

Abstract
While CRISPR-Cas9-mediated genome editing has transformed yeast research, current plasmids and cassettes for Cas9 and guide-RNA expression are species specific. CRISPR tools that function in multiple yeast species could contribute to the intensifying research on non-conventional yeasts. A plasmid carrying a pangenomic origin of replication and two constitutive expression cassettes for Cas9 and ribozyme-flanked gRNAs was constructed. Its functionality was tested by analyzing inactivation of the ADE2 gene in four yeast species. In two Kluyveromyces species, near-perfect targeting (≥96%) and homologous repair (HR) were observed in at least 24% of transformants. In two Ogataea species, Ade− mutants were not observed directly after transformation, but prolonged incubation of transformed cells resulted in targeting efficiencies of 9% to 63% mediated by non-homologous end joining (NHEJ). In an Ogataea parapolymorpha ku80 mutant, deletion of OpADE2 mediated by HR was achieved, albeit at low efficiencies (<1%). Furthermore the expression of a dual polycistronic gRNA array enabled simultaneous interruption of OpADE2 and OpYNR1 demonstrating flexibility of ribozyme-flanked gRNA design for multiplexing. While prevalence of NHEJ prevented HR-mediated editing in Ogataea, such targeted editing was possible in Kluyveromyces. This broad-host-range CRISPR/gRNA system may contribute to exploration of Cas9-mediated genome editing in other Saccharomycotina yeasts.
Keywords: CRISPR/Cas9, ribozymes, Kluyveromyces lactis, Kluyveromyces marxianus, Ogataea polymorpha, Hansenula polymorpha
A novel broad-host-range CRISPR-Cas9 tool to explore genome editing in non-conventional yeasts.
INTRODUCTION
Design and construction of efficient yeast cell factories for industrial production of fuels and a wide range of chemicals is among the key developments in microbial biotechnology in the last 20 years (Nielsen et al.2013; Chen et al.2015; Wang, Huang and Nielsen 2017). Saccharomyces cerevisiae is currently the most intensively used yeast species for production of low-molecular-weight products such as alcohols, organic acids and isoprenoids (Mattanovich, Sauer and Gasser 2014). Its tolerance to high sugar concentrations and low pH, its overall robustness, its ability to grow anaerobically and especially its tractability to genetic manipulation make S. cerevisiae a convenient chassis for various biotechnological purposes (Kavscek et al.2015; Li and Borodina 2015). Furthermore, availability of high-quality genome sequences and well-developed genetic tools (Salazar et al.2017; Stovicek, Holkenbrink and Borodina 2017) facilitate tailoring of S. cerevisiae to specific industrial applications, either by improving existing traits or by expressing heterologous enzymes and pathways. However, its narrow temperature spectrum, limited substrate range and strong tendency toward alcoholic fermentation under aerobic conditions have stimulated interest in studying non-Saccharomyces species, often referred to as ‘non-conventional yeasts’, that exhibit attractive features for further improving sustainability and economic viability of biobased production (Johnson 2013).
Kluyveromyces lactis and K. marxianus are non-conventional yeasts that are generally regarded as safe (GRAS), can utilize lactose as a carbon source and have been applied for bioethanol production from cheese whey, a by-product of the dairy industry (Siso 1996). Kluyveromyces lactis has an excellent capacity for protein secretion, which has been exploited for production of several heterologous proteins (Spohner et al.2016). Kluyveromyces marxianus can grow very fast at temperatures above 40°C and has been used for the production of bioethanol, biomass and flavor compounds (Morrissey et al.2015). The thermotolerant and methylotrophic yeast Ogataea polymorpha (syn. Hansenula polymorpha) is a major established platform for heterologous protein expression thanks to the availability of extremely strong yet tightly controlled promoters (Gellissen 2000). In addition, this yeast has been engineered for high-temperature ethanol fermentation from various carbon sources, including xylose (Kurylenko et al.2014), and glycerol, a by-product originating from biodiesel production (Kata et al.2016). Ogataea polymorpha shares many characteristics with the closely related species O. parapolymorpha, which was taxonomically separated from O. polymorpha in 2010, and now includes the popular former H. polymorpha DL-1 strain (Suh and Zhou 2010).
The unique physiological characteristics of non-conventional yeast species have the potential to reduce production costs of processes that are currently performed with S. cerevisiae. However, analogous to the situation in industrial S. cerevisiae strains, this will require extensive genetic engineering. While obtaining the necessary genome-sequence information is relatively straightforward due to fast developments in whole-genome sequencing (Goodwin, McPherson and McCombie 2016), genetic modification by classical methods is still challenging mostly due to the predominant DNA-repair mechanisms of many non-conventional yeasts (Nonklang et al.2008; Abdel-Banat et al.2010). Unlike S. cerevisiae, these yeasts typically have a very active non-homologous end joining (NHEJ) DNA repair mechanism and use homologous recombination (HR) to a much lesser extent, which makes precise genome editing inefficient as provided linear repair DNA fragments are not efficiently integrated at the targeted genomic locus (Klinner and Schäfer 2004). Introduction of a double-strand break (DSB) in the targeted DNA locus can strongly facilitate genetic engineering, either by introduction of mutations at the cut site or by stimulating the occurrence of HR-mediated DNA repair with co-transformed repair fragments (Jasin and Rothstein 2013; Kuijpers et al.2013b).
Over the past five years, the CRISPR-Cas9 system has emerged as a powerful and versatile tool to engineer the genomes of a wide range of organisms (Hsu, Lander and Zhang 2014). In this system, the endonuclease CRISPR associated protein 9 (Cas9) binds a guide RNA molecule (gRNA) that targets a sequence-specific site in the genome (Jinek et al.2012). The Cas9-gRNA complex then induces a DSB that is lethal unless repaired. Repair of DSBs typically occurs through NHEJ or HR, depending on the presence of a repair DNA fragment and the predominant DSB repair mechanism of the host cell (Shrivastav, De Haro and Nickoloff 2008). CRISPR-Cas9’s ability to induce mutations in the target sequence has been widely exploited in the development of genetic tools for various non-conventional yeasts including K. lactis (Horwitz et al.2015), K. marxianus (Löbs et al.2017; Nambu-Nishida et al.2017), O. polymorpha (Numamoto, Maekawa and Kaneko 2017), Pichia pastoris (Weninger et al.2016), Scheffersomyces stipitis (Cao et al.2017) and Yarrowia lipolytica (Gao et al.2016, Schwartz et al.2016). However, the plasmid and Cas9/gRNA expression systems available today have generally only been designed for and tested in a single yeast species, thus limiting the potential of each system to work on different yeasts with novel characteristics.
In this study, we developed a novel wide-host-range CRISPR/Cas9 system for use across several yeast species, based on plasmid-borne expression of Cas9 and a ribozyme-flanked gRNA that was recently developed for S. pastorianus, a Saccharomyces hybrid highly resilient to genetic modification (Gorter de Vries et al.2017). The system was successfully tested in K. marxianus, K. lactis, O. polymorpha and O. parapolymorpha. The results highlight the potential of cross-species CRISPR-Cas9 tools for genome engineering in yeasts.
MATERIALS AND METHODS
Strains and growth conditions
The K. lactis, K. marxianus, O. polymorpha and O. parapolymorpha strains used in this study are listed in Table 1. For cultivation under non-selective conditions, strains were grown in 500 mL shake flasks containing 100 mL YPD medium (10 g L−1 Bacto yeast extract, 20 g L−1 Bacto peptone, 20 g L−1 glucose, demineralized water), placed in a rotary shaker set to 30°C and 200 rpm. For antibiotic selection, media for cultivation of Kluyveromyces and Ogataea species were supplemented with 200 and 300 μg mL−1 hygromycin B, respectively. Prolonged liquid incubation for generation of gene disruptions in Ogataea species was carried out under selective conditions in 50 mL vented tubes (Cellstar CELLreactor, Greiner Bio-One, Kremsmünster, Austria), containing 25 mL YPD medium and incubated as described above. Solid medium was prepared by addition of 2% (w/v) agar. To verify the disruption of nitrate reductase gene YNR1, O. parapolymorpha was grown on synthetic medium (SM) which contained 20 g L−1 glucose, 3 g L−1 KH2PO4, 0.5 g L−1 MgSO4, 7 H2O, 5 g L−1 (NH4)2SO4, 1 mL L−1 of a trace element solution and of a vitamin solution as previously described in Verduyn et al. (1992) and on synthetic medium with nitrate (SMN) in which (NH4)2SO4 was substituted with 5 g L−1 K2SO4 and 4.3 g L−1 NaNO3. For multiplexed targeting of ADE2 and YNR1 in O. parapolymorpha, SM and SMN media were supplemented with 15 mg L–1 adenine to allow for growth of Ade− mutants. Frozen stock cultures were prepared from exponentially growing shake-flask cultures by addition of 30% (v/v) glycerol, and aseptically stored in 1 mL aliquots at –80°C.
Table 1.
Kluyveromyces and Ogataea strains used in this study.
| Species | Strain | Genotype | Origin |
|---|---|---|---|
| Kluyveromyces lactis | CBS 2359 | Wild type | CBS-KNAWA, van der Walt (1971) |
| IMK829 | Δade2 | This study | |
| Kluyveromyces marxianus | CBS 5795 | Wild type | CBS-KNAWA, van der Walt (1971) |
| NBRC 1777 | Wild type | NBRCB | |
| IMK830 | Δade2 | This study | |
| CBS 397 | Wild type | CBS-KNAWA, van der Walt (1971) | |
| Ogataea polymorpha (syn. Hansenula polymorpha) | CBS 4732 | Wild type | CBS-KNAWA, Yamada et al. (1994) |
| Ogataea parapolymorpha (syn. Hansenula polymorpha) | CBS 11895 (DL-1, ATCC 26012) | Wild type | CBS-KNAWA, Suh and Zhou (2010) |
| IMD001 | ku80 A340AA | This study | |
| IMK828 | ku80 A340AA Δade2 | This study | |
| IMD034 | ade2 C120CA ynr1 G397GT | This study |
CBS 2359, CBS 5795, CBS 397, CBS 4732 and CBS 11895 were obtained from the CBS-KNAW fungal collection (A Westerdijk Fungal Biodiversity Institute, Utrecht, The Netherlands). NBRC 1777 was obtained from the NBRC culture collection (B National Institute of Technology and Evaluation, Tokyo, Japan).
Construction of plasmids and repair DNA fragments
All plasmids used in this study are described in Table 2. The DNA parts harbored by plasmids pUD527, pUD530, pUD531, pUD532 pUD540, pUD555, pUD602 and pUD750 were de novo synthesized by GeneArt (Thermo Fisher Scientific, Waltham, MA, USA).
Table 2.
