

Abstract
We demonstrate for the first time that 4H-1,2,6-thiadiazin-4-one (TDZ) can function as a chemotype for the design of ATP-competitive kinase inhibitors. Using insights from a co-crystal structure of a 3,5-bis(arylamino)-4H-1,2,6-thiadiazin-4-one bound to calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2), several analogues were identified with micromolar activity through targeted displacement of bound water molecules in the active site. Since the TDZ analogues showed reduced promiscuity compared to their 2,4-dianilinopyrimidine counter parts, they represent starting points for development of highly selective kinase inhibitors.
Keywords: thiadiazinone, hinge binder, kinase inhibitor design, kinase water network, CaMKK2
1. Introduction
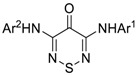
Protein kinases catalyze phosphate transfer from Adenosine Triphosphate (ATP) to tyrosine, threonine or serine residues in specific target proteins. These phosphorylation events occur in almost every signal transduction pathway and provide regulatory points for therapeutic intervention [1]. Kinases have been successfully utilized as drug targets for the past 30 years, with 38 kinase inhibitors approved by the FDA to date [2]. These drugs are predominantly multi-targeted tyrosine kinase inhibitors for the treatment of cancer [3]. However, approval of kinase inhibitors for the treatment on non-oncological diseases, such as rheumatoid arthritis, psoriasis and lung fibrosis has demonstrated their broader utility in treatment of human disease. There are over 500 kinases in the human genome, suggesting that there remains an untapped potential to treat a wide range of human ailments with new classes of inhibitors. Large scale kinome-wide profiling of ATP-competitive kinase inhibitors has also started to uncover the preferred chemotypes for the inhibition of many of the relatively under-studied kinases or dark kinases [4,5,6]. Despite the success in development of kinase inhibitor drugs, there is a still a need for new heterocycles on which to build ATP-competitive inhibitors [7,8]. One chemotype that has not yet been used in kinase inhibitor design is the 4H-1,2,6-thiadiazin-4-one (TDZ, Figure 1) [9,10] that can be prepared from 2,2-dichloromalononitrile [11].
Figure 1.

Representative examples of known dianilino(amino)pyrimidines (highlighted in red) and general (TDZ) core.
Dianilinopyrimidines represent a remarkably common chemotype that is found in ~10% of the clinically approved kinase inhibitor drugs, including ceritinib and palbociclib (Figure 1) [12]. Each of these drugs demonstrates potency and efficacy for its primary kinase target but also has cross activity on a broad range of other kinases. As such, these drugs and many other pyrimidine-based inhibitors have limited use as chemical probes to study the biology of specific kinases. As further testament to the broad activity profile of the dianilinopyrimidine chemotype, the 35 examples that are contained in the chemogenomic inhibitor sets PKIS/PKIS2 showed activity on >400 different protein kinases (excluding mutants) in either enzyme inhibition or affinity capture assays [13,14].
2. Results
2.1. Synthesis
Following analysis of the kinome-wide profiling of the dianilinopyrimidines in PKIS/PKIS2, we selected five R1 and R2 substituent pairs (Table 1 and Figure S1) that showed the broadest range of activity on human kinases. The corresponding dianilino-TDZs (1–5) were synthesized in two-steps from 3,5-dichloro-4H-1,2,6-thiadiazin-4-one (6) [9]. The reason for this strategy is that the first of the two reactive chlorine atoms of dichlorothiadiazinone 6 can be readily displaced by anilines and alkylamines with stoichiometric amounts of the amine (1 equiv.) and 2,6-lutidine (1 equiv.) as base in EtOH, at ca. 0–20 °C. However, more forcing conditions are typically required for the displacement of the remaining chloride. This is owed to electron release by the 3-amino group into the thiadiazine that decreases the electrophilicity of the 3-amino-5-chloro-1,2,6-4H-thiadiazin-4-one [15]. Nevertheless, we were able to use our recently developed Buchwald-Hartwig coupling conditions to overcome this difficulty [16] and enable the efficient synthesis of unsymmetrical 3,5-diamino-thiadiazinones.
Table 1.
Activity of dianilinothiadiazinones (1–5) on a broad range of protein kinases by Differential Scanning Fluorimetry (DSF) assay (see Supporting Information (SI) Table S1).
| ||||
|---|---|---|---|---|
| Entry | Ar1 | Ar2 | Number and Kinases Hit DSF > 2 °C a | |
| 1 |

|
|
1 | TRIB2 |
| 2 |
|

|
16 | CAMK1D/G/K1/K2B, CDC42BPA, CDK2 |
| CHEK2, DYRK2, MAP2K7, PHKG2 | ||||
| PIM1, PKMYT1, RPSKA6, STK3, STK17, TTK | ||||
| 3 |

|
|
9 | CAMK1G/2B, CDC42BPA, |
| CLK1, MAP2K7, PIM1, | ||||
| STK10, TRIB2, TTK | ||||
| 4 |

|

|
0 | |
| - | ||||
| 5 |
|

|
0 | |
| - | ||||
a Average of 4 experiments.
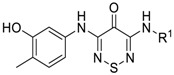
Therefore, treatment of dichlorothiadiazinone 6 with one equivalent of 5-amino-2-methylphenol or 2-amino-N-methylbenzamide in the presence of 2,6-lutidine (1 equiv.), gave the required 3-chloro-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (7) and 2-[(5-chloro-4-oxo-4H-1,2,6-thiadiazin-3-yl)amino]-N-methylbenzamide (8), respectively in good yields with a chromatography-free work-up (Scheme 1). Subsequently, scaffolds 7 and 8 were subjected to a Pd-catalyzed Buchwald-Hartwig coupling to introduce the second aniline. The five desired products (1–5) were obtained in medium to good yields (66–94%, Scheme 1).
Scheme 1.

Synthetic route to dianilino-TDZs 1–5.
After the synthesis of the desired dianilino-TDZs (1–5), the stability of dianilinothiadiazinone in biological systems was assessed: 3,5-bis(phenylamino)-4H-1,2,6-thiadiazin-4-one (9) [9] was subjected to neutral, acidic or slightly basic aqueous conditions (H2O/DMSO 50:50, THF/HCl 2 M 50:50 or THF/H2O 50:50 at pH 9 with a carbonate buffer), presence of amine or thiol nucleophiles (BuNH2 1 equiv., PhNH2 1 equiv., PhSH 1 equiv.), oxidizing [2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) 2 equiv.] and reducing conditions (Sn, 2 equiv.). The dianilinothiadiazine 9 was stable (by TLC) to all the above conditions after 48 h indicating that the thiadiazinone core was stable for potential development.
2.2. Initial Kinase Profiling
The corresponding dianilino-TDZs (1–5) were tested on a panel of 46 protein kinases representing the major kinome branches at a concentration of 10 μM using a Differential Scanning Fluorimetry (DSF) [17]. Only analogues 1–3 induced a significant (>2 °C) thermal shift when incubated with a subset of the proteins (Table 1). Compound 1 showed significant activity only on the pseudokinase TRIB2 (ΔTm = 2.5 °C) [18]. Compound 2 showed the broadest activity profile with ΔTm > 2 °C on 16/46 kinases. Compound 3 showed an intermediate activity profile, ΔTm > 2 °C on 9/46 kinases. Compounds 4 and 5 did not show ΔTm > 2 °C on any of the 46 kinases, although weak activity was detected on TRIB2 (ΔTm ~1 °C).
2.3. CaMKK2 Crystallography
To investigate the molecular details of interaction of the dianilino-TDZs (1–3) with protein kinases, co-crystallization with the corresponding purified proteins was attempted. Diffracting crystals were obtained with compound 2 in complex with CAMKK2 (see Table S2). The structure was solved by molecular replacement. The CAMKK2 kinase domain adopted an active state conformation in which residues of the regulatory and catalytic spines were aligned (Figure 2A); residue Glu236 within α-C helix directly contacted Lys194 (“α-C helix in”); and residues Asp330 and Phe331 within the conserved DFG motif pointed towards and away, respectively, from the ATP-binding site (“DFG -in”).
Figure 2.

The co-crystal structure of CAMKK2 bound to compound 2. (A) Cartoon representation of CAMKK2. (B) Binding interactions between CAMKK2 and compound 2. Dashed lines depict putative hydrogen bonds. Water molecules are shown as red spheres. (C) Electron density (omit) map (shown as a grey mesh contoured at 1.5 σ) for compound 2.
The ligand displayed two direct contact points to the hinge region of the ATP-binding pocket: one involving the oxygen atom of the thiadiazinone moiety and the other through the nitrogen atom of the hydroxymethylaniline moiety (Figure 2B). A water bridge made by the nitrogen atom from the aminobenzamide provided a third contact point to the kinase hinge region. The co-crystal structure revealed that the oxygen atom of the aminobenzamide interacted with the catalytic Lys194 and made a water-bridge with Glu236 of the α-C helix and Asp330 within the conserved DFG motif. Likewise, two water-bridge interactions connected the nitrogen atom from the aminobenzamide moiety and residues Ser175 within P-loop and Asn317 at the bottom of the kinase ATP-binding site. Finally, the ligand aminobenzamide ring made a T-shaped π-π interaction with the Phe267. Compound 2 is bound to CAMKK2 with aniline groups in a twisted conformation relative to the central TDZ ring, as can be seen in the electron density map (Figure 2C).
2.4. CaMKK2 Docking and Water Map Simulation
To further probe the molecular basis of ligand binding to CAMKK2, we compared our co-crystal structure of compound 2 (PDB 5VT1) with the previously published co-crystal structure with STO-609 (PDB:2ZV2) (Figure 3) [19]. We also controlled for hinge contacts in the model by using the 2,4-dianilinopyrimidines as a training set (see Figure S2). We noted that STO-609 can displace a bound water molecule from the ATP-binding site that is still present when compound 2 is bound to the enzyme. The carboxylic acid of STO-609 also forms a tighter interaction with the catalytic lysine than the benzamide of compound 2. To improve its activity, we designed analogues of 2 that would be able to displace the bound water molecule and form stronger interactions with the catalytic lysine.
Figure 3.

Comparison of the interaction of compounds 2 and STO-609 with the hinge region of CAMKK2: (A) PDB:5VT1 with compound 2; (B) PDB:2ZV2 with STO-609.
In order to use molecular docking to guide the design of new analogues, we used the Maestro suite (2017-3) to create a models of compounds STO-609 and 2 bound to CaMKK2 that accurately reflected what was seen in the crystal structures of both [19]. Water Map using a 2 nS simulation was used to populate the hydration sphere of compounds STO-609 and 2. The resulting model correctly identified the crystallographically observed water molecule highlighted in red (Figure 4). In addition, several other water molecules were identified in the ‘back’ pocket of the CaMKK2 that contained the catalytic lysine. Several analogues of compound 2 were designed to directly displace/interact with the key water molecule. In parallel, we designed a series of analogues to strengthen the interaction with the catalytic lysine.
Figure 4.

Hydration sphere of STO-609 (left) and 2 (right) generated by Water Map simulation showing high and low energy waters with graded shading (red—high energy and green—low energy).
The docking simulations showed that strengthening the interaction with the catalytic lysine while retaining the bound water molecule gave better scores than direct displacement of the water (Figure 5). We first optimized the core to see if the weak hit (2) could be a tractable starting point. Molecular simulations of compound 2 (Figure 5A) showed a weaker interaction with the catalytic lysine when the primary amide was switched in compound 10 (Figure 5B); the interaction with the water produced a more effective docking pose with a strong water mediated interaction with the backbone. We were able to boost the proposed lysine interaction with an imidazole substitution (11) (Figure 5C). The carboxylic acid in STO-609 appeared to contribute significantly to the binding affinity to CaMKK2. We designed a switch of the primary amide in compound 2 to a carboxylic acid (12) (Figure 5D) and this gave a 14/15 poses match to where STO-609’s carboxylic acid was directed in the co-crystal structure with CaMKK2 [19].
Figure 5.

