

Abstract
Background
18p deletion syndrome is a rare disorder caused by partial or full monosomy of the short arm of chromosome 18. Clinical symptoms caused by 18p hemizygosity include cognitive impairment, mild facial dysmorphism, strabismus and ptosis. Amongst other genes, SMCHD1 is hemizygous in most patients with 18p deletions. Digenic inheritance of a SMCHD1 mutation and a moderately sized D4Z4 repeat on a facioscapulohumeral muscular dystrophy (FSHD) permissive genetic background of chromosome 4 can cause FSHD type 2 (FSHD2).
Objectives
Since 12% of Caucasian individuals harbor moderately sized D4Z4 repeats on a FSHD permissive background we tested if people with 18p deletions are at risk of developing FSHD.
Methods
To test our hypothesis we studied different cellular systems originating from individuals with 18p deletions not presenting FSHD2 phenotype for transcriptional and epigenetic characteristics of FSHD at D4Z4. Furthermore, individuals with an idiopathic muscle phenotype and an 18p deletion were subjected to neurological examination.
Results
Primary fibroblasts hemizygous for SMCHD1 have a D4Z4 chromatin structure comparable with FSHD2 concomitant with DUX4 expression after transdifferentiation into myocytes. Neurological examination of 18p deletion individuals from 2 independent families with a moderately sized D4Z4 repeat identified muscle features compatible with FSHD.
Conclusions
18p deletions leading to haploinsufficiency of SMCHD1, together with a moderately sized FSHD permissive D4Z4 allele, can associate with symptoms and molecular features of FSHD. We propose that 18p deletion patients should be characterized for their D4Z4 repeat size and haplotype and monitored for clinical features of FSHD.
Keywords: 18p deletion, FSHD, SMCHD1, DUX4
Introduction
Facioscapulohumeral dystrophy (FSHD [OMIM:158900 and 158901]) is the third most common muscular dystrophy, most often inherited in an autosomal dominant fashion. It has an estimated prevalence of 1:8,300–22,000 depending on geographical location[1]. Clinical symptoms of FSHD include facial weakness, often noted by inability to whistle, shoulder girdle weakness recognized by difficulty in raising arms above shoulder level, and upper extremity muscle weakness. Muscle weakness can spread to the lower extremities with disease progression. In 20% of cases, disease progression results in wheelchair dependency. Beside muscle weakness, hearing loss and vascular retinopathy can be present; most often in patients with early onset FSHD. Presentation of initial clinical symptoms can vary from birth to middle age, but generally it starts in the second decade of life. Manifestation of the disease is highly variable between and within families with 12–30% of familial cases with a genetic diagnosis remaining asymptomatic. This high variability in disease presentation suggests a role for other genetic disease modifiers.
The molecular mechanism of FSHD has been largely elucidated and identified a key role for DUX4. DUX4 is a member of the double homeodomain protein family of transcription factors and is normally expressed in luminal cells of the testis[2, 3], in late-differentiating keratinocytes[4] and in thymus[3]. DUX4 was recently identified as key transcriptional regulator in early stage embryos where it initiates zygotic genome activation[5–7]. Inappropriate expression in skeletal muscle leads to an early embryonic transcriptional program, eventually resulting in muscle degeneration and the diagnosis of FSHD[8, 9]. A copy of the DUX4 gene is located within each unit of the D4Z4 macrosatellite repeat at 4q35. This D4Z4 repeat is polymorphic forming arrays of 8–100 units which are in a repressed chromatin state in skeletal muscle of healthy individuals. When the D4Z4 repeat is generally between 1–10 units the chromatin can become relaxed. When this occurs on a genetic background of chromosome 4 that contains a non-canonical DUX4 polyadenylation signal (PAS) distal to the D4Z4 repeat a stable DUX4 transcript is produced in muscle cells[10]. Population studies have shown that the polymorphic DUX4 PAS is present on approximately half of chromosomes 4 in the Caucasian and Asian population[11].
