

Abstract
Soils are reservoirs of antibiotic resistance genes (ARGs), but environmental dynamics of ARGs are largely unknown. Long-term disturbances offer opportunities to examine microbiome responses at scales relevant for both ecological and evolutionary processes and can be insightful for studying ARGs. We examined ARGs in soils overlying the underground coal seam fire in Centralia, PA, which has been burning since 1962. As the fire progresses, previously hot soils can recover to ambient temperatures, which creates a gradient of fire impact. We examined metagenomes from surface soils along this gradient to examine ARGs using a gene-targeted assembler. We targeted 35 clinically relevant ARGs and two horizontal gene transfer-related genes (intI and repA). We detected 17 ARGs in Centralia: AAC6-Ia, adeB, bla_A, bla_B, bla_C, cmlA, dfra12, intI, sul2, tetA, tetW, tetX, tolC, vanA, vanH, vanX and vanZ. The diversity and abundance of bla_A, bla_B, dfra12 and tolC decreased with soil temperature, and changes in ARGs were largely explained by changes in community structure. We observed sequence-specific biogeography along the temperature gradient and observed compositional shifts in bla_A, dfra12 and intI. These results suggest that increased temperatures can reduce soil ARGs but that this is largely due to a concomitant reduction in community-level diversity.
Keywords: thermophile, gene-targeted assembly, metagenome, rplB, coal fire
Targeted assembly of antibiotic resistance genes reveals high diversity and sequence-specific dynamics in response to thermal temperatures
INTRODUCTION
The dissemination of antibiotic resistance genes (ARGs) is a pressing public health concern. The One Health Initiative recognizes the intrinsic link between evolution of bacterial resistance in clinical and environmental settings (Kahn 2016). Clinically relevant ARGs have been detected in ‘pristine environments’ (Lang et al.2010) as well as a variety of marine, plant and soil microbiomes (Fierer et al.2012; Gibson, Forsberg and Dantas 2014; Wang et al.2015a; Fitzpatrick and Walsh 2016). Soil is considered to be an environmental reservoir of ARGs, with greater ARG diversity than the clinic (Nesme and Simonet 2015). Despite that we can easily detect ARGs in soil, dynamics of soil ARGs are not fully understood (Allen et al.2010). Understanding of the dissemination of ARGs in the environment is impeded by our modest understanding of their diversification, maintenance and dissemination (Hiltunen, Virta and Laine 2017).
Understanding the propagation and dissemination of ARGs in soil is difficult because multiple interacting factors influence their fate (Allen et al.2010; Berendonk et al.2015). Perhaps most obviously, ARGs can be selected when there is environmental exposure to antibiotic (Laine, Hiltunen and Virta 2016). Environmental exposure can result from the anthropogenic use of antibiotics, for example in agriculture or via wastewater treatment outputs (Kumar et al.2005; Rizzo et al.2013), or it can result from environmental antibiotic production by microorganisms in situ (Nesme and Simonet 2015). Antibiotic exposure can kill sensitive populations and allow for propagation of resistant strains. Additionally, ARGs can be horizontally transferred (Hiltunen, Virta and Laine 2017) and are often detected on plasmids and other mobile genetic elements (Van Hoek et al.2011; Pal et al.2015). Thus, ARGs on mobile genetic elements may be disseminated more rapidly than through population growth alone. Furthermore, several ARGs are thought to have evolved >2 billion years ago (Aminov and Mackie 2007), and these may be maintained in the absence of selective pressure from antibiotics and transferred vertically. Another complicating factor for understanding ARG dissemination is the influence of the dynamics of soil microbial communities. While interspecies competition can impact ARG abundance, one study of many habitats showed that abiotic soil conditions can be important drivers of ARG profiles (Fierer et al.2012). Anthropogenic influences, such as nitrogen addition to the soil, can also impact ARGs (Forsberg et al.2014). Similarly, studies with changing abiotic conditions, such as increased temperatures, have reported subsequent reductions in ARG abundance (Qian et al.2016; Tian et al.2016). In these examples and others, environmental disturbance can alter soil microbial community structure (Shade et al.2012; Garner et al.2016; Nunes et al.2016), and then can impact local ARGs and their dissemination.
Long-term disturbances that impact multiple microbial generations can provide opportunities to investigate the changes in ARGs in response to environmental stress. One such disturbance is Centralia, PA, the site of an underground coal seam fire that ignited in 1962. As this town was evacuated in 1984, it also represents a post-urban ecosystem of minimal contemporary anthropogenic influence. This fire continues to advance along the coal seam, creating a gradient of contemporary and historical fire impact and allowing for observation of multiple microbial generations’ responses to disturbance and their potential recovery. Surface soil microbial communities in Centralia are exposed to elevated temperatures (21°C–57°C) (Lee et al.2017) and coal combustion pollutants (Janzen and Tobin-Janzen 2008) that include trace elements such as arsenic, copper, aluminum and lead (Janzen and Tobin-Janzen 2008; Melody and Johnston 2015). While temperature increases are large, deposition of coal combustion pollutants occurs at a slow rate and varies based on the subsurface structure and geochemical properties of the burning coal (Janzen and Tobin-Janzen 2008). Depth of the coal seam varies from surface level to 46 m (Elick 2011). Furthermore, surface temperatures cool to ambient levels as the fire progresses, but coal combustion pollutants are not necessarily removed. Previously, we observed changes in bacterial and archaeal community structure with fire history that was well explained by temperature rather than soil properties such as arsenic concentration (Lee et al.2017).