Molecular docking into CaMKK2 of (A) compound 2; (B) compound 10; (C) compound 11; (D) compound 12.
2.5. Optimisation Results on CaMKK2
2.5.1. Outline of Compounds
We proposed a series of modifications of compound 2 relating to the crystal structure and modelling (Figure 6). These included a switch of the para-amide to the ortho-position (10) to better interact with the water and a direct substitution of the para-amide with a carboxylic acid (12) to form a stronger interaction with the water network. A series of mono-substituted ortho-, meta- and para- cyano analogues (13–15, respectively) probed the space available in this pocket and checked conformation constraints. A substitution on the adjacent anilino-nitrogen to the hinge binder to directly replace the water with a 4-methyloxazole (16) was encouraging. A methanol substitution at the ortho position (17) also looked promising and to increase the π-stacking potential of this analogue we added a meta-trifluoromethyl group (18). The use of an imidazole as a hydrogen bond donor/acceptor and the model (Figure 4C) suggested that, though out of plane, it could align with the catalytic lysine. The para-substituted imidazole (11) is about 30–40° out of plane and looked favorable to form a networked interaction between Lys197 and the wider water network. The final analogue was an arylthiadiazinone with a substitution of 2-cyclopentylbenzoic acid (19). This tactic for interaction of the wider water network with the para-carboxylic acid while having an adjacent meta-cyclopentyl to form a π-stacking/lipophilic interaction was previously used to successfully target SGK1, a regulator of epithelial sodium channels (eNaCs) [20].
Figure 6.

Designed compound rationale.
2.5.2. Analogue Synthesis
The designed analogues 10–19 were prepared using 3-chloro-5-[(3-hydroxy-4-methylphenyl)-amino]-4H-1,2,6-thiadiazin-4-one (7) as the substrate for the Buchwald-Hartwig coupling reaction to the relevant aniline. The desired dianilino-TDZs were prepared in medium to good yields (65–94%) except for 4-benzoic acid derivative 19 that gave a low yield (36%) attributed to potential nucleophilic displacements by the carboxylate ion in the reaction conditions (K2CO3). The secondary amine 20 required for the preparation of oxazole derivative 16 was prepared by a reductive amination reaction of 4-aminobenzamide and oxazole-4-carbaldehyde with sodium borohydride (Scheme 2).
Scheme 2.

Synthesis of oxazole amine 20 and dianilino-TDZ 11.
Interestingly, a different route was used to access imidazole derivative 11 as the Buchwald-Hartwig coupling of thiadiazinone 7 with 4-(1H-imidazol-2-yl)aniline led to a complex mixture of products. This was resolved by switching the reaction sequence and performing first the nucleophilic displacement of the 3-chloride of dichlorothiadiazinone 6 with 4-(1H-imidazol-2-yl)aniline to afford anilinothiadiazine 21 and subsequently performing the Buchwald-Hartwig coupling with 5-amino-2-methylphenol (Scheme 2).
The arylthiadiazinone analogue 19 required a different synthetic protocol involving a Suzuki coupling with the relevant arylboronic acid 22. The boronic acid was prepared in two steps from 4-bromo-2-fluorobenzoic acid (Scheme 3). Treatment of 4-bromo-2-fluorobenzoic acid 22 with cyclopentyl magnesium bromide led to 4-bromo-2-cyclopentylbenzoic acid (23). Subsequent lithium-halogen exchange and treatment with triisopropyl borate gave the desired boronic acid 24 albeit in a 35% overall yield. Boronic acid 24 was then reacted with 3-chloro-5-[(3-hydroxy-4-methylphenyl)-amino]-4H-1,2,6-thiadiazin-4-one (7) in the presence of Pd(Ph3P)4 (5 mol %) to yield the aryl-thiadiazinone 19 in 86% yield (Scheme 3) [10]. We note that this reaction order was chosen since it is difficult to perform a mono-arylation Suzuki reaction [10] but easy to mono-displace dichlorothiadiazinone 6 with amine nucleophiles.
Scheme 3.

Synthesis of boronic acid 22 and aryl-anilino-TDZ 19.
2.6. Optimization Results on CaMKK2
To more accurately determine the relative changes in potency of the TDZ analogues and to enable measurement of accurate IC50’s, we developed a Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET) assay. The TR-FRET tracer displacement assays were generated using a protocol derived from the Lanthascreen binding assays (ThermoFisher Scientific, Waltham, MA, USA) [21]. In this assay, we measured the effect of ATP competing compounds that are able to displace a fluorophore-labeled pan-kinase inhibitor (tracer 236) from the ATP binding site. We used ponatinib and staurosporine as internal controls to calibrate the FRET assay. The results of the CAMKK2 FRET assay are shown in Figure 7 and Table 2 (see Figures S3 and S4, SI). Surprisingly, TDZ analogues 1 and 2 showed no measurable activity at a concentration up to 50 μM but analogue 3 gave weak activity with an IC50 34 μM. Nevertheless, several of the structure-optimized analogues showed improvements in potency. TDZ’s 10–12 had the highest affinity for CAMKK2 with IC50 7.8, 3.2 and 10.5 μM, respectively.
Figure 7.

TR-FRET optimization of compound 2 with ponatinib and staurosporine as a control.
Table 2.
CaMKK2 FRET results for advanced TDZ analogues.
| |||||
|---|---|---|---|---|---|
| Compound | R1 | FRET (IC50) a | Compound | R1 | FRET (IC50) a |
| 1 |
|
>50 | 14 |

