended in 1 ml of phosphate-buffered saline (PBS). After washing twice with 1 ml of PBS and twice with 300 μl of 25 mM Tris-HCl (pH 7.4), the cell pellet was resuspended in 100 μl of buffer containing 25 mM Tris-HCl (pH 7.4)–20% sucrose–1 mM EDTA and incubated at room temperature for 10 min. After centrifugation at 4,000 × g for 10 min at room temperature, cells were resuspended in 100 μl of ice-cold 5 mM MgSO4 and incubated on ice for 20 min to generate spheroplasts. The periplasm fraction (retained in the supernatant) and spheroplasts (pellets) were separated by centrifugation at 10,300 × g for 5 min at 4°C. Spheroplasts were resuspended in 100 μl of ice-cold 5 mM MgSO4 and lysed by sonication, and intact spheroplasts and cellular debris were removed by centrifugation at 10,300 × g for 5 min at 4°C. The cytoplasm fraction (retained in the supernatant) was collected after centrifugation at 250,000 × g for 30 min at 4°C. The protein concentration was determined by using a Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Scientific). Twenty micrograms of protein for each fraction sample was separated on 4 to 12% NuPAGE gels (Life Technologies) and immunoblotted with anti-GFP antibody (Roche).
Fluorescent imaging analysis.
H. pylori strains G27/PcagA-HydA-EGFP, G27/PcagA-KapA-EGFP, G27/PcagA-KapA-Δtat-EGFP, and G27/PcagA-TorA-EGFP were grown microaerobically at 37°C on BHI agar plates supplemented with 7% horse serum and 0.25% yeast extract in the presence of 10 μg/ml chloramphenicol. After incubation overnight, bacteria were harvested, washed, and resuspended in fresh BHI medium. These bacteria were loaded onto polylysine-coated glass-bottom microwell dishes (MatTek Corporation) and incubated at 37°C for 10 min to allow the bacteria to settle and adhere to the bottom, followed by gentle washing with fresh HP medium to remove the floating bacteria. Confocal microscopy images were acquired by using a Zeiss LSM 510 confocal laser scanning microscope (Carl Zeiss MicroImaging GmbH, Germany). For EGFP fluorescence detection, excitation was set at 488 nm, and emission was filtered through a green band-pass filter transmitting at between 500 and 545 nm.
Intracellular pH measurement with a fluorimetry assay.
For fluorescence spectroscopy, methods described previously (22) were used, with some modifications. Briefly, 3-ml bacterial suspension aliquots were placed into a fluorimeter cuvette under constant stirring at 37°C and measured with a Fluorolog-3 spectrofluorimeter (Horiba Jobin Yvon). Excitation spectra were collected at 440 to 500 nm by using an emission wavelength of 507 nm. Blank scans of wild-type H. pylori G27 suspensions were used to subtract the nonspecific background signal. Spectra were recorded for three biological replicates at each pH. EGFP excitation was measured at 488 nm, with an emission wavelength of 507 nm. Standard curves for pH as a function of the fluorescence intensity were generated for each strain expressing EGFP (G27/PcagA-KapA-EGFP, G27/PcagA-KapA-Δtat-EGFP, and G27/PcagA-TorA-EGFP) for each individual experiment at pH 4.5, 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, 8.0, and 8.5 with the addition of 10 μM TCS (3,3′,4′,5-tetrachloro-salicylanilide), which collapses the transmembrane ΔpH. For each strain, a regression equation was applied to the standard curve and used to convert time course signal intensities (at an excitation wavelength of 488 nm) to pH units. For time course experiments, continuous fluorescence intensities were recorded every 1 s for an initial 60 s of acquisition before the addition of aliquots of reagents and then for the remaining 3 to 5 min after the addition of the reagents.
ACKNOWLEDGMENTS
This work was supported in part by NIH grants DK46917, DK53462, and DK58333 to G.S.; HL113350 to O.V.; and K08DK100661 and R03DK110579 to E.A.M. and by the UCLA Children's Discovery and Innovation Institute (E.A.M.). This work was also supported with resources and the use of facilities at West Los Angeles Veterans Administration Medical Center.
We declare no competing or financial interests.