We leveraged the long-term disturbance in Centralia to examine ARG biogeography given both the abandonment of human habitation and the presence of multigenerational stressor for the microorganisms. We investigated 12 metagenomes of microbial communities from surface soils along the Centralia temperature gradient for 35 clinically relevant ARGs conferring resistance to eight classes of antibiotics, as well as multidrug efflux pumps and two HGT-relevant genes repA and intI. We used gene-targeted assembly of the metagenomes to capture a breadth of ARG diversity. To examine the potential extent of HGT in Centralia, we asked whether changes in community structure explained any changes in ARG profiles. Because we previously identified changes in community structure along the stressor (Lee et al.2017), we also asked whether functional redundancy (e.g. different ARG sequences belonging to the same resistance class) within the soil microbial community moderated the impact of a disturbance on ARG profiles. Functional redundancy allows that changes in community structure can occur without subsequent change in ARG abundance. Also, because we focused on clinically relevant ARGs rather than potentially novel ARGs from thermophilic lineages, we hypothesized that ARG abundance would decrease with temperature, as observed in other studies (Diehl and Lapara 2010; Qian et al.2016; Tian et al.2016). We were, however, also interested in biogeography of specific gene sequences and hypothesized that they may have unique responses, even within the same resistance class.
METHODS
Reference database construction
Reference gene databases of diverse, near full-length sequences were constructed using selected sequences from FunGene databases (Fish et al.2013) for the following genes: AAC6-Ia, adeB, ANT3, ANT6, ANT9, bla_A, bla_B, bla_C, CAT, cmlA, dfra1, dfra12, ermB, ermC, intI, mexC, mexE, qnr, repA, strA, strB, sul2, tetA, tetD, tetM, tetQ, tetW, tetX, tolC, vanA, vanC, vanH, vanT, vanW, vanX, vanY and vanZ. Seed sequences and Hidden Markov Models (HMMs) for each gene were downloaded from FunGene, and diverse protein and corresponding nucleotide sequences (reference sequences) were selected with gene-specific search parameters (Table S1, Supporting Information). Briefly, minimum size amino acid was set to 70% of the HMM length; minimum HMM coverage was set to 80% as is recommended by Xander software for targeted gene assembly (Wang et al.2015b); and a score cutoff was manually selected based on a notable score reduction between consecutive sequences, as suggested by the Ribosomal Database Project (personal communication). Reference sequences were de-replicated before being used in subsequent analysis, and final sequence numbers are included in Table S1 (Supporting Information).
Sample collection, sequencing and quality control
Study site, soil sampling and soil biogeochemistry were all performed as described (Lee et al.2017). Briefly, surface soils were sampled along a gradient of fire impact that was determined from historical characterizations of the site (Elick 2011): fire affected (n = 6), recovered (n = 5) and reference (n = 1). Fire-affected soils had elevated temperatures due to fire; recovered soils were at ambient temperature but historically had elevated temperatures from the fire; and the reference soil was never impacted by the fire. The reference sample was used as a qualitative control and is not intended as a quantitative and definitive comparison to non-impacted soils. Microbial community DNA was obtained using a phenol chloroform extraction (Cho et al.1996) and purification with MoBio DNEasy PowerSoil kit without vortexing. All samples were sequenced on the Illumina HiSeq 2500 platform with 2 × 150 bp paired-end format at the Joint Genome Institute and quality filtered using BBDuk (https://sourceforge.net/projects/bbmap/). Metagenome coverage was estimated using Nonpareil (Rodriguez-R and Konstantinidis 2014).
Gene-targeted assembly and quality control
A gene-targeted metagenome assembler (Wang et al.2015b) was used to assemble ARGs of interest from quality-filtered metagenomes. For each gene of interest, seed sequences, HMMs and reference gene databases, as described above, were included. The rplB reference gene database, seed sequences and HMMs from the Xander package were used. In most instances, default assembly parameters were used, except to incorporate differences in protein length (i.e. if the protein was shorter than 150 aa (default), as was the case for dfra1, dfra12, AAC6-Ia, ermB, ermC, qnr, vanX and vanZ) (Table S1, Supporting Information). While the assembler includes chimera removal, additional quality control steps were added. Specifically, final assembled sequences (contigs) were searched against the reference gene database as well as the non-redundant database (nr) from NCBI (28 August 2017) using BLAST (v. 2.2.26, Camacho et al.2008). Genes were re-examined if the top hit had an e-value > 10−5 or if top hit descriptors were not the target gene. Genes with low-quality results were re-assembled with adjusted parameters. Aligned sequences from each sample were dereplicated and clustered at 90%, 97% and 99% amino acid identity using the RDP Classifier (Wang et al.2007). Our quality control analyses can be accessed on GitHub (‘assembly_assessments’ repository in https://github.com/ShadeLab/PAPER_Dunivin_Antibiotics_2017/tree/master/assembly_assessments).