Patients diagnosed with FSHD are currently classified into 2 groups based on cis (FSHD1) or trans (FSHD2) genetic variants leading to D4Z4 chromatin relaxation. More than 95% of FSHD patients have a cis mechanism of disease; contraction of the D4Z4 repeat to a size of 1–10 units on a FSHD permissive (i.e. DUX4 PAS containing) chromosomal background, usually 4q35 (4qA; FSHD1). Less than 5% of patients have disease from a trans mechanism; a moderately sized (8–20U in most cases)[12] D4Z4 repeat on a FSHD permissive chromosome 4qA, combined with a heterozygous mutation in genes regulating the chromatin structure at D4Z4 in somatic cells (FSHD2). At least 85% of FSHD2 patients have a mutation in the Structural Maintenance of Chromosomes Flexible Hinge Domain Containing 1 (SMCHD1)[2] gene located on the short arm of chromosome 18 while in two FSHD families mutations in the DNA Methyltransferase 3 Beta (DNMT3B) gene[13] on chromosome 20 have been identified. While in FSHD1 chromatin relaxation is restricted to the shortened D4Z4 repeat, in FSHD2 the epigenetic changes are present on all genomic regions regulated by SMCHD1 or DNMT3B, including both D4Z4 repeats at 4q35 and the highly homologous D4Z4 like repeats at 10q26.
The first epigenetic finding in FSHD was a significant reduction of D4Z4 DNA methylation in patients[14]. Changes in histone modifications were also detected in FSHD samples compared to controls. Somatic cells of controls show the presence of the repressive H3K9me3 and H3K27me3 histone modifications at D4Z4, but unusual for heterochromatin, H3K4me2, a permissive histone mark, is also present at low levels[15]. Patient primary myoblasts, myotubes and fibroblasts[15, 16] have reduced H3K9me3 levels, and a significant increase of H3K27me3 was reported in FSHD2, but not FSHD1 [4]. Furthermore, SMCHD1 mutation carriers have significantly lower amounts of SMCHD1 at D4Z4 than controls.
The mutations in SMCHD1 that result in FSHD2 are distributed over the entire locus. Since about 30% of FSHD2 patients carry a nonsense mutation in SMCHD1, these likely lead to degradation of mutant mRNA by nonsense mediated decay (NMD), resulting in the observed decreased SMCHD1 transcript levels[14]. We therefore hypothesized that conditions leading to SMCHD1 haploinsufficiency are a risk factor for FSHD. We previously described 2 families with a monoallelic 1,2Mb interstitial deletion of 18p, including SMCHD1 and 4 neighboring genes with a classical FSHD phenotype without additional clinical symptoms[12]. Deletions of 18p occur in 1:50,000 individuals and lead to development of 18p deletion (18p-) syndrome. The main clinical features of 18p- syndrome include mental impairment characterized by borderline to low normal IQ, motor and speech delay, global hypotonia, abnormal brain MRI findings, autoimmune disorders and cardiac abnormalities. Hypotonia may cause weakness in the mouth muscles, but there is generally no muscle weakness, unlike the weakness observed in FSHD. Systematic collection and characterization of patients showed that deletions of 18p can vary in size: approximately 50% of patients have a breakpoint in the centromeric region thereby creating hemizygosity of the entire short arm of one of their chromosomes 18, encompassing 66 genes. The remainder of the patients have terminal deletions with breakpoints all along the chromosome arm, with microdeletions rarely described. Recent studies focus on identifying correlations between phenotype and hemizygosity for individual genes as a result of an 18p deletion[17]. Since a heterozygous SMCHD1 deletion together with 4 neighboring genes is associated with FSHD, but not with an 18p- syndrome phenotype[12] we aimed to explore if 18p- patients, missing one copy of SMCHD1 and several to dozens of other genes have clinical or cellular signs of FSHD. Here we describe 2 families with a complex clinical picture with features of 18p- syndrome and FSHD and combine this with molecular studies of the D4Z4 repeat in cell lines of individuals with FSHD or 18p- syndrome.
Materials and Methods
Clinical studies
Samples were obtained with informed consent and the study was approved by the appropriate Institutional Review Boards.
Cell lines, culturing conditions of human primary fibroblast, myoblast and LCLs
Human primary myoblast cell lines were isolated from muscle biopsy of patients Rf1731.103 and Rf1731.201 at University Nice, France. Primary myoblasts were cultured and fused for 72 hours as described previously[4]. Human primary fibroblast cell lines were received from the University of Rochester bio repository (http://www.urmc.rochester.edu/fields-center/), from the University of Texas Health Science Center at San Antonio, or were derived from a lower extremity skin biopsy at the Leiden University Medical Center. Primary fibroblasts were cultured according to a published protocol[16]. LCL cell lines were obtained from the University of Texas Health Science Center at San Antonio and from the Leiden University Medical Center and were cultured according to published protocol[18]. The list of cell lines included in the study, origin, D4Z4 repeat size, genotype and experimental use are listed in Supplemental table 1.