|
>50 |
| 2 |

|
>50 | 15 |
|
>50 |
| 3 |
|
34 | 16 |
|
>50 |
| 10 |

|
7.8 | 17 |

|
>50 |
| 11 |

|
3.2 | 18 |

|
>50 |
| 12 |
|
10.5 | 19 |
|
38 |
| 13 |
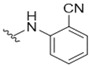
|
43 | - | Ponatinib Staurosporine |
0.4 & 0.0023 |
a Average of 2 experiments for CaMKK2/(μM).
The switch of the amide from the para-position (2) to the ortho-position (10) provided a >8-fold boost likely related to the new more favorable water mediated interaction. The exchange of the para-amide (2) to the para-carboxylic acid (12) led to a >5-fold increase in potency. However, the mono-cyano group substitutions were relatively in-effective. The ortho-cyano (13) showed some activity (43 μM) likely do to the water network interaction but meta-cyano (14) and para-cyano (15) were >50 μM against CaMKK2. This was the same result for the 4-methyloxazole (16), which was surprising but could be related to an inability to be accommodated in the active site. The methanol analogues (17-18) were also effectively inactive, likely due in part to the lack of ability to reach the water interaction. The imidazole (11) preformed well as we expected from our model (Figure 4C) and appeared to form the water network interaction in the back pocket of CaMKK2. The direct carbon-carbon bonded para-carboxylic acid with adjacent meta-cyclopentyl compound (19) was only weakly active (38 μM) and was likely out of position on this scaffold to form the optimal interaction as in STO-609.
2.7. Advanced Enzyme Assay Results on CaMKK2 Demonstrating Functional Inhibition
To further characterize the activity of 10–12 as inhibitors of CAMKK2, the compounds were subjected to an enzyme inhibition assay. CaMKK2 activity was measured by determining the rate of transfer of radiolabeled phosphate from [γ-32P]-ATP to a synthetic peptide substrate [22]. Compounds 10–12, when initially screened at a concentration of 1 µM, showed statistically significant inhibition of CAMKK2 kinase activity. The compounds were then screened at 7 concentrations (see Table S3, SI) to produce moderately potent IC50’s. TDZs 10–12 were demonstrated to be competent inhibitors of the CaMKK2 enzyme with enzymatic IC50’s of 11.9, 6.5 and 4.1 μM, respectively (Table 3).
Table 3.
CaMKK2 Enzyme assay results for advanced TDZ analogues.
| Compound | FRET (IC50) a | CaMKK2 Enzyme Assay b |
|---|---|---|
| IC50 (μM) | ||
| 10 | 7.8 | 11.9 |
| 11 | 10.5 | 6.5 |
| 12 | 3.2 | 4.1 |
| STO-609 | - | 0.04 |
a Average of 2 experiments; b Data are presented as mean ± SEM for 2 experiments
3. Discussion
We demonstrate, for the first time, that the 4H-1,2,6-thiadiazin-4-one (TDZ) chemotype can function as an ATP-competitive kinase inhibitor. TDZ represents a novel hinge binder with the potential to be further optimized into a high quality chemical probe for kinases such as CaMKK2. Furthermore, we report the first protein co-crystallization with this rare heterocycle. The electronics of the TDZ core allows for participation of the sulfur atom to be part of extended conjugated electronic exchanges through the core units to transfer charge [23]. This electronic property, exploited in solar cell applications, can partly explain the general lack of kinome promiscuity compared to the dianilinopyrimidine. The modular synthesis and relative narrow kinome spectrum make the TDZ an attractive chemotype for further development.
CaMKK2 is predominantly expressed in the brain, with trace expression in peripheral tissues such as the testis, spleen and lung [24,25]. In addition to recently being linked to appetite in vivo [26], CAMKK2 is over-expressed in multiple cancers [27,28]. The knockout of CaMKK2 can reduce cell proliferation and tumorigenicity in vivo, making CAMKK2 an attractive drug target. The only reported potent small molecule inhibitor of CaMKK2 is STO-609, which has several liabilities limiting its use as a probe of CaMKK2 activity. These include poor solubility and unfavorable off-target kinome profile with kinases that would cloud the interpretation of a phenotype including ERK8, MNK1 and PIM3 [29,30,31]. In addition, STO-609 is an agonist of the arylhydrocarbon receptor (AhR) [32]. The complicating factors highlight the need for the design and development of high quality inhibitors targeting CAMKK2.
Our results add further credence to the importance of water networks in optimization of kinase inhibitors. The advent of powerful modelling tools such as Water Map and the Schrodinger Maestro platform have made manipulating the water network more accessible [33,34,35]. There are two distinct water network regions that the TDZ core can exploit in binding to CaMKK2. We have shown an ability to exploiting the water network and lysine interactions, we improved on the activity of compound 2 and produced compound 11 that is >15-fold more potent. Our discovery of the TDZ core as a useful chemotype for kinase inhibitor design adds a new a hinge binding heterocycle to the medicinal chemistry tool box and provides another example of the application of sulfur in drug design.
4. Materials and Methods
4.1. Kinase Panel
Kinase selectivity assay—A home-made kinase panel was generated for the following enzymes: AAK1, BMP2K, BMX, BRAF, CAMK1D, CAMK1G, CAMKK1, CAMKK2B, CDC42BPA, CDK2, CDKL1, CHEK2, CLK1, CSNK1G1, CSNK1G3, CSNK2A1, DYRK1A, DYRK2A, EPHA2, GAK, GSG2, MAPK1, MAP2K7, MAPK14B, MAPK3, PHKG2, PIM1, PLK1, PKMYT1, PRPF4B, RPS6KA1A, RPS6KA5A, RPS6KA6A, SLK, SRPK1, SRPK2, STK3, STK6, STK10, STK17A, STK24, STK38L, TRIB2, TTK, VRK1 and VRK2. Proteins were produced in E. coli, purified in a Ni-chelate column, followed by overnight digestion using TEV protease (made in house with an N-terminal 6xHis tag) and dialysis to remove imidazole. To clear samples of uncleaved proteins and the TEV protease, samples were loaded on new Ni-chelate columns. The flow through was collected, concentrated and loaded to a HiLoad Superdex 200 16/600 column (GE Healthcare, Chicago, IL, USA) for final polishing and buffer exchange.
Starting from 100 µM protein stocks, our kinase panel enzymes were diluted to 1 µM in buffer 100 mM K2HPO4 pH 7.5 containing 150 mM NaCl, 10% glycerol and 5X dye (Applied Biosystems catalogue 4461806). The protein/dye mixture was transferred to a 384-well PCR microplate having 20 µL per well. Compounds in DMSO at 10 mM concentration were added in 20 nL volume, using a liquid handling device setup with a pin head, to make 10 µM compound concentration in the assay plate.
Protein thermal shift data was measured in a qPCR instrument (Applied Biosystems QuantStudio 6) programmed to equilibrate the plate at 25 °C for 5 min followed by ramping the temperature to 95 °C at a rate of 0.05 °C/s. Data was processed using Protein Thermal shift software (Applied Biosystems) by fitting experimental curves to a Boltzmann function to calculate differential thermal shifts (dTm) referenced to protein/dye in 0.2% DMSO.
4.2. CaMKK2 Crystallization
4.2.1. Cloning, Protein Expression and Purification
The crystallization of CAMKK2 was performed with a construct of CAMKK2 isoform 7 residues 161-449 (NCBI NP_001257415.1 – SGC construct CAMKK2B-cb002) containing the wild-type kinase domain in vector pNIC28-Bsa4. The construct was transformed into BL21(DE3) Escherichia coli cells that co-express λ-phosphatase and three rare tRNAs (plasmid pACYC-LIC+). Cells were cultured in TB medium containing 50 µg/mL kanamycin and 35 µg/mL chloramphenicol at 37 °C with shaking until the OD600 reached ~3 and then cooled to 18 °C for 1 h. Isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.1 mM and the cultures were left overnight at 18 °C. The cells were collected by centrifugation then resuspended in 2× lysis buffer [1× lysis buffer is 50 mM HEPES buffer, pH 7.5, 0.5 M KOAc, 10% (v/v) glycerol, 50 mM each arginine/glutamate, 10 mM imidazole, 1.0 mM tris(2-carboxyethyl)phosphine (TCEP), Protease Inhibitor Cocktail Set VII (Calbiochem, 1/500 dilution)] and flash-frozen in liquid nitrogen. Cells were lysed by sonication on ice. The resulting proteins were purified using Ni-Sepharose resin (GE Healthcare) and eluted stepwise in 1× lysis buffer with 300 mM imidazole. Removal of the hexahistidine tag was performed at 4 °C overnight using recombinant TEV protease. The protein was further purified using reverse affinity chromatography on Ni-Sepharose followed by gel filtration (Superdex 200 16/60, GE Healthcare). The protein in gel filtration buffer (10 mM HEPES, 500 mM KOAc, 1.0 mM TCEP, 5% (v/v) glycerol, 50 mM each arginine/glutamate) was concentrated to 8.5 mg/mL (measured by UV absorbance in a NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA, USA) using the calculated molecular weight and estimated extinction coefficient) using 30 kDa molecular weight cut-off centrifugal concentrators (Sigma-Aldrich Corp., St. Louis, MO, USA) at 4 °C. The concentrated protein was flash-frozen in a liquid nitrogen bath and stored at −80 °C until use.
4.2.2. Protein Crystallization
Kinase inhibitor (dissolved in 100% DMSO) was added to the protein in 3-fold molar excess and incubated on ice for approximately 30 min. The mixture was centrifuged at 15,000 rpm for 10 min at 4 °C before setting up 150 nL volume sitting drops at three ratios (2:1, 1:1, or 1:2 protein-inhibitor complex to reservoir solution). Crystallization experiments were performed at 20 °C. Crystals were cryoprotected in mother liquor supplemented with 25–30% glycerol before flash-freezing in liquid nitrogen for data collection. Diffraction data were collected at 100 K at Diamond Light Source beamline I03. Crystal optimization used Newman’s buffer system [36].
