

Abstract
Catalytic enantioselective boron–hydride additions to 1,3-enynes, which afford allenyl–B(pin) (pin = pinacolato) products, are disclosed. Transformations are promoted by a readily accessible bis-phosphine–Cu complex and involve commercially available HB(pin). The method is applicable to aryl- and alkyl-substituted 1,3-enynes. Trisubstituted allenyl– B(pin) products were generated in 52–80% yield and, in most cases, in >98:2 allenyl:propargyl and 92:8–99:1 enantiomeric ratio. Utility is highlighted through a highly diastereoselective addition to an aldehyde, and a stereospecific catalytic cross-coupling process that delivers an enantiomerically enriched allene with three carbon-based substitutents. The following key mechanistic attributes are elucidated: (1) Spectroscopic and computational investigations indicate that low enantioselectivity can arise from loss of kinetic stereoselectivity, which, as suggested by experimental evidence, may occur by formation of a propargylic anion generated by heterolytic Cu–C cleavage. This is particularly a problem when trapping of the Cu–allenyl intermediate is slow, namely, when an electron deficient 1,3-enyne or a less reactive boron–hydride reagent (e.g., HB(dan) (dan = naphthalene-1,8-diaminato)) is used or under un-optimal conditions (e.g., lower boron–hydride concentration causing slower trapping). (2) With enynes that contain a sterically demanding ortho-aryl substituent considerable amounts of the propargyl–B(pin) isomer may be generated (25– 96%) because a less sterically demanding transition state for Cu/B exchange becomes favorable. (3) The phosphine ligand can promote isomerization of the enantiomerically enriched allenyl–B(pin) product; accordingly, lower ligand loading might at times be optimal. (4) Catalytic cross-coupling with an enantiomerically enriched allenyl–B(pin) compound might proceed with high stereospecificity (e.g., phosphine–Pd-catalyzed cross-coupling) or lead to considerable racemization (e.g., phosphine–Cu-catalyzed allylic substitution).
TOC image

Introduction
A central advance in organic synthesis relates to catalytic transformations through which an organocopper intermediate is formed enantioselectively by the addition of a Cu–B(pin) (pin = pinacolato) or a Cu–H complex to an alkene. Typically, this is followed by reaction of the resulting organocopper species with an electrophile (e.g., a proton,1 a hydroxyamine,2 an allylic electrophile,3 an enoate,4 or an acyl halide5). A key mechanistic issue is whether kinetic selectivity is partially or entirely lost before the organocopper species is trapped by an electrophile.3g Deeper understanding of factors that impact the stereochemical integrity of the Cu-based intermediates is therefore crucial to the success of future efforts in this emerging area. We recently showed3g that in reactions via Cu–alkyl entity i (Scheme 1a) differences in steric and electronic attributes of the aryl substituent (Ar) and/or the electrophile (E) may engender major differences in product enantiopurity. In transformations involving the less nucleophilic organocopper intermediates or more congested allyl electrophiles, loss of kinetic enantioselectivity was minimized under conditions that allow for faster trapping of a Cu–alkyl intermediate.
Scheme 1.

Relevant Previous Investigations
The focus of the present study is a set of reactions that proceed through a Cu–allenyl complex (iv, Scheme 1b); such entities may arise from addition of a Cu complex to a 1,3-enyne to afford a Cu–propargyl species (iii), which would likely be rapidly converted to the lower energy allenyl isomer.6 We have previously demonstrated that the latter sequence may be utilized in catalytic site- and enantioselective Cu–B(pin) additions to 1,3-enynes followed by a γ–selective reaction with aldehydes (affording v, Scheme 1b).7 More recently, in a noteworthy disclosure,8 Buchwald and co-workers have shown that a similar process may involve a Cu–H complex9 and a ketone (but not an aldehyde, as will be discussed below).
Trisubstituted allenes are valuable axially chiral molecules10 for which only a limited number of catalytic enantioselective methods exist.11,12 To the best of our knowledge, there is no extant catalytic protocol for preparation of enantiomerically enriched trisubstituted allenyl boronates (i.e., α-addition to furnish vi, Scheme 1b).13 An attractive way to synthesize trisubstituted allenyl boronates (viii, Scheme 2a) enantioselectively would be by 1,3-enyne hydroboration,14 which might proceed via Cu–allenyl species vii. We wished to investigate whether, despite the relative robustness of a Cu–allenyl bond (vs the formerly studied Cu–alkyl bonds3g), there might be cases where the stereochemical integrity of a Cu–allenyl intermediate might be jeopardized, and whether conditions may be modified for achieving high enantioselectivity. It remained to be determined the extent to which Cu–B(pin) exchange would be α-selective, affording allenyl product (viii) in favor of the propargyl–B(pin) compound (ix), and precisely what factors can influence this selectivity.
Scheme 2.

Goals of This Study
An efficient and enantioselective method for synthesizing trisubstituted allenyl–B(pin) compounds (Scheme 2b) would provide a direct route to trisubstituted allenyl–B(pin) products that is distinct from more the notable procedures outlined by Ito, Sawamura and Szabó,15 which require enantiomerically enriched propargyl carbonates or phosphates. Moreover, Cu–H complexes react too fast with aldehydes (i.e., rapid reduction of aldehyde substrates is problematic) such that, unlike when the less reactive ketones are involved, the corresponding multicomponent CuH-catalyzed processes8 are low yielding. Allenyl–B(pin) products may be converted by catalytic cross-coupling to other desirable enantiomerically enriched trisubstituted allenes10 (i.e., x→ xii, Scheme 2b).
Herein, we detail our studies aimed at addressing the above questions and the realization of related objectives, including the development of the first catalytic method for enantioselective hydroboration16,17 of 1,3-enynes.
Results and Discussion
1. The method and its scope
a. Reactions with aryl-substituted 1,3-enynes
We began by examining the ability of Cu complexes derived from phosphine or N-heterocyclic carbene (NHC) ligands to promote hydroboration of 1,3-enyne 1a (Scheme 3). We found that (1,2-bis(2,5-diphenylphospholano)ethane (phenyl-bpe),18 either enantiomer of which is commercially available, and Cu(OAc)2 is the optimal combination.19 Allenyl–B(pin) 2a was thus isolated exclusively (i.e., >98:2 viii:ix, Scheme 2a) in 63% yield and 96:4 enantiomeric ratio (er). The same ligand/Cu salt system was employed in the aforementioned transformations involving 1,3-enynes, ketones, and a silyl hydride;8 the stereochemical model proposed in this latter report is applicable here as well.
Scheme 3.

Synthesis of Trisubstituted Allenyl Boronates. Scope I: Aryl-Substituted 1,3-Enynes as Substrates
aReactions were carried out under N2 atm. Conv, >98% in all cases, determined by analysis of 1H spectra of unpurified product mixtures (±2%). Yields for purified allenyl products (±5%). Enantioselectivities were determined by HPLC analysis. bReaction time was 40 h. Experiments were run in duplicate or more. See the Supporting Information for details. pin = pinacolato.
In most – but not all – instances, reactions of aryl-substituted enynes afforded the allenyl–B(pin) with reasonable efficiency (2a-j, 51–80% yield; Scheme 3), without observable amounts of the propargyl isomer formation (<2%), and in up to 98:2 er. This included enynes that contain 1,2-disubstituted alkenyl moiety (2k-l; Scheme 3), although longer reaction times were necessary.
Processes leading to para-trifluoromethylphenyl-substituted 2e and o-fluorophenyl-substituted 2i were less enantioselective (80:20 and 90:10 er, respectively). This is in contrast to transformations of the corresponding meta-substituted isomers: 2g and 2h were generated in 93:7 and 94:6 er, respectively. Reactions that generate para-bromo- or meta-trifluoromethyl-substituted 2d and 2g (93:7–95:5 er) were less enantioselective than those delivering para-tolyl- or para-methoxyphenyl- or meta-tolyl-substituted 2b, 2c and 2f (97:3–98:2 er). Thus, although less pronounced, these data further indicate that enantioselectivity can be lower with electron-deficient enynes. We note that in the previously reported catalytic Cu–H addition/ketone trapping strategy,8 there were no examples regarding reactions with enynes that bear an electron withdrawing or an ortho-substituted aryl group. Another key finding is that only in the transformation leading to ortho-fluorophenyl-substituted 2i was the propargyl addition byproduct formed (25%; see below for further discussion).
b. Reactions with alkyl-substituted enynes
Transformations with alkyl-substituted enynes were for the most part efficient and exceptionally enantioselective (4a-i, 48–80% yield and 95:5–99:1 er; Scheme 4). Among suitable substrates were those containing a bulky cyclohexyl group (e.g., 4h, in 65% yield and 98:2 er). A carboxylic ester or an alkene unit was tolerated, as these moieties were not subject to competitive reduction or hydroboration, respectively. Nevertheless, methyl ketone 4f was formed less efficiently (67% conv, 22% recovered 3f, 48% yield).20 Based on a previous hypothesis, presented in connection to a set of NHC–Cu-catalyzed allylic substitutions,21 internal Cu–carbonyl chelation might lead to reduced reaction rates. Consistent with the suggested scenario, 4g, containing a larger iso-propylketone unit, was obtained in higher yield (67% vs 48%, Scheme 4).