Transdifferentiation of human primary fibroblasts into myogenic cell cultures
Transdifferentiation of fibroblasts was achieved by ectopic expression of MyoD, or GFP as control, as described previously[13]. Every transdifferentiation experiment was performed twice independently and figures show the average of these 2 experiments.
RNA isolation, cDNA synthesis and qRT-PCR
Samples originating from different cell cultures used in the study were harvested in QIAzol lysis reagent (#79306 Qiagen N.V., Venlo, The Netherlands) for RNA isolation. RNA isolation, cDNA synthesis and qRT-PCR reactions were performed as described previously[4].
Chromatin Immunoprecipitation
Histone ChIP-qPCR experiments were carried out as described before[16] using 3μg chromatin per reaction and antibodies against H3 (ab1791, 2μl/reaction, Abcam, Cambridge, UK), H3K9me3 (39161, 5 μl/reaction, Active Motif, Carlsbad, USA), H3K27me3 (17-622, 5 μl/reaction, Merck-Millipore, Amsterdam Zuid-Oost, The Netherlands), H3K4me2 (39141, 5 μl/reaction Active Motif, Carlsbad, USA) and total IgG (5 μl/reaction, Merck-Millipore, Amsterdam Zuid-Oost, The Netherlands). SMCHD1 ChIPs were performed as described before[2] using 60 μg chromatin per reaction and antibody against SMCHD1 (ab31865, 5 μg/rxn, Abcam, Cambridge, UK). qPCR was performed as described previously[4]. Every ChIP-qPCR experiment was performed 2 independent times and figures show the average values.
Western blots
Patient primary myoblasts and primary fibroblasts were lysed in Radio-Immunoprecipitation Assay (RIPA) Buffer (0.1% SDS, 1% NP40, 0.5% sodiumdeoxycholine, 150mM NaCl, 5mM EDTA and 1× Complete protease inhibitor cocktail (Roche, Basel, Switzerland) in 20 mM Tris pH 7.4. The lysate was sonicated using a Bioruptor Pico (Diagenode, Liege, Belgium) and centrifuged for 10 minutes at 13.500 RPM to pellet the cell debris. Protein concentration was determined using Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA) and equal protein amounts were boiled in 1× Laemmli Sample Buffer (2% SDS, 10% glycerol, 2% β-mercaptoethanol and 0.01% bromophenol blue in 60 mM Tris pH6.8). Samples were loaded on Criterion TGX 4–20% gels (Bio-Rad, Hercules, CA) and transferred to an Immobilon-FL PVDF membrane (EMD Millipore, Billeria, MA). The membrane was subsequently blocked in 4% skim milk in PBS and probed for SMCHD1 (ab176731, Abcam, Cambridge, UK), α-Tubulin (T6199, Sigma Aldrich, St. Louis, MO) or HSP90 (#4874, Cell Signaling Technology, Danvers, MA) in Immunobooster (Takara Bio Inc. Kusatsu, Japan). Membranes were incubated with donkey-α-rabbit IRDye-800 and donkey-α-goat IRDye-680 (Li-cor, Lincoln, NE) followed by visualization of immunocomplexes using an Odyssey Infrared Imaging System (Li-cor, Lincoln, NE). Protein quantification was performed using the accompanying software package.
Immunofluorescence detection of DUX4 and high-throughput microscopy analysis
Cells were fixed with 2% paraformaldehyde and permeabilized with 1% Triton in PBS. Primary antibody against DUX4 (Abcam ab124699) was diluted 1:2000 in PBS and primary antibody detecting MYH1E (MF20, Hybridoma Bank, Iowa University) was diluted 1:1000 in PBS. Secondary antibodies Alexa 488 conjugated anti-rabbit (A21206, Thermo Fisher Scientific, Waltham, MA, USA) and Alexa 594 conjugated anti-mouse (A21203, Thermo Fisher Scientific, Waltham, MA, USA) were diluted 1:500 in PBS. Visualization of nuclei was achieved by incubation with DAPI (4′,6-Diamidine-2′-phenylindole dihydrochloride, Sigma-Aldrich, 268298) diluted 1:1000 in PBS. Transdifferentiated fibroblasts (Figure 5C) were imaged with Thermo Cellomics Arrayscan@ VTI HCS Reader and 500 images per cell line were taken at 20× magnification. Number of nuclei in myotubes were determined by analyzing images with a custom made colocalization program in HCS studio software. Number of DUX4 positive nuclei was determined by manual screening of all images. Primary myoblast cultures shown in Figure 2E were imaged with Nicon Eclipse Ti microscope equipped with A1 confocal detector at 20× magnification.
