

Abstract
Background
Rhinovirus (RV) can exacerbate allergen-driven asthma. However, it has been suggested that serial infections with RV may also lead to asthma-like features in childhood without prior allergen exposure.
Aim
We sought to test the effects of RV infection in the absence of allergen challenge on lung tissue remodeling and to understand whether RV induced factors in common with allergen that promote remodeling.
Methods
We infected C57BL/6 mice multiple times with RV in the absence or presence of allergen to assess airway remodeling. We used knockout mice and blocking reagents to determine the participation of LIGHT (TNFSF14), as well as IL-1β and TGF-β, each previously shown to contribute to lung remodeling driven by allergen.
Results
Recurrent RV infection without allergen challenge induced an increase in peribronchial smooth muscle mass and subepithelial fibrosis. Rhinovirus (RV) induced LIGHT expression in mouse lungs after infection, and alveolar epithelial cells and neutrophils were found to be potential sources of LIGHT. Accordingly, LIGHT-deficient mice, or mice where LIGHT was neutralized, displayed reduced smooth muscle mass and lung fibrosis. Recurrent RV infection also exacerbated the airway remodeling response to house dust mite allergen, and this was significantly reduced in LIGHT-deficient mice. Furthermore, neutralizing IL-1β or TGF-β also limited subepithelial fibrosis and/or smooth muscle thickness induced by RV.
Conclusion
Rhinovirus can promote airway remodeling in the absence of allergen through upregulating common factors that also contribute to allergen-associated airway remodeling.
Keywords: asthma, collagen, rhinovirus, smooth muscle, TNFSF14
1. | Introduction
The development of asthma is linked not only to respiratory allergen sensitization but also to viral respiratory tract infections. Viral infections are thought to trigger 80% of asthma exacerbations in children and nearly 50% in adults, with human rhinovirus (RV) being the most common virus identified.1,2 However, in addition to exacerbating preexisting asthma, it has been suggested that viral infection may predispose to development of asthma or induce symptoms similar to those seen in allergic asthmatics. Clinical studies have shown that children at an early age who wheeze because of RV infections are at significantly increased risk of developing asthma.3,4 Moreover, longitudinal analyses found that preschool children can have more than six RV infections per year and serial infections can lead to episodes of wheezing reminiscent of asthma.5,6 This has then led to the hypothesis that recurrent RV infection may not only be a stimulus for asthma progression but could also play a significant role in the development of asthmatic inflammatory features in the lung in childhood without an allergen trigger.7 In line with this, a number of mouse studies have shown that RV infection can lead to rapid transient influx of inflammatory cells into the lungs, including neutrophils and T cells, which results in acute asthmalike symptoms.8-11
Airway remodeling is an important feature in severe asthma that manifests as subepithelial thickening, extracellular matrix deposition or fibrosis, and an increase in airway smooth muscle (ASM) mass.12-14 Originally, it was thought that chronic airway inflammation precedes airway structural changes, as remodeling features were suggested to be absent in symptomatic infants with an airflow limitation.15 However, it has been suggested more recently that components of an airway remodeling response can also be present in preschool children with airflow limitation even before any diagnosis of asthma.16,17 Thus, remodeling may also begin in early childhood due to extrinsic influences such as viral infections that could be unrelated to allergen exposure.7 Substantiating this idea, several studies have shown that RV infection of cultured lung epithelial cells can upregulate several soluble inflammatory molecules linked to airway remodeling in asthma including amphiregulin, vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and matrix metalloproteinase 9 (MMP-9).18-21
In this context, previous work from our laboratory showed that another inflammatory molecule, tumor necrosis factor (TNF) superfamily member 14 (TNFSF14), also termed LIGHT or CD258, is a strong driver of airway remodeling in models of allergen-induced severe asthma.22 Moreover, we recently also showed that LIGHT can contribute to fibrosis in the lungs and skin in models of scleroderma.23,24 This led us to question whether RV might also upregulate the expression of LIGHT and then whether recurrent RV infection could promote airway remodeling that involved this cytokine regardless of allergen exposure. Indeed, here we show that infection of naïve mice with RV1B alone promotes expression of LIGHT in the lungs, and that LIGHT, together with TGF-β and IL-1β, is active in promoting airway fibrosis and increases in smooth muscle mass when mice are infected multiple times with RV1B. The data suggest that even without allergen sensitization RV can drive production of inflammatory factors that can contribute to airway remodeling and that RV may enhance asthma-related remodeling through upregulating common factors that are also active with allergen.
2. | Methods
2.1 | Mice
WT and LIGHT–/– C57BL/6 mice22 were either purchased from The Jackson Laboratory (Bar Harbor, ME) or bred in-house. All experiments were in compliance with the regulations of the La Jolla Institute for Allergy and Immunology Animal Care Committee in accordance with guidelines of the Association for the Assessment and Accreditation of Laboratory Animal Care.
2.2 | RV generation and experimental protocols
RV1B stocks were generated as described elsewhere.25 Mice were infected recurrently with 50 μl of RV1B (∼5 × 107 PFU/mL) intranasally (i.n.) twice a week for a period of 3 weeks and then killed 24 hours after the last i.n. infection. For blocking LIGHT signaling or neutralizing TGF-β and IL-1β, control rat IgG (100 μg; Millipore Sigma, Darmstadt, Germany), LTβR.Fc (100 μg; made in-house), α-TGF-β (300 μg; Bio X Cell, West Lebanon, NH), or α-IL-1β (100 μg; R&D Systems Inc. Minneapolis, MN) was administered i.p. 24 hours before every RV challenge throughout the study period. For HDM experiments, mice were exposed i.n. to 50 μg of HDM extract on days 0, 1, 7, and 8, and then to 25 μg of HDM extract on days 21-23. The next week, HDM extract (25 μg) was given alone, or with concomitant i.n. infection with RV1B (∼5 × 107 PFU/mL), twice a week for another 4 weeks.