4.2.3. Structure Solution and Refinement
Diffraction data were integrated using XDS [37] and scaled using AIMLESS from the CCP4 software suite (version 7.0.057, London, UK) [38]. Molecular replacement (MR) was performed with Phaser [19] using the CAMKK2 bound to STO-609 co-structure (PDB ID 2ZV2) [19]. Automated refinement was performed with Refmac [39,40]. Coot [41] was used for manual model building and refinement. Structure validation was performed using MolProbity [42]. Structure factors and coordinates have been deposited in the PDB (see Table S2, SI).
4.3. Molecular Modelling
4.3.1. Molecular Modelling
Molecular modelling was performed using Schrödinger Maestro software package (version 2018-1, Schrödinger, Mannheim, Germany) [43]. Structures of small molecules were prepared using and the LigPrep module of Schrodinger suite employing OPLS3 force for all computations. X-ray crystal structure for the CaMMKK2 (PDB:5VT1/2ZV2) was pre-processed using the protein preparation wizard of Schrödinger suite to optimize the hydrogen bonding network [43].
Prior to Glide docking, the grid box was centered using corresponding x-ray ligand as template. The ligand docking was performed using default SP settings of Schrodinger Glide with softened vdw’s potential (0.6) and additional hydrogen bond constraints to NH of V270 (hinge residue). Graphical illustrations were generated using Schrödinger Maestro software (version 2018-1, Schrödinger, Mannheim, Germany).
4.3.2. Hydration Site Analysis
Hydration site analysis calculated with Water Map (Schrödinger Release 2017-3: Water Map, Schrödinger, LLC, New York, NY, 2017). The 5VT1 structure was prepared with Protein Preparation Wizard (as above). Waters were analyzed within 6 Å of the co-crystallized ligand and the 2 ns simulation was conducted with OPLS3 force field.
4.4. Biochemical Assays
4.4.1. CaMKK2B TR-FRET Assay
CAMKK2 kinase domain (132–470) was cloned in a pNIC-Bio2 vector in fusion with N-terminal 10xHis tag followed by a TEV protease cleavage site and a C-terminal biotin ligase recognition sequence. This construct was used in the expression of CAMKK2 in E. coli BL21(DE3)-R3-BirA [44]. Protein was purified in a Ni-NTA column (Thermo Scientific, Waltham, MA, USA) followed TEV digestion overnight, dialysis to remove imidazole and re-purification in Ni-NTA to remove undigested samples and TEV protease (made in house with an N-terminal 6xHis tag). As a last step, this sample was loaded to a HiLoad Superdex 200 16/600 column (GE Healthcare, Chicago, IL, USA) for final polishing and buffer exchange.
Tracer displacement assay was measured in 15 µL volume containing 5 nM of our C-terminal biotinylated CAMKK2 kinase domain, 2 nM of Europium-labeled streptavidin in buffer 50 mM HEPES pH 7.5, 10 mM MgCl2, 1 mM EGTA, 0.01% Brij-35 and 8 nM of tracer 236 (measured KD of 8.13 ± 0.9 nM) as described [45].
4.4.2. CaMKK2 Enzyme Assay
CaMKK2 activity was measured as described previously [22]. A standard 30 μL assay, 1 ng of recombinant bacterial expressed human CaMKK2 (residues 50–588) was added to assay buffer (50 mM HEPES [pH 7.4], 1 mM DTT, 0.02% (v/v) Brij-35) containing 200 μM CaMKKtide peptide substrate, 50 μM CaCl2, 1 μM calmodulin (Sigma-Aldrich Corp., St. Louis, MO, USA), 50 μM [γ-32P]-ATP (Perkin Elmer) and 5 mM MgCl2, in the presence and absence of different concentrations of small-molecule inhibitors. Reactions were incubated for 10 min at 30 °C, after which they were terminated by spotting 15 μL onto P81 phosphocellulose paper (Whatman, GE Healthcare, Chicago, IL, USA) and washing extensively in 1% phosphoric acid. Radioactivity was quantified by scintillation counting.
4.5. Chemistry Experimental Section
4.5.1. General Methods and Materials
All chemicals were commercially available except those whose synthesis is described. Anhydrous Na2SO4 was used for drying organic extracts and all volatiles were removed under reduced pressure. 1,4-Dioxane was dried by refluxing over CaH2. All reaction mixtures and column eluents were monitored by TLC using commercial glass backed thin layer chromatography (TLC) plates (Merck Kieselgel 60 F254) [46]. The plates were observed under UV light at 254 and 365 nm. The technique of dry flash chromatography was used throughout for all prep scale chromatographic separations using Merck Silica Gel 60 (less than 0.063 mm). Melting points were determined using a PolyTherm-A, Wagner & Munz, Koefler-Hotstage Microscope apparatus or were determined using a TA Instruments DSC Q1000 with samples hermetically sealed in aluminum pans under an argon atmosphere; using heating rates of 5 °C/min (DSC m.p. listed by onset and peak values). Solvents used for recrystallization are indicated after the melting point. UV spectra were obtained using a Perkin-Elmer Lambda-25 UV/vis spectrophotometer and inflections are identified by the abbreviation “inf.” IR spectra were recorded on a Shimadzu FTIR-NIR Prestige-21 spectrometer with Pike Miracle Ge ATR accessory and strong, medium and weak peaks are represented by s, m and w, respectively. 1H and 13C-NMR spectra were recorded on a Bruker Avance 300 (at 300 and 75 MHz, respectively), or a 500 machine (at 500 and 125 MHz, respectively). Deuterated solvents were used for homonuclear lock and the signals are referenced to the deuterated solvent peaks. APT NMR studies identified quaternary and tertiary carbons, which are indicated by (s) and (d) notations, respectively. MALDI-TOF mass spectra were recorded on a Bruker Autoflex III Smartbeam instrument. Low resolution (EI) mass spectra were recorded on a Shimadzu Q2010 GC-MS with direct inlet probe. 3,5-Dichloro-4H-1,2,6-thiadiazin-4-one (6) was prepared according to the reported procedure [9].
4.5.2. Preparation of Aniline Starting Materials
4-[(Oxazol-4-ylmethyl)amino]benzamide (20). To a stirred solution of 4-aminobenzamide (136 mg, 1.00 mol) in EtOH (5 mL), at ca. 20 °C, was added oxazole-4-carbaldehyde (97 mg, 1.00 mmol) in one portion and the mixture was stirred at this temperature for 12 h. Then NaBH4 (75.6 mg, 2.00 mmol) and the mixture was stirred for a further 6 h. H2O (10 mL) was then added and the mixture stirred for 30 min. The colorless solid formed was then filtered under reduced pressure and washed with EtOH (2 mL), DCM (5 mL) and n-hexane (5 mL) to give the title compound 20 (153.6 mg, 71%) as colorless plates, m.p. 161–162 °C (from EtOH); Rf 0.44 (DCM/MeOH, 90:10); (found: C, 60.78; H, 5.03; N, 19.26. C11H11N3O2 requires C, 60.82; H, 5.10; N, 19.34%); λmax(EtOH)/nm 216 (log ε 3.86), 292 (4.34); υmax/cm−1 3381 m, 3273 m, 3169 m, 3129 w, 1639 m, 1599s , 1530 s, 1422 m, 1391 s, 1385 m, 1342 m, 1278 w, 1267 m, 1242 w, 1204 m, 1186 m, 1150 s, 1126 m, 1109 s, 1086 m, 1061 s, 1003 m, 922 m, 874 w, 842 m, 828 m, 804 m, 789 m, 775 m, 762 m, 727 m, 702 m; δH(500 MHz; CDCl3) 8.33 (1H, d, J 0.7), 7.97 (1H, d, J 0.9), 7.63 (2H, d, J 8.7, Ar H), 7.55 (1H, br s, NH), 6.86 (1H, br s, NH), 6.61 (2H, d, J 8.8, Ar H), 6.57 (1H, dd, J 5.9, 5.9, Ar H), 4.20 (2H, d, J 5.8, CH2); δC(125 MHz; CDCl3) 167.9 (s), 152.1 (s), 150.7 (s), 137.7 (s), 136.1 (d), 128.9 (d), 121.3 (s), 111.0 (d), 38.4 (t); m/z (APCI+) 218 (MH+, 59%), 201 (93), 175 (32), 137 (100), 120 (55).
4-Borono-2-cyclopentylbenzoic acid (24). To a stirred solution of 4-bromo-2-fluorobenzoic acid (22) (2.00 g, 9.13 mmol) in THF, at ca. 0 °C, under a N2 atmosphere, was added a solution of 2 M cyclopentyl magnesium bromide (16 mL, 32 mmol) and the mixture stirred at this temperature for 4.5 h. Then was added slowly 2 M HCl (25 mL) followed by EtOAc (40 mL). The two layers were separated and the organic layer was washed with H2O (2 × 20 mL) and then dried (MgSO4). The solvent was removed under vacuum to give 4-bromo-2-cyclopentylbenzoic acid (23) as a colorless solid (2.10 g, 85%) that was used directly in next step without further purification.
4-Bromo-2-cyclopentylbenzoic acid (23) (2.10 g, 7.80 mmol) was dissolved in THF (50 mL) and the mixture cooled to −78 °C with stirring. Triisopropyl borate (6.30 mL, 27.3 mmol, 3.5 equiv.) was then added, followed by the slow addition of a solution of n-BuLi (hexanes) 2.5 M (13 mL, 31.2 mmol, 4 equiv.). The reaction mixture was slowly warmed to room temperature and stirred for 5 h. Then a solution of 2 M HCl (20 mL) was added and the mixture stirred for 10 min. The mixture was extracted with EtOAc (2 × 25 mL) and the combined organic layers were then stirred with 2.5 M NaOH (30 mL) for 1 h. The layers were separated and the aqueous layer acidified to pH 2-3 with concentrated HCl. The mixture was then extracted by EtOAc (2 × 25 mL), the organic layer dried (Na2SO4) and the solvent was removed under vacuum. The crude colorless solid was stirred in DCM (10 mL) and filtered to give the title compound 24 (750 mg, 35% overall yield) as a colorless solid, m.p. 162–165 °C; Rf 0.38 (n-hexane/Et2O, 50:50); υmax/cm−1 3215 br (O-H), 2955 w, 2947 w and 2870 w (alkyl C-H), 1692 s, 1678 s, 1503 w, 1441 w, 1366 s, 1333 m, 1302 m, 1248 m, 1213 m, 1188 m, 1144 m, 1113 m, 1072 w, 1044w, 1011 w, 932 w, 903 w, 849 w, 791 m, 716 s; δH(500 MHz; DMSO-d6) 12.86 (1H, br s, OH), 8.19 (2H, s, OH), 7.87 (1H, s, Ar H), 7.62 (1H, d, J 7.6, Ar H), 7.54 (1H, d, J 7.6, Ar H), 3.67–3.60 (1H, m, alkyl H), 1.98 (2H, br s, alkyl H), 1.79 (2H, br s, alkyl H), 1.66-1.53 (4H, m, alkyl H); δC(125 MHz; DMSO-d6) 169.9 (s), 144.1 (s), 137.1 (s), 133.4 (s), 132.3 (d), 131.0 (d), 127.6 (d), 41.1 (d), 34.4 (t), 25.3 (t); mass spectrometry and elemental analysis data could not be obtained due to compound instability.
4.5.3. Preparation of 3-Amino-Substituted-4H-1,2,6-Thiadiazines
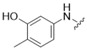