Scheme 4.
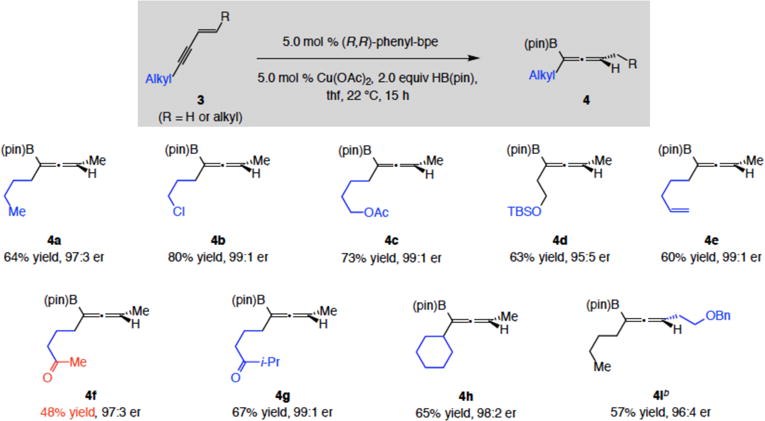
Synthesis of Trisubstituted Allenyl Boronates. Scope II: Alkyl-Substituted 1,3-Enynes as Substrates
aReactions were carried out under N2 atm. Conv, >98% in all cases except for 4f (67%), determined by analysis of 1H spectra of unpurified product mixtures (±2%). Yields for purified products (±5%). Enantioselectivities were determined by HPLC analysis. bReaction time was 40 h. Experiments were run in duplicate or more. See the Supporting Information for details.
2. Utility
a. Efficiency of allenyl–B(pin) synthesis
1,3-Enynes may be prepared by catalytic Sonogashira cross-coupling22 between a terminal alkyne and an alkenyl bromide. This compares favorably with the alternative multistep sequence needed for synthesis of trisubstituted allenylboronates, where the following steps are required: deprotonation of the alkyne substrate with a strong base (e.g., n-BuLi), addition to an aldehyde, oxidation to the ynone, enantioselective reduction, alcohol activation (i.e., carbonate generation), and phosphine–Cu-catalyzed boryl allylic substitution.15 Enantiomerically enriched propargyl alcohols can be prepared by the addition of a terminal alkyne to an aldehyde.23 Nonetheless, reactions with acetaldehyde, which afford especially important building blocks, proceed with low enantioselectivity,24 and carbonate formation and an ensuing boryl addition must still be carried out.
b. Diastereoselective addition to aldehydes
As noted earlier, a key advantage of the present approach is that it allows for diastereoselective additions to aldehydes and formation of fragments. A representative case, entailing catalytic enantioselective synthesis of trisubstituted allenyl–B(pin) 4j and its conversion to alkyne 5,25 is presented in Scheme 5. As expertly demonstrated by Roush and Chen, who originally developed this widely applicable addition reaction, use of the other phosphoric acid enantiomer will lead to the formation of the alternative diastereomer with high selectivity.25a The reaction leading to the formation of 4j, obtained in 68% yield and 95:5 er, was carried out on 0.5 gram of the enyne 3j at reduced catalyst loading (2.5 mol %). This approach complements the strategies of Krische and co-workers,26 in regards to efficient and enantioselective segphos–Ir-catalyzed coupling of alcohols and 1,3-enynes, which must bear a specific protecting group. The protocol is also applicable to a wider range of aryl- and alkyl-substituted substrates (Schemes 3–4).
Scheme 5.

Efficient and Diastereoselective Addition to an Aldehydea
aCarried out under N2 atm. Conv (>98% in all cases) determined by analysis of 1H spectra of unpurified product mixtures (±2%). Yields for purified products (±5%). Enantioselectivities were determined by HPLC analysis. Experiments were run in duplicate or more. See the Supporting Information for details.
c. Catalytic cross-coupling and fluctuations in er
Another important mode of functionalization for trisubstituted allenyl–B(pin) compounds involves catalytic cross-coupling.27 A key issue here is that loss of enantiopurity of the metal–allenyl intermediate must not be competitive with C–C bond formation. The examples in Scheme 6 reveal that the initial enantiomeric purity may be retained or vanished, depending on the coupling process. The dppe– Pd-catalyzed reaction of 4d with phenyl iodide delivered 6 in 70% yield without any detectable alteration in enantiomeric purity (95:5 er). In contrast, attempts to carry out enantioselective allylic substitution under conditions developed by us for NHC–Cu-catalyzed allylic substitutions involving monosubstituted allenyl–B(pin) and allylic phosphate28 led only to formation of a mixture of unidentifiable byproducts. Additionally, (phenyl)bpe–Cu-catalyzed multicomponent allylic substitution with allylphosphate afforded 7 in 70% yield, but only in the racemic form (see below for analysis).
Scheme 6.

Retention/Loss of Kinetic Selectivity Depends on the Type of Reactiona
aCarried out under N2 atm; >98% conv in both cases (determined by analysis of 1H spectra of unpurified product mixtures (±2%)). Yields are for purified products (±5%). Enantioselectivity was determined by HPLC analysis. Experiments were run in duplicate or more. See the Supporting Information for details.
3. Influence of reaction conditions and enyne structure on allenyl:propargyl selectivity and enantioselectivity
To gain further insight as to why, depending on electronic and/or steric attributes of an enyne substrate or reaction conditions, there might be significant variations in allenyl:propargyl ratio or er, the following investigations were performed.
a. Influence of enyne structure on differences in allenyl:propargyl selectivity
As was noted and illustrated in entries 1–2 of Table 1, unlike other aryl- or alkyl-substituted enynes, significant amounts of the propargyl–B(pin) isomer was generated during the reaction of an ortho-fluorophenyl-substituted enyne. Moreover, in the transformation involving ortho- bromophenyl-substituted 1m (entry 3, Table 1) propargyl–B(pin) 8m was formed preferentially (20:80 allenyl:propargyl ratio),29 and propargyl–B(pin) 8n was generated with 4:96 allenyl:propargyl selectivity, in 48% yield and 92:8 er (entry 4).
Table 1.
Effect of o-Substituted Aryl Enyne Group on Allenyl:Propargyl Selectivitya

| |||||
|---|---|---|---|---|---|
| entry | G | 2:8b | yield (%)c | erd | |
| 1 |
|
<b>>98:2 | 63 (2a) | 96:4 | |
| 2 | F (1i) | 7525 | 55 (2i) | 90:10(2i) | |
| 3 |
|
<o>20:80 | 69 (2m & 8m)e | nd | |
| 4 |
|
<o>4:96 | 48 (8n) | 92:8 (8n) | |
Reactions were carried out under N2 atm. Conv (>98% in all cases) was determined by analysis of 1H NMR spectra of the unpurified product mixtures (±2%).
Determined by analysis of 1H NMR spectra of unpurified product mixtures.
Yields are for isolated and purified allenyl boronate products (±5%).
Er was determined by HPLC analysis (±1%).