Plasmids used in this study.
| Name (Addgene Plasmid #) | Relevant characteristics | Origin |
|---|---|---|
| pYTK079 | Template for hph (HygR) open reading frame | Lee et al. (2015) |
| pUD423 | Template for AaTEF1p-Spcas9D147Y P411T-ScPHO5t cassette | Gorter de Vries et al. (2017) |
| pUD527 | ori kanR SHRA AgTEF1p-amdS-AgTEF1t SHRB | GeneArt |
| pUD530 | ori kanR panARS(OPT) | GeneArt |
| pUD531 | ori kanR SHRC ScTDH3p-BsaI BsaI-ScCYC1t SHRI | GeneArt |
| pUD532 | ori kanR SHRI ori bla SHRA | GeneArt |
| pUD540 | ori ampR BsaIHH-gRNAOpADE2-HDVBsaI | GeneArt |
| pUD555 | ori ampR BsaIHH-gRNAKlADE2-HDVBsaI | GeneArt |
| pUD602 | ori ampR BsaIHH-gRNAOpKU80-HDVBsaI | GeneArt |
| pUD750 | ori ampR BsaIHH-gRNAOpADE2-HDV-HH-gRNAOpYNR1-HDVBsaI | This study |
| pUDP002 (plasmid #103872) | ori ampR panARS(OPT) AgTEF1p-hph-AgTEF1t ScTDH3pBsaI BsaIScCYC1t AaTEF1p-Spcas9D147Y P411T-ScPHO5t | This study |
| pUDP013 (plasmid #103873) | ori ampR panARS(OPT) AgTEF1p-hph-AgTEF1t ScTDH3p-HH-gRNAOpADE2-HDV- ScCYC1t AaTEF1p-Spcas9D147Y P411T-ScPHO5t | This study |
| pUDP025 (plasmid #103874) | ori ampR panARS(OPT) AgTEF1p-hph-AgTEF1t ScTDH3p-HH-gRNAKlADE2-HDV- ScCYC1t AaTEF1p-Spcas9D147Y P411T-ScPHO5t | This study |
| pUDP046 (plasmid #107062) | ori ampR panARS(OPT) AgTEF1p-hph-AgTEF1t ScTDH3p-HH-gRNAOpKU80-HDV- ScCYC1t AaTEF1p-Spcas9D147Y P411T-ScPHO5t | This study |
| pUDP082 (plasmid #103875) | ori ampR panARS(OPT) AgTEF1p-hph-AgTEF1t ScTDH3p-HH-gRNAKmADE2-HDV- ScCYC1t AaTEF1p-Spcas9D147Y P411T-ScPHO5t | This study |
| pUDP123 (plasmid # 107269) | ori ampR panARS(OPT) AgTEF1p-hph-AgTEF1t ScTDH3p-HH-gRNAOpADE2-HDV-HH-gRNAOpYNR1-HDV-ScCYC1t AaTEF1p-Spcas9D147Y P411T-ScPHO5t | This study |
Restriction enzyme sites are indicated in superscript and gRNA target sequences are indicated in subscript. SHRs represent specify synthetic homologous recombination sequences used for plasmid assembly. Aa: Arxula adeninivorans; Sp: Streptococcus pyogenes; Ag: Ashbya gossypii; Sc: Saccharomyces cerevisiae; Op: Ogataea (para)polymorpha; Kl: Kluyveromyces lactis; Km: Kluyveromyces marxianus; HH: hammerhead ribozyme; HDV: hepatitis delta virus ribozyme. pUDP013 (gRNAOpADE2) targets ADE2 in both O. polymorpha and O. parapolymorpha. The addgene plasmid code (when relevant) is indicated next to the plasmid name between brackets.
For the construction of pUDP002, an intermediate plasmid containing the Klebsiella pneumoniae hph (HygR) open reading frame (ORF), expressed from the TEF1 promoter and terminator from the yeast Ashbya gossypii (Eremothecium gossypii), was constructed by ‘Gibson’ assembly (Gibson et al.2009) from plasmids pYTK079 (Lee et al.2015) and pUD527: the hph ORF was amplified from pYTK079 using primers 9837 and 9838, and inserted into a plasmid backbone which was generated by PCR amplification of pUD527 using primers 9839 and 9840. Plasmid pUDP002 was constructed by Gibson assembly from five overlapping fragments, using synthetic homologous recombination sequences (Kuijpers et al.2013a): (i) the AgTEF1p-hph-AgTEF1t hygromycin resistance cassette was amplified from the intermediate plasmid using primers 9841 and 9842 and sequence-verified before further use; (ii) the AaTEF1p-cas9D147Y P411T-ScPHO5t expression cassette (Bao et al.2015; Gorter de Vries et al.2017) was obtained by amplification of pUD423 using primers 3841 and 9393; (iii) the pangenomic yeast replication origin panARS(OPT) (Liachko and Dunham 2014) was amplified from pUD530 using primers 9663 and 3856; (iv) the empty ScTDH3p-ScCYC1t gRNA expression cassette was obtained by digestion of pUD531 with SmaI; (v) the Escherichia coli pBR322 origin and bla gene for ampicillin selection were obtained by digestion of pUD532 with SmaI. The five fragments were gel-purified and 0.1 pmol of each fragment was assembled in a Gibson assembly reaction. An E. coli clone containing the correctly assembled plasmid (verified by digestion with PdmI) was stocked and pUDP002 (Addgene Plasmid #103872) was used as gRNA entry plasmid for subsequent plasmid construction.
The gRNA expression plasmids pUDP013 (plasmid #103873), pUDP025 (plasmid #103874), pUDP046 (plasmid #107062) and pUDP123 (plasmid #107269) were assembled in a one-pot ‘Golden Gate’ assembly (Engler, Kandzia and Marillonnet 2008) by BsaI digestion of pUDP002 and a synthesized donor plasmid containing the respective gRNA sequences flanked by ribozymes and BsaI restriction sites. pUDP013, pUDP025, pUDP046 and pUDP123 were constructed using pUD540, pUD555, pUD602 and pUD750, respectively. The Golden Gate assemblies were carried out in a final volume of 20 μL, using T4 DNA ligase buffer (Thermo Fisher Scientific), 10 U BsaI (New England Biolabs), 1 U T4 DNA ligase (Thermo Fisher Scientific) and 10 ng of both pUDP002 and the respective donor plasmid. For the construction of pUDP082 (plasmid #103875), the partially overlapping primers Km-ade2-F and Km-ade2-R were annealed, extended and amplified by PCR to yield a 233-bp double-stranded DNA fragment containing the KmADE2 gRNA target sequence flanked by ribozymes and BsaI sites, which was integrated into pUDP002 by Golden Gate assembly as described above. All constructed gRNA-harboring plasmids were verified by digestion with PdmI. Plasmids pUDP002, pUDP013, pUDP025, pUDP046, pUDP082 and pUDP123 were deposited at Addgene (https://www.addgene.org/).
The ADE2 repair DNA fragments of K. lactis (962 bp), K marxianus (959 bp), O. polymorpha (960 bp) and O. parapolymorpha (960 bp) were constructed from strains CBS 2359, CBS 5795, CBS 4732 and CBS 11895, respectively, using primer sets 10723 & 10724 and 10725 & 10726 (K. lactis), 10727 & 10728 and 10729 & 10730 (K. marxianus), 10346 & 10347 and 10348 & 10349 (O. polymorpha) and 10354 & 10355 and 10356 & 10357 (O. parapolymorpha) to amplify the homology regions flanking ADE2. Both regions were then joined by overlap extension PCR using primer sets 10723 & 10726, 10727 & 10730, 10346 & 10349 and 10354 & 10357 in the case of K. lactis, K. marxianus, O. polymorpha and O. parapolymorpha, respectively. In all cases, the final repair fragment was gel-purified and further amplified to obtain quantities required for transformation, using primer sets 10723 & 10726, 10727 & 10730, 10346 & 10349, and 10354 & 10357 for K. lactis, K. marxianus, O. polymorpha and O. parapolymorpha, respectively. The primers pairs were designed to amplify the 480 bp (±1 or 2 bp in some cases) upstream the ATG or the 480-bp terminator region downstream the stop codon of the interrupted/deleted ORF.
Design of gRNA target sequences and BsaI-flanked entry constructs
All 20-bp Cas9 gRNA target sequences used in this study are described in Table 3. Approximately 10 candidate target sequences were chosen from the first third of each targeted ORF, based on the presence of an NGG protospacer adjacent motif (PAM) site. Any target sequence with off-targets (defined as a sequence with NGG or NAG PAM and more than 15 nucleotides identical to the candidate sequence) was excluded, based on a blastn homology search against the respective yeast genome (https://blast.ncbi.nlm.nih.gov). The remaining target sequences were ranked based on AT content (‘AT score’; 0 being the lowest and 1 being the highest possible AT content) and secondary structure as predicted with the complete gRNA sequence, using minimum free energy prediction by RNAfold (Lorenz et al.2011) (‘RNA score’; 0 being the lowest and 1 being the highest possible number of unpaired target sequence nucleotides). Finally, the target sequences with the highest cumulative AT and RNA score that did not exceed an AT score of 0.8 were chosen for use in this study.
Table 3.
gRNA target sequences used in this study.
| Locus | Target sequence (5΄-3΄) | Position in ORF (bp) | AT score | RNA score |
|---|---|---|---|---|
| KlADE2 | TTTCAATACCTCAAGTGCTTCGG | 508/1710 | 0.65 | 0.70 |
| KmADE2 | GCCCATTTTTCTGCGTATAGCGG | 537/1710 | 0.55 | 0.70 |
| OpADE2* | CTGGAATTGATCTGCTTGGCCGG | 120/1704 | 0.50 | 0.35 |
| OpKU80 | CATCGTTCTGCAGAAGATCATGG | 340/2076 | 0.55 | 0.55 |
| OpYNR1 | AGCACAGACCATAGTAACTGGGG | 397/2580 | 0.55 | 0.55 |
Target sequences are shown including PAM sequence (underlined). The gRNA for OpADE2 targets the respective genes at the same position in both O. polymorpha and O. parapolymorpha. Position in ORF indicates the base pair after which the Cas9-mediated DSB is expected, out of the total length of the ORF. AT score indicates AT content of the 20-bp target sequence. RNA score indicates the fraction of unpaired nucleotides of the 20-bp target sequence, predicted with the complete gRNA sequence using minimum free energy prediction by RNAfold (Lorenz et al.2011).
*The same sequence was used for single (pUDP013) and for double (pUDP123) editing.
To integrate ribozyme-flanked gRNAs into pUDP002, the synthetic 233-bp dsDNA gRNA entry constructs were flanked by inward-facing BsaI sites generating sticky ends (underlined) ‘GGTCTCGCAAA’ and ‘ACAGCGAGACC’ at their 5΄ and 3΄ ends, respectively, compatible with BsaI-digested pUDP002. The sequence between the BsaI sites consisted of (i) the hammerhead ribozyme with the first six nucleotides being the reverse complement (c) of the first six nucleotides of the gRNA spacer ‘cN6cN5cN4cN3cN2cN1CUGAUGAGUCCGUGAGGACGAAACGAGUA AGCUCGUC’, (ii) followed by the 20-nucleotide gRNA spacer, followed by the structural gRNA ‘GUUUUAGAGCUAGAAAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCUUUU’, (iii) followed by the hepatitis delta virus ribozyme ‘GGCCGGCAUGGUCCCAGCCUCCUCGCUGGCGCCGGCUGGGCAACAUGCUUCGGCAUGGCGAAUGGGAC’ (Gao and Zhao 2014; Gorter de Vries et al.2017). For multiplexed targeting of O. parapolymorpha ADE2 and YNR1 using a single expression cassette, two ribozyme-flanked gRNAs were connected in a tandem array using 20-bp linker ‘GTGTAATGTCCAGAGTTGTG’, and otherwise constructed as described previously (Gorter de Vries et al.2017).