Figure 5.
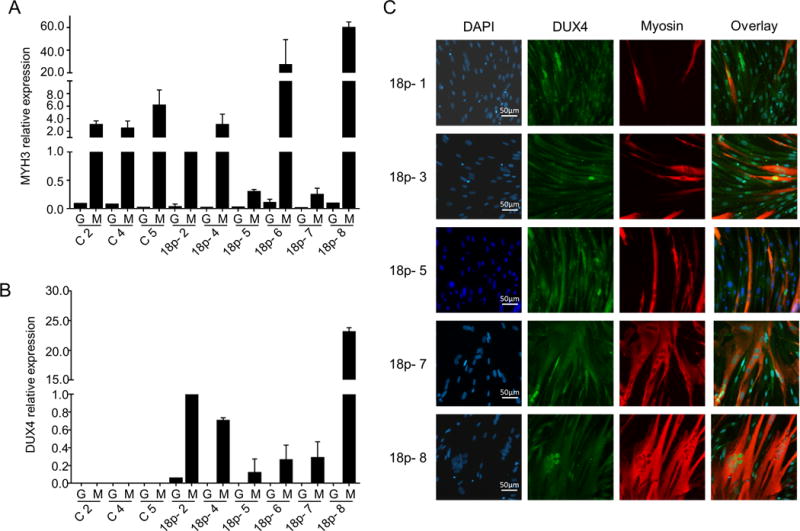
18p- fibroblasts express DUX4 after myogenic transdifferentiation. (A and B) Relative expression of MYH3 and DUX4 in MyoD (M) or GFP (G) transduced control (C) and 18p- primary fibroblasts (18p-). Expression values were normalized to GUSB and value of 18p-2 was set to one. Bar diagrams show the mean and SD of 2 independent experiments. (C) Representative images of DUX4 protein detection by immunofluorescence staining at 20× magnification. Cells were stained with DAPI (nuclei; blue), DUX4 (green), and myogenic differentiation marker MYH1E (red) High content screening was performed by CellomicsArrayscan@VTI HCS instrument at 20× magnification and representative images are shown.
Figure 2. Primary myoblasts originating from 18p- individuals presenting with a combined 18p-and FSHD phenotype express DUX4.
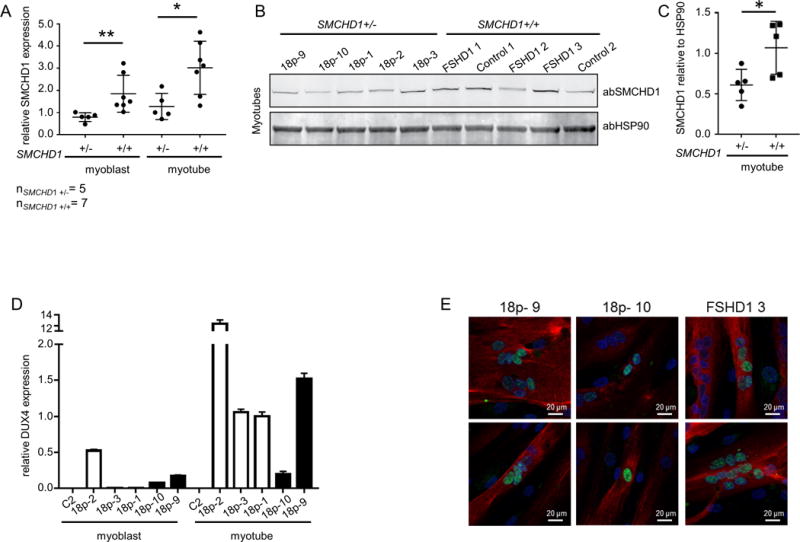
(A) Relative expression of SMCHD1 normalized to GUSB in control and 18p- myoblasts and myotubes. Values of 18p- 3 were set to 1. (B) Western blot analysis of SMCHD1 and HSP90 in myotubes having one or two copies of SMCHD1. (C) Quantification of western blot signals by normalizing SMCHD1 signal intensities to HSP90 signal intensities. P value was calculated by unpaired t-test followed by Mann-Whitney test. * represents p<0.05 and ** represents p<0.01. (D) Expression levels of DUX4 normalized to GUSB in undifferentiated and differentiated primary myoblast cultures originating from control, from 3 patients with FSHD phenotype and a microdeletion on chromosome 18p without presenting any 18p- phenotype (open bars) and from 18p- 9 and18p- 10 (solid black bars). (E) DUX4 (green signal) immunofluorescence analysis in differentiated myotubes with myosin (red signal) as differentiation marker in primary myotubes originating from 18p- 9, 18p- 10 and FSHD1 3. High content screening was performed using a Nikon confocal microscope at 20× magnification and 2 representative images are shown for every cell culture.