Based on analysis of the 1H NMR spectra of the unpurified product mixture with diphenylmethane as the internal standard. See the Supporting Information for details. nd = not determined.
b. Fluctuations in er based on boron–hydride identity and concentration
The lower er for reactions of enynes containing a relatively electron-deficient aryl group might be because of the slower reaction between the Cu–allenyl complex and the boron–hydride. Indeed, er decreased as H–B(pin) concentration was diminished and/or when the less reactive H–B(dan) was used (Figure 1). Whereas enyne 1a was converted to allenyl–B(pin) 2a in 91:9 er with 0.5 equiv HB(pin) (square symbol, Figure 1), er improved significantly (96:4 er) with 1.0 equiv of the hydride; further increase in boron–hydride concentration was inconsequential. In transformation of 1a with the less reactive HB(dan) (dan = naphthalene-1,8-diaminato; diamond symbol, Figure 1), as the hydride amount was increased from 0.5 to 1.0 to 2.0 to 4.0 equiv, so did the er (from 77:23 to 81:19 to 89:11 to 92:8). With the more electron-deficient para-trifluoromethylphenyl-substituted enyne 1e (triangle symbol) and ortho-fluorophenyl-substituted enyne 1i (circle symbol), enantioselectivity improved with 0.5 equiv HB(pin) when 4.0 equiv hydride was present (from 52:48 and 70:30 er to 81:19 and 91:9 er, respectively). Faster trapping of the enantiomerically enriched Cu–allenyl intermediates seems to allow for more effective preservation of kinetic selectivity. The fact that a plateau is reached with >2.0 equiv HB(pin) may be attributed to the lower kinetic selectivity of reactions involving the boron–hydride reagent (vs PMHS).30 Similar to what was observed in a recently disclosed study,3g er values are higher at lower alkene concentration; this suggests that, when HB(pin) is used as the reagent, a Cu–H complex that reacts with 1,3-enyne with lower enantioselectivity may be formed. For instance, when the amount of 1e is reduced from 1.0 to 0.25 equiv, while maintaining constant of HB(pin) concentration, er increases from 62:38 (Figure 1) to 75:25 er.50
Figure 1.

Influence of electrophile concentration and/or identity on er. Reactions were carried out under N2 atm; >98% conv in all cases (determined by analysis of 1H spectra of unpurified product mixtures (±2%)). Yields (in parentheses) are for isolated and purified allenyl boronate products, and are based on the limiting reagent (±5%). Enantioselectivity was determined through HPLC analysis (±1%). See the Supporting Information for details. dan = naphthalene-1,8-diaminato.
c. Influence of bis-phosphine:Cu salt ratio on er
We carried out selected transformations with different amounts of the bis-phosphine ligand (5.0 mol % Cu(OAc)2; Table 2). For 2e and 2i, er was higher at lower catalyst loading, and there was no additional improvement when less than 2.5 mol % ligand was present. Moreover, when equal amounts of the chiral ligand and Cu salt were used, er values remained constant throughout the transformation (e.g., with 1e, 80:20 er after 0.5 h and 15 h),50 further suggesting that diminished enantioselectivity likely originates from reduced kinetically nonselective Cu–H addition. The diminution in er was not observed with electron-neutral or electron-rich enynes. This implies that, with electron-deficient allenes, free phosphine can lead to lowering of er. If so, er could be improved by properly adjusting ligand concentration (see below for discussion).
Table 2.
Effect of Bis-phosphine Amount on Enantioselectivitya

| ||||
|---|---|---|---|---|
| entry | Ar | mol % ligand | yield (%)b | erc |
| 1 | p-F3CC6H4 (1e) | <b>2.5 | 48 | <b>80:20 |
| 2 | p-F3CC6H4 (1e) | 5.0 | 51 | 80:20 |
| 3 | p-F3CC6H4 (1e) | <o>10 | 45 | <o>65:35 |
|
| ||||
| 4 | o-FC6H4 (1i) | <b>2.5 | 48 | <b>92:8 |
| 5 | o-FC6H4 (1i) | 5.0 | 55 | 90:10 |
| 6 | o-FC6H4 (1i) | <o>10 | 55 | <o>80:20 |
Reactions were carried out under N2 atm; >98% conv in all cases (determined by analysis of 1H spectra of unpurified product mixtures (±2%)).
Yields are for isolated and purified allenyl boronate products (±5%).
Enantioselectivity was determined through HPLC analysis (±1%). See the Supporting Information for details.
4. DFT studies
The er fluctuations might be a consequence of the fluxional attributes of the metal–allenyl intermediates31, a possibility, which, despite some available evidence,32 has not been rigorously investigated.33,34 Similar considerations have been applied to metal–alkenyl complexes involved in alkyne hydroboration35 (or hydrosilylation36 or hydrogenation37), where unimolecular rearrangements via metallacyclopropenes38 or zwitterionic metal carbenes39,40 have been proposed to cause selectivity variations. However, it seems unlikely that such resonance contributions are strong enough to cause the same type of process to occur with organocopper compounds.41 As several transition metal complexes (e.g., Cu-, Au-, or Pd-based) have been reported to promote isomerization of chiral allenes,42,43 we chose to use density functional theory (DFT) and high level ab initio DLPNO-CCSD(T)44 calculations as the means to investigate these mechanistic issues.
Recent advances in computational chemistry have rendered high level ab initio CCSD(T) calculations significantly more affordable, while maintaining high accuracy, through the use of localized orbitals (DLPNO-CCSD(T)).45 These latter calculations are intended to serve as the internal standard in the present investigation, rather than relying solely on density functionals, which have been calibrated against external databases. Density functional MN1546 showed the best overall agreement with the reference and, accordingly, the corresponding values will be used.
Specifically, we set out to address the following questions:
What might be the mechanism of unimolecular racemization (i.e., interconversion of Cu–allenyl diastereomers), and how might these processes compare energetically to the more frequently invoked π-allyl isomerization?
How facile is the bimolecular trapping of Cu–allenyl intermediates with boron– hydride in comparison to unimolecular isomerization, which leads to diminished er?
What electronic factors govern the racemization process, and is there more than one pathway that leads to lowering of er?
Why do catalytic cross-coupling reactions involving a Pd–allenyl complex proceed with minimal loss of enantioselectivity but not in the case of phosphine–Cu-catalyzed allylic substitution reactions (see Scheme 6)?
a. Fluxional processes with Cu–allenyl intermediates
Computational studies indicate that Cu–H addition leading to generation of the major Cu–allenyl diastereomer (Cu–amajor) via ts(CuHa)major (29.0 kcal/mol; Figure 2) is exergonic by 21.3 kcal/mol (i.e., largely irreversible Cu–H addition; Cu(I) is likely generated by facile in situ reduction9). The barrier to formation of the minor diastereomer (Cu–aminor) is predicted to be 6.2 kcal/mol higher in energy, which is in agreement with a previous study.8 The initial Cu–propargyl species (Cu–pmajor and Cu–pminor) were found to reside in a shallow minimum on the potential energy surface (e.g., 5.5 kcal/mol for Cu–pmajor), readily transforming to allenyl isomers (e.g., via ts(iso)major; free energy = 6.6 kcal/mol).6,30 In contrast, we find that the transition state for interconversion of the Cu–allenyl diasteromers (Cu–amajor and Cu–aminor) is energetically more demanding (17.0 kcal/mol for ts(rac)). This barrier is sufficiently high for the initial stereochemistry to be preserved if subsequent trapping following Cu–H addition is competitive, yet low enough for ts(rac) to be accessible (i.e., low reactivity and/or concentration of electrophile). The transition state (ts(rac)) is best described as a Cu–propargyl anion complex, wherein the alkynyl π-system, orthogonal to the extended π cloud, is transition metal bound. The C1–C2 bond contraction from 1.313 Å in Cu–amajor to 1.297 Å in ts(rac) and elongation of the C2–C3 bond from 1.325 to 1.359 Å support this proposal. The geometric parameters for ts(rac) are similar to bond lengths in the corresponding free propargyl anion (1.259 and 1.356 Å for C1– C2 and C2–C3, respectively), but considerably different compared to the ones in the neutral alkyne after protonation at C3 (1.220 and 1.453 Å, respectively). The latter comparison underscores the importance of resonance contributions in stabilizing the negative charge. Furthermore, the Cu–C1 bond in ts(rac) is relatively ionic, and natural bond orbital (NBO) analysis indicates 84% contribution from C1 (vs 16% from Cu; more on this below).
Figure 2.