Strain construction
Yeast transformations
Kluyveromyces lactis and K. marxianus were transformed using the LiAc/single-strand carrier DNA/polyethylene glycol method (Gietz and Schiestl 2007). Overnight pre-cultures in YPD medium were used to inoculate a shake flask containing YPD medium to an initial OD660nm of 0.5. Cultures were then incubated at 30 °C until an OD660nm of 2.0 was reached, harvested and transformed with 200 ng of plasmid DNA and 300 or 1000 ng of repair DNA fragment in the case of K. lactis or K. marxianus, respectively. After heat shock, cells were recovered in 1 mL YPD medium for 3 h and plated on selective YPD medium. Plates were typically kept for 5 days at 30°C and then incubated at 4°C for 2 h before assessing the percentage of red Ade− colonies. Ogataea polymorpha and O. parapolymorpha were transformed using the procedure for preparation of competent cells and electroporation described by Saraya et al. (2014), with modifications. All steps were performed at 30°C, OD660nm of all cultures harvested for transformation was normalized to OD660nm 1.2 by dilution with sterile demineralized water, all centrifugation steps were carried out for 3 min at 3000 g, the DTT incubation step was done for 20 min, and the washing step with STM buffer was performed twice with 50 mL. Electroporation was carried out with 40 μL of a freshly prepared competent cell suspension in 2 mm gap cuvettes (Bio-Rad, Hercules, CA, USA), using 200 ng of plasmid DNA and 1 μg of repair fragment. A Micropulser Electroporator (Bio-Rad) set to the ‘Sc2’ preset (1.5 kV) was used for pulse delivery. After electroporation, cells were recovered in 1 mL YPD medium for 1 h at 30°C before plating onto selective YPD medium. For the direct inoculation of prolonged liquid incubation cultures, 100 μL of recovered cell suspension was used as inoculum. Selection plates were typically kept for 4 days at 30°C and then incubated at 4°C for a minimum of 24 h before assessing the percentage of red Ade− colonies.
Molecular diagnosis of yeast mutants
For molecular analysis of K. lactis and K. marxianus mutants, colonies were grown overnight in YPD medium. Genomic DNA was extracted using the method described by Singh and Weil (2002), and used as template for PCR reactions targeting the ADE2 locus. Primer sets 10909 & 10910, and 10911 & 10912 were used for K. lactis and K. marxianus, respectively. For Sanger sequencing of putative NHEJ-corrected mutants, PCR products were purified and then sequenced using primers 10737 and SeqADE2 for K. lactis and K. marxianus, respectively. For O. polymorpha and O. parapolymorpha, genomic DNA was directly isolated from colonies, using the LiAc-sodium dodecyl sulfate method (Looke, Kristjuhan and Kristjuhan 2011). Primer sets 10380 & 10381, and 10915 & 10916 were used to check for deleted ADE2 loci in O. polymorpha and O. parapolymorpha, respectively, while sequencing of NHEJ-corrected mutants was done using primer sets 10378 & 10379, and 10386 & 10387, respectively. The deleted ADE2 locus in strain IMK828 was amplified and Sanger-sequenced using primers 10915 and 10916. Additionally for O. parapolymorpha primer pair 12257 & 12266 (Table 4) was used to amplify OpYNR1 to verify editing of this locus by Sanger sequencing.
Table 4.
Primers used in this study.
| Name | Sequence (5΄ - 3΄) | Purpose |
|---|---|---|
| 3841 | CACCTTTCGAGAGGACGATG | Construction of pUDP002 |
| 3856 | CTAGCGTGTCCTCGCATAGTTC | Construction of pUDP002 |
| 9393 | TGCCGAACTTTCCCTGTATGAAGCGATCTGACCAATCCTTTGCC GTAGTTTCAACGTATGTTTTCATTTTTGCGATGCCAG | Construction of pUDP002 |
| 9663 | CATACGTTGAAACTACGGCAAAGGATTGGTCAGATCGCTTCAT ACAGGGAAAGTTCGGCATCAACATCTTTGGATAATATCAGAATGAG | Construction of pUDP002 |
| 9837 | ATACAGTTCTCACATCACATCCGAACATAAACAAGGATCCATG GGTAAAAAGCCTGAACTC | Construction of pUDP002 |
| 9838 | ACAAGTTCTTGAAAACAAGAATCTTTTTATTGTCCTCGAG TTATTCCTTTGCCCTCGGAC | Construction of pUDP002 |
| 9839 | CTCGAGGACAATAAAAAGATTCTTG | Construction of pUDP002 |
| 9840 | GGATCCTTGTTTATGTTCGGATG | Construction of pUDP002 |
| 9841 | ACTATATGTGAAGGCATGGC | Construction of pUDP002 |
| 9842 | GTTGAACATTCTTAGGCTGG | Construction of pUDP002 |
| Km-ade2-F | GGTCTCGCAAAGTCAAGCTGATGAGTCCGTGAGGACGAAACG AGTAAGCTCGTCGCCCATTTTTCTGCGTATAGGTTTT AGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCC GTTATCAACTTGAAAAAGTGGCACC | Construction of pUDP082 |
| Km-ade2-R | GGTCTCGCTGTGTCCCATTCGCCATGCCGAAGCATGTTGCCC AGCCGGCGCCAGCGAGGAGGCTGGGACCATGCCGGCCA AAAGCACCGACTCGGTGCCACTTTTTCAA GTTGATAACGGACTAGCCTTATTTTAACTTGC | Construction of pUDP082 |
| 10723 | GTAGTACCGACCTTATCCGTG | Construction of K. lactis ADE2 repair fragment |
| 10724 | GTTGTCTTAGTGAAGAAGGTGAAC | Construction of K. lactis ADE2 repair fragment |
| 10725 | TATATAATAACATCACGTTCACCTTCTTCACTAAGACAACAGCTGC CAAATTAGAAACTATCG | Construction of K. lactis ADE2 repair fragment |
| 10726 | TGTGCGTTGATATATGCCAAC | Construction of K. lactis ADE2 repair fragment |
| 10727 | ATCATAGACAGTCAGTTAGTTCCC | Construction of K. marxianus ADE2 repair fragment |
| 10728 | TTCTTTGGTCCATGATTAACAAGG | Construction of K. marxianus ADE2 repair fragment |
| 10729 | ACTACAACAATATAAACCTTGTTAATCATGGACCAAAGAAGTATTC AACTACCTCCAACAAGAAG | Construction of K. marxianus ADE2 repair fragment |
| 10730 | CAAATTTATGAAGTTTGTGCCATTTG | Construction of K. marxianus ADE2 repair fragment |
| 10346 | TGTGCACTCAATTGCAACC | Construction of O. polymorpha ADE2 repair fragment |
| 10347 | TTCCAACGACCTTTGAGTCC | Construction of O. polymorpha ADE2 repair fragment |
| 10348 | TAATTTAATTTAATTTACATGGACTCAAAGGTCGTTGGAAGTTGGC TATGAGGAATACCTTAAC | Construction of O. polymorpha ADE2 repair fragment |
| 10349 | GGGACGTTTACTGGACGG | Construction of O. polymorpha ADE2 repair fragment |
| 10354 | CCTGATGTGCACTCAATTGC | Construction of O. parapolymorpha ADE2 repair fragment |
| 10355 | CAACGACCTTCGAGTCCATC | Construction of O. parapolymorpha ADE2 repair fragment |
| 10356 | TATTAATTTAATTTAATTTAGATGGACTCGAAGGTCGTTGCTCTG TTGGCTATGAAGAATACC | Construction of O. parapolymorpha ADE2 repair fragment |
| 10357 | GTTTATTGGATGGCAATCTCG | Construction of O. parapolymorpha ADE2 repair fragment |
| 10737 | AATTGCATCTCTTTGTGATGTC | Sanger sequencing of K. lactis ADE2 disruptions |
| SeqADE2 | CTGCAACTGCTTGTTCAGCC | Sanger sequencing of K. marxianus ADE2 disruptions |
| 10378 | CCAATTACAAGTACTACTTCGAG | Sanger sequencing of O. polymorpha ADE2 disruptions fw |
| 10379 | CTAGCTCCTTGGTGAAAGG | Sanger sequencing of O. polymorpha ADE2 disruptions rv |
| 10386 | ACAAGTACTACTTCGAGGAC | Sanger sequencing of O. parapolymorpha ADE2 disruptions fw |
| 10387 | CTAGCTCCTTGGTAAAGGG | Sanger sequencing of O. parapolymorpha ADE2 disruptions rv |
| 10751 | GGACGCCTGCTTAGACTTG | Sanger sequencing of O. parapolymorpha KU80 disruptions fw |
| 10752 | AGCACGGTATATTCGCACAG | Sanger sequencing of O. parapolymorpha KU80 disruptions rv |
| 12257 | CACCATGGTCGGAAGAACC | Sanger sequencing of O. parapolymorpha YNR1 disruptions fw |
| 12266 | ATGTAATTCCTCACGAACTTTGG | Sanger sequencing of O. parapolymorpha YNR1 disruptions rv |
| 10909 | TCTTCGTCGCCATTTATTGTTGAG | Diagnosis of K. lactis ADE2 deletions fw |
| 10910 | CTATTGCGGTTCGCTCTTCC | Diagnosis of K. lactis ADE2 deletions rv |
| 10911 | ATTCGCCGAATCTGACGTG | Diagnosis of K. marxianus ADE2 deletions fw |
| 10912 | TGGTGTGCAGACGGATAAAC | Diagnosis of K. marxianus ADE2 deletions rv |
| 10380 | AGGTGCTCAAACACAAAGAG | Diagnosis of O. polymorpha ADE2 deletions fw |
| 10381 | TCGTATCTCGTAAGTTGATTTAGG | Diagnosis of O. polymorpha ADE2 deletions rv |
| 10915 | CCGTCTGAACGGAATGATGTC | Diagnosis and Sanger sequencing of O. parapolymorpha ADE2 deletions fw |
| 10916 | CCCTCAACTGCAGACACATAG | Diagnosis and Sanger sequencing of O. parapolymorpha ADE2 deletions rv |
Construction of IMK829, IMK830, IMD001, IMK828 and IMD034
To construct IMK829 (K. lactis Δade2), strain CBS 2359 was co-transformed with 200 ng of pUDP025 (harboring gRNAKlADE2) and 300 ng of a 962-bp repair DNA fragment as described above. The resulting transformants were analyzed by diagnostic PCR using primers 10909 and 10910, and a mutant exhibiting a PCR product compatible with deletion of the ADE2 ORF was isolated and renamed IMK829. To construct IMK830 (K. marxianus Δade2), strain NBRC 1777 was co-transformed with 200 ng of pUDP082 (harboring gRNAKmADE2) and 1 μg of a 959-bp repair DNA fragment as described above. The resulting transformants were analyzed by diagnostic PCR using primers 10911 and 10912, and a mutant exhibiting a PCR product compatible with deletion of the ADE2 ORF was isolated and renamed IMK830. For the construction of IMD001 (O. parapolymorpha ku80A340AA), the O. parapolymorpha KU80 ORF (accession XM_014078010.1) was identified by a tblastn homology search (https://blast.ncbi.nlm.nih.gov) using the S. cerevisiae YKU80 protein sequence and O. parapolymorpha CBS 11895 (DL-1) RefSeq assembly (accession GCF_000187245.1) (Ravin et al.2013). Strain CBS 11895 was transformed with 200 ng of pUDP046 (harboring gRNAOpKU80), and a single transformant was picked and used to inoculate a prolonged liquid incubation culture as described above. After 96 h, cells were plated on selective YPD medium, genomic DNA was isolated from randomly picked colonies, and the KU80 locus was amplified and Sanger-sequenced using primers 10751 and 10752. A mutant containing a single adenine nucleotide inserted between position 340 and 341 of the KU80 ORF was restreaked three times subsequently on non-selective YPD medium to remove pUDP046, and renamed IMD001. To construct IMK828 (O. parapolymorpha ku80A340AAΔade2), strain IMD001 was co-transformed with 200 ng of pUDP013 (harboring gRNAOpADE2) and 1 μg of a 960-bp repair DNA fragment as described above. After recovery, 100 μL of transformation cell suspension was directly used for inoculation of a prolonged liquid incubation culture. After 48 h, cells were plated on selective medium and the resulting colonies inspected for occurrence of the red Ade− phenotype. A mutant which exhibited a PCR product compatible with deletion of the ADE2 ORF when analyzed by diagnostic PCR using primers 10915 and 10916 was restreaked thrice subsequently on non-selective YPD medium to remove pUDP013, and renamed IMK828. For the construction of IMD034 (O. parapolymorpha ade2C120CA ynr1G397GT), the O. parapolymorpha HPODL_02384/YNR1 ORF (accession XM_014082012.1) was identified in O. parapolymorpha CBS 11895 (DL-1) RefSeq assembly (accession GCF_000187245.1) as previously described for OpKU80. Strain CBS 11895 was transformed with 200 ng of pUDP123 (harboring dual gRNAOpADE2-OpYNR1), and a single transformant was picked and used to inoculate a prolonged liquid incubation culture as described above. After 96 h, cells were plated on selective YPD medium. A set of 94 Ade− mutants were picked and grown overnight on non-selective YPD and then replica plated on SM and SMN media. A set of five transformants exhibiting both Ade− (red colonies) and Nit− (absence of growth on nitrate) phenotypes were randomly picked, genomic DNA was isolated and the OpADE2 and OpYNR1 loci were amplified and Sanger-sequenced using primer pairs 10386 & 10387 and 12257 & 12266 respectively. One sequence-confirmed double interruption mutant was restreaked three times subsequently on non-selective YPD medium to remove pUDP123, and renamed IMD034 (Table 1).