Ecological analyses
Phylum-level rplB relative abundance was used to examine differences in community structure. Relative abundance for each site was averaged among samples of the same fire classification (i.e. fire-affected, recovered, reference) and compared to 16S rRNA gene sequence data from a previous work (Lee et al.2017). For subsequent ecological analyses, the RDP Classifier was used to generate an OTU table from 90%, 97% and 99% amino acid identities. We refer to contigs clustered at 99% identity as ‘ARG sequences’ throughout the remainder of the text. The OTU tables were analyzed in R (R Development Core Team 2008). OTU tables were separated based on the gene(s) of interest (rplB and ARGs). Due to Nonpareil-estimated differences in coverage, rplB and ARG OTU tables were rarefied to an even sampling depth (258 and 180 assembled sequences, respectively) using the vegan package (Oksanen et al.2017). Pieluo's evenness was calculated, and richness was estimated using PhyloSeq (McMurdie and Holmes 2013). The Psych package was used to calculate Spearman's rank correlations between alpha diversity (richness and evenness) and soil temperature for both rplB and ARGs. Bray-Curtis distance was used to obtain dissimilarity matrices, and principal component analysis was used to visualize beta diversity. Distance matrices of rarefied, relativized data were analyzed using Mantel tests with Spearman's rank correlations. Mantel tests were performed on rplB, ARG and spatial distance matrices of sample locations.
Resistance gene comparison
We assessed ARG biogeography at the gene, taxonomic class and sequence levels. To compare the abundance of ARGs among data sets, total counts of rplB were used to normalize the abundance of each ARG sequence. Total counts of each ARG were calculated as the sum of the relative abundance of each ARG sequence. The Psych package (Revelle 2017) was used to calculate Spearman's rank correlations between soil geochemical properties and total gene counts for each ARG. Pairwise correlations for the total abundance of each resistance gene were also calculated. For taxonomic analysis of each ARG, the top BLAST result and the taxize package (Chamberlain et al.2017) were used to assign taxonomy to each ARG sequence. When the top hit was an uncultured bacterium, the second or third hit was used, and when all three top hits were unknown, the taxonomy was labeled unknown. Total counts of each taxonomic class were summed for each ARG, and Spearman's rank correlations were used to test for correlations between class abundance and temperature for all ARGs with representatives from at least three taxonomic groups. Spearman's rank correlations were performed on normalized and relativized abundance information, but only relativized abundance is shown because it agreed with normalized data and also had unique features. Furthermore, we examined biogeography of individual ARG sequences. A Venn analysis was performed between ARGs in fire affected and recovered samples using the VennDiagram package (Chen and Boutros 2011). The mean normalized abundance for each ARG sequence among samples was plotted against the number of sites it was observed in (occurrence). ARG sequences present in only one site were subsequently removed, and we used hierarchical cluster and heatmap analysis with the pheatmap package (Kolde 2015) to examine similar sequence biogeography along the temperature gradient.
Reproducibility, code and data
Our computing workflows and R script can be accessed on GitHub (https://github.com/ShadeLab/PAPER_Dunivin_Antibiotics_2017). Metagenomes are available from IMG/GOLD study ID: Gs0114513.
RESULTS AND DISCUSSION
Soil samples and gene-targeted assembly
We previously collected soils along the Centralia temperature gradient (Lee et al.2017). We submitted DNA extracted from 12 soils (temperature range = 12.1°C–54.2°C) to the Joint Genome Institute for small-scale Community Science Project; we did not submit all 18 originally collected samples because there was a 12-sample limit with the small-scale award, and so we chose samples for sequencing that were representative of the thermal gradient. We sequenced metagenomes from soils that had elevated temperatures due to the fire (fire-affected, n = 6), those that were historically impacted (recovered, n = 5) and those with no documented impact (reference, n = 1) (Fig. S1, Supporting Information). Quality-filtered metagenome size ranged from 21 to 51 Gbp, and Nonpareil-estimated coverage (Rodriguez-R and Konstantinidis 2014) varied from 29.12% to 89.96% (Table S2, Supporting Information). Though we measured a suite of geochemical data (Table S3, Supporting Information), our previous work found temperature to be the strongest driver of community structure (Lee et al.2017), and we found that ARGs only correlated with temperature (Table S4, Supporting Information).
We used a gene-targeted metagenome assembler to probe Centralia metagenomes for ARGs. While this gene-centric methodology does not permit analysis of entire gene cassettes or flanking regions, it improves detection of low abundance genes, increases the length of assembled gene sequences and is capable of detecting strain-level sequence variation (Wang et al.2015b). In addition to assembling ARGs of interest, we assembled rplB, a single copy gene and phylogenetic marker. We found that rplB assembled using these methods was comparable 16S rRNA gene data (Supplementary results; Fig. S2, Supporting Information), showing that gene-targeted assembly produced results consistent with previous work.