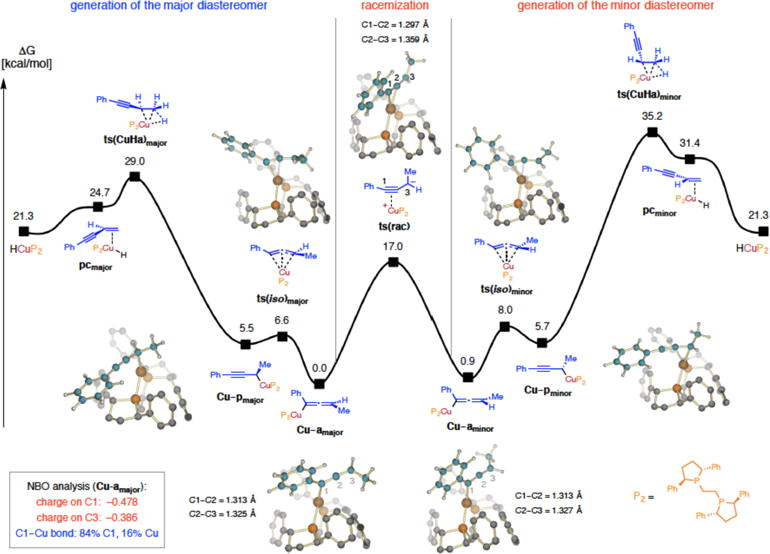
Fluxional nature of Cu–allenyl complexes (enantioselective Cu–H addition, propargyl-to-allenyl isomerization and racemization). Free energy values were obtained at the MN15/Def2TZVPPthf(SMD)//M06L/DF-Def2SVPthf(PCM) level; pc, π-complex; ts(CuHa), transition state for copper hydride addition; ts(rac), transition state for racemization; ts(iso), transition state for π-allyl isomerization; Cu–p, Cu–propargyl intermediate; Cu–a, Cu–allenyl intermediate. See the Supporting Information for details.
b. Competition between unimolecular isomerization and bimolecular trapping; impact of bulky substituents on allenyl:propargyl ratios
We calculated the relative barriers between ts(rac) and tsα/tsγ, leading to allenyl (2a and 2n) and propargyl products (8a and 8n) via intermediates BHaα and BHaγ (Figure 3). Transition states ts(rac), tsα, and tsγ were found to be similar in energy when functionals that account for dispersion were applied (e.g., MN15). What is more, in agreement with the experimental data, with a phenyl-substituted substrate, tsα (15.7 kcal/mol) was found to be favored over tsγ (18.2 kcal/mol).47 The finding that the computed relationship between tsα and tsγ is reversed in the case of an o-tolyl-substituted enyne (18.8 vs 16.6 kcal/mol, respectively) is congruent with the experimental observation (Table 1). That ts(rac) (16.4 kcal/mol; cf. Figure 3) is predicted to be slightly lower in energy than tsγ (by 0.2 kcal/mol) still supports the observed high enantioselectivity and is consistent with expected uncertainties in DFT energies.48 The energy needed to reach tsα in case of the para- trifluoromethylphenyl-substituted substrate is 16.1 kcal/mol (not shown in Figure 3; see the Supporting Information), whereas it is 15.7 kcal/mol for the phenyl-substituted species (cf. Figure 3). Although the difference between these latter values is smaller than the error bar in computational accuracy, it is consistent with the scenario that a Cu–allenyl species bearing an electron-withdrawing moiety is likely less nucleophilic.3g
Figure 3.

Competition between racemization and trapping with H–B(pin); free energy values were obtained at the MN15/Def2TZVPPthf(SMD)//M06L/DF-Def2SVPthf(PCM) level; ts(rac), transition state for racemization; Cu–a, Cu–allenyl intermediate; tsα, transition state for α-addition to H–B(pin); tsγ, transition state for α-addition to HB(pin); BHa, borohydride adduct. See the Supporting Information for details.
Structural analysis offers a clue for the reversal in allenyl:propargyl product selectivity. The short CMe ⋯ Cβ distance of 2.98 Å in tsα for ortho-tolyl-substituted enynes points to destabilizing A(1,3) strain, which is absent in the case of phenyl-substituted substrates. In tsγ the aryl ring is able to rotate such that it is in conjugation with the Cu–C1 bond and A(1,3) strain49 can be avoided (CMe ⋯ Cβ distance = 3.61 Å).
c. Transition states leading to isomerization and impact of excess phosphine
We then investigated the influence of electronic modification on the barrier to loss of kinetic enantioselectivity (e.g., effect of an electron withdrawing unit; Figure 4). Based on our calculations, a para-trifluoromethylphenyl group can stabilize the propargyl anion en route to ts(rac) (15.8 kcal/mol vs 17.0 kcal/mol for a phenyl moiety), and the corresponding barrier is higher for the methyl-substituted intermediate (21.1 kcal/mol). The decrease in resonance stabilization is reflected in the contraction of the C1–C2 bond in the methyl-substituted complex (1.291 Å) compared to the one that contains a para-trifluoromethylphenyl group (1.300 Å). Nonetheless, the ease with which ts(rac) can be accessed supports the notion that isomerization of metal–alkenyl species via the aforementioned zwitterionic metal carbene intermediates is more challenging.41
Figure 4.
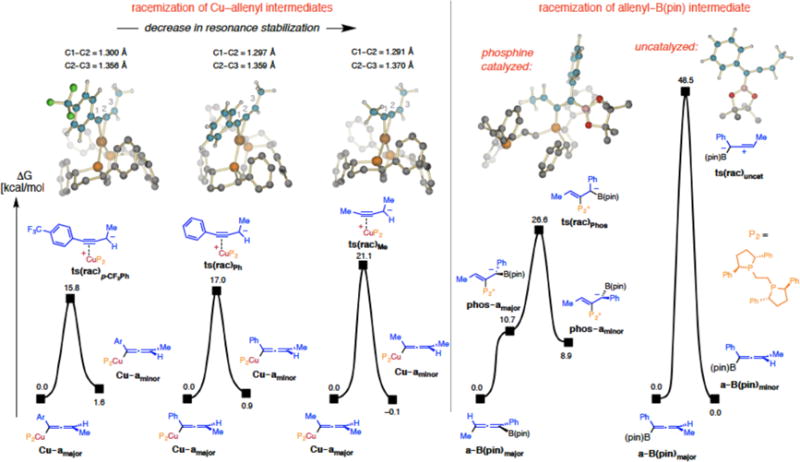
Comparison of various modes of racemization; free energy values were obtained at the MN15/Def2TZVPPthf(SMD)//M06L/DF-Def2SVPthf(PCM) level; ts(rac), transition state for racemization; Cu–a, Cu–allenyl intermediate; a–B(pin), allenyl–B(pin) product; phos-a, phosphine adduct with allenyl–B(pin) product; Ar = p-F3CC6H4; uncat = uncatalyzed. See the Supporting Information for details.
In search of additional evidence vis-á-vis the proposed unimolecular racemization (Figure 4), we synthesized Cu–allenyl complex Cu–amajor diastereoselectively and monitored its conversion to the corresponding minor isomer (Cu–amajor) by low temperature 1H NMR spectroscopy. Addition of enyne 3c (at −50 °C) to a sample of the bis-phosphine–Cu–H complex, generated from a mixture of CuOt-Bu, bisphosphine and a silyl hydride (tetramethyldisiloxane), led to the formation of 1:99 ratio of diastereomers (Figure 5a). The alternative route to access the major Cu-allenyl diastereomer through transmetalation of enantiomerically enriched allenyl– B(pin) products to a bisphosphine CuOt-Bu complex was not stereospecific and inapplicable.50 After 2.75 h (at −50 °C) the ratio was reduced to 4.5:95.5 (Figure 5b). Accordingly, a free energy of activation (ΔG‡) of 18.5 kcal/mol was measured (at −50 °C),50 which is in agreement with the value calculated for the model system (ts(rac)Me = 21.1 kcal/mol; Figure 4). When equilibration was allowed to be established at 22 °C, followed by cooling to −50 °C, the diastereomeric ratio plummeted (52:48; Figure 5c). Subsequent trapping with HB(pin) afforded racemic allenyl– B(pin), ruling out a step other than Cu–H addition as the stereochemistry-determining step.51
Figure 5.