Molecular biology techniques
PCR amplification with Phusion High Fidelity Polymerase (Thermo Fisher Scientific) was performed according to the manufacturer's instructions using PAGE-purified oligonucleotide primers (Sigma-Aldrich, St. Louis, MO, USA). Diagnostic PCR was done using DreamTaq polymerase (Thermo Fisher Scientific) and desalted primers (Sigma-Aldrich). All primer sequences are shown in Table 4. DNA fragments obtained by PCR were separated by gel electrophoresis. Gel purification was carried out using the Zymoclean Gel DNA Recovery Kit (Zymo Research, Irvine, CA, USA). PCR purification was performed using either the GenElute PCR Clean-Up Kit (Sigma-Aldrich) or GeneJET PCR purification kit (Thermo Fisher Scientific). Gibson assembly was done using the NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs, Ipswich, MA, USA) according to the manufacturer's recommendations. Restriction digest with PdmI and SmaI was performed using FastDigest enzymes (Thermo Fisher Scientific), according to the manufacturer's instructions. Escherichia coli strain XL1-blue was used for plasmid transformation, amplification and storage. Plasmids were isolated from E. coli with the GenElute Plasmid Miniprep Kit (Sigma-Aldrich).
Whole-genome sequencing of O. parapolymorpha IMD001
Genomic DNA of O. parapolymorpha IMD001 was isolated with the QIAGEN Genomic-tip 100/G kit (Qiagen, Hilden, Germany) from a stationary-phase overnight shake-flask culture grown on YPD medium, according to the manufacturer's instructions. Genomic DNA was quantified using a Qubit® 2.0 Fluorometer (Thermo Fisher Scientific). Ten micrograms of genomic DNA was sequenced by Novogene Bioinformatics Technology (Yuen Long, Hong Kong, China) on a HiSeq 2500 (Illumina, San Diego, CA, USA) with 150-bp paired-end reads using TruSeq PCR-free library preparation (Illumina). In order to verify complete absence of plasmid pUDP046, sequencing reads were mapped to the sequence of pUDP046 and to the genome of O. parapolymorpha CBS 11895 (accession GCF_000187245.1) (Ravin et al.2013) using the Burrows–Wheeler Alignment tool and further processed using SAMtools (Li et al.2009; Li and Durbin 2010). The absence of sequences from pUDP046 was confirmed by visualizing the generated .bam files in the Integrative Genomics Viewer software (Robinson et al.2011). Sequencing data are available at NCBI (https://www.ncbi.nlm.nih.gov/) under BioProject PRJNA413643.
RESULTS
pUDP: a plasmid-based wide-host-range yeast CRISPR/Cas9 system
The pUDP system was designed to enable Cas9-mediated genome editing in different yeast species by simple transformation with a single plasmid. To this end, DNA parts encoding the plasmid origin of replication, cas9 expression cassette, selection marker and gRNA expression cassette were chosen to function in a wide range of yeast species (Fig. 1). To ensure replication of pUDP plasmids, the pangenomic yeast origin of replication panARS(OPT) was used. This origin of replication, which was inspired by a K. lactis chromosomal ARS, has been shown to function in at least 10 different species of ascomycetous yeasts, including the species used in this study (Liachko and Dunham 2014). The cas9D147Y P411T nuclease variant (Bao et al.2015) was expressed under control of the TEF1 promoter from Arxula adeninivorans (Blastobotrys adeninivorans), which enabled strong constitutive expression in various yeasts (Steinborn et al.2006; Terentiev et al.2004). Similarly, the K. pneumoniae hph gene encoding hygromycin B phosphotransferase, which conferred hygromycin resistance in a wide range of microorganisms, was expressed under control of the Ashbya gossypii (Eremothecium gossypii) TEF1 promoter (Wach et al.1994) that showed activity in a range of yeasts (Mazzoni, Serafini and Falcone 2005; Kim et al.2010). To avoid RNA modifications that interfere with biological function, transcription of gRNAs for genome editing is commonly controlled by RNA polymerase III promoters. By analogy with expression of gRNA in mammalian cells (Cong et al.2013; Mali et al.2013), the RNA polymerase III SNR52 promoter was used in S. cerevisiae (DiCarlo et al.2013; Mans et al.2015), but accurate annotation and characterization of this promoter might not be available for other yeast species. Therefore, self-processing ribozyme-flanked gRNAs were expressed from an RNA polymerase II promoter (Gao and Zhao 2014; Ryan et al.2014). This concept has already been successfully applied in several different organisms (Gao et al.2016; Gorter de Vries et al.2017; Weninger et al.2016). In this system, the gRNA is flanked by a hammerhead and hepatitis delta virus ribozyme at its 5΄ and 3΄ end, respectively, resulting in precise release of mature gRNA after self-cleavage. To ensure sufficient gRNA transcription, the strong glycolytic TDH3 promoter from S. cerevisiae was used. These elements were combined in plasmid pUDP002, which could subsequently serve as entry plasmid to insert any desired gRNA. To streamline integration of ribozyme-flanked gRNAs, pUDP002 contained two different BsaI restriction sites between ScTDH3p and ScCYC1t, enabling directional insertion of synthetic BsaI-flanked gRNA constructs (Gorter de Vries et al.2017).
Figure 1.

Components of the pUDP genome editing system. (A) Map of pUDP002 (Addgene plasmid #103872), a wide-host-range gRNA entry plasmid. pUDP002 is composed of a hph (HygR) selection marker cassette under control of the TEF1 promoter from Ashbya gossypii conferring hygromycin resistance, Spcas9D147Y P411T under control of the TEF1 promoter from Arxula adeninivorans, the pangenomic yeast replication origin panARS(OPT), a BsaI cloning site for entry of gRNA constructs, and a pBR322-derived E. coli origin and bla gene for ampicillin selection. A, B, F, C and I indicate 60 bp synthetic homologous recombination sequences used for ‘Gibson’ assembly of the plasmid. (B) Representation of the ribozyme-flanked gRNA expression cassette design. Guide RNAs (represented by gRNAADE2) were flanked on their 5΄ by a hammerhead ribozyme (HH represented in orange) and on their 3΄ by a hepatitis delta virus ribozyme (HDV represented in bronze). When integrated into pUDP002, this construct is under control of the RNA polymerase II promoter TDH3 and the CYC1 terminator from S. cerevisiae. Upon ribozyme self-cleavage, a mature gRNA comprised of the guiding protospacer (in blue) and the structural gRNA fragment (in green) is released.
To test the genome-editing efficiency of the pUDP system, the ADE2 gene was targeted in all four non-conventional yeasts used in this study: K. lactis, K. marxianus, O. polymorpha and O. parapolymorpha. The ADE2 gene encodes a phosphoribosylaminoimidazole carboxylase, also referred to as AIR carboxylase, involved in adenine biosynthesis. Besides causing adenine auxotrophy, loss-of-function mutations in ADE2 result in a red-color phenotype due to accumulation of the oxidized form of 5-amino imidazole ribonucleotide (AIR). This allows detection of ADE2 targeting by simple visual inspection of transformation plates (Roman 1956). Therefore, gRNAs were designed targeting ADE2 in each species based on available genome data (Table 3), and gRNA-harboring plasmids pUDP025, pUDP082 and pUDP013 for deletion of ADE2 in K. lactis, K. marxianus and O. polymorpha/O. parapolymorpha, respectively, were constructed.