Detected ARGs and changes in their abundance with temperature
We examined a suite of genes encoding resistance to aminoglycosides, beta-lactams, chloramphenicol, sulfonamides, tetracyclines, trimethoprim and vancomycin, as well as plasmid-related and genes encoding multidrug efflux pumps (Table 1). From Centralia metagenomes, we assembled 1165 unique ARGs clustered at 99% amino acid identity. Though we targeted 35 distinct types of ARGs and two HGT-related genes, only 17 of these could be assembled from Centralia metagenomes. The genes ANT3, ANT6, ANT9, CAT, dfra1, ermB, ermC, mexC, mexE, qnr, repA, strA, strB, tetD, tetM, tetQ, vanC, vanT, vanW and vanY were not observed, suggesting that they were either below detection or absent. For detected ARGs, we found positive correlations between vanA, H and X genes and between tolC and dfra12 (Fig. S3, Supporting Information). vanAHX genes are known to be associated with one another in VanA-type operons (Périchon and Courvalin 2009), and genes tolC and dfra12 have previously been observed in isolates (Wannaprasat, Padungtod and Chuanchuen 2011). While sul2 and intI1 have been previously shown to be correlated (Johnson et al.2016), we did not observe a significant correlation between these genes. This discrepancy could be because our analysis does not distinguish between integron classes. Several ARGs in Centralia were negatively correlated with soil temperature (Fig. 1; Table S4, Supporting Information), but no ARGs were correlated with other measured soil geochemical properties (results not shown; Table S3, Supporting Information). The most abundant ARGs detected in Centralia were adeB, bla_B and dfra12 (Fig. 1; Fig. S4, Supporting Information). We note that the highest ARG normalized abundance was typically in Cen04 (13.3°C) but that this is due to low rplB abundance in the sample.
Table 1.
Resistance genes tested in this study.
| Antibiotic specificity | Gene |
|---|---|
| Aminoglycoside | AAC6-Ia, ANT3, ANT6, ANT9, strA,B |
| β-Lactams | bla_a, b, c |
| Chloramphenicol | CAT, chloramphenicol efflux pump |
| Macrolide | ermB,C, qnr |
| Multidrug efflux | adeB, mexC,E, tolC |
| Plasmid | intI, repA |
| Sulfonamide | sul2 |
| Tetracycline | tetA,D,M,Q,W,X |
| Trimethoprim | dfra1, dfra12 |
| Vancomycin | vanA,C,H,T,W,X,Y,Z |
Figure 1.

Negative correlations between normalized abundance of ARGs and soil temperature. Coverage-adjusted abundance for bla_A, bla_B, tolC and dfra12 was normalized to total abundance of the single copy gene rplB. Normalized abundance is plotted against soil temperature. Note the differences in y-axes. The linear trend line and P-value corresponding to the Spearman's rank correlation are shown. Shape indicates soil classification based on fire history.
Our results are generally in agreement with other studies of ARGs in soils. For example, Fitzpatrick and Walsh (2016) also reported low abundance or absence of qnr, tet and van genes in soil. Several studies also reported that genes encoding dihydrofolate reductases and/or beta-lactamases were abundant in soils (Forsberg et al.2014; Fitzpatrick and Walsh 2016; Li, Xia and Zhang 2017). Previous studies reported reductions in clinically relevant ARGs with increased temperatures in digesters and compost (Diehl and Lapara 2010; Qian et al.2016; Tian et al.2016). Diehl and Lapara (2010) observed a negative relationship between temperature and genes encoding tetracycline resistance and class 1 integrons in anaerobic digesters, but not aerobic ones. This may be further relevant to Centralia soils, as there likely are pockets of anaerobic activity in hot soils, especially at venting sites, which have measurably higher % moisture content due to steam escaping (Table S3, Supporting Information). To our knowledge, this is the first description of a reduction in ARG abundances with temperature in situ with soil. These results suggest that ARGs may be reduced in soil environments by increasing temperature. Thus, we speculate that increases in temperatures expected to reduce microbial community diversity may result in decreased clinically relevant ARGs in the environment.