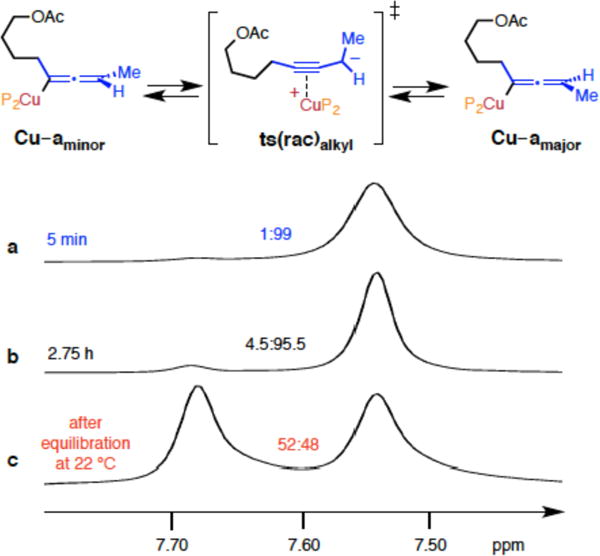
Low temperature 31P NMR experiment to investigate the isomerization of Cu–amajor to Cu–aminor; all measurements where performed at −50 °C in thf–d8. See the Supporting Information for details.
For further insight regarding a possible adverse influence of free phosphine on enantioselectivity (see Table 2), we probed the feasibility of a mechanism that is reminiscent of what has formerly been proposed for Morita-Baylis-Hillman transformations.52 Indeed, when an enantiomerically enriched sample of 2i (90:10 er) was subjected to 5.0 mol % of the bis- phosphine ligand (22 °C, thf, 15 h to emulate the reaction conditions), the enantiomeric purity of recovered 2i was found to be measurably lower (83:17 er; eq 1). The same experiment with allenyl–B(pin) compounds that lack an electron-withdrawing aryl unit (e.g., 2a) did not lead to any observable change in er. Calculations suggest that reversible addition of phosphine53 to the central/sp-hybridized carbon of the chiral allenyl B(pin) product might lead to racemization (right panel, Figure 4). The 26.6 ± 2.0 kcal/mol estimate for ts(rac)Phos is a reasonable value for a process at ambient temperature.
 |
(1) |
d. Variations in loss of er in different catalytic reactions of allenyl–B(pin) products
The above considerations raise another intriguing question: Why is there complete retention of stereochemistry during phosphine–Pd-catalyzed cross-coupling whereas with a phosphine–Cu-catalyzed allylic substitution it is entirely lost (see Scheme 6)? DFT studies show that the barrier for unimolecular transition state for racemization of an allenyl Pd–allenyl complex ts(rac)Pd (Figure 6) is 26.8 kcal/mol, which is markedly higher than those that correspond to a Cu–allenyl species (see Figure 4). In addition, we find that reductive elimination, also a unimolecular process, can outcompete racemization (22.7 kcal/mol for ts(re)major,Pd). We have previously shown that allylic substitution involving Cu-alkyl intermediates bearing a bis-phosphine ligand can be energetically demanding,3g rendering isomerization problematic. Comparison of the electronic features of Pd–allenyl species (Pd–amajor) with those of the corresponding organocopper entities (Cu–amajor, see Figure 2) offers further support for the increased configurational stability of the former four-coordinate systems. While the Cu–C1 bond is relatively polar (84% C1, 16% Cu), NBO analysis predicts a greater degree of covalent character for the Pd–C1 bond (65% C1, 35% Pd), implying that more energy is required for formation of the corresponding propargyl anion transition state in the case of organopalladium complexes.
Figure 6.

Relevance of metal–allene racemization to bis-phosphine–Pd-catalyzed cross-coupling; free energy values have been obtained at the MN15/Def2TZVPPthf(SMD)//M06L/DF-Def2SVPthf(PCM) level; P2 = (phenyl)bpe; ts(rac), transition state for racemization; ts(iso), transition state for π-allyl isomerization; Pd–p, Pd–propargyl intermediate; Pd–a, Pd–allenyl intermediate; ts(re), transition state for reductive elimination; pc, π-complex. See the Supporting Information for details.
Conclusions
We have developed the first enantioselective method for preparation of trisubstituted allenyl–B(pin) compounds, obtained by catalytic hydroboration of 1,3-enynes; the method is efficient and boradly applicable. We demonstrate that strategies involving the use of an allenyl– B(pin) compound complements the related multicomponent strategies where the enantiomerically enriched Cu–allenyl intermediate reacts in situ with an electrophile. For example, in cases where the electrophile might react competitively with a Cu–H complex (e.g., an aldehyde; Scheme 5), or if certain subsequent cross-coupling reactions are desired, it is preferable to utilize an allenyl–B(pin) species.
The present studies shed light on factors that impact allenyl:propargyl ratios and/or enantioselectivity:
With enynes bearing an electron-deficient substituent, where the Cu–allenyl intermediates are more stabilized (i.e., are less reactive), loss of er might arise from competitive racemization. This can be minimized by the use of larger amounts of and/or more reactive boron–hydride reagents (i.e., HB(pin)).
Particularly with enynes bearing an electron-deficient substituent, a Lewis basic phosphine ligand can promote isomerization of an enantiomerically enriched allenyl–B(pin) product.
Depending on the type of transformation, functionalization of a trisubstituted allenyl–B(pin) compound might take place with high stereospecificity or lead to major loss of enantiomeric purity.
The insights provided by the present investigations are applicable to other sets of transformations. The examples provided in Scheme 7 are illustrative. It can now be more easily understood why whereas 9a and 9b were obtained in 97% and 99% enantiomeric excess (ee; ≥98.5:15 er), respectively,8 synthesis of para-trifluoromethylphenyl-substituted 9c, formed via a less nucleophilic Cu–allenyl intermediate, is notably less enantioselective [84% ee (92:8 er)]. It is probably for similar reasons that enantioselectivity for 9c decreases further to 80% ee (90:10er) when the relative amount of the ketone is lowered (3:1 enyne:acetophenone, slower Cu–allene trapping), but improves to 88% ee (94:6 er) with excess ketone (1:3 enyne:acetophenone, faster trapping).
Scheme 7.
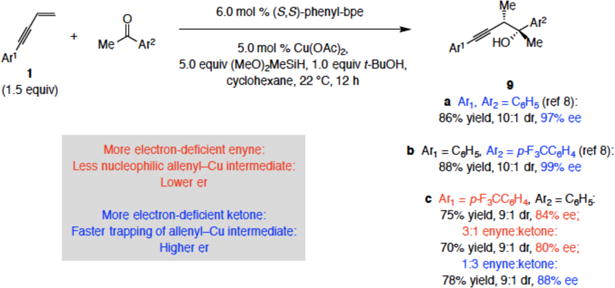
Relevance to a Previously Reported Methoda
aReactions to obtain were carried out under N2 atm; >98% conv in both cases (determined by analysis of 1H spectra of unpurified product mixtures (±2%)). Yields are for purified products (±5%). Enantioselectivity was determined by HPLC analysis. Experiments were run in duplicate or more. See the Supporting Information for details.
Development of additional catalytic enantioselective methods, the associated mechanistic investigations, and applications to synthesis of biologically active molecules continue in these laboratories.
Supplementary Material
Acknowledgments
This research was supported by a grant from the NIH (GM-47480) and postdoctoral fellowships to Y.H. by the Shanghai Institute of Organic Chemistry, Zhejiang Medicine Co., Pharmaron, and to J. d. P. by Alfonso Martin Escudero Foundation.