Efficient CRISPR/Cas9 targeting enables gene deletion in Kluyveromyces species
To test the effectiveness of the pUDP system in K. lactis, gRNAKlADE2 was inserted into pUDP002 and the resulting plasmid pUDP025 was used to target ADE2 in K. lactis CBS 2359. Transformation with pUDP025 without a repair DNA fragment yielded a total of 35 transformants of which 19 (54%) exhibited a red Ade− phenotype. In the presence of a 962-bp repair fragment which targeted the ADE2 promoter and terminator (Fig. 2A), the transformation of pUDP025 yielded 26 red transformants out of a total 27 (96%) (Fig. 2B). In comparison, transformation of the backbone plasmid pUDP002, which does not express a gRNA, generated a number of transformants that was ca. 35-fold higher, none of which displayed a red phenotype. This difference in transformation efficiency already provided information about the quality of the gRNA and the editing. Unless repaired a chromosomal DSB should be lethal, therefore this 35-fold drop in transformation efficiency would represent the fraction of transformants that did not successfully repair the CRISPR-Cas9 induced break and subsequently died from it. Out of the 26 Ade− transformants obtained after co-transformation with pUDP025 and repair fragment, 13 red colonies were randomly picked and analyzed by diagnostic PCR, revealing two distinct groups. Nine colonies exhibited a normally sized ADE2 locus, while four colonies (31%) showed a PCR-fragment size compatible with complete deletion of the KlADE2 ORF (Fig. 2C). Subsequent Sanger sequencing of the amplified fragments derived from the nine clones exhibiting an ADE2 wild-type size band identified the presence of indels at the targeted site. These indels reflected imperfect repair of the Cas9-induced DSB via NHEJ, resulting in introduction of loss-of-function mutations and disruption of the Cas9 target site (Fig. 2D). These results demonstrated that the pUDP system could be used for efficient targeting of the ADE2 gene in K. lactis CBS 2359, resulting in repair by either HR or NHEJ DNA repair mechanisms.
Figure 2.

Efficient gRNA targeting in K. lactis CBS 2359 enables marker-free gene deletion. (A) Schematic representation of ADE2 editing upon transformation of CBS 2359 with pUDP025 (gRNAKlADE2) and a repair DNA fragment. The primers for diagnostic PCR of transformants are indicated. (B) Colony morphology of CBS 2359 upon transformation with pUDP025 and a marker-free 962 bp repair fragment. (C) Diagnosis of 13 randomly picked red Ade− transformants of CBS 2359 upon transformation with pUDP025 and a 962-bp marker-free repair fragment. Four transformants (HR mutants 1–4) showed a PCR product of 1177 bp corresponding to the deleted allele. The control labeled CBS 2359 and nine transformants (NHEJ mutants 5–13) showed a PCR product of 2838 bp corresponding to the wild-type allele. (D) Sanger sequencing results of purified PCR fragments from nine Ade− mutants (corresponding to mutants 5–13 in panel C) derived from the transformation of CBS 2359 with pUDP025 and repair fragment.
The pUDP system was similarly tested in K. marxianus by targeting ADE2 in the haploid strain NBRC 1777 and the diploid strain CBS 397, using plasmid pUDP082 expressing gRNAKmADE2. The transformation of K. marxianus NBRC 1777 with pUDP082, without and with a 959-bp repair DNA fragment resulted in 13 and 30 transformants, respectively. Of these, 13 out of 13 and 29 out of 30 transformants were red, indicating successful disruption of ADE2 in both cases. To determine which repair mechanism contributed to the repair of the Cas9-induced DSB in the presence of the repair fragment, 13 randomly picked red transformants were subjected to diagnostic PCR. Similar to the results obtained in K. lactis, two groups of transformants were identified. Three transformants (24%) showed a genotype corresponding to the repair of the DSB with the repair fragment (Fig. 3A and B), while the remaining transformants exhibited a PCR result that was undistinguishable from that of the NBRC 1777 ADE2 wild-type locus. Sanger sequencing of the Cas9 target site again revealed the presence of indels, leading to nonsense mutations in the ADE2 coding sequence (Fig. 3C).
Figure 3.

Efficient gRNA targeting enables marker-free gene deletion in haploid K. marxianus NBRC 1777 and gene disruption in diploid K. marxianus CBS 397. (A) Schematic representation of the ADE2 editing upon transformation of NBRC 1777 with pUDP082 (gRNAKmADE2) and a repair DNA fragment. The primers for diagnostic PCR of transformants are indicated. (B) Diagnosis of 13 randomly picked red Ade− transformants of NBRC 1777 upon transformation with pUDP082 and a 959-bp marker-free repair fragment. Three transformants (HR mutants 1–3) showed a PCR product of 1218 bp corresponding to the deleted allele. The control labeled NBRC 1777 and 10 transformants (NHEJ mutants 4–13) showed a PCR product of 2915 bp corresponding to the wild-type allele. (C) Sanger sequencing results of purified PCR fragments from 10 Ade− mutants (corresponding to mutants 4–13 in panel B) derived from the transformation of NBRC 1777 with pUDP082 and repair fragment. (D) Sanger sequencing results of purified PCR fragments of 10 randomly picked red Ade− mutants derived from the transformation of CBS 397 with pUDP082 and repair fragment.
When the diploid strain CBS 397 was transformed with pUDP082 without repair DNA fragment, 106 out of 133 (80%) of the colonies were red, while 117 out of 143 (82%) colonies showed the red phenotype when a 959-bp repair fragment was provided. In comparison, a control transformation with pUDP002 yielded 262 transformants without any red phenotypes. From the transformation with pUDP082 and repair fragment, 10 red colonies were randomly picked and analyzed by diagnostic PCR, resulting in PCR products that were identical in size to the native ADE2 locus in all cases. Sanger sequencing of the same mutants confirmed the link between the red-colored phenotype and occurrence of small indels at the Cas9 cut site, which likely introduced loss of function mutations in ADE2 (Fig. 3D). Interestingly, Sanger sequencing also indicated that the editing occurred identically at both KmADE2 alleles as a clear and continuous sequence was observed over the cut site. These results demonstrated that the ADE2 gene can be efficiently targeted by the pUDP system in both K. marxianus strains, with DNA repair mediated by HR or NHEJ mechanisms in the haploid strain NBRC 1777 and NHEJ in the diploid strain CBS 397.
CRISPR/Cas9 editing enables gene deletion in Ogataea species
To test the effectiveness of the pUDP system in O. parapolymorpha, plasmid pUDP013 harboring gRNAOpADE2 was used to transform strain CBS 11895 (DL-1). Transformations were performed with or without a 960-bp repair DNA fragment designed to mediate HR at the promoter and terminator regions of the OpADE2 gene (Fig. 4A). In presence of the repair fragment, an average of 298 ± 50 colonies per transformation were obtained, while a lower average number of 64 ± 9 colonies were counted after transformation without the repair fragment. In contrast to the situation in Kluyveromyces species, none of these colonies exhibited a red Ade− phenotype, although the ability of the transformants to grow on selective medium indicated that the plasmid was present. The heterologous origin of genetic parts on pUDP002 might result in a suboptimal expression of the necessary components of the CRISPR-Cas9 machinery in O. parapolymorpha. To allow more time for expression of Cas9 and gRNA, two randomly picked transformants (CBS 11895 + pUDP013) from independent experiments were incubated for a longer period in selective YPD medium. These prolonged liquid incubation cultures were sampled after 48, 96 and 192 h, and samples taken at each of these time points were plated on selective YPD medium. Already after 48 h of incubation, 31 ± 7% of the plated colonies exhibited the red Ade− phenotype. This fraction increased to 61 ± 2% and 63 ± 2% after 96 and 192 h of incubation, respectively (Fig. 4B). From a plate obtained after 96 h of liquid incubation, 10 red colonies were randomly picked and subjected to diagnostic PCR. All 10 transformants showed a band compatible with the size of the wild-type ADE2 locus (Fig. 4C). As previously found in K. lactis and K. marxianus, Sanger sequencing of the ADE2 gene in these 10 transformants (NHEJ #1 to #10) confirmed the presence of short indels at the Cas9 target site, which introduced nonsense mutations within ADE2 (Fig. 4D). The presence of the repair fragment did not change this outcome: prolonged liquid incubation cultures that were started from colonies obtained by co-transformation of pUDP013 and the repair fragment gave rise to identical proportions of Ade− mutants as those described above. For the closely related species O. polymorpha, the obtained results were qualitatively comparable. Transformation of strain O. polymorpha CBS 4732 with pUDP013 did not result in colonies with a red phenotype, neither in the presence nor in the absence of a 960-bp repair DNA fragment. Also, no mutations in ADE2 were detected as evaluated by Sanger sequencing of nine randomly picked (white) colonies from a transformation without the repair fragment. When two independent transformants (CBS 4732 + pUDP013) were incubated in liquid medium, plating after 192 h yielded 9 ± 1% of red colonies. These results indicated that the pUDP system promoted less efficient genome editing in the O. polymorpha strain CBS 4732 than in O. parapolymorpha CBS 11895, although the system could be utilized for NHEJ-mediated disruption of the ADE2 gene in both yeasts.
Figure 4.

Prolonged liquid incubation enables gene disruption in O. parapolymorpha CBS 11895 (DL-1). (A) Schematic representation of the potential ADE2 editing upon transformation of CBS 11895 with pUDP013 (gRNAOpADE2) and a repair DNA fragment. (B) Percentage of red Ade− colonies observed based on phenotypic screening of CBS 11895 + pUDP013 (gRNAOpADE2) transformants when plated directly after transformation (0 h), or after prolonged liquid incubation of established transformants in selective medium (for 48, 96 and 192 h) and subsequent plating. Mean and standard deviation were calculated with a total of 4411, 4710 and 4301 colonies (obtained after 48, 96 and 192 h, respectively) from two liquid incubation cultures started with single CBS 11895 transformants which were obtained in independent transformations. No red Ade− mutants were observed when cells were plated directly after transformation. (C) Diagnosis of 10 randomly picked red Ade− colonies of CBS 11895 after transformation with pUDP013 and 96 h of subsequent liquid incubation under selective conditions. All 10 transformants (NHEJ mutants 1–10) and the control labeled CBS 11895 showed a PCR product of 2877 bp corresponding to the wild-type allele. (D) Sanger sequencing results of purified PCR fragments from 10 red Ade− colonies (corresponding to mutants 1–10 in panel B) derived from the transformation of CBS 11895 with pUDP013 and 96 h of subsequent liquid incubation
In contrast to S. cerevisiae (Baudin et al.1993) and Kluyveromyces species (this study), the HR-mediated DNA repair mechanism in Ogataea species is relatively inefficient (Klinner and Schäfer 2004). The removal of KU80 in O. polymorpha has been reported to result in a strong reduction of NHEJ in return favoring the occurrence of HR-mediated DNA repair (Saraya et al.2012). To delete KU80 in O. parapolymorpha, plasmid pUDP046 harboring gRNAOpKU80 was constructed and used to transform strain CBS 11895 (Fig. 5A). Following the liquid incubation procedure established for disruption of OpADE2, a single transformant was picked, incubated in selective YPD medium for 96 h and plated on selective YPD plates. Of eight colonies randomly picked and subjected to Sanger sequencing of the KU80 locus, five (NHEJ #3 to #7) contained a single adenine nucleotide inserted at the Cas9 targeting site between position 340 and 341 of the OpKU80 coding sequencing (A340AA), resulting in a loss-of-function mutation (Fig. 5B). One of these transformants was isolated by restreaking three times on non-selective medium, and renamed IMD001 (Opku80A340AA).
Figure 5.