Diversity of ARGs
We also examined the amino acid-level diversity of ARGs in Centralia metagenomes. We tested sequence cutoffs of 90%, 97% and 99% amino acid identity, but overarching patterns did not vary based on sequence cutoff (results not shown). Thus, our subsequent diversity analysis applied the most stringent cutoff (99% amino acid identity), as was applied in the original gene targeted assembly paper (Wang et al.2015b). ARG richness was negatively correlated with temperature (ρ = −0.57; P < 0.05), but evenness had a variable response with temperature (ρ = −0.47; P > 0.05) (Fig. 2B and D). ARG alpha diversity (within-sample) trends were thus similar to rplB and 16S rRNA gene diversity trends (Supplementary results; Fig. 2A and C), highlighting the influence of community structure on soil ARG profiles. In addition, overall differences in the composition of ARGs among sites were related to differences in rplB community structure (Mantel's r = 0.54; P < 0.05 on 999 permutations; Fig. S5, Supporting Information). This result also supports that compositional shifts in membership among Centralia sites were driving the observed differences in ARGs, not propagation of ARGs by gene transfer. These results agree with a recent analysis that reported congruence between community structure and ARG profiles in soils (Forsberg et al.2014). Similar to patterns in rplB and 16S rRNA genes, ARG profiles could not be explained by distance between sample sites (Mantel's r = 0.01, P > 0.05 on 999 permutations). This suggests that local dispersal of ARGs, which could be indicative of HGT, is not a common mechanism of ARG dissemination in this system. However, when we considered fire-affected and recovered metagenomes separately, we found that rplB community structure explained ARG composition in fire-affected soils (Mantel's r = 0.71; P < 0.05 on 719 permutations), but not in recovered soils (Mantel's r = 0.30; P > 0.05 on 119 permutations). We determined that this result was not driven by one anomalous sample by performing iterative ‘leave-one-out’ Mantel tests with four of five recovered soils, and all tests showed no correlation between rplB and ARGs (results not shown). The reason for no relationship between rplB and ARG in recovered soils is unclear (one hypothesis is that there is no signal given higher diversity), but this observation very indirectly suggests a potential larger influence of HGT in recovered soils than fire-affected soils that could be explored in future work.
Figure 2.

Observed richness (A, B) and evenness (C, D) of rplB (A, C) and ARG (B, D) along the Centralia temperature gradient. Assembled sequences were clustered at 99% amino acid identity and rarefied to an even sampling depth. Observed number of sequences (richness) and Pielou's evenness are plotted against soil temperature. Shape indicates soil classification based on fire history.
ARG distribution and sequence-specific biogeography
Only 12 ARG sequences were shared between fire-affected and recovered soils (Fig. 3A). On one hand, this is expected because soils are heterogeneous and have high ARG diversity (Fitzpatrick and Walsh 2016). Forsberg et al. (2014) observed 2895 ARG sequences in a functional antibiotic resistance screen from 18 agricultural and grassland soils. Of these, only 2.6% were present in two or more soils, which is comparable to our data (1.1%). Similarly, the distinction between fire-affected and recovered soil in our study is in part explained by generally high ARG diversity, with minimal overlap of ARG sequences detected between all sites. Furthermore, most ARG sequences (94.16%), whether they were rare (<1.5% normalized abundance to rplB) or prevalent, were detected only in one metagenome (Fig. 3B). Though the gene-targeted assembly approach maximizes observation of diversity given metagenome coverage, it is possible that even greater coverage of these metagenomes could result in detection of more shared ARG sequences between samples. There were 13 distinct biogeographical dynamics that indicated genes sensitive to the fire, and these were classified into two categories based on their prevalence and patterns of detection: abundant-transient and rare-transient sequences (Fig. 4). Abundant-transient ARG sequences belonged to genes adeB, bla_B, dfra12, intI, sul2 and vanZ. These sequences had a rplB-normalized abundance of ≥1.5% of the total community within at least one metagenome. Rare-transient biogeographic patterns were observed for ARG sequences belonging to adeB, bla_A, bla_B, CEP, dfra12, intI, tolC, vanA, vanX and vanH. Rare-transient sequences represented those with ≤1.5% of the total community. However, stepwise relationships with temperature were observed for several ARG sequences, suggesting the potential enrichment by fire for microbes harboring these ARG sequences. Two clusters of rare-transient sequences with no temperature relationship were observed based on differences in normalized abundance (Fig. 4), suggesting that they had no relationship with fire or temperature. Thus, we observed sequence-specific biogeography for ARG sequences along the temperature gradient, showing that the average changes in ARG abundance do not always fully explain the biogeography of each unique resistance gene sequence detected within that gene family.
Figure 3.
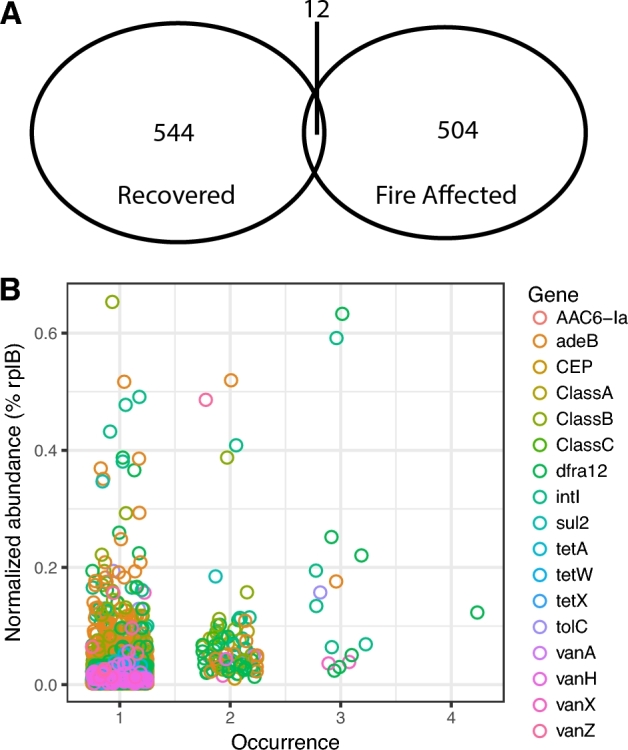
Presence of ARG sequences in Centralia metagenomes. (A) Venn diagram of ARG sequences observed in recovered and fire-affected soils. (B) ARG abundance-occurrence patterns in Centralia metagenomes. Percent normalized abundance of ARG sequences was averaged among 12 metagenomes and plotted against the number of sites each sequence occurs in. Each point represents one cluster, and color indicates gene.