Footnotes
SUPPORTING INFORMATION
This Supporting Information is available free of charge via the Internet at DOI:10.1021/jacs.132966 Experimental details for all reactions and analytic details for all products (PDF)
ORCID
Youming Huang: 0000-0002-5148-459X; Juan del Pozo: 0000-0003-4366-7983; Sebastian Torker: 0000-0002-2892-4411; Amir H. Hoveyda: 0000-0002-1470-6456
References
- 1.For representative reports, see:; (a) Lee Y, Hoveyda AH. J Am Chem Soc. 2009;131:3160–3161. doi: 10.1021/ja809382c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee Y, Jang H, Hoveyda AH. J Am Chem Soc. 2009;131:18234–18235. doi: 10.1021/ja9089928. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sasaki Y, Zhong C, Sawamura M, Ito H. J Am Chem Soc. 2010;132:1226–1227. doi: 10.1021/ja909640b. [DOI] [PubMed] [Google Scholar]; (d) Jang H, Zhugralin AR, Lee Y, Hoveyda AH. J Am Chem Soc. 2011;133:7859–7871. doi: 10.1021/ja2007643. [DOI] [PubMed] [Google Scholar]; (e) Corberán R, Mszar NW, Hoveyda AH. Angew Chem, Int Ed. 2011;50:7079–7082. doi: 10.1002/anie.201102398. [DOI] [PubMed] [Google Scholar]; (f) Sasaki Y, Horita Y, Zhong C, Sawamura M, Ito H. Angew Chem, Int Ed. 2011;50:2778–2782. doi: 10.1002/anie.201007182. [DOI] [PubMed] [Google Scholar]; (g) Meng F, Jang H, Hoveyda AH. Chem Eur J. 2013;19:3204–3214. doi: 10.1002/chem.201203803. [DOI] [PubMed] [Google Scholar]; (h) Kubota K, Yamamoto E, Ito H. Adv Synth Catal. 2013;355:3527–3531. [Google Scholar]; (i) Jang H, Jung B, Hoveyda AH. Org Lett. 2014;16:4658–4661. doi: 10.1021/ol5022417. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Wang Z, He X, Zhang R, Zhang G, Xu G, Zhang Q, Xiong T, Zhang Q. Org Lett. 2017;19:3067–3070. doi: 10.1021/acs.orglett.7b01135. [DOI] [PubMed] [Google Scholar]
- 2.For representative reports, see:; (a) Matsuda N, Hirano K, Satoh T, Miura M. J Am Chem Soc. 2013;135:4934–4937. doi: 10.1021/ja4007645. [DOI] [PubMed] [Google Scholar]; (b) Zhu S, Niljianskul N, Buchwald SL. J Am Chem Soc. 2013;135:15746–15749. doi: 10.1021/ja4092819. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Miki Y, Hirano K, Satoh T, Miura M. Angew Chem, Int Ed. 2013;52:10830–10834. doi: 10.1002/anie.201304365. [DOI] [PubMed] [Google Scholar]; (d) Zhu S, Buchwald SL. J Am Chem Soc. 2014;136:15913–15916. doi: 10.1021/ja509786v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Shi SL, Buchwald SL. Nat Chem. 2015;7:38–44. doi: 10.1038/nchem.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Sakae R, Hirano K, Satoh T, Miura M. Angew Chem, Int Ed. 2015;54:613–617. doi: 10.1002/anie.201409104. [DOI] [PubMed] [Google Scholar]; (g) Niljianskul N, Zhu S, Buchwald SL. Angew Chem, Int Ed. 2015;54:1638–1641. doi: 10.1002/anie.201410326. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Yang Y, Shi SL, Niu D, Liu P, Buchwald SL. Science. 2015;349:62–66. doi: 10.1126/science.aab3753. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Niu D, Buchwald SL. J Am Chem Soc. 2015;137:9716–9721. doi: 10.1021/jacs.5b05446. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Nishikawa D, Hirano K, Miura M. J Am Chem Soc. 2015;137:15620–15623. doi: 10.1021/jacs.5b09773. [DOI] [PubMed] [Google Scholar]; (k) Pirnot MT, Wang YM, Buchwald SL. Angew Chem, Int Ed. 2016;55:48–57. doi: 10.1002/anie.201507594. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Kato K, Hirano K, Miura M. Angew Chem, Int Ed. 2016;55:14400–14404. doi: 10.1002/anie.201608139. [DOI] [PubMed] [Google Scholar]; (m) Zhu S, Niljianskul N, Buchwald SL. Nat Chem. 2016;8:144–150. doi: 10.1038/nchem.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Shi SL, Wong ZL, Buchwald SL. Nature. 2016;532:353–356. doi: 10.1038/nature17191. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Wang H, Yang JC, Buchwald SL. J Am Chem Soc. 2017;139:8428–8431. doi: 10.1021/jacs.7b04816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For representative reports, see:; (a) Jia T, Cao P, Wang B, Lou Y, Yin X, Wang M, Liao J. J Am Chem Soc. 2015;137:13760–13763. doi: 10.1021/jacs.5b09146. [DOI] [PubMed] [Google Scholar]; (b) Wang YM, Buchwald SL. J Am Chem Soc. 2016;138:5024–5027. doi: 10.1021/jacs.6b02527. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Han JT, Jang WJ, Kim N, Yun J. J Am Chem Soc. 2016;138:15146–15149. doi: 10.1021/jacs.6b11229. [DOI] [PubMed] [Google Scholar]; (d) Lee J, Torker S, Hoveyda AH. Angew Chem, Int Ed. 2017;56:821–826. doi: 10.1002/anie.201611444. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Radomkit S, Liu Z, Closs A, Mikus MS, Hoveyda AH. Tetrahedron. 2017;73:5011–5017. doi: 10.1016/j.tet.2017.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Xu G, Zhao H, Fu B, Cang A, Zhang G, Zhang Q, Xiong T, Zhang Q. Angew Chem, Int Ed. 2017;56:13130–13134. doi: 10.1002/anie.201707070. [DOI] [PubMed] [Google Scholar]; (g) Lee J, Radomkit S, Torker S, del Pozo J, Hoveyda AH. Nat Chem. 2018;10:99–108. doi: 10.1038/nchem.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Kim N, Han JT, Ryu DH, Yun J. Org Lett. 2017;19:6144–6147. doi: 10.1021/acs.orglett.7b03022. [DOI] [PubMed] [Google Scholar]
- 4.For example, see:; Zhou Y, Bandar JS, Buchwald SL. J Am Chem Soc. 2017;139:8126–8129. doi: 10.1021/jacs.7b04937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For example, see:; Huang Y, Smith KB, Brown MK. Angew Chem, Int Ed. 2017;56:13314–13318. doi: 10.1002/anie.201707323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mszar NW, Haeffner F, Hoveyda AH. J Am Chem Soc. 2014;136:3362–3365. doi: 10.1021/ja500373s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meng F, Haeffner F, Hoveyda AH. J Am Chem Soc. 2014;136:11304–11307. doi: 10.1021/ja5071202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Y, Perry IB, Lu G, Liu P, Buchwald SL. Science. 2016;353:144–150. doi: 10.1126/science.aaf7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For a review on CuH-catalyzed reactions, see:; Deutsch C, Krause N, Lipshutz BH. Chem Rev. 2008;108:2916–2927. doi: 10.1021/cr0684321. [DOI] [PubMed] [Google Scholar]
- 10.For a review on preparation of trisubstituted allenes, see:; Ye J, Ma S. Org Chem Front. 2014;1:1210–1224. [Google Scholar]