Construction of IMD001, an O. parapolymorpha ku80 strain that enables low efficiency gene deletion. (A) Schematic representation of the OpKU80 editing upon transformation of CBS11895 with pUDP046 (gRNAOpKU80). The primers for diagnostic PCR and sequencing of transformants are indicated. (B) Sanger sequencing results of purified PCR fragments from eight randomly picked colonies derived from the transformation of CBS 11895 with pUDP046 (gRNAOpKU80) and 96 h of subsequent liquid incubation. The transformants NHEJ #1,NHEJ#2 and NHEJ#8 displayed a wild-type sequence, while mutants NHEJ #3 to #7 included frameshift mutations (C) Schematic representation of the ADE2 editing upon transformation of O. parapolymorpha IMD001 (CBS 11895 ku80) with pUDP013 (gRNAOpADE2) and a repair DNA fragment. The primers for the diagnostic PCR of transformants are indicated. (D) Diagnosis of all seven red Ade− colonies obtained (from a total of ca. 1900 colonies) upon transformation of IMD001 with pUDP013 and a 960-bp marker-repair fragment, and subsequent liquid incubation in selective conditions for 48 h, started directly from the transformation recovery culture. All mutants showed a PCR product of 1226 bp corresponding to the deleted allele. The control labeled IMD001 showed a PCR product of 2915 bp corresponding to the wild-type allele.
While strain IMD001 was not able to grow on selective medium indicating loss of pUDP046, complete curation of the plasmid had to be verified in order to use IMD001 for subsequent genetic interventions with pUDP plasmids. Circular plasmids with limited homology to the nuclear DNA have been reported to integrate into the genome of Ogataea (Hansenula) yeasts, despite the presence of an origin of replication on the plasmid (Kunze, Kang and Gellissen 2009). In particular, upon cultivation under selective conditions, plasmids were found to have integrated into the nuclear genome with high variations in copy number in strain CBS 11895 (DL-1) (Kang et al.2001). To verify that no pUDP046 plasmid sequences had recombined into the chromosomal DNA of strain IMD001, its genome was sequenced using Illumina 150-bp pair-end short reads that were mapped onto a reference comprised of an O. parapolymorpha CBS 11895 genome assembly (accession number: GCF_000187245.1) and the sequence of pUDP046. No reads were found to map onto the pUDP046 sequence, thus confirming the absence of unwanted integration of the transformed plasmid into the nuclear genome and the suitability of O. parapolymorpha IMD001 as a host strain for further genetic engineering.
To test the impact of the KU80 disruption on the pUDP system in O. parapolymorpha, plasmid pUDP013 (gRNAOpADE2) was used to transform strain IMD001 together with the 960-bp repair DNA fragment. As previously observed for the wild-type strain O. parapolymorpha CBS 11895, no red Ade− transformants were observed on the transformation plates. In an attempt to improve occurrence of homology-mediated repair, a liquid incubation culture was directly inoculated with the transformation mix, grown for 48 h and then plated on selective YPD medium. On these plates, only 7 out of ∼1900 colonies exhibited a red Ade− phenotype, representing a <1% targeting efficiency. Diagnostic PCR performed on these seven transformants revealed that all harbored a deleted ADE2 version compatible with HR-mediated repair of the CRISPR-Cas9-induced DSB (Fig. 5C and D). One of the obtained mutants was further analyzed by Sanger sequencing, which confirmed scarless integration of the repair fragment. Longer liquid incubation times of up to 192 h did not increase the incidence of red colonies after plating. These results demonstrated that the KU80 disruption abolished NHEJ-mediated DSB repair in O. parapolymorpha IMD001, and that the CBS 11895 strain background possesses a basal HR-mediated DNA repair activity that can be utilized for precise genome engineering. However, the low incidence of HR-mediated repair precludes efficient genome editing unless the desired phenotype is easily screenable.
Multiplexed gene targeting in O. parapolymorpha by expression of double ribozyme-flanked gRNA array
As recently demonstrated, HH-HDV ribozyme-flanked gRNAs can be concatenated into polycistronic arrays enabling multiplexing gene editing in S. pastorianus (Gorter de Vries et al.2017) and transcriptional interference in S. cerevisiae (Deaner, Mejia and Alper 2017). To explore this possibility in O. parapolymorpha, a gRNA expression plasmid pUDP123 carrying spacers targeting OpADE2 and OpYNR1 was designed. The additionally targeted YNR1 gene encodes a nitrate reductase, which is involved in utilization of nitrate as N-source (Brito et al.1996; Navarro et al.2003). Ogataea strains harboring a non-functional nitrate reductase are unable to grow on media with nitrate as sole N-source.
A tandem array of [HH-gRNA-HDV] targeting OpADE2 and OpYNR1 spaced with a 20-bp linker and expressed under control of the ScTDH3 promoter as previously described (Gorter de Vries et al.2017) was carried by the recombinant plasmid pUDP123 (hph cas9 ScTDH3p-HH-gRNAOpADE2-HDV-HH-gRNAOpYNR1-HDV-ScCYC1t) that was transformed in O. parapolymorpha CBS 11895 (Fig. 6A). Following the liquid incubation procedure established for disruption of OpADE2 and OpKU80, a transformant was picked, incubated in selective YPD medium for 96 h and then plated onto selective YPD medium. Of the resulting colonies, 32% were exhibiting a red Ade− phenotype. To verify whether these Ade– mutants were able or not to grow on nitrate, 94 red transformants were randomly picked and grown on non-selective medium (YPD). After full growth, 10 μl of cell suspension was replicated on non-selective (SM) and selective (SMN) media supplemented with 15 mg L−1 adenine to complement the Ade− phenotype. About 10% of the red-phenotype transformants gradually returned white. While one cannot exclude a reversion of the mutation, the pattern observed would suggest that the replicated red colonies were not pure. Growth of the transformant on SMN was disturbed by the addition of adenine which might also serve as nitrogen source as O. parapolymorpha is equipped with an adenine deaminase. However, since the adenine concentration remained ∼300-fold lower than the nitrate concentration, a significant difference between Nit+ and Nit− strains should be noticeable. Indeed out of the 94 red transformants, 17 showed a strongly reduced growth on SMN supplemented with adenine when compared to the control CBS 11895, and 5 of these potential double mutants were further analyzed by Sanger sequencing. While all five were confirmed to harbor a frameshift around the CRISPR cut site in OpADE2, only three transformants were concomitantly exhibiting a frameshift in OpYNR1 (Fig. 6C). Transformant #2 was isolated by restreaking three times on non-selective medium, and renamed IMD034 (Opade2C120CAOpynr1G397GT). Assuming that all other red clones that were also growing slowly on SMN medium shared this genotype, double editing would have occurred at a rate of 2%–5%.
Figure 6.

Simultaneous deletion of OpADE2 and OpYNR1 alleles using a single ribozyme flanked gRNA array in O. parapolymorpha CBS11895. (A) Representation of the gRNA array expression cassette in pUDP123. The dual gRNA array was under the control of the RNA polymerase II promoter ScTDH3 and ScCYC1 terminator. Each gRNA was flanked on its 5΄ by a hammerhead ribozyme (HH represented in orange) and on its 3΄ by a hepatitis delta virus (HDV represented in bronze) ribozyme which were separated by a 20-bp linker. Upon ribozyme self-cleavage, the mature gRNAs are released. The OpADE2 guiding spacer (in purple), the OpYNR1 guiding spacer (in yellow) and the constant structural gRNA fragment (in green) are indicated. (B) Schematic representation of the OpADE2 and OpYNR1 loci of CBS11895. The primers for the validation of transformants are indicated. (C) Sanger sequencing results of OpADE2 and OpYNR1 editing site of five randomly picked red Ade– mutants that have lost ability to grow on nitrate. Transformant labeled Transf#2 was renamed IMD034.
DISCUSSION
The pUDP system, which combines generic DNA parts with expression of self-processing gRNAs driven by RNA polymerase II, was shown to enable genome editing in different yeasts belonging to the Saccharomycotina subphylum. The pUDP002 entry plasmid only required insertion of a functional gRNA to enable successful single deletion or disruption of the ADE2 gene in four different species of Kluyveromyces and Ogataea yeasts (Fig. 7) but also double editing at two different chromosomal loci in O. parapolymorpha. The panARS(OPT)-harboring pUDP plasmids can be cured simply by growth in non-selective medium (Liachko and Dunham 2014), as illustrated by whole-genome sequencing of engineered O. parapolymorpha strain IMD001, enabling sequential rounds of genetic modifications with the same system in rapid succession.
Figure 7.
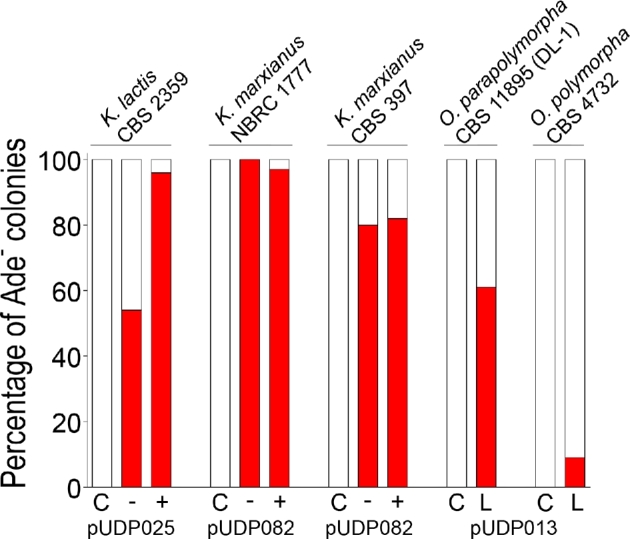
Wide-host-range applicability of the pUDP CRISPR/Cas9 system. The depicted data summarize the ADE2 targeting efficiency (red colonies/total colonies) of the pUDP system in the four yeast species used in this study. The pUDP plasmids differ only in their gRNA target specificity: pUDP025, pUDP082 and pUDP013 harbor gRNAKlADE2, gRNAKmADE2 and gRNAOpADE2, respectively. The results shown for Kluyveromyces species were obtained directly on transformation plates either with (+) or without (−) co-transformation of a ca. 960-bp marker-free repair DNA fragment. The results for O. parapolymorpha and O. polymorpha were obtained after 96 and 192 h of prolonged liquid incubation (L), respectively, started from colonies that were transformed without a repair fragment. Control transformations (C) with pUDP002 did not result in the occurrence of Ade− mutants.
In Kluyveromyces species, near-perfect targeting (≥96%) and HR-mediated repair DNA integration occurred at practicable rates in K. lactis CBS 2359 (31%) and the haploid K. marxianus NBCR 1777 (24%), while highly efficient gene disruption was achieved in the diploid K. marxianus strain CBS 397. In Ogataea species, mutants could be obtained after prolonged liquid incubation, allowing relatively straightforward and marker-free disruption of genes in the two industrially relevant O. (para)polymorpha strains CBS 4732 and CBS 11895 (DL-1). The observed delay in targeting activity suggests that the heterologous origin of functional parts of the pUDP system might reflect suboptimal expression of gRNA and/or Cas9 in Ogataea species. The expression levels of gRNA and Cas9 may have narrow optima for efficient genome editing, as recently demonstrated in P. pastoris, a related methylotrophic yeast (Weninger et al.2016). While the A. adeninivorans TEF1 promoter employed for Cas9 expression in this study has been demonstrated to enable strong constitutive expression in O. polymorpha (Terentiev et al.2004), optimization of the gRNA expression presently under the control of the S. cerevisiae TDH3 promoter might be envisaged to eliminate the observed delay in the occurrence of mutants.