3-Chloro-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (7) (General procedure). To a stirred solution of 3,5-dichloro-4H-1,2,6-thiadiazin-4-one (6) (366.0 mg, 2.000 mmol) in EtOH (4 mL), at ca. 20 °C, was added 5-amino-2-methylphenol (246.3 mg, 2.000 mmol) in one portion followed by 2,6-lutidine (233 μL, 4.00 mmol) and the mixture was stirred at this temperature until complete consumption of the starting material (TLC, 1 h). The yellow solid formed was then filtered under reduced pressure and washed with EtOH (2 mL), DCM (5 mL) and n-hexane (5 mL) to give the title compound 7 (477.1 mg, 77%) as orange needles, m.p. 248–250 °C (from EtOH/THF); Rf 0.53 (n-hexane/t-BuOMe, 50:50); (found: C, 44.45; H, 2.97; N, 15.56. C10H8ClN3O2S requires C, 44.53; H, 2.99; N, 15.58%); λmax(DCM)/nm 241 (log ε 3.88), 343 (4.34), 408 (3.67); υmax/cm−1 3383br (O-H), 3341 m and 2922 w (C-H), 1584 s, 1562 s, 1557 s, 1553 s, 1503 m, 1450 w, 1427 m, 1421 m, 1406 w, 1366 w, 1327 w, 1312 w, 1263 m, 1238 m, 1194 w, 1153 m, 1123 s, 999 m, 972 m, 874 m, 862 m, 851 s, 802s , 737 m, 727 s; δH(500 MHz; CDCl3) 9.91 (1H, s), 9.36 (1H, s), 7.32 (1H, d, J 2.0, Ar H), 7.06 (1H, dd, J 8.1, 2.0, Ar H), 7.00 (1H, d, J 8.2, Ar H), 2.08 (3H, s, CH3); δC(125 MHz; CDCl3) 157.0 (s), 155.1 (s), 150.0 (s), 140.7 (s), 136.2 (s), 130.1 (d), 120.0 (s), 111.8 (d), 107.4 (d), 15.5 (q); m/z (MALDI-TOF) 272 (MH+ + 2, 42%), 270 (MH+, 94), 252 (100), 234 (32), 180 (42).
2-[(5-Chloro-4-oxo-4H-1,2,6-thiadiazin-3-yl)amino]-N-methylbenzamide (8). Similar treatment of 3,5-dichloro-4H-1,2,6-thiadiazin-4-one (6) (183 mg, 1.00 mmol) in EtOH (1 mL), with 2-amino-N-methyl-benzamide (150 mg, 1.00 mmol) and 2,6-lutidine (116 μL, 2.00 mmol) after 48 h gave the title compound 8 (228 mg, 77%) as yellow needles, m.p. 252–255 °C (from benzene); Rf 0.22 (n-hexane/t-BuOMe, 50:50); (found: C, 44.45; H, 2.92; N, 18.74. C11H9ClN4O2S requires C, 44.53; H, 3.06; N, 18.88%); λmax(DCM)/nm 240 inf (log ε 4.47), 300 (4.64), 336 (4.82), 402 (4.25); υmax/ cm−1 3310 m, 3111 w, 1626 m, 1595 m, 1585 m, 1541 s, 1537 s, 1452 m, 1435 m, 1406 m, 1329 m, 1308 m, 1285 w, 1238 w, 1178 m, 1169 m, 1150 w, 1107 w, 1053 w, 1001 w, 947 w, 885 m, 858 m, 841 m, 773 m, 752 m, 727 m; δH(500 MHz; CDCl3) 12.36 (1H, br s, NH), 8.76 (1H, s, NH), 8.47 (1H, d, J 8.2, Ar H), 7.78 (1H, d, J 7.4, Ar H), 7.55 (1H, dd, J 7.4, 7.4, Ar H), 7.20 (1H, dd, J 7.2, 7.2, Ar H), 2.81 (3H, d, J 3.5, CH3); δC(125 MHz; CDCl3) 168.3 (s), 157.3 (s), 149.5 (s), 141.5 (s), 137.9 (s), 131.8 (d), 128.2 (d), 122.9 (d), 121.3 (s), 119.1 (d), 26.2 (q); m/z (MALDI-TOF) 298 (M+ + 2, 25%), 296 (M+, 100%), 265 (42).
3-{[4-(1H-Imidazol-2-yl)phenyl]amino}-5-chloro-4H-1,2,6-thiadiazin-4-one (21). Similar treatment of 3,5-dichloro-4H-1,2,6-thiadiazin-4-one (6) (91.5 mg, 0.500 mmol) in MeCN (2 mL), with 4-(1H-imidazol-2-yl)aniline dihydrochloride (116 mg, 0.500 mmol) and Hünig’s base (261 μL, 1.50 mmol) after 2 h gave the title compound 21 (63.3 mg, 42%) as orange needles, m.p. 298-300 °C (from EtOH/THF); Rf 0.45 (t-BuOMe); λmax(DCM)/nm 268 (log ε 4.00), 342 (4.53), 403 (3.73); υmax/cm−1 3293 m, 2768 br, 1630 s, 1589s, 1547 s, 1537 s, 1512 s, 1445 m, 1402 w, 1331 w, 1296 w, 1248 w, 1182 m, 1107 m, 1005 w, 949 m, 885m, 868 m, 849 s, 779 m, 733 s, 717 s; δH(500 MHz; CDCl3) 12.51 (1H, br s, NH), 10.21 (1H, s, NH), 7.90 (2H, d, J 8.9, Ar H), 7.87 (2H, d, J 8.4, Ar H), 7.12 (2H, s, Ar H); δC(125 MHz; CDCl3) 157.1 (s), 149.9 (s), 145.2 (s), 145.2 (s), 137.6 (s), 126.8 (s), 126.5 (s), 126.3 (d), 125.0 (d), 120.6 (d), 113.5 (d); m/z (ESI+) 306 (MH+, 15%), 160 (33), 153 (19), 130 (38), 62 (100).
4.5.4. Preparation of 3,5-Diaminosubstituted Thiadiazines
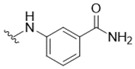
3-({5-[(3-Hydroxy-4-methylphenyl)amino]-4-oxo-4H-1,2,6-thiadiazin-3-yl}amino)benzamide (1) (General procedure). To a mixture of 3-chloro-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (7) (53.9 mg, 0.200 mmol), Pd[3,5-(F3C)2C6H3]3 (5.3 mg, 1.25 mol %), DPEPhos (5.3 mg, 5 mol %), powdered dry K2CO3 (66.4 mg, 0.480 mmol) and 3-aminobenzamide (30.0 mg, 0.220 mmol) was added dioxane (5 mL). The stirred suspension was then deaerated by bubbling of Ar through it for 5 min and then heated at reflux under Ar until complete consumption of the starting thiadiazine (TLC, 2 h). The mixture was cooled to ca. 20 °C, then adsorbed onto silica and chromatographed (n-hexane/acetone, 50:50) to give the title compound 1 (63.1 mg, 85%) as orange needles, m.p. 297–298 °C (from THF); Rf 0.30 (n-hexane/acetone, 50:50); (found: C, 55.11; H, 4.15; N, 18.83. C17H15N5O3S requires C, 55.28; H, 4.09; N, 18.96%); λmax(EtOH)/nm 207 (log ε 4.62), 338 (4.), 453 (3.80); υmax/cm−1 3447 w, 3373 w, 3345 m, 3329 m, 3177 w, 1641 m, 1628 m, 1614 m, 1582 s, 1537 s, 1510 s, 1485 m, 1477 m, 1435 m, 1422 m, 1341 w, 1327 w, 1310 w, 1294 w, 1275 w, 1234 m, 1196 w, 1180 m, 1124 m, 1070 w, 999 m, 869 m, 860 m, 787 m; δH(500 MHz; DMSO-d6) 9.63 (1H, s, NH), 9.38 (1H, s, NH), 9.29 (1H, s, OH), 8.30 (1H, s, NH), 7.92 (1H, s, NH), 7.89 (1H, dd, J 8.1, 1.8, Ar H), 7.53 (1H, d, J 7.8, Ar H), 7.41–7.36 (3H, m, Ar H and NH), 7.05 (1H, dd, J 8.1, 1.9, Ar H), 6.97 (1H, d, J 8.2, Ar H), 2.07 (3H, s, CH3); δC(125 MHz; DMSO-d6) 167.8 (s), 155.1 (s), 154.6 (s), 147.1 (s), 146.8 (s), 139.0 (s), 137.3 (s), 134.9 (s), 130.1 (d), 128.3 (d), 122.2 (d), 121.4 (d), 118.8 (d), 118.4 (s), 110.6 (d), 106.1 (d), 15.4 (q); m/z (ESI+) 370 (MH+, 100%), 369 (M+, 7), 214 (14); HRMS found for MH+ 370.09675, C17H16N5O3S requires 370.09684.
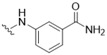
4-({5-[(3-Hydroxy-4-methylphenyl)amino]-4-oxo-4H-1,2,6-thiadiazin-3-yl}amino)benzamide (2). Similar treatment of 3-chloro-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (7) (53.9 mg, 0.200 mmol) with 4-aminobenzamide (30.0 mg, 0.220 mmol) after 1 h gave after chromatography (n-hexane/acetone, 50:50) the title compound 2 (69.2 mg, 94%) as orange needles, m.p. > 300 °C (from MeOH/THF); Rf 0.27 (n-hexane/acetone, 50:50); (found: C, 55.39; H, 4.25; N, 18.78. C17H15N5O3S requires C, 55.28; H, 4.09; N, 18.96%); λmax(EtOH)/nm 208 (log ε 4.60), 235 inf (4.28), 347 (4.82), 451 (3.94); υmax/cm−1 3645 w, 3362 m, 3325 m, 3188 w, 2955 m, 2918 w, 2870 w, 1667 m, 1607 m, 1593 m, 1531 m, 1510 s, 1485 m, 1429 m, 1416 m, 1402 m, 1337 m, 1323 m, 1242 m, 1190 m, 1177 m, 1159 m, 1126 m, 1055 m, 1001 w, 955 w, 924 w, 889 m, 860 m, 849 m, 802 m, 785 m; δH(500 MHz; DMSO-d6) 9.76 (1H, s, NH), 9.39 (1H, s, NH), 9.28 (1H, s, OH), 7.88–7.84 (5H, m, Ar H and NH), 7.40 (1H, d, J 2.0, Ar H), 7.22 (1H, br s, NH), 7.05 (1H, dd, J 8.2, 2.1, Ar H), 6.98 (1H, d, J 8.2, Ar H), 2.08 (3H, s, CH3); δC(125 MHz; DMSO-d6) 167.3 (s), 155.1 (s), 154.8 (s), 147.3 (s), 146.5 (s), 141.8 (s), 137.2 (s), 130.1 (d), 128.1 (d), 127.8 (s), 118.5 (s), 118.1 (d), 110.7 (d), 106.2 (d), 15.5 (q); m/z (ESI+) 370 (MH+, 100%); HRMS found for MH+ 370.09655, C17H16N5O3S requires 370.09684.
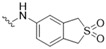
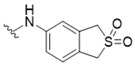
3-[(2,2-Dioxido-1,3-dihydrobenzo[c]thiophen-5-yl)amino]-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (3). Similar treatment of 3-chloro-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (7) (53.9 mg, 0.200 mmol) with 5-amino-1,3-dihydrobenzo[c]thiophene 2,2-dioxide (40.3 mg, 0.220 mmol) after 4 h gave after chromatography (n-hexane/acetone, 50:50) the title compound 3 (56.9 mg, 68%) as orange needles, m.p. > 300 °C (from EtOH/THF); Rf 0.74 (n-hexane/acetone, 50:50); (found: C, 52.06; H, 3.92; N, 13.37. C18H16N4O4S2 requires C, 51.91; H, 3.87; N, 13.45%); λmax(EtOH)/nm 208 (log ε 4.62), 338 (4.73), 454 (3.89); υmax/cm−1 3335 w, 3316 w, 2970 w, 2949 w, 2924 w, 2870 w, 1614 m, 1587 m, 1530 m, 1504 s, 1487 m, 1460 m, 1454 m, 1433 m, 1381 m, 1300 m, 1263 m, 1217 m, 1186 m, 1177 m, 1165 m, 1123 m, 1105 m, 1091 m, 1047 w, 1001 w, 980 w, 910 w, 806 m, 731 w; δH(500 MHz; DMSO-d6) 9.69 (1H, s, NH), 9.36 (1H, s, NH), 9.27 (1H, s, OH), 7.89 (1H, s, Ar H), 7.72 (1H, d, J 8.2, Ar H), 7.39 (1H, s, Ar H), 7.32 (1H, d, J 8.4, Ar H), 7.04 (1H, d, J 8.2, Ar H), 6.98 (1H, d, J 8.2, Ar H), 4.50 (2H, s, CH2), 4.43 (2H, s, CH2), 2.07 (3H, s, CH3); δC(125 MHz; DMSO-d6) 155.1 (s), 154.7 (s), 147.2 (s), 146.7 (s), 138.9 (s), 137.3 (s), 132.5 (s), 130.1 (d), 126.1 (d), 126.0 (s), 119.5 (d), 118.5 (s), 116.1 (d), 110.7 (d), 106.2 (d), 56.3 (t), 55.7 (t), 15.4 (q); m/z (ESI+) 417 (MH+, 21%), 391 (100), 214 (24); HRMS found for MH+ 417.06829, C18H17N4O4S2 requires 417.06857.
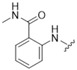
N-Methyl-2-([5-({3-[(methylsulfonyl)methyl]phenyl}amino)-4-oxo-4H-1,2,6-thiadiazin-3-yl]-amino}-benzamide (4). Similar treatment of 2-[(5-chloro-4-oxo-4H-1,2,6-thiadiazin-3-yl)amino]-N-methyl-benzamide (8) (59.3 mg, 0.200 mmol) with 3-[(methylsulfonyl)methyl]aniline (40.8 mg, 0.220 mmol) after 3 h gave after chromatography (t-BuOMe) the title compound 4 (58.6 mg, 66%) as orange needles, m.p. > 300 °C (from DMA); Rf 0.23 (t-BuOMe); (found: C, 50.98; H, 4.25; N, 15.56. C19H19N5O4S2 requires C, 51.22; H, 4.30; N, 15.72%); λmax(THF)/nm 240 (log ε 4.54), 267 (4.37), 354 (4.78), 443 (4.19); υmax/cm−1 3347 w, 3287 w, 2936 w, 1628 m, 1614 m, 1605 m, 1593 m, 1582 m, 1530 s, 1518 s, 1493 m, 1489 m, 1450 m, 1429 m, 1410 m, 1333 m, 1302 m, 1292 m, 1287 m, 1242 m, 1227 w, 1169 m, 1148 w, 1117 m, 1088 w, 972 m, 945 w, 851 w, 789 m, 777 m, 750 m, 729 w; δH(300 MHz; DMSO-d6) 12.09 (1H, s, NH), 9.72 (1H, s, NH), 8.68 (1H, d, J 4.4, Ar H), 8.52 (1H, d, J 8.3, Ar H), 7.89 (1H, s, Ar H), 7.75 (2H, dd, J 7.4, 7.4, Ar H), 7.52 (1H, dd, J 7.7, 7.7, Ar H), 7.36 (1H, dd, J 7.8, 7.8, Ar H), 7.10 (2H, dd, J 7.8, 7.8, Ar H), 4.47 (2H, s, CH2), 2.94 (3H, s, CH3), 2.82 (3H, d, J 4.4, CH3); δC(125 MHz; DMSO-d6) 168.5 (s), 155.1 (s), 147.1 (s), 146.6 (s), 139.0 (s), 131.7 (d), 129.4 (s), 128.6 (d), 128.1 (d), 125.4 (d), 121.6 (d), 121.4 (d), 120.5 (s), 119.5 (d), 118.1 (d), 59.5 (t), 26.1 (q), one C (q) resonance missing; m/z (ESI+) 446 (MH+, 17%), 391 (100); HRMS found for MH+ 446.09443, C19H20N5O4S2 requires 446.09512.