REFERENCES
- 1.Marshall BJ, Warren JR. 1984. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet i:1311–1315. [DOI] [PubMed] [Google Scholar]
- 2.Sachs G, Weeks DL, Wen Y, Marcus EA, Scott DR, Melchers K. 2005. Acid acclimation by Helicobacter pylori. Physiology 20:429–438. doi: 10.1152/physiol.00032.2005. [DOI] [PubMed] [Google Scholar]
- 3.Mobley HL, Island MD, Hausinger RP. 1995. Molecular biology of microbial ureases. Microbiol Rev 59:451–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rektorschek M, Buhmann A, Weeks D, Schwan D, Bensch KW, Eskandari S, Scott D, Sachs G, Melchers K. 2000. Acid resistance of Helicobacter pylori depends on the UreI membrane protein and an inner membrane proton barrier. Mol Microbiol 36:141–152. doi: 10.1046/j.1365-2958.2000.01835.x. [DOI] [PubMed] [Google Scholar]
- 5.Scott DR, Marcus EA, Weeks DL, Lee A, Melchers K, Sachs G. 2000. Expression of the Helicobacter pylori ureI gene is required for acidic pH activation of cytoplasmic urease. Infect Immun 68:470–477. doi: 10.1128/IAI.68.2.470-477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weeks DL, Eskandari S, Scott DR, Sachs G. 2000. A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science 287:482–485. doi: 10.1126/science.287.5452.482. [DOI] [PubMed] [Google Scholar]
- 7.Marcus EA, Moshfegh AP, Sachs G, Scott DR. 2005. The periplasmic alpha-carbonic anhydrase activity of Helicobacter pylori is essential for acid acclimation. J Bacteriol 187:729–738. doi: 10.1128/JB.187.2.729-738.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith KS, Ferry JG. 2000. Prokaryotic carbonic anhydrases. FEMS Microbiol Rev 24:335–366. doi: 10.1111/j.1574-6976.2000.tb00546.x. [DOI] [PubMed] [Google Scholar]
- 9.Pflock M, Finsterer N, Joseph B, Mollenkopf H, Meyer TF, Beier D. 2006. Characterization of the ArsRS regulon of Helicobacter pylori, involved in acid adaptation. J Bacteriol 188:3449–3462. doi: 10.1128/JB.188.10.3449-3462.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pflock M, Kennard S, Delany I, Scarlato V, Beier D. 2005. Acid-induced activation of the urease promoters is mediated directly by the ArsRS two-component system of Helicobacter pylori. Infect Immun 73:6437–6445. doi: 10.1128/IAI.73.10.6437-6445.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pflock M, Kennard S, Finsterer N, Beier D. 2006. Acid-responsive gene regulation in the human pathogen Helicobacter pylori. J Biotechnol 126:52–60. doi: 10.1016/j.jbiotec.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 12.Wen Y, Feng J, Scott DR, Marcus EA, Sachs G. 2006. Involvement of the HP0165-HP0166 two-component system in expression of some acidic-pH-upregulated genes of Helicobacter pylori. J Bacteriol 188:1750–1761. doi: 10.1128/JB.188.5.1750-1761.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wen Y, Feng J, Scott DR, Marcus EA, Sachs G. 2007. HP0165-HP0166 two-component system (ArsRS) regulates the acid-induced expression of HP1186 alpha-carbonic anhydrase in Helicobacter pylori by activating the pH-dependent promoter. J Bacteriol 189:2426–2434. doi: 10.1128/JB.01492-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott DR, Marcus EA, Wen Y, Singh S, Feng J, Sachs G. 2010. Cytoplasmic histidine kinase (HP0244)-regulated assembly of urease with UreI, a channel for urea and its metabolites, CO2, NH3, and NH4+, is necessary for acid survival of Helicobacter pylori. J Bacteriol 192:94–103. doi: 10.1128/JB.00848-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wen Y, Feng J, Scott DR, Marcus EA, Sachs G. 2009. The pH-responsive regulon of HP0244 (FlgS), the cytoplasmic histidine kinase of Helicobacter pylori. J Bacteriol 191:449–460. doi: 10.1128/JB.01219-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong W, Sano K, Morimatsu S, Scott DR, Weeks DL, Sachs G, Goto T, Mohan S, Harada F, Nakajima N, Nakano T. 2003. Medium pH-dependent redistribution of the urease of Helicobacter pylori. J Med Microbiol 52:211–216. doi: 10.1099/jmm.0.05072-0. [DOI] [PubMed] [Google Scholar]