Recently, species-specific CRISPR-Cas9 systems have been published for a range of non-conventional yeasts including K. lactis, K. marxianus and O. polymorpha (Horwitz et al.2015; Löbs et al.2017; Nambu-Nishida et al.2017; Numamoto et al.2017). To our knowledge, the study by Horwitz et al. (2015) is the only published application of CRISPR-Cas9 in K. lactis. The authors report Cas9-mediated integration at three genomic loci simultaneously, which occurred at a rate of 2% in a NHEJ-deficient strain with an integrated Cas9 cassette using ca. 1000-bp homology flanks. However, data on targeting efficiency were not provided, preventing a meaningful comparison with the results obtained by the pUDP system in this study (96% targeting efficiency, 31% HR-rate, wild-type K. lactis strain, ca. 480-bp homology flanks, single locus). However, the absolute quantification of the HR-mediated repair left no doubt regarding pUDP tool efficacy for gene editing in K. lactis. In K. marxianus NBRC 1777, the pUDP system achieved higher targeting efficiencies (97% vs 65%) and similar efficiencies of marker-free HR (22% vs 38%) compared to the most efficient CRISPR system for this yeast reported to date (Nambu-Nishida et al.2017). However, in this comparison it should be taken into account that, in this study, longer homology flanks were used (480 bp vs 50 bp), which may have facilitated HR-mediated repair. The published CRISPR-Cas9 tool for O. polymorpha (Numamoto et al.2017) used strain BY4330, a mutant derived from the O. polymorpha NCYC495 background which is closely related to strain CBS 4732 employed in this study. Compared to this tool, the pUDP system achieved similar efficiencies of NHEJ-mediated gene disruption when compared with O. parapolymorpha CBS 11895 (61% vs 71%), but was less effective when compared with O. polymorpha CBS 4732 (9% vs 71%). In contrast to our findings, the authors were able to obtain gene-disrupted and marker-integrated transformants directly on transformation plates, indicating either a more suitable expression of gRNA and/or Cas9 or a different genetic tractability of the strain lineages. Compared to the pUDP system, all three discussed CRISPR/Cas9 tools (partially) rely on species-specific genetic parts. The pKD1 stability element was employed for plasmid replication in K. lactis (Horwitz et al.2015), while the KmSNR52 promoter was used for gRNA expression in the case of K. marxianus (Nambu-Nishida et al.2017), and an OptRNALeu promoter for gRNA expression in O. polymorpha (Numamoto et al.2017). These RNA polymerase III-dependent promoters are unlikely to function in other species or genera.
RNA polymerase III-driven SpCas9-based editing strategies, such as those based on chimeric gRNA expression systems, are not easily compatible with multiplexed genome editing. Hitherto, the maximum number of gRNAs shown to enable simultaneous DSBs at different loci in S. cerevisiae is six. However, this result required an elaborate construction scheme using three plasmids for the individual expression of each gRNA (Mans et al.2015). In contrast, ribozyme-flanked gRNA expression, as previously reported in S. pastorianus (Gorter de Vries et al.2017), allowed to achieve the first double gene editing in O. parapolymorpha using a polycistronic gRNA array including two ribozyme-flanked spacers. The RNA polymerase II expression certainly facilitated to increase and to a greater extend modulate the expression of the gRNA. The expression of multiple gRNAs could be used as well to enhance gene deletion, even in absence of efficient HR, by providing the possibility to induce double cuts around the target gene which could result in a complete gene deletion even when repaired by NHEJ. Exploring and increasing the efficiency of multiplexed editing ability will be instrumental to further unlock genetic tractability of non-conventional yeasts.
In addition to the challenge of introducing DSBs quickly and efficiently, CRISPR-Cas9-mediated genome engineering in non-conventional yeasts is limited by their intrinsic ability for HR-mediated DNA repair, as illustrated in this study and by other previously developed CRISPR tools (Nambu-Nishida et al.2017; Numamoto et al.2017; Weninger et al.2016). While low HR activity can often be compensated for by working in a NHEJ-deficient background, such strains may have undesirable properties, e.g. higher stress sensitivity (Nielsen, Nielsen and Mortensen 2008; Takahashi, Masuda and Koyama 2006) and reduction of cellular fitness (Snoek et al.2009), which might only emerge after prolonged cultivation. For example, the commonly targeted KU70/80 complex is involved in telomere maintenance, and disruption of the complex results in telomere shortening (Boulton and Jackson 1996), altered position of telomeric DNA in the nucleus (Laroche et al.1998) and promotes telomere degradation and recombination (Polotnianka, Li and Lustig 1998). As an alternative to inactivating NHEJ, Rad52, a highly conserved protein which plays an important role in HR-mediated DNA repair in yeasts and other organisms (Symington 2002), has been exploited to improve the efficiency of HR. Expression (Di Primio et al.2005) or protein delivery (Kalvala et al.2010) of S. cerevisiae derived Rad52 as well as the utilization of a recently described Cas9-ScRad52 fusion protein (Shao et al.2017) increased HR rates in mammalian cells. However, overexpression of KlRad52 in K. lactis did not have a beneficial effect (Kooistra, Hooykaas and Steensma 2004). Another approach to optimize HR could be cell cycle synchronization and transformation of cells in S-phase, during which cells exhibit the highest ratio of HR over NHEJ (Barlow and Rothstein 2010; Chapman, Taylor and Boulton 2012, Karanam et al.2012). Cell arrest in S-phase with hydroxyurea indeed led to increased rates of HR in various (non-conventional) yeasts (Tsakraklides et al.2015).
To conclude, the pUDP CRISPR/Cas9 system presented here has been shown to work in four different species of yeasts belonging to the Saccharomycotina, limited by host-dependent targeting efficiency and intrinsic HR capability. We expect the pUDP system to be also applicable for Cas9-mediated genome engineering in other industrially relevant Saccharomycotina yeasts. The approach presented in this study demonstrates the potential of wide host-range tools for genome editing. Although highly efficient genome editing is likely to require species-specific characteristics, the pUDP system can be used for rapid introduction of screenable mutations (e.g. auxotrophic markers) and, as demonstrated for O. parapolymorpha, the elimination of NHEJ. Moreover, due to its broad host range, the pUDP plasmid can serve as a starting point for optimization of Cas9-mediated gene targeting in individual yeast species by promoter and terminator replacement studies.
Acknowledgements
We thank Marcel van den Broek (Delft University of Technology) for the bioinformatics analysis and Philip de Groot (Delft University of Technology) for his contribution to the pUDP system.
FUNDING
HJ, ARGdV and RM were supported by the BE-Basic R&D Program, which was granted an FES subsidy from the Dutch Ministry of Economic Affairs, Agriculture and Innovation (EL&I). JAV was a fellow in the YEASTCELL training network, which received funding from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007-2013/under REA grant agreement n° 606795. JAV, ASR, JPM and J-MGD were supported by the European Union project CHASSY, which received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 720824. JTP acknowledges support by an Advanced Grant (project no. # 694633) awarded by the European Research Council. TP was a fellow in Pacmen which received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 722287.
Conflict of interest. None declared.
REFERENCES
- Abdel-Banat BM, Nonklang S, Hoshida H et al. . Random and targeted gene integrations through the control of non-homologous end joining in the yeast Kluyveromyces marxianus. Yeast 2010;27:29–39. [DOI] [PubMed] [Google Scholar]
- Bao Z, Xiao H, Liang J et al. . Homology-integrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth Biol 2015;4:585–94. [DOI] [PubMed] [Google Scholar]
- Barlow JH, Rothstein R. Timing is everything: cell cycle control of Rad52. Cell Div 2010;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin A, Ozier-Kalogeropoulos O, Denouel A et al. . A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res 1993;21:3329–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton SJ, Jackson SP. Identification of a Saccharomyces cerevisiae Ku80 homologue: roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res 1996;24:4639–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito N, Avila J, Perez MD et al. . The genes YNI1 and YNR1, encoding nitrite reductase and nitrate reductase respectively in the yeast Hansenula polymorpha, are clustered and co-ordinately regulated. Biochem J 1996;317 (Pt 1):89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, Gao M, Lopez-Garcia CL et al. . Centromeric DNA facilitates nonconventional yeast genetic engineering. ACS Synth Biol 2017;6:1545–53. [DOI] [PubMed] [Google Scholar]
- Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 2012;47:497–510. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhou YJ, Siewers V et al. . Enabling technologies to advance microbial isoprenoid production. Adv Biochem Eng Biot 2015;148:143–60. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D et al. . Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339:819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaner M, Mejia J, Alper HS. Enabling graded and large-scale multiplex of desired genes using a dual-mode dCas9 activator in Saccharomyces cerevisiae. ACS Synth Biol 2017;6:1931–43. [DOI] [PubMed] [Google Scholar]
- Di Primio C, Galli A, Cervelli T et al. . Potentiation of gene targeting in human cells by expression of Saccharomyces cerevisiae Rad52. Nucleic Acids Res 2005;33:4639–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo JE, Norville JE, Mali P et al. . Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 2013;41:4336–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS One 2008;3:e3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Tong Y, Wen Z et al. . Multiplex gene editing of the Yarrowia lipolytica genome using the CRISPR-Cas9 system. J Ind Microbiol Biot 2016;43:1085–93. [DOI] [PubMed] [Google Scholar]
- Gao Y, Zhao Y. Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing. J Integr Plant Biol 2014;56:343–9. [DOI] [PubMed] [Google Scholar]
- Gellissen G. Heterologous protein production in methylotrophic yeasts. Appl Microbiol Biot 2000;54:741–50. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY et al. . Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 2009;6:343–5. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2007;2:31–4. [DOI] [PubMed] [Google Scholar]
- Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 2016;17:333–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter de Vries AR, de Groot PA, van den Broek M et al. . CRISPR-Cas9 mediated gene deletions in lager yeast Saccharomyces pastorianus. Microb Cell Fact 2017;16:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz AA, Walter JM, Schubert MG et al. . Efficient multiplexed integration of synergistic alleles and metabolic pathways in yeasts via CRISPR-Cas. Cell Syst 2015;1:88–96. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014;157:1262–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M, Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 2013;5:a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I et al. . A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012;337:816–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EA. Biotechnology of non-Saccharomyces yeasts—the ascomycetes. Appl Microbiol Biot 2013;97:503–17. [DOI] [PubMed] [Google Scholar]