N-Methyl-2-({5-[(4-morpholinophenyl)amino]-4-oxo-4H-1,2,6-thiadiazin-3-yl}amino)benzamide (5). Similar treatment of 2-[(5-chloro-4-oxo-4H-1,2,6-thiadiazin-3-yl)amino]-N-methylbenzamide (8) (59.3 mg, 0.200 mmol) with 4-morpholinoaniline (39.2 mg, 0.220 mmol) after 3 h gave after chromatography (n-hexane/acetone, 50:50) the title compound 5 (60.9 mg, 69%) as red plates, m.p. 285–287 °C (from EtOH/THF); Rf 0.42 (n-hexane/acetone, 50:50); (found: C, 57.25; H, 4.91; N, 19.33. C21H22N6O3S requires C, 57.52; H, 5.06; N, 19.17%); λmax(DCM)/nm 356 (log ε 4.61), 457 (3.82); υmax/cm−1 3329 w, 2963 w, 2916 w, 2851 w, 1614 m, 1593 m, 1585 m, 1526 m, 1511 s, 1504 s, 1449 m, 1435 m, 1412 m, 1402 m, 1317 m, 1287 m, 1267 m, 1240 m, 1167 w, 1123 m, 1088 w, 1070 w, 1053 w, 932 m, 810 m, 748 m; δH(500 MHz; DMSO-d6) 12.04 (1H, s, NH), 9.49 (1H, s, NH), 8.67 (1H, s, NH), 8.50 (1H, d, J 8.3, Ar H), 7.73 (1H, d, J 7.6, Ar H), 7.63 (2H, d, J 8.8, Ar H), 7.51 (1H, dd, J 7.6, 7.6, Ar H), 7.09 (1H, dd, J 7.4, 7.4, Ar H), 6.93 (2H, d, J 8.8, Ar H), 3.74 (4H, dd, J 3.9, 3.9, CH2), 3.06 (4H, dd, J 3.6, 3.6, CH2), 2.81 (3H, d, J 4.2, CH3); δC(125 MHz; DMSO-d6) 168.5 (s), 155.0 (s), 147.3 (s), 147.1 (s), 146.1 (s), 139.1 (s), 131.7 (d), 130.9 (s), 128.1 (d), 121.3 (d), 120.8 (d), 120.4 (s), 117.9 (d), 115.3 (d), 66.0 (t), 48.8 (t), 26.1 (q); m/z (ESI+) 439 (MH+, 100%); HRMS found for MH+ 439.15424, C21H23N6O3S requires 439.15469.
2-({5-[(3-Hydroxy-4-methylphenyl)amino]-4-oxo-4H-1,2,6-thiadiazin-3-yl}amino)benzamide (10). Similar treatment of 3-chloro-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (7) (53.9 mg, 0.200 mmol) with 2-aminobenzamide (30.0 mg, 0.220 mmol) after 3 h gave after filtration of the reaction mixture and washing with H2O (5 mL) and EtOH (5 mL) the title compound 10 (67.6 mg, 91%) as orange needles, m.p. 290 °C (decomp., from EtOH/THF); Rf 0.44 (DCM/Et2O, 90:10); (found: C, 55.42; H, 4.16; N, 18.77. C17H15N5O3S requires C, 55.28; H, 4.09; N, 18.96%); λmax(EtOH)/nm 232 (log ε 4.14), 262 inf (3.94), 326 inf (4.42), 352 (4.59), 453 (3.85); υmax/cm−1 3411 br, 1643 m, 1582 s, 1530 s, 1518 s, 1510 s, 1503 s, 1452 m, 1400 m, 1310 m, 1231 m, 1177 m, 1124 m, 999 w, 833 w, 750 m; δH(500 MHz; DMSO-d6) 12.36 (1H, s, NH), 9.45 (1H, s, OH), 8.56 (1H, d, J 8.4, Ar H), 8.21 (1H, br s, NH), 7.83 (1H, d, J 7.9, Ar H), 7.67 (1H, br s, NH), 7.51 (1H, dd, J 7.7, 7.7, Ar H), 7.39 (1H, d, J 1.7, Ar H), 7.09-7.05 (2H, m, Ar H), 6.97 (1H, d, J 8.1, Ar H), 2.07 (3H, s, CH3), one NH resonance missing; δC(125 MHz; DMSO-d6) 170.6 (s), 155.1 (s), 155.0 (s), 147.2 (s), 146.4 (s), 139.8 (s), 137.3 (s), 132.0 (d), 130.1 (d), 128.8 (d), 121.1 (d), 119.3 (s), 118.5 (s), 117.8 (d), 110.5 (d), 106.1 (d), 15.4 (q); m/z (ESI+) 370 (MH+, 100%), 369 (M+, 25); HRMS found for MH+ 370.09656, C17H16N5O3S requires 370.09684.
3-{[4-(1H-Imidazol-2-yl)phenyl]amino}-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (11). Similar treatment of 5-chloro-3-{[4-(1H-imidazol-2-yl)phenyl]amino}-4H-1,2,6-thiadiazin-4-one (21) (61.1 mg, 0.200 mmol) with 5-amino-2-methylphenol (27.1 mg, 0.220 mmol) after 18 h gave after filtration of the reaction mixture and washing with H2O (5 mL) and EtOH (5 mL) the title compound 11 (63.0 mg, 80%) as orange plates, m.p. > 300 °C (from EtOH/THF); Rf 0.62 (DCM/THF, 50:50); (found: C, 58.19; H, 4.31; N, 21.26. C19H16N6O2S requires C, 58.15; H, 4.11; N, 21.42%); λmax(THF)/nm 351 (log ε 4.50), 456 (3.56); υmax/cm−1 3358 w, 3314 w, 3167 w, 1593 m, 1579 m, 1526 m, 1508 m, 1504 s, 1445 m, 1422 m, 1319 m, 1248 m, 1231 w, 1126 w, 1101 w, 1049 w, 947 w, 926 w, 887 w, 858 w, 829 m, 814 w, 772 w, 760 w, 708 m; δH(300 MHz; DMSO-d6) 12.36 (1H, s, NH), 9.65 (1H, s, NH), 9.36 (1H, s, NH), 9.28 (1H, s, OH), 7.87 (4H, s, Ar H), 7.40 (1H, d, J 1.9, Ar H), 7.21 (1H, s, Ar H), 7.05 (1H, dd, J 8.1, 1.9, Ar H), 6.98 (2H, d, J 7.7, Ar H), 2.08 (3H, s, CH3); δC(75 MHz; DMSO-d6) 155.1 (s), 154.7 (s), 147.1 (s), 146.8 (s), 145.5 (s), 138.7 (s), 137.3 (s), 130.1 (d), 128.6 (d), 125.3 (s), 125.0 (d), 119.1 (d), 118.4 (s), 117.1 (d), 110.6 (d), 106.1 (d), 15.4 (q); m/z (ESI+) 393 (MH+, 100%); HRMS found for MH+ 393.11204, C19H17N6O2S requires 393.11282.
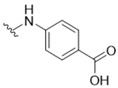
4-({5-[(3-Hydroxy-4-methylphenyl)amino]-4-oxo-4H-1,2,6-thiadiazin-3-yl}amino)benzoic acid (12). Similar treatment of 3-chloro-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (7) (53.9 mg, 0.200 mmol) with 4-aminobenzoic acid (30.2 mg, 0.220 mmol) after 30 min gave after filtration of the reaction mixture and washing with H2O (5 mL) and EtOH (5 mL) the title compound 12 (26.8 mg, 36%) as orange needles, m.p. > 300 °C (from dioxane); Rf 0.56 (DCM/Et2O, 90:10); (found: C, 54.98; H, 3.90; N, 15.16. C17H14N4O4S requires C, 55.13; H, 3.81; N, 15.13%); λmax(MeOH)/nm 212 (log ε 4.35), 235 (4.04), 269 (3.75), 349 (4.55), 447 (3.68); υmax/cm−1 3362 w, 3327 w, 2924 w, 1591 s, 1547 s, 1530 s, 1414 m, 1479 m, 1421 m, 1391 s, 1385 s, 1315 m, 1248 m, 1229 m, 1206 m, 1179 m, 1152 s, 1128 m, 1111 m, 1001 w, 860 w, 837 w, 822 w, 785 m, 752w , 737 w, 710 w; δH(500 MHz; DMSO-d6) 10.25 (1H, br, CO2H), 9.46 (1H, s, NH), 9.25 (1H, s, OH), 7.83 (2H, d, J 8.0, Ar H), 7.66 (2H, d, J 8.1, Ar H), 7.16 (1H, s, Ar H), 6.95 (2H, s, Ar H), 2.08 (3H, s, CH3), one NH resonance missing; δC(125 MHz; DMSO-d6) 168.5 (s), 155.7 (s), 154.5 (s), 146.9 (s), 146.7 (s), 138.9 (s), 137.3 (s), 135.9 (s), 129.9 (d), 129.4 (d), 118.4 (s), 117.9 (d), 110.1 (d), 106.1 (d), 15.6 (q); m/z (ESI+) 371 (MH+, 17%), 370 (M+, 100); HRMS found for M+ 370.07236, C17H14N4O4S requires 370.07358.
2-({5-[(3-Hydroxy-4-methylphenyl)amino]-4-oxo-4H-1,2,6-thiadiazin-3-yl}amino)benzonitrile (13). Similar treatment of 3-chloro-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (7) (53.9 mg, 0.200 mmol) with 2-aminobenzonitrile (26.0 mg, 0.220 mmol) after 4 h gave after chromatography (DCM/Et2O, 90:10) the title compound 13 (57.9 mg, 82%) as orange needles, m.p. 229–230 °C (from benzene/MeCN); Rf 0.67 (DCM/Et2O, 90:10); (found: C, 58.09; H, 3.57; N, 19.82. C17H13N5O2S requires C, 58.11; H, 3.73; N, 19.93%); λmax(DCM)/nm 264 (log ε 3.90), 332 inf (4.36), 345 (4.53), 443 (3.82); υmax/cm−1 3383 br, 3341 w, 2218 w (C≡N), 1587 s, 1562 s, 1557 s, 1503 m, 1454 w, 1427 m, 1365 w, 1312 m, 1263 m, 1238 m, 1211 m, 1196 m, 1153 s, 112 3s, 999 m, 972 m, 874 m, 862 m, 853 m, 802 m, 727 m; δH(300 MHz; DMSO-d6) 9.53 (1H, s, NH), 9.51 (1H, s, NH), 9.28 (1H, s, OH), 7.99 (1H, d, J 8.3, Ar H), 7.84 (1H, dd, J 7.8, 1.4, Ar H), 7.71 (1H, ddd, J 7.6, 7.6, 1.5, Ar H), 7.37 (1H, d, J 2.0, Ar H), 6.98 (1H, d, J 8.2, Ar H), 2.08 (3H, s, CH3); δC(75 MHz; DMSO-d6) 155.1 (s), 154.5 (s), 147.5 (s), 146.7 (s), 140.8 (s), 137.1 (s), 134.0 (d), 133.0 (d), 130.1 (d), 124.2 (d), 122.1 (d), 118.7 (s), 116.8 (s), 110.8 (d), 106.4 (d), 104.9 (s), 15.4 (q); m/z (ESI+) 352 (MH+, 100%); HRMS found for MH+ 352.08600, C17H14N5O2S requires 352.08627.
3-({5-[(3-Hydroxy-4-methylphenyl)amino]-4-oxo-4H-1,2,6-thiadiazin-3-yl}amino)benzonitrile (14). Similar treatment of 3-chloro-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (7) (53.9 mg, 0.200 mmol) with 3-aminobenzonitrile (26.0 mg, 0.220 mmol) after 1 h gave after chromatography (DCM/Et2O, 90:10) the title compound 14 (65.8 mg, 94%) as yellow needles, m.p. 258–259 °C (from EtOH/THF); Rf 0.61 (DCM/Et2O, 90:10); (found: C, 58.05; H, 3.71; N, 19.85. C17H13N5O2S requires C, 58.11; H, 3.73; N, 19.93%); λmax(THF)/nm 281 (log ε 4.09), 341 (4.76), 447 (3.09); υmax/cm−1 3345 w, 2237 w (C≡N), 1578 m, 1574 m, 1535 s, 1510 s, 1505 s, 1487 m, 1476 m, 1325 m, 1312 m, 1296 m, 1244 m, 1165 m, 1123 m, 999 m, 858 m, 789 m; δH(300 MHz; DMSO-d6) 9.90 (1H, s, NH), 9.40 (1H, s, NH), 9.28 (1H, s, OH), 8.25 (1H, s, Ar H), 8.13 (1H, d, J 8.1, Ar H), 7.53 (1H, dd, J 7.8, 7.8, Ar H), 7.46 (1H, d, J 7.6, Ar H), 7.39 (1H, d, J 1.9, Ar H), 7.05 (1H, dd, J 8.2, 1.9, Ar H), 6.98 (1H, d, J 8.3, Ar H), 2.08 (3H, s, CH3); δC(75 MHz; DMSO-d6) 155.1 (s), 154.7 (s), 147.4 (s), 146.4 (s), 140.0 (s), 137.1 (s), 130.1 (d), 129.9 (d), 125.7 (d), 123.8 (d), 121.6 (d), 118.8 (s), 118.6 (s), 111.3 (s), 110.7 (d), 106.3 (d), 15.4 (q); m/z (ESI+) 352 (MH+, 100%), 351 (M+, 27); HRMS found for MH+ 352.08583, C17H14N5O2S requires 352.08627.
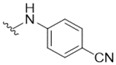
4-({5-[(3-Hydroxy-4-methylphenyl)amino]-4-oxo-4H-1,2,6-thiadiazin-3-yl}amino)benzonitrile (15). Similar treatment of 3-chloro-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (7) (53.9 mg, 0.200 mmol) with 4-aminobenzonitrile (26.0 mg, 0.220 mmol) after 1 h gave after chromatography (DCM/Et2O, 90:10) the title compound 15 (61.5 mg, 88%) as yellow needles, m.p. 280 °C (decomp., from EtOH/THF); Rf 0.61 (DCM/Et2O, 90:10); (found: C, 58.00; H, 3.62; N, 19.85. C17H13N5O2S requires C, 58.11; H, 3.73; N, 19.93%); λmax(THF)/nm 355 (log ε 4.46), 445 (3.65); υmax/cm−1 3366 w, 3323 w, 2974 w, 2220 w (C≡N), 1620 w, 1614 w, 1601 m, 1580 m, 1535 m, 1531 m, 1518 s, 1510 s, 1487 m, 1414 m, 1325 m, 1248 w, 1231 w, 1177 m, 1123 m, 1092 w, 1049 m, 999 m, 964 w, 910 w, 853 w, 837 m, 797 m, 729 w; δH(500 MHz; DMSO-d6) 10.02 (1H, s, NH), 9.44 (1H, s, NH), 9.30 (1H, s, OH), 8.02 (1H, d, J 8.9, Ar H), 7.76 (1H, d, J 8.9, Ar H), 7.38 (1H, d, J 2.1, Ar H), 7.05 (1H, dd, J 8.1, 2.1, Ar H), 6.98 (1H, d, J 8.3, Ar H), 2.08 (3H, s, CH3); δC(125 MHz; DMSO-d6) 155.1 (s), 154.9 (s), 147.8 (s), 146.1 (s), 143.5 (s), 137.1 (s), 132.9 (d), 130.2 (d), 119.3 (s), 118.77 (d), 118.72 (s), 110.8 (d), 106.4 (d), 103.5 (s), 15.5 (q); m/z (ESI+) 352 (MH+, 100%), 351 (M+, 56); HRMS found for MH+ 352.08593, C17H14N5O2S requires 352.08627.