- 11.(a) Hayashi T, Tokunaga N, Inoue K. Org Lett. 2004;6:305–307. doi: 10.1021/ol036309f. [DOI] [PubMed] [Google Scholar]; (b) Li CY, Wang XB, Sun XL, Tang Y, Zheng JC, Xu ZH, Zhou YG, Dai LX. J Am Chem Soc. 2007;129:1494–1495. doi: 10.1021/ja068642v. [DOI] [PubMed] [Google Scholar]; (c) Zhang W, Zheng S, Liu N, Werness JB, Guzei IA, Tang W. J Am Chem Soc. 2010;132:3664–3665. doi: 10.1021/ja100173w. [DOI] [PubMed] [Google Scholar]; (d) Nishimura T, Makino H, Nagaosa M, Hayashi T. J Am Chem Soc. 2010;132:12865–12867. doi: 10.1021/ja1066509. [DOI] [PubMed] [Google Scholar]; (e) Wang Y, Zhang W, Ma S. J Am Chem Soc. 2013;135:11517–11520. doi: 10.1021/ja406135t. [DOI] [PubMed] [Google Scholar]; (f) Qian H, Yu X, Zhang J, Sun J. J Am Chem Soc. 2013;135:18020–18023. doi: 10.1021/ja409080v. [DOI] [PubMed] [Google Scholar]; (g) Wang M, Liu ZL, Zhang X, Tian PP, Xu YH, Loh TP. J Am Chem Soc. 2015;137:14830–14833. doi: 10.1021/jacs.5b08279. [DOI] [PubMed] [Google Scholar]; (h) Chu WD, Zhang L, Zhang Z, Zhou Q, Mo F, Zhang Y, Wang J. J Am Chem Soc. 2016;138:14558–14561. doi: 10.1021/jacs.6b09674. [DOI] [PubMed] [Google Scholar]; (i) Yao Q, Liao Y, Lin L, Lin X, Ji J, Liu X, Feng X. Angew Chem, Int Ed. 2016;55:1859–1863. doi: 10.1002/anie.201509455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For additional examples (also see ref 9) of trisubstituted allenes prepared through modification of enantiomerically enriched substrates, see:; a) Hung SC, Wen YF, Chang JW, Liao CC, Uang BJ. J Org Chem. 2002;67:1308–1313. doi: 10.1021/jo016201i. [DOI] [PubMed] [Google Scholar]; (b) Molander GA, Sommers EM, Baker SR. J Org Chem. 2006;71:1563–1568. doi: 10.1021/jo052201x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dieter RK, Chen N, Gore VK. J Org Chem. 2006;71:8755–8760. doi: 10.1021/jo061442h. [DOI] [PubMed] [Google Scholar]; (d) Liu Z, Wasmuth AS, Nelson SG. J Am Chem Soc. 2006;128:10352–10353. doi: 10.1021/ja0629110. [DOI] [PubMed] [Google Scholar]; (e) Kobayashi K, Naka H, Wheatley AEH, Kondo Y. Org Lett. 2008;10:3375–3377. doi: 10.1021/ol801249w. [DOI] [PubMed] [Google Scholar]; (f) Cérat P, Gritsch PJ, Goudreau SR, Charette AB. Org Lett. 2010;12:564–567. doi: 10.1021/ol902766f. [DOI] [PubMed] [Google Scholar]; (g) Dabrowski JA, Haeffner F, Hoveyda AH. Angew Chem, Int Ed. 2013;52:7694–7699. doi: 10.1002/anie.201303501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For a recent report that includes synthesis of a racemic trisubstituted allenyl–B(pin), see:; Zhao J, Szabó KJ. Angew Chem, Int Ed. 2016;55:1502–1506. doi: 10.1002/anie.201510132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For hydroboration of enynes by Pd-based complexes, see:; Matsumoto Y, Naito M, Hayashi T. Organometallics. 1992;11:2732–2734. [Google Scholar]
- 15.(a) Ito H, Sasaki Y, Sawamura M. J Am Chem Soc. 2008;130:15774–15775. doi: 10.1021/ja806602h. [DOI] [PubMed] [Google Scholar]; (b) Sasaki Y, Sawamura M, Ito H. Chem Lett. 2011;40:1044–1046. [Google Scholar]; (c) Zhao TSN, Yang Y, Lessing T, Szabó KJ. J Am Chem Soc. 2014;136:7563–7566. doi: 10.1021/ja502792s. [DOI] [PubMed] [Google Scholar]
- 16.For selected reports on catalytic enantioselective hydroboration of acyclic alkenes, see:; (a) Hayashi T, Matsumoto Y, Ito Y. J Am Chem Soc. 1989;111:3426–3428. [Google Scholar]; (b) Moteki SA, Takacs JM. Angew Chem, Int Ed. 2008;47:894–897. doi: 10.1002/anie.200703127. [DOI] [PubMed] [Google Scholar]; (c) Noh D, Chea H, Ju J, Yun J. Angew Chem, Int Ed. 2009;48:6062–6064. doi: 10.1002/anie.200902015. [DOI] [PubMed] [Google Scholar]; (d) Mazet C, Gérard D. Chem Commun. 2011;47:298–300. doi: 10.1039/c0cc01547d. [DOI] [PubMed] [Google Scholar]; (e) Feng X, Jeon H, Yun J. Angew Chem, Int Ed. 2013;52:3989–3992. doi: 10.1002/anie.201208610. [DOI] [PubMed] [Google Scholar]; (f) Chen J, Xi T, Lu Z. Org Lett. 2014;16:6452–6455. doi: 10.1021/ol503282r. [DOI] [PubMed] [Google Scholar]; (g) Zhang L, Zuo Z, Wan X, Huang Z. J Am Chem Soc. 2014;136:15501–15504. doi: 10.1021/ja5093908. [DOI] [PubMed] [Google Scholar]; (h) Zhang H, Lu Z. ACS Catal. 2016;6:6596–6600. [Google Scholar]; For reviews on catalytic enantioselective hydroboration, see:; (i) Crudden C, Edwards D. Eur J Org Chem. 2003:4695–4712. [Google Scholar]; (j) Carroll A-M, O’Sullivan TP, Guiry PJ. Adv Synth Catal. 2005;347:609–631. [Google Scholar]
- 17.For catalytic enantioselective proto-boryl additions to alkenes and allenes, see: Ref 1a-b, e-h.
- 18.Pilkington CJ, Zanotti-Gerosa A. Org Lett. 2003;5:1273–1275. doi: 10.1021/ol0341952. [DOI] [PubMed] [Google Scholar]
- 19.See the Supporting Information for data regarding ligand screening.
- 20.None of the byproduct derived from reduction of the methyl ketone group could be detected (1H NMR analysis), and extended reaction times (e.g., 40 h) results in the formation of a complex mixture of unidentifiable compounds.
- 21.Meng F, McGrath KP, Hoveyda AH. Nature. 2014;513:367–374. doi: 10.1038/nature13735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.For a recent review on Sonogashira cross-coupling, see:; Thomas AM, Sujatha A, Anilkumar G. RCS Adv. 2014;4:21688–21698. [Google Scholar]
- 23.(a) Frantz DE, Fässler R, Carreira EM. J Am Chem Soc. 2000;122:1806–1807. [Google Scholar]; (b) Boyall D, López F, Sasaki H, Frantz DE, Carreira EM. Org Lett. 2000;2:4233–4236. doi: 10.1021/ol006791r. [DOI] [PubMed] [Google Scholar]; (c) Anand NK, Carreira EM. J Am Chem Soc. 2001;123:9687–9688. doi: 10.1021/ja016378u. [DOI] [PubMed] [Google Scholar]; (d) Molina YS, Ruchti J, Carreira EM. Org Lett. 2017;19:743–745. doi: 10.1021/acs.orglett.6b03692. [DOI] [PubMed] [Google Scholar]
- 24.Dominguez B. WO 2004/092098 (Great Britain) 2004 [Google Scholar]
- 25.For studies regarding the use of chiral phosphoric acids in diastereo- and enantioselective synthesis of propargyl alcohols, involving allenyl–B(pin) compounds prepared by an alternative approach, see:; (a) Chen M, Roush WR. J Am Chem Soc. 2012;134:10947–10952. doi: 10.1021/ja3031467. [DOI] [PMC free article] [PubMed] [Google Scholar]; For related studies, see:; (b) Sasaki Y, Sawamura M, Ito H. Chem Lett. 2011;40:1044–1046. [Google Scholar]
- 26.Geary LM, Woo SK, Leung JC, Krische MJ. Angew Chem, Int Ed. 2012;51:2972–2976. doi: 10.1002/anie.201200239. [DOI] [PubMed] [Google Scholar]
- 27.de Meijre A, Brase S, Oestreich M, editors. Metal-Catalyzed Cross-Coupling Reactions and More. Wiley–VCH; Weinheim, Germany: 2014. [Google Scholar]
- 28.Jung B, Hoveyda AH. J Am Chem Soc. 2012;134:1490–1493. doi: 10.1021/ja211269w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Repeated attempts to measure er for inseparable mixtures of 2j and 8j were not successful.
- 30.The phenomenon of er plateau also applies to reactions with phenyl- (→2a) and p-trifluoromethylphenyl-substituted (→2e) enynes. With PHMS, enantioselectivity for Cu–H addition to the abovementioned 1,3-enynes is 99.5:0.5 and 94:6 er, respectively, as judged by in situ trapping with acetophenone (see ref 8 and Scheme 7). However, when a boron–hydride is involved, selectivities range from 96:4 er to 81:19 er (see Figure 1).