Figure 4.

Normalized abundance of ARG sequences in Centralia metagenomes. Abundance of each gene sequence (clustered at 99% amino acid identity) present in ≥2 metagenomes was normalized to rplB. Complete-linkage clustering was calculated with the rplB-normalized abundance of each ARG sequence. Heatmap shows normalized abundance on a blue scale. Soil sites (column) are ordered by increasing soil temperature. Each row represents one ARG sequence, and ARG is noted by color.
ARG compositional shifts
We examined both rplB-normalized and relativized abundance patterns to compare changes in composition of ARGs and changes in proportional contributions of ARGs. For this analysis, composition was considered at the phylum or Proteobacteria class levels based on top BLAST hits. For ARGs that represented more than three phyla or Proteobacteria classes (bla_A, bla_B, dfra12, intI) (Tables S5 and S6, Supporting Information), we explored for correlations with temperature. We observed changes in ARG composition with temperature for bla_A, dfra12 and intI (Fig. 5).
Figure 5.

Relative abundance of taxonomically similar ARGs. Phylum-level taxonomy for bla_A, bla_B, dfra12, intI and rplB for each site is shown. Color indicates phylum- and Proteobacteria class-level taxonomy of ARGs, and sites are ordered by increasing soil temperature. dfra12 was not detected in Cen01.
Generally, community structure was associated with ARG composition. rplB-level reduction in Betaproteobacteria corresponded with reductions in Betaproteobacteria-related ARG. Betaproteobacteria-related bla_A and dfra12 genes decreased with temperature (Fig. 5; Table S6, Supporting Information). Thus, reductions in total bla_A and dfra12 counts are largely explained by a reduction in Betaproteobacteria. This pattern does not extend to bla_B since Betaproteobacteria-related bla_B genes were only detected in one soil (Cen16). We did not detect changes in Gammaproteobacteria based on rplB. This corresponded with consistent relative abundances of Gammaproteobacteria-related bla_A, bla_B, dfra12 and intI (Table S6, Supporting Information). Gammaproteobacteria-related dfra12 increased in relative abundance with soil temperature (ρ = 0.95, P < 0.05), further highlighting that a reduction in total dfra12 relative abundance is not due to changes in Gammaproteobacteria-related sequences. Phylum-level community structure, therefore, corresponded with compositional changes in ARGs, highlighting the influence of the underlying community on soil ARGs.
We observed evidence for functional redundancy of ARGs in Centralia through compositional shifts along the temperature gradient. Total bla_A relative abundance decreased with temperature (Fig. 1); however, taxonomic groups of bla_A were differentially impacted along the temperature gradient (Fig. 5; Table S6, Supporting Information). Both normalized and relativized abundance of Actinobacteria-related bla_A genes increased (ρ > 0.6, P < 0.05) while Betaproteobacteria-related bla_A genes decreased (ρ < 0.6, P < 0.05) with temperature (Table S6, Supporting Information). Thus, fire impacted the abundance and composition of bla_A. A decrease in total bla_A (Fig. 1) was accompanied by an increase in Actinobacteria-related bla_A. This asymmetric response with temperature suggests an impact of functional redundancy on soil ARG profiles. We also observed a shift in intI composition despite consistent intI abundance along the temperature gradient. The relative abundance of Beta- and Gammaproteobacteria-related intI decreased with temperature (ρ < 0.6, P < 0.05), but the relative abundance of Nitrospirae-related intI increased with temperature (ρ > 0.6, P < 0.05) (Fig. 5; Table S6, Supporting Information). We therefore observed changes in composition of intI with fire despite a lack of change in total intI abundance. Notably, previous studies have described Nitrospirae-related intI. Oliveira-Pinto et al. (2016) isolated an intI gene cassette related to Nitrospirae from a metal-rich stream, and Goltsman et al. (2009) identified both integrase and ARGs on chromosomes of Nitrospirae strains isolated from acid mine drainage. It is unclear, however, whether Nitrospirae-related intI genes are associated with ARG transfer. As intI encodes for a DNA integrase, this result suggests that Nitrospirae might contribute more to HGT in fire-affected soils, but we cannot determine whether this putative gene transfer would include ARGs. We posit that reductions in ARG abundance due to increased temperature could increase subsets of clinically relevant ARGs, and studies using temperature as a control for ARGs should consider sequence-level ARG dynamics within the system.