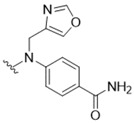
4-({5-[(3-Hydroxy-4-methylphenyl)amino]-4-oxo-4H-1,2,6-thiadiazin-3-yl}(oxazol-4-ylmethyl)amino)-benzamide (16). Similar treatment of 3-chloro-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (7) (53.9 mg, 0.200 mmol) with 4-[(oxazol-4-ylmethyl)amino]benzamide (20) (47.3 mg, 0.220 mmol) after 3 h gave after chromatography (DCM/t-BuOMe, 50:50) the title compound 16 (66.9 mg, 74%) as yellow needles, m.p. 235–237 °C (from EtOH/THF); Rf 0.43 (DCM/t-BuOMe, 90:10); (found: C, 56.12; H, 3.97; N, 18.59. C21H18N6O4S requires C, 55.99; H, 4.03; N, 18.66%); λmax(THF)/nm 312 inf (log ε 4.54), 347 (4.81), 422 (4.01); υmax/cm−1 3404 w, 3377 w, 3358 w, 3120 w, 1678 m, 1593 s, 1547 m, 1524 m, 1479 s, 1470 s, 1422 m, 1342 m, 1269 m, 1238 m, 1182 m, 1177 m, 1124 m, 1113 m, 1059 m, 1051 m, 997 m, 984 m, 920 m, 848 m, 831 m, 826 m, 761 m, 756 m; δH(300 MHz; DMSO-d6) 10.03 (1H, s, NH), 9.69 (1H, s, NH), 9.33 (1H, s, OH), 8.34 (1H, d, J 0.8, NH), 8.01 (1H, d, J 0.8, NH), 7.71 (1H, d, J 8.8, Ar H), 7.37 (1H, d, J 1.9, Ar H), 7.06 (1H, dd, J 8.2, 2.0, Ar H), 7.00 (1H, d, J 8.3, Ar H), 6.92 (1H, dd, J 5.8, 5.8, Ar H), 6.72 (1H, d, J 8.8, Ar H), 4.25 (2H, d, J 5.7, CH2), 2.08 (3H, s, CH3); δC(75 MHz; DMSO-d6) 163.7 (s), 155.4 (s), 155.1 (s), 152.1 (s), 151.9 (s), 149.9 (s), 145.0 (s), 137.4 (s), 136.6 (s), 136.1 (d), 130.1 (d), 129.4 (d), 119.8 (s), 119.3 (s), 111.3 (d), 106.9 (d), 38.2 (t), 15.5 (q), one C (d) resonance missing; m/z (ESI+) 451 (MH+, 100%); HRMS found for MH+ 451.11757, C21H19N6O4S requires 451.11830.
3-[(3-Hydroxy-4-methylphenyl)amino]-5-{[2-(hydroxymethyl)phenyl]amino}-4H-1,2,6-thiadiazin-4-one (17). Similar treatment of 3-chloro-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (7) (53.9 mg, 0.200 mol) with (2-aminophenyl)methanol (27.1 mg, 0.220 mol) after 7 h gave after filtration of the reaction mixture and washing with H2O (5 mL) and EtOH (5 mL) the title compound 17 (46.1 mg, 65%) as orange needles, m.p. 242–243 °C (from EtOH/THF); Rf 0.29 (DCM/t-BuOMe, 90:10); (found: C, 57.17; H, 4.59; N, 15.82. C17H16N4O3S requires C, 57.29; H, 4.53; N, 15.72%); λmax(THF)/nm 265 (log ε 4.03), 323 (4.71), 334 inf (4.62), 441 (3.89); υmax/cm−1 3453 w, 3370 w, 3319 w, 3312 w, 2907 w, 1601 w, 1589 m, 1568 m, 1535 m, 1530 m, 1526 m, 1497 s, 1460 m, 1454 m, 1422 m, 1416 m, 1310 m, 1252 m, 1231 w, 1213 w, 1202 w, 1186 w, 1175 w, 1132 w, 1003 m, 934 w, 844 m, 806 m, 748s; δH(300 MHz; DMSO-d6) 9.94 (1H, s, NH), 9.38 (1H, s, NH), 9.28 (1H, s, OH), 8.08 (1H, d, J 7.8, Ar H), 7.39–7.29 (3H, m, Ar H), 7.06–6.96 (3H, m, Ar H), 5.72 (1H, s, OH), 4.60 (2H, s, CH2), 2.08 (3H, s, CH3); δC(75 MHz; DMSO-d6) 155.1 (s), 154.7 (s), 146.9 (s), 146.8 (s), 137.6 (s), 137.3 (s), 130.9 (s), 130.1 (d), 128.4 (d), 127.7 (d), 122.7 (d), 119.5 (d), 118.4 (s), 110.5 (d), 106.0 (d), 62.3 (t), 15.4 (q); m/z (ESI+) 357 (MH+, 98%), 356 (M+, 100); HRMS found for MH+ 357.10121, C17H17N4O3S requires 357.10159.
3-[(3-Hydroxy-4-methylphenyl)amino]-5-{[2-(hydroxymethyl)-3-(trifluoromethyl)phenyl]amino}-4H-1,2,6-thiadiazin-4-one (18). Similar treatment of 3-chloro-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (7) (53.9 mg, 0.200 mol) with [2-amino-6-(trifluoromethyl)phenyl]methanol (42.1 mg, 0.220 mol) after 4 h gave after filtration of the reaction mixture and washing with H2O (5 mL) and EtOH (5 mL) the title compound 18 (55.5 mg, 65%) as orange needles, m.p. 241–242 °C (from EtOH/c-hexane); Rf 0.60 (DCM/t-BuOMe, 80:20); (found: C, 51.23; H, 3.17; N, 13.26. C18H15F3N4O3S requires C, 50.94; H, 3.56; N, 13.20%); λmax(THF)/nm 278 (log ε 4.31), 284 (4.30), 340 (4.96), 351 inf (4.89), 447 (4.16); υmax/ cm−1 3537 w, 3366 w, 3171 w, 1582 m, 1541 m, 1537 s, 1508 m, 1504 s, 1483 m, 1443 m, 1323 m, 1306 m, 1287 m, 1273 w, 1175 m, 1134 m, 1123 m, 1107 m, 1092 m, 1013 w, 997 m, 978 w, 851 w, 789 m; δH(500 MHz; DMSO-d6) 10.20 (1H, s, NH), 9.43 (1H, s, NH), 9.28 (1H, s, OH), 8.38 (1H, d, J 8.2, Ar H), 7.54 (1H, dd, J 8.0, 8.0, Ar H), 7.43 (1H, d, J 7.8, Ar H), 7.37 (1H, d, J 2.1, Ar H), 7.05 (1H, dd, J 8.1, 2.1, Ar H), 6.98 (1H, d, J 8.3, Ar H), 6.07 (1H, dd, J 4.5, 4.5, Ar H), 4.71 (2H, d, J 3.9, CH2), 2.08 (3H, s, CH3); δC(125 MHz; DMSO-d6) 155.1 (s), 154.7 (s), 147.1 (s), 146.7 (s), 140.2 (s), 137.2 (s), 130.2 (d), 128.5 (s), 128.2 (d), 127.2 (q, 2JCF 30.1), 124.1 (q, 1JCF 274.2), 123.8 (d), 119.4 (q, 3JCF 5.8), 118.6 (s), 110.6 (d), 106.2 (d), 56.9 (t), 15.5 (q); δF(282 MHz; DMSO-d6) -56.5 (s, CF3); m/z (ESI+) 425 (MH+, 100%), 424 (M+, 9); HRMS found for MH+ 425.08829, C18H16F3N4O3S requires 425.08897.
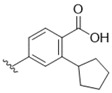
2-Cyclopentyl-4-{5-[(3-hydroxy-4-methylphenyl)amino]-4-oxo-4H-1,2,6-thiadiazin-3-yl}-benzoic acid (19). A stirred mixture of 3-chloro-5-[(3-hydroxy-4-methylphenyl)amino]-4H-1,2,6-thiadiazin-4-one (7) (53.9 mg, 0.200 mol), 4-borono-2-cyclopentylbenzoic acid (24) (51.5 mg, 0.220 mol), Na2CO3 (21.2 mg, 0.200 mol) and Pd(Ph3P)4 (11.6 mg, 0.0100 mol, 5 mol %), in dioxane/H2O 5:3 (0.8 mL) was deaerated by bubbling of Ar through it for 5 min and then heated to ca. 100 °C under Ar until complete consumption of the starting thiadiazine (TLC, 3 h). The mixture was then cooled to ca. 20 °C, diluted with DCM (10 mL) and extracted with saturated Na2CO3 (2 × 10 mL). The combined aqueous phase was then acidified with 2 M HCl to a pH of 3 and then extracted with DCM (3 × 10 mL), the organic phase dried (Na2SO4), filtered and evaporated under reduced pressure to give the title compound 19 (72.9 mg, 86%) as yellow plates, m.p. 277–278 °C (from c-hexane); Rf 0.40 (DCM/t-BuOMe, 80:20); (found: C, 62.67; H, 4.91; N, 9.85. C22H21N3O4S requires C, 62.40; H, 5.00; N, 9.92%); λmax(THF)/nm 278 (log ε 4.02), 358 (4.37), 422 inf (3.92); υmax/ cm−1 3455 w, 3321 w, 2955 w, 2866 w, 1694 m, 1620 m, 1595 m, 1547 s, 1422 m, 1310 m, 1267 m, 1234 w, 1175 m, 1119 m, 999 w, 941 w, 901 w, 858 w, 804 w, 797 w, 733 w; δH(500 MHz; DMSO-d6) 13.04 (1H, br, COOH), 10.00 (1H, s, NH), 9.38 (1H, s, OH), 8.23 (1H, dd, J 1.5, Ar H), 7.87 (1H, dd, J 8.2, 1.6, Ar H), 7.72 (1H, d, J 8.2, Ar H), 7.41 (1H, d, J 2.0, Ar H), 7.11 (1H, dd, J 8.1, 2.1, Ar H), 7.01 (1H, d, J 8.2, Ar H), 3.81–3.76 (1H, m, CH), 2.09 (3H, s, CH3), 2.08-2.04 (2H, m, CH2), 1.76–1.82 (2H, m, CH2), 1.70–1.61 (2H, m, CH2), 1.60–1.52 (2H, m, CH2); δC(75 MHz; DMSO-d6) 169.2 (s), 159.6 (s), 155.1 (s), 152.2 (s), 150.4 (s), 145.4 (s), 137.6 (s), 136.4 (s), 132.4 (s), 130.2 (d), 128.8 (d), 126.5 (d), 125.0 (d), 119.8 (s), 111.7 (d), 107.3 (d), 41.0 (d), 34.4 (t), 35.2 (t), 15.5 (q); m/z (ESI+) 424 (MH+, 100%), 423 (M+, 4); HRMS found for MH+ 424.13167, C22H22N3O4S requires 424.13255.
Acknowledgments
The authors thank the University of Cyprus, the Cyprus Research Promotion Foundation, the National Cancer Institute of the National Institute of Health and the following organizations and companies in Cyprus for generous donations of chemicals and glassware: The State General Laboratory; the Agricultural Research Institute; the Ministry of Agriculture; MedoChemie, Ltd.; Medisell, Ltd.; and Biotronics, Ltd. Furthermore, we thank the A. G. Leventis Foundation for helping to establish the NMR facility at the University of Cyprus. In addition, we thank Brandie M. Ehrmann for LC-MS/HRMS support provided by the Mass Spectrometry Core Laboratory at the University of North Carolina at Chapel Hill, and thank Yi Liang and Opher Gileadi for support and assistance. We also thank the Biocenter Finland/DDCB for financial support towards the goals of our work and the CSC-IT Center for Science Ltd. (Finland) for the allocation of computational resources. We thank Diamond Light Source for access to beamline I03 (MX14664) that contributed to the results presented here.
We also acknowledge-The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA), Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda and Wellcome Trust.
Supplementary Materials
The following are available online, Figure S1: Design of TDZs 1–5, Table S1: DSF kinome selectivity panel, Table S2: X-ray crystallography data, Figure S2: Validation of modelling docking poses showing the same hinge contacts as standard 2,4-dianilinopyrimidines, Figure S3 and S4: CaMKK2 FRET results for advanced thiadiazinone analogues, Table S3: CaMKK2 Enzyme assay raw data results for TDZs 10–12 and STO-609, 1H and 13C-NMR spectra of all new compounds.
Author Contributions
C.R.M.A./W.J.Z./D.H.D./P.A.K./A.S.K. conceived and designed the study; C.R.M.A., P.H.G., R.M.C., A.S.K., T.L., C.G.L. and J.S.O. performed the experiments; C.R.M.A, P.G, R.M.C., C.G.L., J.S.O and A.K analyzed the data; C.R.M.A., A.S.K., J.W.S., W.J.Z., D.H.D., T.M.W. and P.A.K. edited the paper; C.R.M.A. wrote the paper.
Funding
This research was funded by the University of Cyprus grant number [postdoctoral funding (A.S.K.)]; the Cyprus Research Promotion Foundation grant number [NEAYPODOMH/NEKYP/0308/02)], the National Cancer Institute of the National Institute of Health grant number [R01CA218442]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 1–5 and 10–19 are available from the authors.
References
- 1.Brognard J., Hunter T. Protein Kinase Signalling Networks in Cancer. Curr. Opin. Genet. Dev. 2011;21:4–11. doi: 10.1016/j.gde.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.U.S. Food & Drug Administration New drugs at FDA: CDER’s new molecular entities and new therapeutic biological products. [(accessed on 23 April 2018)]; Available online: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/ DrugInnovation/default.htm.