2.3 | Bronchoalveolar lavage and lung histology
Bronchoalveolar lavage (BAL) was performed 24 hours after the last RV1B challenge. Part of the lung was kept in RNAlater solution (Invitrogen, San Diego, CA) for RNA isolation, and the other part was snap-frozen and homogenized for analysis of total lung collagen using Sircol™ Assay kit (Biocolor Assay, UK). For lung histology, 5 to 6-μm sections were cut and stained with hematoxylin and eosin (H&E) for examining cell infiltration. Magnification ×200 was used for histologic scoring, and at least 5 fields were scored to obtain the average for each mouse. For airway remodeling analysis, paraformaldehyde-fixed lung sections were stained with Masson's Trichrome blue or anti–alpha-smooth muscle actin (Sigma) and scored with an image analysis system (Image-Pro Plus, Media Cybernetics, Rockville, Md).22,23 In some experiments, smooth muscle thickness was also evaluated using immunofluorescence staining as described elsewhere.23 Results using gene-deficient mice or blocking reagents are expressed as induced responses above background levels in naïve unmanipu-lated mice.
2.4 | Lung cell isolation
For sorting of cell populations where indicated, lungs were digested using 1× collagenase/hyaluronidase solution and DNase (StemCell Technologies). After incubation with mouse Fc-blocking antibody (2.4G2), neutrophils (CD45+CD11b+Ly6G+), Ly6G– cells (CD45+CD11b+Ly6G–), and CD45– cells were isolated using a FACSAria cell sorter (BD Biosciences).
2.5 | RT-PCR and real-time PCR
Mouse lung tissue or sorted cells, or human cell lines A549 and BEAS-2B, were homogenized using TRIzol reagent (Invitrogen, Carlsbad, CA) with gentleMACS Dissociator (Miltenyi Biotec, San Diego, Ga) for RNA isolation. An aliquot of total RNA (1 μg) from whole lung or 0.4 μg of total RNA from sorted cells was reverse-transcribed to cDNA using Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics Corporation, Indianapolis, IN). RV1B viral RNA expression in the lung was conducted using RV1B-specific primers. Data are presented as normalized to ribosomal protein housekeeping gene L32 for lung samples or GAPDH for A549 or BEAS-2B cell lines.
2.6 | Statistical analyses
All results are presented as mean ± SEM. Statistical analyses were performed with GraphPad Prism software (GraphPad Software, La Jolla, CA). Mann-Whitney U test was performed for analysis of two groups, where indicated. Nonparametric Kruskal-Wallis test was used along with Dunn's post hoc analysis when more than two groups were compared. *P < .05, **P < .01, ***P < .001.
3. | Results
3.1 | Recurrent rhinovirus infection induces airway remodeling in vivo
The minor-group rhinovirus serotype, RV1B, that infects both humans and mice by binding low-density lipoprotein receptor, induces airway neutrophilia in mice reminiscent of the response in humans.8 In this context, acute RV1B infection of mice can lead to inflammatory factors that participate in the initial influx of neutrophils, as well as lymphocytes and other lymphoid cell types, and can contribute to exacerbation of acute lung inflammation driven by allergen.8-11,26 However, little attention has been given to whether RV1B infection can promote airway remodeling. As multiple exposures to RV in children have been associated with wheezing and possible development of lung remodeling before asthma diagnosis, we asked whether recurrent RV1B challenge was able to drive airway fibrosis and lung remodeling in the mouse in the absence of allergen sensitization. RV is thought to largely infect lung epithelial cells, and the proportion of infectible upper and lower epithelial cells increases with the inoculum but does not exceed 10% even when using different strains of RVs.27,28 Furthermore, with a single RV1B dose given via the lungs to mice, viral load is maximal at 1-day postinfection, and RV1B is not detectable thereafter.29 Thus, mice were exposed i.n. to RV1B twice a week for a period of 3 weeks to allow increased infection in the lower airways and attempt to create a model that potentially mimics inflammatory activity associated with multiple exposures in children (Figure 1).
Figure 1.
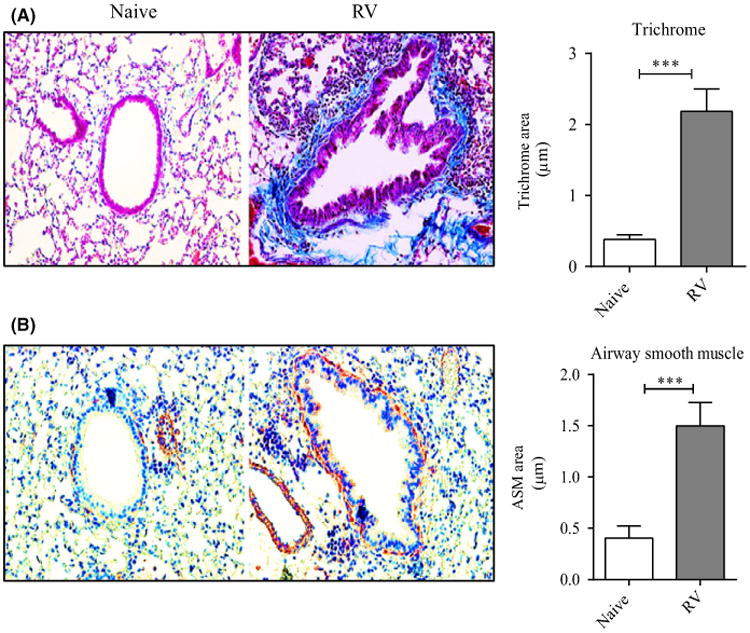
Recurrent RV1B infection induces airway remodeling in vivo. Naïve mice were infected i.n. with RV1B, twice a week for 3 wk and killed 24 h after the last challenge. A, Lung sections stained with Masson's trichrome blue, and scored for the extent of fibrosis in the right panel. B, IHC of lung sections stained for α-smooth muscle actin (brown), and scored for the extent of smooth muscle mass in the right panel. Results are means ± SEM from 3-4 mice/group and representative of 3 experiments. ***P < .001
Significantly, we observed that multiple infections with RV1B led to collagen deposition in the subepithelial regions (Figure 1A), and total lung collagen was significantly increased (Figure S1A). Furthermore, recurrent RV1B challenge also led to an increase in the thickness of peribronchial smooth muscle (Figure 1B). As expected, we found marked bronchovascular inflammation, and significantly elevated numbers of BAL neutrophils but not eosinophils compared to control mice (Figure S1B,C). Thus, mice exposed multiple times to RV display salient features of airway remodeling without concomitant allergen sensitization or a primary Th2 environment as shown by the lack of eosinophilia.