- 17.Scott DR, Marcus EA, Weeks DL, Sachs G. 2002. Mechanisms of acid resistance due to the urease system of Helicobacter pylori. Gastroenterology 123:187–195. doi: 10.1053/gast.2002.34218. [DOI] [PubMed] [Google Scholar]
- 18.Okada Y, Inouye A. 1976. pH-sensitive glass microelectrodes and intracellular pH measurements. Biophys Struct Mech 2:21–30. doi: 10.1007/BF00535650. [DOI] [PubMed] [Google Scholar]
- 19.Zilberstein D, Agmon V, Schuldiner S, Padan E. 1984. Escherichia coli intracellular pH, membrane potential, and cell growth. J Bacteriol 158:246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slonczewski JL, MacNab RM, Alger JR, Castle AM. 1982. Effects of pH and repellent tactic stimuli on protein methylation levels in Escherichia coli. J Bacteriol 152:384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olsen KN, Budde BB, Siegumfeldt H, Rechinger KB, Jakobsen M, Ingmer H. 2002. Noninvasive measurement of bacterial intracellular pH on a single-cell level with green fluorescent protein and fluorescence ratio imaging microscopy. Appl Environ Microbiol 68:4145–4147. doi: 10.1128/AEM.68.8.4145-4147.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilks JC, Slonczewski JL. 2007. pH of the cytoplasm and periplasm of Escherichia coli: rapid measurement by green fluorescent protein fluorimetry. J Bacteriol 189:5601–5607. doi: 10.1128/JB.00615-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer-Rosberg K, Scott DR, Rex D, Melchers K, Sachs G. 1996. The effect of environmental pH on the proton motive force of Helicobacter pylori. Gastroenterology 111:886–900. doi: 10.1016/S0016-5085(96)70056-2. [DOI] [PubMed] [Google Scholar]
- 24.Scott DR, Weeks D, Hong C, Postius S, Melchers K, Sachs G. 1998. The role of internal urease in acid resistance of Helicobacter pylori. Gastroenterology 114:58–70. doi: 10.1016/S0016-5085(98)70633-X. [DOI] [PubMed] [Google Scholar]
- 25.Wen Y, Marcus EA, Matrubutham U, Gleeson MA, Scott DR, Sachs G. 2003. Acid-adaptive genes of Helicobacter pylori. Infect Immun 71:5921–5939. doi: 10.1128/IAI.71.10.5921-5939.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sachs G, Kraut JA, Wen Y, Feng J, Scott DR. 2006. Urea transport in bacteria: acid acclimation by gastric Helicobacter spp. J Membr Biol 212:71–82. doi: 10.1007/s00232-006-0867-7. [DOI] [PubMed] [Google Scholar]
- 27.Kneen M, Farinas J, Li Y, Verkman AS. 1998. Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys J 74:1591–1599. doi: 10.1016/S0006-3495(98)77870-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. 1994. Green fluorescent protein as a marker for gene expression. Science 263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 29.Feilmeier BJ, Iseminger G, Schroeder D, Webber H, Phillips GJ. 2000. Green fluorescent protein functions as a reporter for protein localization in Escherichia coli. J Bacteriol 182:4068–4076. doi: 10.1128/JB.182.14.4068-4076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Craggs TD. 2009. Green fluorescent protein: structure, folding and chromophore maturation. Chem Soc Rev 38:2865–2875. doi: 10.1039/b903641p. [DOI] [PubMed] [Google Scholar]
- 31.Wickner W, Schekman R. 2005. Protein translocation across biological membranes. Science 310:1452–1456. doi: 10.1126/science.1113752. [DOI] [PubMed] [Google Scholar]
- 32.Thomas JD, Daniel RA, Errington J, Robinson C. 2001. Export of active green fluorescent protein to the periplasm by the twin-arginine translocase (Tat) pathway in Escherichia coli. Mol Microbiol 39:47–53. doi: 10.1046/j.1365-2958.2001.02253.x. [DOI] [PubMed] [Google Scholar]