CONCLUSIONS
This case study of ARG biogeography over a long-term, severe thermal disturbance demonstrates the importance of community structure on soil ARG abundance and composition. Despite the stressor and the withdrawal of human activity, the diversity of ARG observed in Centralia is comparable to other soil systems (Forsberg et al.2014; Fitzpatrick and Walsh 2016). For several clinically relevant ARGs, we observed a reduction in total abundance with increased temperature. While this has been reported in anthropogenic systems (Diehl and Lapara 2010; Qian et al.2016; Tian et al.2016), we further probed Centralia datasets for compositional and sequence-specific ARG biogeography and found nuanced results. Generally, the reduction in ARG abundance could be explained by indirect effects (i.e. compositional shifts in the community). We posit that increased temperatures could result in a reduction in the diversity and abundance of ARGs in the environment, but our data also suggest that this reduction will not impact all ARG sequences similarly. ARG biogeographical dynamics in soil are thus largely dependent on community structure, which may also drive observed fine-scale abundance-occurrence patterns.
Supplementary Material
Supplementary data are available at FEMSEC online.
Acknowledgements
This research was supported through computational resources provided by the Institute for Cyber-Enabled Research. We thank the Ribosomal Database Project for their advice on reference gene database construction. We thank Trevor Grady for graphic design assistance in creating Fig. S1.
SUPPLEMENTARY DATA
Supplementary data are available at FEMSEC online.
FUNDING
Metagenome sequencing was supported by the Joint Genome Institute Community Science Project #1834. The work conducted by the U.S. Department of Energy (DOE) Joint Genome Institute, a DOE Office of Science User Facility, is supported under Contract No. DE-AC02-05CH11231. TKD was supported by the Department of Microbiology and Molecular Genetics Ronald and Sharon Rogowski Fellowship for Food Safety and Toxicology Graduate Fellowship.
Conflict of interest. None declared.
REFERENCES
- Allen HK, Donato J, Wang HH et al. Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Microbiol 2010;8:251–9. [DOI] [PubMed] [Google Scholar]
- Aminov RI, Mackie RI. Evolution and ecology of antibiotic resistance genes. FEMS Microbiol Lett 2007;271:147–61. [DOI] [PubMed] [Google Scholar]
- Berendonk TU, Manaia CM, Merlin C et al. Tackling antibiotic resistance: the environmental framework. Nat Rev Microbiol 2015;13:310–7. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V et al. BLAST+: architecture and applications. BMC Bioinformatics 2009;10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain S, Szoecs E, Foster Z et al. Taxanomic information from around the web. 2017. cran.r-project.org/package=taxize. (20 October 2017, date last accessed).
- Chen H, Boutros PC. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 2011;12:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J, Lee D, Cho Y et al. Direct extraction of DNA from soil for amplification of 16S rRNA gene sequences by polymerase chain reaction. J Microbiol 1996;34:229–35. [Google Scholar]
- Diehl DL, Lapara TM. Effect of temperature on the fate of genes encoding tetracycline resistance and the integrase of class 1 integrons within anaerobic and aerobic digesters treating municipal wastewater solids. Environ Sci Technol 2010;44:9128–33. [DOI] [PubMed] [Google Scholar]
- Elick JM. Mapping the coal fire at Centralia, Pa using thermal infrared imagery. Int J Coal Geol 2011;87:197–203. [Google Scholar]
- Fierer N, Leff JW, Adams BJ et al. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. P Natl Acad Sci USA 2012;109:21390–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish JA, Chai B, Wang Q et al. FunGene: The functional gene pipeline and repository. Front Microbiol 2013;4:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick D, Walsh F. Antibiotic resistance genes across a wide variety of metagenomes. FEMS Microbiol Ecol 2016;92:1–8. [DOI] [PubMed] [Google Scholar]
- Forsberg KJ, Patel S, Gibson MK et al. Bacterial phylogeny structures soil resistomes across habitats. Nature 2014;509:612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner E, Wallace JS, Argoty GA et al. Metagenomic profiling of historic Colorado Front Range flood impact on distribution of riverine antibiotic resistance genes. Sci Rep 2016;6:38432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson MK, Forsberg KJ, Dantas G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J 2014;9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goltsman DSA, Denef VJ, Singer SW et al. Community genomic and proteomic analyses of chemoautotrophic iron-oxidizing "Leptospirillum rubarum" (group II) and "Leptospirillum ferrodiazotrophum" (group III) bacteria in acid mine drainage biofilms. Appl Environ Microb 2009;75:4599–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiltunen T, Virta M, Laine A-L. Antibiotic resistance in the wild: an eco-evolutionary perspective. Philos T Roy Soc B 2017;372:20160039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janzen C, Tobin-Janzen T. Microbial communities in fire-affected soils. In: Dion P, Nautiyal CS (eds). Microbiology of Extreme Soils. Berlin Heidelberg: Springer, 2008, 299–316. [Google Scholar]
- Johnson TA, Stedtfeld RD, Wang Q et al. Clusters of antibiotic resistance genes enriched together stay together in swine agriculture. mBio 2016;7:e02214–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn LH. Antimicrobial resistance: A One Health perspective. Trans R Soc Trop Med Hyg. 2017;111:255–60. [DOI] [PubMed] [Google Scholar]
- Kolde R. Package “pheatmap.” 2015. cran.r-project.org/package=pheatmap. (20 October 2017, date last accessed).