3.2 | Blockade of LIGHT reduces features of airway remodeling induced by RV
We then assessed whether the TNF family molecule LIGHT might play a role in lung remodeling driven by RV infection. Our previous studies in mice challenged intranasally with HDM allergen, or intratracheally with the antibiotic bleomycin, showed that LIGHT was upregulated in the lungs and was a major participant in driving maximal airway fibrosis and tissue remodeling.22,23 Because there is no reliable reagent to detect LIGHT protein in the mouse, we measured LIGHT mRNA in whole lung tissue and found its expression was induced within 24 hours of initial infection (Figure 2A). There are several possible sources of LIGHT, including T cells, dendritic cells, and macrophages. However, given the early expression in vivo, the fact that RV primarily infects and replicates in lung epithelial cells, and that RV also induces rapid lung neutrophilia in the initial 24 hours of infection, we tested whether these two cell types might also be sources of LIGHT. Due to the difficulty of isolating mouse lung epithelial cells, we infected the human alveolar epithelial cell line, A549, in vitro and found significant upregulation of LIGHT mRNA (Figure 2B). We then sorted CD45+CD11b+Ly6G+ neutrophils from the lungs and compared LIGHT expression in these cells with that in CD11b+Ly6G– cells, both in naïve mice or 24 hours after infection with RV1B. Interestingly, in naïve lungs, LIGHT mRNA was detectable in Ly6G– cells, which represent a mixture of dendritic cells and macrophages, but stronger expression was seen in Ly6G+ neutrophils and this expression was substantially enhanced after infection (Figure 2C). In contrast, CD45– cells had little/no expression of LIGHT. These data suggest that neutrophils and myeloid cells may primarily produce LIGHT during RV infection, although given the complications in extracting CD45– cells, this does not exclude epithelial cells as also being a source. To further implicate neutrophils, we injected mice with a depleting anti-Ly6G/Gr-1 antibody and found that expression of LIGHT mRNA was reduced in total lung extracts (Figure 2D), albeit not to background levels, again implying that there are several sources of LIGHT.
Figure 2.

RV1B infection induces LIGHT expression. A, LIGHT mRNA expression in lungs of mice infected a single time with RV1B after 14 h compared to naïve animals, normalized to L32. B, LIGHT mRNA expression in RV1B-infected alveolar epithelial cells (A549). Fold increase normalized to GAPDH. C, LIGHT mRNA expression in sorted lung cell populations from 6 to 8 naïve and RV1B-infected mice. Fold increase normalized to L32. D, LIGHT mRNA expression in lungs of mice treated with control IgG or anti-Ly6G and infected with RV1B for 14 h, normalized to L32. Results are means ± SEM from 3-4 mice/group or replicates of 2 cell cultures, and representative of at least 2 experiments each. *P < .05
Next, we challenged LIGHT–/– mice with RV1B as in Figure 1. Gene-deficient mice displayed significantly less fibrosis compared to WT mice, as measured by peribronchial trichrome staining (Figure 3A) and assay for total lung collagen (Figure S2A), although this phenotype was not reduced completely. Staining and quantification of α-SMA expression also revealed a decrease in peribronchial smooth muscle mass (Figure 3B), although again not to baseline levels. Unlike the airway remodeling features, LIGHT–/– mice had relatively unaffected lung inflammation as measured by histologic scoring of infiltrates around the bronchioles, albeit analysis of BAL fluid showed a significant decrease in neutrophilia and slightly elevated lymphocyte levels (Figure S2B-D). Airway hyperresponsiveness (AHR), as measured by response to methacholine, was elevated in RV-challenged WT mice but was not reproducibly reduced in LIGHT–/– mice (data not shown), correlating with significant levels of inflammation still remaining in the lungs of these mice. This implies that in this scenario, AHR was primarily a feature of the inflammatory cell response rather than the remodeling response. The viral load was also similar in LIGHT–/– mice as measured by qPCR 24 hours after the last infection (Figure S2E), suggesting the reduced remodeling response in the absence of LIGHT was not related to altered viral load.
Figure 3.
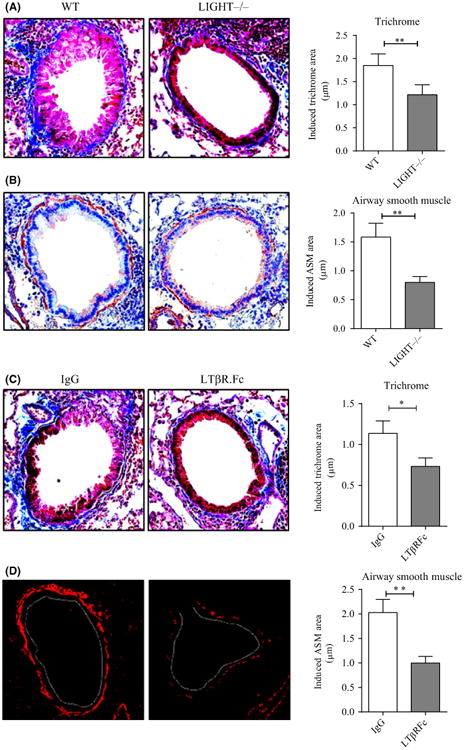
Neutralizing LIGHT reduces lung remodeling following recurrent RV1B infection. A,B, WT and LIGHT–/–mice were infected with RV1B twice a week for 3 wk as in Figure 1 and killed 24 h after the last i.n exposure. A, Lung sections stained with Masson's trichrome blue, and scored for the extent of fibrosis. B, IHC of lung sections stained for α-smooth muscle actin (brown), and scored for smooth muscle mass. C,D, WT mice were infected with RV1B as above and treated with control IgG or LTβR.Fc, given 24 h before each RV challenge. C, Lung sections stained with Masson's trichrome blue, and scored for the extent of fibrosis. D, IF of lung sections stained for α-smooth muscle actin (red), and scored for smooth muscle mass (white dashed line, bronchial lumen). Data are induced responses above background levels in naïve unmanipulated mice. Results are means ± SEM from 3- 4 mice/group, and representative of 2-3 experiments. *P < .05, **P < .01
To further confirm these results, we treated WT mice with a lymphotoxin beta receptor fusion protein (LTβR.Fc) that can block the interaction between LIGHT and its two receptors LTβR and HVEM.22 Similar to LIGHT–/– mice, WT mice treated with LTβR.Fc had less fibrosis (Figure 3C) and smooth muscle mass (Figure 3D). LTβR–/– mice lack lymph nodes and display other developmental issues, and thus cannot be used to effectively assess the contribution of this receptor. However, we carried out similar experiments in HVEM–/– mice and observed no significant difference in terms of RV-induced collagen accumulation and α-SMA expression (data not shown) suggesting LTβR is the primary LIGHT receptor that is active. Collectively, these data show that LIGHT is expressed during infection with RV in the absence of allergen exposure and can contribute partially to airway remodeling that occurs during repetitive infections, although it is not the only factor driving this phenotype.