- 3.Cohen P., Alessi D.R. Kinase drug discovery—What’S next in the field? ACS Chem. Biol. 2013;8:96–104. doi: 10.1021/cb300610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klaeger S., Heinzlmeir S., Wilhelm M., Polzer H., Vick B., Koenig P.-A., Reinecke M., Ruprecht B., Petzoldt S., Meng C., et al. The target landscape of clinical kinase drugs. Science. 2017;358:eaan4368. doi: 10.1126/science.aan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fabian M.A., Biggs W.H., Treiber D.K., Atteridge C.E., Azimioara M.D., Benedetti M.G., Carter T.A., Ciceri P., Edeen P.T., Floyd M., et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 6.Anastassiadis T., Deacon S.W., Devarajan K., Ma H., Peterson J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011;29:1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fedorov O., Müller S., Knapp S. The (un)targeted cancer kinome. Nat. Chem. Biol. 2010;6:166–169. doi: 10.1038/nchembio.297. [DOI] [PubMed] [Google Scholar]
- 8.Knapp S., Arruda P., Blagg J., Burley S., Drewry D.H., Edwards A., Fabbro D., Gillespie P., Gray N.S., Kuster B., et al. A public-private partnership to unlock the untargeted kinome. Nat. Chem. Biol. 2013;9:3–6. doi: 10.1038/nchembio.1113. [DOI] [PubMed] [Google Scholar]
- 9.Geevers J., Trompen W.P. Synthesis and reactions of 3,5-dichloro-4H-1,2,6-thiadiazin-4-one. Recl. Trav. Chim. Pays-Bas. 1974;93:270–272. doi: 10.1002/recl.19740930911. [DOI] [Google Scholar]
- 10.Ioannidou H.A., Kizas C., Koutentis P.A. Palladium Catalyzed C–C Coupling Reactions of 3,5-Dichloro-4H-1,2,6-thiadiazin-4-one. Org. Lett. 2011;13:3466–3469. doi: 10.1021/ol201212b. [DOI] [PubMed] [Google Scholar]
- 11.Kalogirou A.S., Koutentis P.A. A Qualitative Comparison of the Reactivities of 3,4,4,5-Tetrachloro-4H-1,2,6-thiadiazine and 4,5-Dichloro-1,2,3-dithiazolium Chloride. Molecules. 2015;20:14576–14594. doi: 10.3390/molecules200814576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu P., Nielsen T.E., Clausen M.H. Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discov. Today. 2016;21:5–10. doi: 10.1016/j.drudis.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 13.Elkins J.M., Fedele V., Szklarz M., Abdul Azeez K.R., Salah E., Mikolajczyk J., Romanov S., Sepetov N., Huang X.P., Roth B.L., et al. Comprehensive characterization of the Published Kinase Inhibitor Set. Nat. Biotechnol. 2016;34:95–103. doi: 10.1038/nbt.3374. [DOI] [PubMed] [Google Scholar]
- 14.Drewry D.H., Wells C.I., Andrews D.M., Angell R., Al-Ali H., Axtman A.D., Capuzzi S.J., Elkins J.M., Ettmayer P., Frederiksen M., et al. Progress towards a public chemogenomic set for protein kinases and a call for contributions. PLoS ONE. 2017;12:e0181585. doi: 10.1371/journal.pone.0181585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koutentis P.A., Rees C.W. Cyclisation chemistry of 4H-1,2,6-thiadiazines. J. Chem. Soc. Perkin Trans. 1. 2000:2601–2607. doi: 10.1039/b004113k. [DOI] [Google Scholar]
- 16.Kalogirou A.S., Koutentis P.A. Pd-catalyzed C-N Coupling of Primary (Het)arylamines with 5-Substituted 3-Chloro-4H-1,2,6-thiadiazin-4-ones. Tetrahedron Lett. 2018 submitted. [Google Scholar]
- 17.Niesen F.H., Berglund H., Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 18.Hill R., Madureira P.A., Ferreira B., Baptista I., Machado S., Colaço L., dos Santos M., Liu N., Dopazo A., Ugurel S., et al. TRIB2 confers resistance to anti-cancer therapy by activating the serine/threonine protein kinase AKT. Nat. Commun. 2017;8:14687. doi: 10.1038/ncomms14687. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Kukimoto-Niino M., Yoshikawa S., Takagi T., Ohsawa N., Tomabechi Y., Terada T., Shirouzu M., Suzuki A., Lee S., Yamauchi T., et al. Crystal structure of the Ca²⁺/calmodulin-dependent protein kinase kinase in complex with the inhibitor STO-609. J. Biol. Chem. 2011;286:22570–22579. doi: 10.1074/jbc.M111.251710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hammond M., Washburn D.G., Hoang H.T., Manns S., Frazee J.S., Nakamura H., Patterson J.R., Trizna W., Wu C., Azzarano L.M., et al. Design and synthesis of orally bioavailable serum and glucocorticoid-regulated kinase 1 (SGK1) inhibitors. Bioorg. Med. Chem. Lett. 2009;19:4441–4445. doi: 10.1016/j.bmcl.2009.05.051. [DOI] [PubMed] [Google Scholar]
- 21.LanthaScreen® Kinase Binding Assay User Guide. [(accessed on 23 April 2018)]; Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/LanthaScreen_KinaseBinding_Assay_man.pdf.
- 22.Scott J.W., Park E., Rodriguiz R.M., Oakhill J.S., Issa S.M.A., O’Brien M.T., Dite T.A., Langendorf C.G., Wetsel W.C., Means A.R., et al. Autophosphorylation of CaMKK2 generates autonomous activity that is disrupted by a T85S mutation linked to anxiety and bipolar disorder. Sci. Rep. 2015;5:14436. doi: 10.1038/srep14436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hermerschmidt F., Kalogirou A.S., Min J., Zissimou G.A., Tuladhar S.M., Ameri T., Faber H., Itskos G., Choulis S.A., Anthopoulos T.D., et al. 4H-1,2,6-Thiadiazin-4-one-containing small molecule donors and additive effects on their performance in solution-processed organic solar cells. J. Mater. Chem. C. 2015;3:2358–2365. doi: 10.1039/C4TC02931C. [DOI] [Google Scholar]
- 24.Racioppi L., Means A.R. Calcium/Calmodulin-dependent Protein Kinase Kinase 2: Roles in Signaling and Pathophysiology. J. Biol. Chem. 2012;287:31658–31665. doi: 10.1074/jbc.R112.356485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin F., Marcelo K.L., Rajapakshe K., Coarfa C., Dean A., Wilganowski N., Robinson H., Sevick E., Bissig K.-D., Goldie L.C., et al. The camKK2/camKIV relay is an essential regulator of hepatic cancer. Hepatology. 2015;62:505–520. doi: 10.1002/hep.27832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price D.J., Drewry D.H., Schaller L.T., Thompson B.D., Reid P.R., Maloney P.R., Liang X., Banker P., Buckholz R.G., Selley P.K., et al. Bioorg. Med. Chem. Lett. 2018 doi: 10.1016/j.bmcl.2018.03.034. [DOI] [PubMed] [Google Scholar]
- 27.Racioppi L. CaMKK2: A novel target for shaping the androgen-regulated tumor ecosystem. Trends Mol Med. 2013;19:83–88. doi: 10.1016/j.molmed.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Brien M.T., Oakhill J.S., Ling N.X.Y., Langendorf C.G., Hoque A., Dite T.A., Means A.R., Kemp B.E., Scott J.W. Impact of Genetic Variation on Human CaMKK2 Regulation by Ca2+-Calmodulin and Multisite Phosphorylation. Sci. Rep. 2017;7:43264. doi: 10.1038/srep43264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levine Y.C., Li G.K., Michel T. Agonist-modulated Regulation of AMP-activated Protein Kinase (AMPK) in Endothelial Cells. Evidence for an AMPK→Rac1→Akt→Endothelial Nitric-oxide synthase pathway. J. Biol. Chem. 2007;282:20351–20364. doi: 10.1074/jbc.M702182200. [DOI] [PubMed] [Google Scholar]
- 30.Tokumitsu H., Inuzuka H., Ishikawa Y., Kobayashi R. A Single Amino Acid Difference between α and β Ca2+/Calmodulin-dependent Protein Kinase Kinase Dictates Sensitivity to the Specific Inhibitor, STO-609. J. Biol. Chem. 2003;278:10908–10913. doi: 10.1074/jbc.M213183200. [DOI] [PubMed] [Google Scholar]
- 31.Kinase Profiling Inhibitor Database. [(accessed on 23 April 2018)]; Available online: http://www.kinase-screen.mrc.ac.uk/screening-compounds/348780.
- 32.Monteiro P., Gilot D., Langouet S., Fardel O. Activation of the aryl hydrocarbon receptor by the calcium/calmodulin-dependent protein kinase kinase inhibitor 7-oxo-7H-benzimidazo[2,1-a]benz[de]isoquinoline-3-carboxylic acid (STO-609) Drug Metab. Dispos. 2008;36:556–563. doi: 10.1124/dmd.108.023333. [DOI] [PubMed] [Google Scholar]
- 33.Robinson D.D., Sherman W., Farid R. Understanding kinase selectivity through energetic analysis of binding site waters. Chem. Med. Chem. 2010;5:618–627. doi: 10.1002/cmdc.200900501. [DOI] [PubMed] [Google Scholar]
- 34.Kohlmann A., Zhu X., Dalgarno D. Application of MM-GB/SA and WaterMap to SRC Kinase Inhibitor Potency Prediction. ACS Med. Chem. Lett. 2012;3:94–99. doi: 10.1021/ml200222u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Higgs C., Beuming T., Sherman W. Hydration Site Thermodynamics Explain SARs for Triazolylpurines Analogues Binding to the A2A Receptor. ACS Med. Chem. Lett. 2010;1:160–164. doi: 10.1021/ml100008s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newman J. Novel buffer systems for macromolecular crystallization. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:610–612. doi: 10.1107/S0907444903029640. [DOI] [PubMed] [Google Scholar]
- 37.Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A., et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murshudov G.N., Skubák P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Small-Molecule Drug Discovery Suite 2018-1. Schrödinger, LLC; New York, NY, USA: 2018. [Google Scholar]
- 44.Keates T., Cooper C.D.O., Savitsky P., Allerston C.K., Phillips C., Hammarström M., Daga N., Berridge G., Mahajan P., Burgess-Brown N.A., Müller S., Gräslund S., Gileadi O. Expressing the human proteome for affinity proteomics: Optimising expression of soluble protein domains and in vivo biotinylation. N. Biotechnol. 2012;29:515–525. doi: 10.1016/j.nbt.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.LanthaScreen™ Eu kinase binding assay for CAMKK2. [(accessed on 23 April 2018)]; Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/CAMKK2_LanthaScreen_Binding.pdf.
- 46.Harwood L.M. “Dry-Column” Flash Chromatography. Aldrichimica Acta. 1985;18:25. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.