- Kumar K, Gupta SC, Chander Y et al. Antibiotic use in agriculture and its impact on the terrestrial environment. Adv Agron 2005;87:1–54. [Google Scholar]
- Laine A-L, Hiltunen T, Virta M. Antibiotic resistance in the wild: an eco- evolutionary perspective. Philos T Roy Soc B 2016;372:20160039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang KS, Anderson JM, Schwarz S et al. Novel florfenicol and chloramphenicol resistance gene discovered in alaskan soil by using functional metagenomics. Appl Environ Microb 2010;76:5321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-H, Sorensen JW, Grady KL et al. Divergent extremes but convergent recovery of bacterial and archaeal soil communities to an ongoing subterranean coal mine fire. ISME J 2017;11:1447–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L-G, Xia Y, Zhang T. Co-occurrence of antibiotic and metal resistance genes revealed in complete genome collection. ISME J 2017;11:651–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 2013;8;e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melody SM, Johnston FH. Coal mine fires and human health: What do we know?? Int J Coal Geol 2015;152:1–14. [Google Scholar]
- Nesme J, Simonet P. The soil resistome: a critical review on antibiotic resistance origins, ecology and dissemination potential in telluric bacteria. Environ Microbiol 2015;17:913–30. [DOI] [PubMed] [Google Scholar]
- Nunes I, Jacquiod S, Brejnrod A et al. Coping with copper: legacy effect of copper on potential activity of soil bacteria following a century of exposure. FEMS Microbiol Ecol 2016;92, DOI: 10.1093/femsec/fiw175. [DOI] [PubMed] [Google Scholar]
- Oksanen J, Guillaume Blanchet F, Friendly M et al. Community Ecology Package, Package “vegan.” 2017. cran.r-project.org/package=vegan. (20 October 2017, date last accessed).
- Oliveira-Pinto C, Costa PS, Reis MP et al. Diversity of gene cassettes and the abundance of the class 1 integron-integrase gene in sediment polluted by metals. Extremophiles 2016;20:283–9. [DOI] [PubMed] [Google Scholar]
- Pal C, Bengtsson-Palme J, Kristiansson E et al. Co-occurrence of resistance genes to antibiotics, biocides and metals reveals novel insights into their co-selection potential. BMC Genomics 2015;16:964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Périchon B, Courvalin P. VanA-type vancomycin-resistant Staphylococcus aureus. Antimicrob Agents Ch 2009;53:4580–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Sun W, Gu J et al. Reducing antibiotic resistance genes, integrons, and pathogens in dairy manure by continuous thermophilic composting. Bioresource Technol 2016;220:425–32. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. http://www.r-project.org/. [Google Scholar]
- Revelle W. Procedures for Psychological, Psychometric, and Personality Research. 2017. cran.r-project.org/package=psych. (20 October 2017, date last accessed).
- Rizzo L, Manaia C, Merlin C et al. Urban wastewater treatment plants as hotspots for antibiotic resistant bacteria and genes spread into the environment: A review. Sci Total Environ 2013;447:345–60. [DOI] [PubMed] [Google Scholar]
- Rodriguez-R LM, Konstantinidis KT. Nonpareil: A redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 2014;30:629–35. [DOI] [PubMed] [Google Scholar]
- Shade A, Peter H, Allison SD et al. Fundamentals of microbial community resistance and resilience. Front Microbiol 2012;3:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Z, Zhang Y, Yu B et al. Changes of resistome, mobilome and potential hosts of antibiotic resistance genes during the transformation of anaerobic digestion from mesophilic to thermophilic. Water Res 2016;98:261–9. [DOI] [PubMed] [Google Scholar]
- Van Hoek AHAM, Mevius D, Guerra B et al. Acquired antibiotic resistance genes: an overview. Front Microbiol 2011;2:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang FH, Qiao M, Chen Z et al. Antibiotic resistance genes in manure-amended soil and vegetables at harvest. J Hazard Mater 2015a;299:215–21. [DOI] [PubMed] [Google Scholar]
- Wang Q, Fish JA, Gilman M et al. Xander: employing a novel method for efficient gene-targeted metagenomic assembly. Microbiome 2015b;3:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microb 2007;73:5261–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wannaprasat W, Padungtod P, Chuanchuen R. Class 1 integrons and virulence genes in Salmonella enterica isolates from pork and humans. Int J Antimicrob Ag 2011;37:457–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data are available at FEMSEC online.