3.3 | LIGHT plays a role in promoting collagen and smooth muscle thickness with concomitant rhinovirus and allergen challenge
We then tested whether LIGHT was involved in airway remodeling when rhinovirus infection was combined with allergen exposure. We sensitized and challenged mice with HDM extract alone over a period of 3 weeks to develop acute airway inflammation and then exposed mice to either HDM alone or HDM with RV1B twice a week for another 4 weeks. Co-exposure of mice to RV1B resulted in an approximate doubling of the remodeling response in terms of peribronchial collagen and smooth muscle mass (Figure 4A,B) over that seen with HDM. We previously reported that LIGHT contributed to remodeling in the lungs of HDM-challenged mice, but eosinophil numbers, the overall extent of peribronchial cellular infiltrates, and the overall Th2 response were largely unaltered,22 and this observation was reproduced here. Moreover, LIGHT still contributed to remodeling driven by HDM given together with RV1B (Figure 4A,B), although a substantial level of remodeling was evident in the absence of LIGHT.
Figure 4.
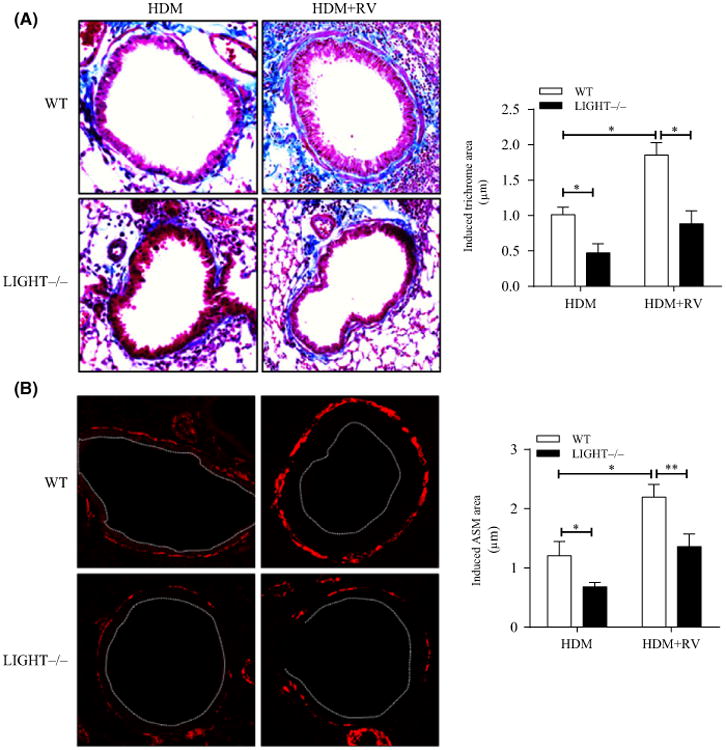
Airway remodeling with co-exposure to rhinovirus and allergen is dependent on LIGHT. WT and LIGHT–/– mice were sensitized and acutely challenged with HDM for a period of 3 wk and then chronically challenged with HDM over the next 4 wk with or without co-exposure to RV1B. A, Lung sections stained with Masson's trichrome blue, and scored for the extent of fibrosis. B, IF of lung sections stained for α-smooth muscle actin (red), and scored for smooth muscle mass (white dashed line, bronchial lumen). Data are induced responses above background levels in naïve unmanipulated mice. Results are means ± SEM from 4 mice/group, and representative of 2 experiments. *P < .05, **P < .01
3.4 | IL-1β and TGF-β also participate in airway remodeling induced by recurrent rhinovirus exposure
As remodeling was not completely negated by neutralizing the activity of LIGHT, we then sought other molecules that might be upregulated by RV and could contribute to the remodeling process. IL-1β is one candidate as revealed by induction of lung remodeling in mice where this cytokine was overexpressed in the lungs using a transgenic30 or adenoviral approach.31 RV1B has been shown to induce IL-1β in the lungs in vivo,8 and we confirmed an early increase in mRNA for IL-1β in lung tissue of RV1B-infected mice. Expression of IL-1β mRNA was primarily in sorted Ly6G+ neutrophils and to a lesser extent Ly6G– cells from naïve mouse lungs, and this was upregulated after RV1B infection (Figure S3A,B). Further suggesting that neutrophils were a primary source of this cytokine, depletion of these cells with anti-Ly6G antibody significantly reduced RV1B-induced IL-1β expression (Figure S3C). We did not see a strong signal for IL-1β in lung CD45– cells, again with the caveats discussed above, but observed significant upregulation of IL-1β mRNA after infection of human bronchial epithelial cells (BEAS-2B), but not in A549 alveolar epithelial cells (Figure S3D, and data not shown). This is consistent with a previous study of IL-1β expression in isolated bronchial epithelial cells infected with RV.20
We then treated mice with a neutralizing antibody to IL-1 given 24 hours before each recurrent RV1B challenge. Similar to inhibiting LIGHT activity, blocking IL-1β significantly reduced fibrosis (Figure 5A) as well as smooth muscle mass (Figure 5B). This was also accompanied by a significant decrease in BAL neutrophils but comparable lymphocytes numbers, reminiscent of the effect when LIGHT was neutralized (Figure S4A,B). Viral load was again comparable in both groups (Figure S4C).
Figure 5.