- 33.Benoit SL, Maier RJ. 2014. Twin-arginine translocation system in Helicobacter pylori: TatC, but not TatB, is essential for viability. mBio 5:e01016-13. doi: 10.1128/mBio.01016-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palmer T, Sargent F, Berks BC. 2005. Export of complex cofactor-containing proteins by the bacterial Tat pathway. Trends Microbiol 13:175–180. doi: 10.1016/j.tim.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 35.Robinson C, Matos CF, Beck D, Ren C, Lawrence J, Vasisht N, Mendel S. 2011. Transport and proofreading of proteins by the twin-arginine translocation (Tat) system in bacteria. Biochim Biophys Acta 1808:876–884. doi: 10.1016/j.bbamem.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 36.Bagos PG, Nikolaou EP, Liakopoulos TD, Tsirigos KD. 2010. Combined prediction of Tat and Sec signal peptides with hidden Markov models. Bioinformatics 26:2811–2817. doi: 10.1093/bioinformatics/btq530. [DOI] [PubMed] [Google Scholar]
- 37.Bendtsen JD, Nielsen H, Widdick D, Palmer T, Brunak S. 2005. Prediction of twin-arginine signal peptides. BMC Bioinformatics 6:167. doi: 10.1186/1471-2105-6-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rose RW, Bruser T, Kissinger JC, Pohlschroder M. 2002. Adaptation of protein secretion to extremely high-salt conditions by extensive use of the twin-arginine translocation pathway. Mol Microbiol 45:943–950. doi: 10.1046/j.1365-2958.2002.03090.x. [DOI] [PubMed] [Google Scholar]
- 39.Sargent F, Bogsch EG, Stanley NR, Wexler M, Robinson C, Berks BC, Palmer T. 1998. Overlapping functions of components of a bacterial Sec-independent protein export pathway. EMBO J 17:3640–3650. doi: 10.1093/emboj/17.13.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pavlasova E, Harold FM. 1969. Energy coupling in the transport of beta-galactosides by Escherichia coli: effect of proton conductors. J Bacteriol 98:198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Vliet AH, Kuipers EJ, Waidner B, Davies BJ, de Vries N, Penn CW, Vandenbroucke-Grauls CM, Kist M, Bereswill S, Kusters JG. 2001. Nickel-responsive induction of urease expression in Helicobacter pylori is mediated at the transcriptional level. Infect Immun 69:4891–4897. doi: 10.1128/IAI.69.8.4891-4897.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weeks DL, Gushansky G, Scott DR, Sachs G. 2004. Mechanism of proton gating of a urea channel. J Biol Chem 279:9944–9950. doi: 10.1074/jbc.M312680200. [DOI] [PubMed] [Google Scholar]
- 44.Maier RJ, Fu C, Gilbert J, Moshiri F, Olson J, Plaut AG. 1996. Hydrogen uptake hydrogenase in Helicobacter pylori. FEMS Microbiol Lett 141:71–76. doi: 10.1111/j.1574-6968.1996.tb08365.x. [DOI] [PubMed] [Google Scholar]
- 45.Harris AG, Hazell SL. 2003. Localisation of Helicobacter pylori catalase in both the periplasm and cytoplasm, and its dependence on the twin-arginine target protein, KapA, for activity. FEMS Microbiol Lett 229:283–289. doi: 10.1016/S0378-1097(03)00850-4. [DOI] [PubMed] [Google Scholar]
- 46.Barrett CM, Ray N, Thomas JD, Robinson C, Bolhuis A. 2003. Quantitative export of a reporter protein, GFP, by the twin-arginine translocation pathway in Escherichia coli. Biochem Biophys Res Commun 304:279–284. doi: 10.1016/S0006-291X(03)00583-7. [DOI] [PubMed] [Google Scholar]
- 47.Santini CL, Bernadac A, Zhang M, Chanal A, Ize B, Blanco C, Wu LF. 2001. Translocation of jellyfish green fluorescent protein via the Tat system of Escherichia coli and change of its periplasmic localization in response to osmotic up-shock. J Biol Chem 276:8159–8164. doi: 10.1074/jbc.C000833200. [DOI] [PubMed] [Google Scholar]
- 48.Athmann C, Zeng N, Kang T, Marcus EA, Scott DR, Rektorschek M, Buhmann A, Melchers K, Sachs G. 2000. Local pH elevation mediated by the intrabacterial urease of Helicobacter pylori cocultured with gastric cells. J Clin Invest 106:339–347. doi: 10.1172/JCI9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N. 1993. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc Natl Acad Sci U S A 90:5791–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cormack BP, Valdivia RH, Falkow S. 1996. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 51.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]