- 31.For a review on η3-allenyl/propargyl transition metal complexes, see:; (a) Chen JT. Coord Chem Rev. 1999;190–192:1143–1168. [Google Scholar]; For early reports on allenyl-to-propargyl isomerization:; (b) Chen MC, Keng RS, Lin YC, Wang Y, Cheng MC, Lee GH. J Chem Soc, Chem Commun. 1990:1138–1140. [Google Scholar]; (c) Ogoshi S, Fukunishi Y, Tsutsumi K, Kurosawa H. Inorg Chim Acta. 1997;265:9–15. [Google Scholar]
- 32.For a review regarding transfer an/or loss of stereochemical information during propargylic substitution, see ref 9. For selected reports invoking the fluxional nature of Pd–allenyl intermediates, see:(a) Ref 11b.; (b) Yoshida M, Hayashi M, Shishido K. Org Lett. 2007;9:1643–1646. doi: 10.1021/ol070224n. [DOI] [PubMed] [Google Scholar]; (c) Yoshida M, Okada T, Shishido K. Tetrahedron. 2007;63:6996–7002. [Google Scholar]; (d) Wang Y, Zhang W, Ma S. J Am Chem Soc. 2013;135:11517–11520. doi: 10.1021/ja406135t. [DOI] [PubMed] [Google Scholar]; (e) Wang Y, Zhang W, Ma S. Org Chem Front. 2014;1:807–811. [Google Scholar]
- 33.For an experimental study regarding racemization of an enantiomerically enriched Pd–allenyl complex, see:; Ogoshi S, Nishida T, Shinagawa T, Kurosawa H. J Am Chem Soc. 2001;123:7164–7165. doi: 10.1021/ja010583s. [DOI] [PubMed] [Google Scholar]
- 34.For a computational study on propargylic substitution promoted by a Pd-based catalyst, see:; Vaz B, Pereira R, Pérez M, Alvarez R, de Lera AR. J Org Chem. 2008;73:6534–6541. doi: 10.1021/jo800756b. [DOI] [PubMed] [Google Scholar]
- 35.For trans-selective hydroboration of internal alkynes, see:; Sundararaju B, Fürstner A. Angew Chem, Int Ed. 2013;52:14050–14054. doi: 10.1002/anie.201307584. [DOI] [PubMed] [Google Scholar]; For a related example involving a Pd complex, see:; Xu S, Zhang Y, Li B, Liu SY. J Am Chem Soc. 2016;138:14566–14569. doi: 10.1021/jacs.6b09759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.For trans-selective hydrosilylation or hydrosilylation/desilylation sequences through the use of cationic Ru complexes, see:; (a) Trost BM, Ball ZT. J Am Chem Soc. 2001;123:12726–12727. doi: 10.1021/ja0121033. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Ball ZT, Jöge T. J Am Chem Soc. 2002;124:7922–7923. doi: 10.1021/ja026457l. [DOI] [PubMed] [Google Scholar]; (c) Fürstner A, Radkowski K. Chem Commun. 2002:2182–2183. doi: 10.1039/b207169j. [DOI] [PubMed] [Google Scholar]
- 37.For trans-selective hydrogenation of internal alkynes catalyzed by cationic Ru complexes, see:; Radkowski K, Sundararaju B, Fürstner A. Angew Chem, Int Ed. 2013;52:355–360. doi: 10.1002/anie.201205946. [DOI] [PubMed] [Google Scholar]; For a related report involving reactions promoted by Ru-based olefin metathesis catalysts, proceeding through a mechanistically distinct reduction/isomerization sequence, see:; Kusy R, Grela K. Org Lett. 2016;18:6196–6199. doi: 10.1021/acs.orglett.6b03254. [DOI] [PubMed] [Google Scholar]
- 38.(a) Chung LW, Wu Y-O, Trost BM, Ball ZT. J Am Chem Soc. 2003;125:11578–11582. doi: 10.1021/ja034833b. [DOI] [PubMed] [Google Scholar]; (b) Song LJ, Wang T, Zhang X, Chung LW, Wu YD. ACS Catal. 2017;7:1361–1368. [Google Scholar]; (c) Rosca DA, Radkowski K, Wolf LM, Wagh M, Goddard R, Thiel W, Fürstner A. J Am Chem Soc. 2017;139:2443–2455. doi: 10.1021/jacs.6b12517. [DOI] [PubMed] [Google Scholar]
- 39.Jang WJ, Lee WL, Moon JH, Lee JY, Yun J. Org Lett. 2016;18:1390–1393. doi: 10.1021/acs.orglett.6b00325. [DOI] [PubMed] [Google Scholar]
- 40.For a recent mechanistic investigation regarding hydroboration of terminal alkynes promoted by Co-based complexes, see:; Obligacion JV, Neely JM, Yazdani AN, Pappas I, Chirik PJ. J Am Chem Soc. 2015;137:5855–5858. doi: 10.1021/jacs.5b00936. [DOI] [PubMed] [Google Scholar]
-
41.Preliminary calculations imply that isomerization of a Cu–alkenyl intermediate, as proposed previously (ref 39; see Scheme below) is less likely. With MN15/def2-TZVPPthf(SMD) the barrier is estimated to be approximately 36–38 kcal/mol (see the Supporting Information for details). The following scheme is illustrative of how isomerization has been proposed to take place via zwitterionic metal carbene complex.

- 42.For racemization of enantiomerically enriched allenes by organometallic complexes, see:Cu-based complexes:; (a) Claesson A, Olsson L-I. J Chem Soc, Chem Comm. 1979:524–525. [Google Scholar]; Pd-based complexes:; (b) Horváth A, Bäckvall J-E. Chem Commun. 2004:964–965. doi: 10.1039/b316482a. [DOI] [PubMed] [Google Scholar]; Au-based complexes:; (c) Li H, Harris RJ, Nakufuku K, Widenhoefer RA. Organometallics. 2016;35:2242–2248. [Google Scholar]
- 43.For cases where facile racemization has been utilized in the stereoconvergent transformations with racemic allenes, see:; (a) Zhang Z, Bender CF, Widenhoefer RA. J Am Chem Soc. 2007;129:14148–14149. doi: 10.1021/ja0760731. [DOI] [PubMed] [Google Scholar]; (b) Wang YM, Kuzniewski CN, Rauniyar V, Hoong C, Toste FD. J Am Chem Soc. 2011;133:12972–12975. doi: 10.1021/ja205068j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Butler KL, Tragni M, Widenhoefer RA. Angew Chem, Int Ed. 2012;51:5175–5178. doi: 10.1002/anie.201201584. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Khrakovsky DA, Tao C, Johnson MW, Thornbury RT, Shevick SL, Toste FD. Angew Chem, Int Ed. 2016;55:6079–6083. doi: 10.1002/anie.201601550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liakos DG, Neese F. J Chem Theory Comput. 2015;11:4054–4063. doi: 10.1021/acs.jctc.5b00359. [DOI] [PubMed] [Google Scholar]
-
45.DLPNO-CCSD(T)/def2-TZVPPthf(SMD) calculations through applying the NormalPNO setting have been performed for a selected set of geometries, which seem to support the MN15 values (see diagram below). Other investigated density functionals (M06, M06L, ωB97XD and PBE0–D3BJ) have been found to exhibit larger errors (see the Supporting Information for further details). These predictions are valid only if the M06L/def2-SVP optimized structures are a valid representation of the geometry in solution. (Abbreviations: MSD, mean signed deviation; MUD, mean unsigned deviation; energy values are given in kcal/mol.)

- 46.Yu HS, He X, Li SL, Truhlar DG. Chem Sci. 2016;7:5032–5051. doi: 10.1039/c6sc00705h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barriers of comparable magnitude were calculated with M06, M06L, ωB97XD and PBE0-D3BJ functionals, but not when those that do not account for dispersion were used (i.e., PBE0). For example, PBE0 predicted a value of 16.0 kcal/mol for ts(rac) with tsα having an energy of 27.7 kcal/mol. Dispersion effects, therefore, seem to be crucial when modeling unimolecular processes that compete with bimolecular events. Moreover, we find that only MN15 predicts tsα to be lower in energy than tsγ (Figure 2), a fact that is also supported by the DLPNO-CCSD(T) values. In contrast, the transition states are of similar energy with the other investigated density functionals. See the Supporting Information for details.
- 48.Even the most accurate density functionals available show mean absolute deviations >2.0 kcal/mol in reasonably large benchmark studies (for example, see: ref 46). Nonetheless, the MN15 energies are significantly more accurate compared to the values obtained with PBE0, which predict tsγ (27.3 kcal/mol) at significantly higher energy compared to ts(rac) (14.6 kcal/mol).
- 49.Hoffman RW. Chem Rev. 1989;89:1841–1860. [Google Scholar]
- 50.See the Supporting Information for details.
- 51.Computational uncertainties preclude a more definitive answer. However, we did not identify obvious structural reasons that would support high er if trapping with HB(pin) was stereochemistry-determining.
- 52.For a recent review on Morita-Baylis-Hillman reactions, see:; Pellissier H. Tetrahedron. 2017;73:2831–2861. [Google Scholar]
- 53.For a summary of related observations (phosphine conjugate addition/elimination), see:; Yu S, Ma S. Angew Chem, Int Ed. 2012;51:3074–3112. doi: 10.1002/anie.201101460. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.