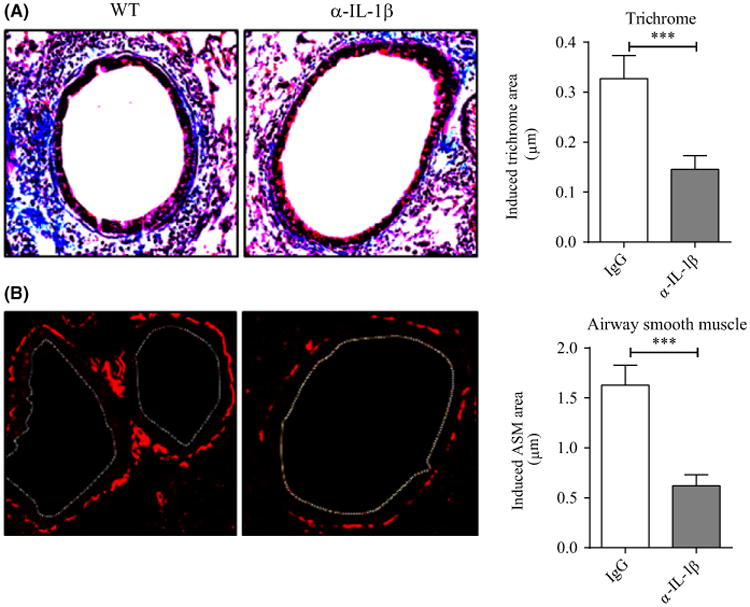
Blocking IL-1β reduces RV1B-induced airway remodeling. WT mice were infected with RV1B twice a week for 3 wk and treated with control IgG and α-IL-1β antibody given 24 h before each RV challenge. A, Lung sections stained with Masson's trichrome blue, and scored for the extent of fibrosis. B, IF of lung sections stained for α-smooth muscle actin, and scored for smooth muscle mass (white dashed line, bronchial lumen). Data are induced responses above background levels in naïve unmanipulated mice. Results are means ± SEM from 3-4 mice/group, and representative of 2 experiments. *** P < .001
Lastly, we assessed any contribution of TGF-β, a cytokine thought to be central to lung remodeling driven by many stimuli. TGF-β activation has been suggested to be modulated in part by IL-1β,31 and we previously found that TGF-β expression could be upregulated in lung macrophages by LIGHT,22 although many other factors are likely to determine its production. We did not find a significant change in total lung TGF-β1 levels in mice acutely exposed to RV1B, but we did observe increased TGF-β1 mRNA expression from sorted Ly6G+ neutrophils after infection (Figure S5A). We then treated mice with a TGF-β1 neutralizing antibody. TGF-β has been primarily associated with collagen production. However, a significant decrease in α-SMA expression was seen in the treated group whereas collagen accumulation in the subepithelial region was comparable with the control group (Figure 6A,B). Neutralizing TGF-β also significantly decreased BAL neutrophils, but comparable lymphocyte numbers were observed (Figure S5B,C). Similar to the prior results, the viral load was not significantly affected by neutralizing TGF-β (Figure S5D). Taken together, these data show that recurrent RV1B infection of the lungs results in airway remodeling that is dependent on LIGHT and IL-1β in terms of smooth muscle hyperplasia and collagen deposition and TGF-β also contributes to the smooth muscle mass.
Figure 6.
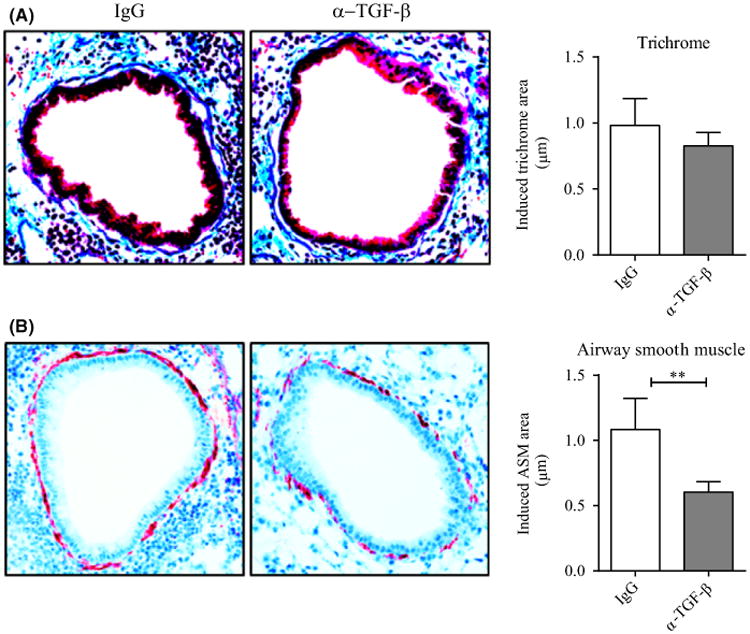
Neutralizing TGF-β inhibits smooth muscle mass but not collagen deposition induced by RV1B. A, Lung sections stained with Masson's trichrome blue, and scored for the extent of fibrosis. B, IHC of lung sections stained with α-smooth muscle actin and scored for smooth muscle mass. Data are induced responses above background levels in naïve unmanipulated mice. Results are means ± SEM from 3-4 mice/group and representative of 2 experiments. **P < .01
4. | Discussion
Rhinovirus has long been associated with allergen-induced asthma exacerbations in humans and transient lung inflammation,7,32 but how much RV can promote lung remodeling in the absence of allergen has not been clear. Here, we show that repetitive challenges with RV1B in mice without allergen sensitization can drive lung remodeling features that are similar to those in allergen-induced severe asthma. Moreover, we show that several inflammatory factors, LIGHT, IL-1β, and TGF-β, which have been linked to allergen-driven lung tissue remodeling, are induced or active in the lungs during RV1B infection. This provides a mechanistic explanation of why rhinovirus infection might promote remodeling in the absence of allergen as well as how rhinovirus could augment specific asthma symptoms that result from allergen exposure.