- Kalvala A, Rainaldi G, Di Primio C et al. . Enhancement of gene targeting in human cells by intranuclear permeation of the Saccharomyces cerevisiae Rad52 protein. Nucleic Acids Res 2010;38:e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HA, Kang W, Hong WK et al. . Development of expression systems for the production of recombinant human serum albumin using the MOX promoter in Hansenula polymorpha DL-1. Biotechnol Bioeng 2001;76:175–85. [DOI] [PubMed] [Google Scholar]
- Karanam K, Kafri R, Loewer A et al. . Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell 2012;47:320–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kata I, Semkiv MV, Ruchala J et al. . Overexpression of the genes PDC1 and ADH1 activates glycerol conversion to ethanol in the thermotolerant yeast Ogataea (Hansenula) polymorpha. Yeast 2016;33:471–8. [DOI] [PubMed] [Google Scholar]
- Kavscek M, Strazar M, Curk T et al. . Yeast as a cell factory: current state and perspectives. Microb Cell Fact 2015;14:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DU, Hayles J, Kim D et al. . Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol 2010;28:617–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinner U, Schäfer B. Genetic aspects of targeted insertion mutagenesis in yeasts. FEMS Microbiol Rev 2004;28:201–23. [DOI] [PubMed] [Google Scholar]
- Kooistra R, Hooykaas PJ, Steensma HY. Efficient gene targeting in Kluyveromyces lactis. Yeast 2004;21:781–92. [DOI] [PubMed] [Google Scholar]
- Kuijpers NG, Solis-Escalante D, Bosman L et al. . A versatile, efficient strategy for assembly of multi-fragment expression vectors in Saccharomyces cerevisiae using 60 bp synthetic recombination sequences. Microb Cell Fact 2013;12:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers NGA, Chroumpi S, Vos T et al. . One-step assembly and targeted integration of multigene constructs assisted by the I-SceI meganuclease in Saccharomyces cerevisiae. FEMS Yeast Res 2013;13:769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunze G, Kang HA, Gellissen G. Hansenula polymorpha (Pichia angusta): biology and applications. In: Satyanarayana T, Kunze G (eds.) Yeast Biotechnology: Diversity and Applications. Dordrecht: Springer Netherlands, 2009, 47–64. [Google Scholar]
- Kurylenko OO, Ruchala J, Hryniv OB et al. . Metabolic engineering and classical selection of the methylotrophic thermotolerant yeast Hansenula polymorpha for improvement of high-temperature xylose alcoholic fermentation. Microb Cell Fact 2014;13:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laroche T, Martin SG, Gotta M et al. . Mutation of yeast Ku genes disrupts the subnuclear organization of telomeres. Curr Biol 1998;8:653–7. [DOI] [PubMed] [Google Scholar]
- Lee ME, DeLoache WC, Cervantes B et al. . A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth Biol 2015;4:975–86. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A et al. . The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Borodina I. Application of synthetic biology for production of chemicals in yeast Saccharomyces cerevisiae. FEMS Yeast Res 2015;15:1–12. [DOI] [PubMed] [Google Scholar]
- Liachko I, Dunham MJ. An autonomously replicating sequence for use in a wide range of budding yeasts. FEMS Yeast Res 2014;14:364–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löbs A-K, Engel R, Schwartz C et al. . CRISPR–Cas9-enabled genetic disruptions for understanding ethanol and ethyl acetate biosynthesis in Kluyveromyces marxianus. Biotechnol Biofuels 2017;10:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looke M, Kristjuhan K, Kristjuhan A. Extraction of genomic DNA from yeasts for PCR-based applications. BioTechniques 2011;50:325–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C et al. . ViennaRNA Package 2.0. Algorithms Mol Biol 2011;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM et al. . RNA-guided human genome engineering via Cas9. Science 2013;339:823–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mans R, van Rossum HM, Wijsman M et al. . CRISPR/Cas9: a molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res 2015;15:fov004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattanovich D, Sauer M, Gasser B. Yeast biotechnology: teaching the old dog new tricks. Microb Cell Fact 2014;13:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni C, Serafini A, Falcone C. The inactivation of KlNOT4, a Kluyveromyces lactis gene encoding a component of the CCR4-NOT complex, reveals new regulatory functions. Genetics 2005;170:1023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey JP, Etschmann MM, Schrader J et al. . Cell factory applications of the yeast Kluyveromyces marxianus for the biotechnological production of natural flavour and fragrance molecules. Yeast 2015;32:3–16. [DOI] [PubMed] [Google Scholar]
- Nambu-Nishida Y, Nishida K, Hasunuma T et al. . Development of a comprehensive set of tools for genome engineering in a cold- and thermo-tolerant Kluyveromyces marxianus yeast strain. Sci Rep 2017;7:8993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro FJ, Perdomo G, Tejera P et al. . The role of nitrate reductase in the regulation of the nitrate assimilation pathway in the yeast. FEMS Yeast Res 2003;4:149–55. [DOI] [PubMed] [Google Scholar]
- Nielsen J, Larsson C, van Maris A et al. . Metabolic engineering of yeast for production of fuels and chemicals. Curr Opin Biotechnol 2013;24:398–404. [DOI] [PubMed] [Google Scholar]
- Nielsen JB, Nielsen ML, Mortensen UH. Transient disruption of non-homologous end-joining facilitates targeted genome manipulations in the filamentous fungus Aspergillus nidulans. Fungal Genet Biol 2008;45:165–70. [DOI] [PubMed] [Google Scholar]
- Nonklang S, Abdel-Banat BM, Cha-aim K et al. . High-temperature ethanol fermentation and transformation with linear DNA in the thermotolerant yeast Kluyveromyces marxianus DMKU3-1042. Appl Environ Microb 2008;74:7514–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numamoto M, Maekawa H, Kaneko Y. Efficient genome editing by CRISPR/Cas9 with a tRNA-sgRNA fusion in the methylotrophic yeast Ogataea polymorpha. J Biosci Bioeng 2017;124:487–92. [DOI] [PubMed] [Google Scholar]
- Polotnianka RM, Li J, Lustig AJ. The yeast Ku heterodimer is essential for protection of the telomere against nucleolytic and recombinational activities. Curr Biol 1998;8:831–5. [DOI] [PubMed] [Google Scholar]
- Ravin NV, Eldarov MA, Kadnikov VV et al. . Genome sequence and analysis of methylotrophic yeast Hansenula polymorpha DL1. BMC Genomics 2013;14:837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W et al. . Integrative genomics viewer. Nat Biotechnol 2011;29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman H. A system selective for mutations affecting the synthesis of adenine in yeast. CR Trav Lab Carls P 1956;26:299–314. [Google Scholar]
- Ryan OW, Skerker JM, Maurer MJ et al. . Selection of chromosomal DNA libraries using a multiplex CRISPR system. Elife 2014;3:e03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar AN, Gorter de Vries AR, van den Broek M et al. . Nanopore sequencing enables near-complete de novo assembly of Saccharomyces cerevisiae reference strain CEN.PK113-7D. FEMS Yeast Res 2017, DOI 10.1093/femsyr/fox074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraya R, Gidijala L, Veenhuis M et al. . Tools for genetic engineering of the yeast Hansenula polymorpha. Methods Mol Biol 2014;1152:43–62. [DOI] [PubMed] [Google Scholar]
- Saraya R, Krikken AM, Kiel JA et al. . Novel genetic tools for Hansenula polymorpha. FEMS Yeast Res 2012;12:271–8. [DOI] [PubMed] [Google Scholar]
- Schwartz CM, Hussain MS, Blenner M et al. . Synthetic RNA polymerase iii promoters facilitate high-efficiency CRISPR-Cas9-mediated genome editing in Yarrowia lipolytica. ACS Synth Biol 2016;5:356–9. [DOI] [PubMed] [Google Scholar]
- Shao S, Ren C, Liu Z et al. . Enhancing CRISPR/Cas9-mediated homology-directed repair in mammalian cells by expressing Saccharomyces cerevisiae Rad52. Int J Biochem Cell Biol 2017;92:43–52. [DOI] [PubMed] [Google Scholar]
- Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res 2008;18:134–47. [DOI] [PubMed] [Google Scholar]
- Singh MV, Weil PA. A method for plasmid purification directly from yeast. Anal Biochem 2002;307:13–17. [DOI] [PubMed] [Google Scholar]
- Siso MIG. The biotechnological utilization of cheese whey: A review. Bioresource Technol 1996;57:1–11. [Google Scholar]
- Snoek IS, van der Krogt ZA, Touw H et al. . Construction of an hdfA Penicillium chrysogenum strain impaired in non-homologous end-joining and analysis of its potential for functional analysis studies. Fungal Genet Biol 2009;46:418–26. [DOI] [PubMed] [Google Scholar]
- Spohner SC, Schaum V, Quitmann H. et al. Kluyveromyces lactis: an emerging tool in biotechnology. J Biotechnol 2016;222:104–16. [DOI] [PubMed] [Google Scholar]
- Steinborn G, Boer E, Scholz A et al. . Application of a wide-range yeast vector (CoMed) system to recombinant protein production in dimorphic Arxula adeninivorans, methylotrophic Hansenula polymorpha and other yeasts. Microb Cell Fact 2006;5:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stovicek V, Holkenbrink C, Borodina I. CRISPR/Cas system for yeast genome engineering: advances and applications. FEMS Yeast Res 2017;17:fox030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh SO, Zhou JJ. Methylotrophic yeasts near Ogataea (Hansenula) polymorpha: a proposal of Ogataea angusta comb. nov. and Candida parapolymorpha sp. nov. FEMS Yeast Res 2010;10:631–8. [DOI] [PubMed] [Google Scholar]
- Symington LS. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol R 2002;66:630–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Masuda T, Koyama Y. Enhanced gene targeting frequency in ku70 and ku80 disruption mutants of Aspergillus sojae and Aspergillus oryzae. Mol Genet Genomics 2006;275:460–70. [DOI] [PubMed] [Google Scholar]
- Terentiev Y, Pico AH, Boer E et al. . A wide-range integrative yeast expression vector system based on Arxula adeninivorans-derived elements. J Ind Microbiol Biot 2004;31:223–8. [DOI] [PubMed] [Google Scholar]
- Tsakraklides V, Brevnova E, Stephanopoulos G et al. . Improved gene targeting through cell cycle synchronization. PLoS One 2015;10:e0133434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt JP. New Combinations in the genera Brettanomyces, Kluyveromyces, Lodderomyces and Wingea. Bothalia 1971;10:417–8. [Google Scholar]
- Verduyn C, Postma E, Scheffers WA et al. . Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast 1992;8:501–17. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pohlmann R et al. . New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 1994;10:1793–808. [DOI] [PubMed] [Google Scholar]
- Wang G, Huang M, Nielsen J. Exploring the potential of Saccharomyces cerevisiae for biopharmaceutical protein production. Curr Opin Biotechnol 2017;48:77–84. [DOI] [PubMed] [Google Scholar]
- Weninger A, Hatzl AM, Schmid C et al. . Combinatorial optimization of CRISPR/Cas9 expression enables precision genome engineering in the methylotrophic yeast Pichia pastoris. J Biotechnol 2016;235:139–49. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Maeda K, Mikata K. The phylogenetic relationships of the hat-shaped ascospore-forming, nitrate-assimilating Pichia species, formerly classified in the genus Hansenula Sydow et Sydow, based on the partial sequences of 18S and 26S ribosomal RNAs (Saccharomycetaceae): the proposals of three new genera, Ogataea, Kuraishia, and Nakazawaea. Biosci Biotech Bioch 1994;58:1245–57. [DOI] [PubMed] [Google Scholar]