Although RV infection has been primarily linked to exacerbations of asthma, RVs have the potential to induce asthma-like changes in the lungs before allergen exposure.3,4,28,33 In line with this, several clinical studies have recognized that multiple infections with RVs can often occur in nonasthmatic preschool children which lead to recurrent wheezing episodes.3,6 While one argument is that the clinical symptoms would simply reflect the acute cellular infiltrate in the lung induced by RV infection, which includes neutrophils, T cells, and other lymphoid cells, it is possible that airway remodeling also contributes to lung dysfunction seen with repeated RV insults. Supporting this idea, several in vitro studies reported that infection of cultured lung epithelial cells could lead to production of molecules that have been associated with severe asthma-associated airway remodeling, including amphiregulin, VEGF, FGF, and MMP-9.18-21 One publication also reported RV could induce collagen V expression in bronchial epithelial cells, although whether this was a direct activity was not clear.34 The latter study additionally observed in mice that a single dose of RV1B could enhance fibronectin and collagen I gene expression within 2 days of infection.34 Our data further add to these findings and suggest that RV is capable of inducing lung remodeling without allergen exposure. Moreover, we found that an appreciable increase in airway remodeling was observed in the lungs only with repeated rhinovirus infection, implying that the clinical studies related to wheezing in children following recurrent infections may indeed be partially related to airway remodeling.
Which molecules promote airway remodeling downstream of allergen or in this case RV is still being elucidated. Our data highlight three molecules, LIGHT, IL-1β, and TGF-β, as important for the RV-induced response, although others may also be active. We previously showed that LIGHT is a strong mediator of airway remodeling induced by repetitive intranasal exposure of mice to HDM allergen,22 and we demonstrate here that LIGHT is also upregulated and active during RV infection. Furthermore, LIGHT still significantly contributed to airway remodeling features in mice concomitantly challenged with RV and allergen. As suggested from the previous literature on rhinovirus that relates to lung epithelial cells, it is likely that any induction of asthma-like symptoms is due to the production of inflammatory molecules in these cells, and/or production of molecules in lung-infiltrating cells such as neutrophils that are recruited by chemokines made as a direct consequence of the infection. We now substantiate these ideas. There may not be a single cell type that makes LIGHT,35 but based on in vitro and in vivo studies, we suggest that neutrophils are a likely primary source during RV infection, with epithelial cells and lung myeloid cells also capable of contributing to its production. LIGHT can bind two receptors, HVEM and LTβR, which are expressed on hematopoietic cells such as macrophages and dendritic cells as well as nonhematopoietic stromal cells including epithelial cells and fibroblasts. As HVEM and LTβR are constitutively expressed at moderate/low levels on the above cell types, this suggests the limiting factor in determining whether these molecules participate in driving lung remodeling is the availability of LIGHT and how strongly it is induced.
Exactly how LIGHT promotes lung tissue remodeling is still being determined. LIGHT can in some circumstances control T-cell responses via HVEM expressed on these cells, and a minor possibility exists that reduced T-cell activity to RV1B could have accounted for the phenotype we describe in LIGHT-deficient animals. We did not assess this in our experiments due to the lack of reagents to track RV-specific T cells and the weak replicative capacity of RV1B that likely results in poor T-cell responses. However, others found no difference in the T-cell or antibody response to lung infection with influenza A virus.36 Rather we think LIGHT primarily induces remodeling through activities on other inflammatory cells and on structural cells. We have previously shown that it can induce several inflammatory molecules in lung macrophages and epithelial cells, such as TGF-β, TSLP, and MMP-9, and thus it may enhance the lung remodeling response indirectly.22,23,37 Moreover, LIGHT is likely to further amplify the remodeling phenotype by inducing various chemokines including CXCL1, 3, and 5 that we have previously demonstrated are targets of LIGHT in bronchial epithelial cells. In this regard, although overall lung inflammation in RV-infected WT and LIGHT–/– mice was comparable, we observed a partial but significant decrease in BAL neutrophils in LIGHT–/– mice, which is likely related to an altered chemokine environment. Correlating with this, we did find a significant decrease in mouse lung CXCL1 gene expression in the absence of LIGHT (data not shown) that could explain this phenotype. Additional direct effects of LIGHT are also likely to be important. In vitro studies have found that LIGHT can promote some aspects of epithelial-mesenchymal transition in lung epithelial cells such as downregulation of E-cadherin and upregulation of fibronectin.3739 Moreover, our recent studies suggest it can also drive proliferation of lung fibroblasts/myofibroblasts that could contribute to increases in smooth muscle mass (da Silva Antunes and Croft, unpublished). Therefore, LIGHT has the possibility to contribute to lung remodeling in several ways.
Along with LIGHT, we found that RV infection also induced IL-1β gene expression and again at least in part this was a consequence of the neutrophilia that accompanied infection. IL-1β was also active in driving airway remodeling, although similar to LIGHT its targets and mechanisms of action in this regard are not clear. IL-1β has been suggested to control αvβ8-mediated activation of TGF-β in fibroblasts,31 and thus, neutralization of IL-1β in mice infected with RV1B could have led to reduced TGF-β activity, although its likely range of activities linked to remodeling may be numerous. Correlating with the former, we did find that TGF-β was also required for features of the remodeling response induced by RV infection, and like blocking IL-1β or LIGHT, this was accompanied by decreased neutrophilia. Again, the latter is likely related to migration as TGF-β has previously been found to be capable of directly stimulating neutrophil adhesion and chemotaxis.40,41 However, although TGF-β is universally considered to be central to airway fibrosis by directly promoting ECM proteins from epithelial cells or fibroblasts,42 we observed that TGF-β neutralization only decreased smooth muscle thickness but did not affect collagen deposition around the bronchioles, which was in contrast to blocking either LIGHT or IL-1β that reduced both features. Thus, the interplay between LIGHT, IL-1β, and TGF-β is complex, and a requirement for all three proteins in the remodeling response induced by RV may not simply reflect that one molecule is upstream or downstream of another molecule. Production of LIGHT, IL-1β, and TGF-β may be directly or indirectly dependent on TLR3 and/or RIG-I and MDA5 that have been implicated in mediating responses to RV in lung epithelial cells.43 We found a dose-dependent increase in LIGHT mRNA expression when bronchial epithelial cells (BEAS-2B) were stimulated with the TLR3 agonist poly I:C (data not shown). However, LIGHT, IL-1β, and TGF-β might be primary products of neutrophils and possibly other inflammatory cells (Figures 2, and S4 and 5), and thus, whether these cytokines are directly downstream of the pattern recognition receptor signals induced by RV is perhaps unlikely given that RV primarily infects epithelial cells. This is of interest to understand in future studies.
In summary, this study extends our present knowledge of how RV alone or in combination with an allergen might contribute to airway dysfunction and specifically to fibrosis and lung tissue remodeling. We identify three molecules that contribute to these processes, although it is likely that other factors will also be induced directly or indirectly by RV that additionally are active. Importantly, two of these molecules in LIGHT and IL-1β might be amenable to targeting that could minimize the effects of multiple RV infections in early life that could contribute to wheezing in nonasthmatics, as well as could reduce exacerbations of wheezing in asthmatics that have previously responded to allergen.
Supplementary Material
Acknowledgments
Funding information: This work was supported by NIH grant AI070535 to M.C.
Abbreviations
- BAL
bronchoalveolar lavage
- HVEM
herpesvirus entry mediator
- LIGHT
homologous to lymphotoxins, exhibits inducible expression, competes with HSV glycoprotein D for HVEM, a receptor expressed by T lymphocytes
- LTβR
lymphotoxin beta receptor
- Mch
methacholine
- TNFSF
tumor necrosis factor superfamily protein
- WT
wild type
- α-SMA
alpha-smooth muscle actin
Footnotes
Conflicts sof Interests: The authors declare that they have no conflict of interests.
Author Contributions: A.K.M. contributed to conception and design of the work, data acquisition and analysis, interpretation, and writing of the manuscript. T.A.D. performed data acquisition and analysis and reviewing the manuscript. D.B. performed interpretation and reviewing the manuscript. M.C. contributed to conception and design of the work, interpretation, and writing of the manuscript.
Supporting Information: Additional Supporting Information may be found online in the supporting information tab for this article.
References
- 1.Johnston SL, Pattemore PK, Sanderson G, et al. Community study of role of viral infections in exacerbations of asthma in 9-11 year old children. BMJ. 1995;310:1225–1229. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asthma in adults. BMJ. 1993;307:982–986. doi: 10.1136/bmj.307.6910.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lemanske RF, Jr, Jackson DJ, Gangnon RE, et al. Rhinovirus illnesses during infancy predict subsequent childhood wheezing. J Allergy Clin Immunol. 2005;116:571–577. doi: 10.1016/j.jaci.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 4.Jackson DJ, Gangnon RE, Evans MD, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med. 2008;178:667–672. doi: 10.1164/rccm.200802-309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winther B, Hayden FG, Hendley JO. Picornavirus infections in children diagnosed by RT-PCR during longitudinal surveillance with weekly sampling: association with symptomatic illness and effect of season. J Med Virol. 2006;78:644–650. doi: 10.1002/jmv.20588. [DOI] [PubMed] [Google Scholar]
- 6.Jartti T, Lee WM, Pappas T, Evans M, Lemanske RF, Jr, Gern JE. Serial viral infections in infants with recurrent respiratory illnesses. Eur Respir J. 2008;32:314–320. doi: 10.1183/09031936.00161907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leigh R, Proud D. Virus-induced modulation of lower airway diseases: pathogenesis and pharmacologic approaches to treatment. Pharmacol Ther. 2015;148:185–198. doi: 10.1016/j.pharmthera.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartlett NW, Walton RP, Edwards MR, et al. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat Med. 2008;14:199–204. doi: 10.1038/nm1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glanville N, Peel TJ, Schroder A, et al. Tbet deficiency causes T helper cell dependent airways Eosinophilia and Mucus hypersecretion in response to rhinovirus infection. PLoS Pathog. 2016;12:e1005913. doi: 10.1371/journal.ppat.1005913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han M, Chung Y, Young Hong J, et al. Toll-like receptor 2-expressing macrophages are required and sufficient for rhinovirus-induced airway inflammation. J Allergy Clin Immunol. 2016;138:1619–1630. doi: 10.1016/j.jaci.2016.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Girkin JL, Hatchwell LM, Collison AM, et al. TRAIL signaling is proinflammatory and proviral in a murine model of rhinovirus 1B infection. Am J Physiol Lung Cell Mol Physiol. 2017;312:L89–L99. doi: 10.1152/ajplung.00200.2016. [DOI] [PubMed] [Google Scholar]
- 12.Jeffery PK. Remodeling and inflammation of bronchi in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2004;1:176–183. doi: 10.1513/pats.200402-009MS. [DOI] [PubMed] [Google Scholar]
- 13.Pepe C, Foley S, Shannon J, et al. Differences in airway remodeling between subjects with severe and moderate asthma. J Allergy Clin Immunol. 2005;116:544–549. doi: 10.1016/j.jaci.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Kaminska M, Foley S, Maghni K, et al. Airway remodeling in subjects with severe asthma with or without chronic persistent airflow obstruction. J Allergy Clin Immunol. 2009;124:45–51. doi: 10.1016/j.jaci.2009.03.049. [DOI] [PubMed] [Google Scholar]
- 15.Saglani S, Malmstrom K, Pelkonen AS, et al. Airway remodeling and inflammation in symptomatic infants with reversible airflow obstruction. Am J Respir Crit Care Med. 2005;171:722–727. doi: 10.1164/rccm.200410-1404OC. [DOI] [PubMed] [Google Scholar]
- 16.O'Reilly R, Ullmann N, Irving S, et al. Increased airway smooth muscle in preschool wheezers who have asthma at school age. J Allergy Clin Immunol. 2013;131:1024–1032. doi: 10.1016/j.jaci.2012.08.044. [DOI] [PubMed] [Google Scholar]
- 17.Saglani S, Payne DN, Zhu J, et al. Early detection of airway wall remodeling and eosinophilic inflammation in preschool wheezers. Am J Respir Crit Care Med. 2007;176:858–864. doi: 10.1164/rccm.200702-212OC. [DOI] [PubMed] [Google Scholar]
- 18.Leigh R, Oyelusi W, Wiehler S, et al. Human rhinovirus infection enhances airway epithelial cell production of growth factors involved in airway remodeling. J Allergy Clin Immunol. 2008;121:1238–1245. doi: 10.1016/j.jaci.2008.01.067. [DOI] [PubMed] [Google Scholar]
- 19.Skevaki CL, Psarras S, Volonaki E, et al. Rhinovirus-induced basic fibroblast growth factor release mediates airway remodeling features. Clin Transl Allergy. 2012;2:14. doi: 10.1186/2045-7022-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bochkov YA, Hanson KM, Keles S, Brockman-Schneider RA, Jarjour NN, Gern JE. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol. 2010;3:69–80. doi: 10.1038/mi.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tacon CE, Wiehler S, Holden NS, Newton R, Proud D, Leigh R. Human rhinovirus infection up-regulates MMP-9 production in airway epithelial cells via NF-{kappa}B. Am J Respir Cell Mol Biol. 2010;43:201–209. doi: 10.1165/rcmb.2009-0216OC. [DOI] [PubMed] [Google Scholar]
- 22.Doherty TA, Soroosh P, Khorram N, et al. The tumor necrosis factor family member LIGHT is a target for asthmatic airway remodeling. Nat Med. 2011;17:596–603. doi: 10.1038/nm.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herro R, Da Silva Antunes R, Aguilera AR, Tamada K, Croft M. Tumor necrosis factor superfamily 14 (LIGHT) controls thymic stromal lymphopoietin to drive pulmonary fibrosis. J Allergy Clin Immunol. 2015;136:757–768. doi: 10.1016/j.jaci.2014.12.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herro R, Antunes Rda S, Aguilera AR, Tamada K, Croft M. The tumor necrosis factor superfamily molecule LIGHT promotes keratinocyte activity and skin fibrosis. J Invest Dermatol. 2015;135:2109–2118. doi: 10.1038/jid.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehta AK, Duan W, Doerner AM, et al. Rhinovirus infection interferes with induction of tolerance to aeroantigens through OX40 ligand, thymic stromal lymphopoietin, and IL-33. J Allergy Clin Immunol. 2016;137:278–288. doi: 10.1016/j.jaci.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phan JA, Kicic A, Berry LJ, et al. Rhinovirus exacerbates house-dust-mite induced lung disease in adult mice. PLoS One. 2014;9:e92163. doi: 10.1371/journal.pone.0092163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mosser AG, Brockman-Schneider R, Amineva S, et al. Similar frequency of rhinovirus-infectible cells in upper and lower airway epithelium. J Infect Dis. 2002;185:734–743. doi: 10.1086/339339. [DOI] [PubMed] [Google Scholar]
- 28.Gern JE, Galagan DM, Jarjour NN, Dick EC, Busse WW. Detection of rhinovirus RNA in lower airway cells during experimentally induced infection. Am J Respir Crit Care Med. 1997;155:1159–1161. doi: 10.1164/ajrccm.155.3.9117003. [DOI] [PubMed] [Google Scholar]
- 29.Song DJ, Miller M, Beppu A, et al. Rhinovirus infection of ORMDL3 transgenic mice is associated with reduced rhinovirus viral load and airway inflammation. J Immunol. 2017;199:2215–2224. doi: 10.4049/jimmunol.1601412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Inter-leukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 2005;32:311–318. doi: 10.1165/rcmb.2004-0309OC. [DOI] [PubMed] [Google Scholar]
- 31.Kitamura H, Cambier S, Somanath S, et al. Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin alphavbeta8-mediated activation of TGF-beta. J Clin Invest. 2011;121:2863–2875. doi: 10.1172/JCI45589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Proud D, Leigh R. Epithelial cells and airway diseases. Immunol Rev. 2011;242:186–204. doi: 10.1111/j.1600-065X.2011.01033.x. [DOI] [PubMed] [Google Scholar]
- 33.Rakes GP, Arruda E, Ingram JM, et al. Rhinovirus and respiratory syncytial virus in wheezing children requiring emergency care. IgE and eosinophil analyses. Am J Respir Crit Care Med. 1999;159:785–790. doi: 10.1164/ajrccm.159.3.9801052. [DOI] [PubMed] [Google Scholar]
- 34.Kuo C, Lim S, King NJ, et al. Rhinovirus infection induces expression of airway remodelling factors in vitro and in vivo. Respirology. 2011;16:367–377. doi: 10.1111/j.1440-1843.2010.01918.x. [DOI] [PubMed] [Google Scholar]
- 35.Croft M, Duan W, Choi H, Eun SY, Madireddi S, Mehta A. TNF superfamily in inflammatory disease: translating basic insights. Trends Immunol. 2012;33:144–152. doi: 10.1016/j.it.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sedgmen BJ, Dawicki W, Gommerman JL, Pfeffer K, Watts TH. LIGHT is dispensable for CD4+ and CD8+ T cell and antibody responses to influenza A virus in mice. Int Immunol. 2006;18:797–806. doi: 10.1093/intimm/dxl016. [DOI] [PubMed] [Google Scholar]
- 37.da Silva Antunes R, Madge L, Soroosh P, Tocker J, Croft M. The TNF family molecules LIGHT and lymphotoxin alphabeta induce a distinct steroid-resistant inflammatory phenotype in human lung epithelial cells. J Immunol. 2015;195:2429–2441. doi: 10.4049/jimmunol.1500356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hung JY, Chiang SR, Tsai MJ, et al. LIGHT is a crucial mediator of airway remodeling. J Cell Physiol. 2015;230:1042–1053. doi: 10.1002/jcp.24832. [DOI] [PubMed] [Google Scholar]
- 39.Mikami Y, Yamauchi Y, Horie M, et al. Tumor necrosis factor super-family member LIGHT induces epithelial-mesenchymal transition in A549 human alveolar epithelial cells. Biochem Biophys Res Commun. 2012;428:451–457. doi: 10.1016/j.bbrc.2012.10.097. [DOI] [PubMed] [Google Scholar]
- 40.Brandes ME, Mai UE, Ohura K, Wahl SM. Type I transforming growth factor-beta receptors on neutrophils mediate chemotaxis to transforming growth factor-beta. J Immunol. 1991;147:1600–1606. [PubMed] [Google Scholar]
- 41.Gagliardo R, Chanez P, Gjomarkaj M, et al. The role of transforming growth factor-beta1 in airway inflammation of childhood asthma. Int J Immunopathol Pharmacol. 2013;26:725–738. doi: 10.1177/039463201302600316. [DOI] [PubMed] [Google Scholar]
- 42.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slater L, Bartlett NW, Haas JJ, et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 2010;6:e1001178. doi: 10.1371/journal.ppat.1001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.