Statistical analysis
All statistical analysis was done using GraphPad Prism 6 software and the statistical method applied in different experiments is described in the results section and figure legends. Scatterplots and bar diagrams show the mean and standard deviation of the mean.
Results
18p- individuals can show clinical signs of FSHD
Clinical diagnosis of individuals with contiguous gene syndromes can be challenging because of their complex phenotype and are therefore more often diagnosed through genetic analysis. Identification of a FSHD phenotype in individuals with SMCHD1 monosomy as a result of an 18p deletion combined with a FSHD permissive moderately sized D4Z4 repeat is challenging because of the wide spectrum of symptoms associated with 18p- syndrome and the well documented inter- and intra-familial clinical variability in FSHD. From our patient cohort we identified 3 individuals from 2 families having an interstitial deletion of 18p and clinical signs of FSHD. These individuals were subjected to detailed clinical examination. Array CGH was performed to define the 18p deletion. The FSHD diagnosis was confirmed based on the identification of a FSHD permissive 4qA haplotype, D4Z4 repeat sizing and by DNA methylation analysis[14].
Family 1
The index case (Rf1731.102, 18p-9, Figure 1A, B) was first examined at age 26 when she started having difficulty in lifting her arms overhead. Physical examination showed classical features of FSHD such as asymmetrical facial and shoulder girdle weakness with scapular winging, mild paravertebral and abdominal muscle involvement, and asymmetrical muscle wasting of the anterior compartment of the leg. Total CPK blood levels were increased up to 245 UI/L (normal range < 200 UI/L). Cardiac screening did not show major abnormalities. At age 50 she was wheelchair dependent with a clinical severity score (CSS)[19] of 8. Besides FSHD-like symptoms mild intellectual disability (IQ 65), short statue, ocular ptosis and strabismus were also observed. She also had micrognathia, low-set ears, hyperlordosis and bilateral pes cavus. The MRI of the brain showed white matter abnormalities and the muscle biopsy examination revealed non-specific dystrophic changes consistent with FSHD. D4Z4 repeat sizing identified one D4Z4 repeat of 12 units and one of 13 units, each on a FSHD permissive 4qA chromosome. Based on D4Z4 DNA methylation testing in genomic DNA from peripheral blood mononuclear cells (PBMCs) a delta1 value of -24% was established which is in the range of FSHD2 methylation values (<-21%) (Figure 1A). Array Comparative Genomic Hybridization (aCGH) analysis showed a 1.2 Mb deletion (arr[hg19] 18p11.32p11.31(1,852,498–3,039,186)x1) affecting 5 protein coding genes including SMCHD1 (METTL4, NDC80, SMCHD1, EMILIN2, LPIN2). Furthermore, an interstitial duplication of 151 kb proximal to the deletion was detected (arr[hg19] 18p11.31(3,553,032–3,703,842)×3) affecting exons 5, 6 and 7 of DLGAP1 and the uncharacterized antisense transcripts DLGAP1-AS1 and DLGAP1-AS2 (BC094703) (Supplemental figure 1A). This figure also shows the 18p deletion in the previously described FSHD2 families[12].
Figure 1. Combined 18p- and FSHD phenotype in two independent families.
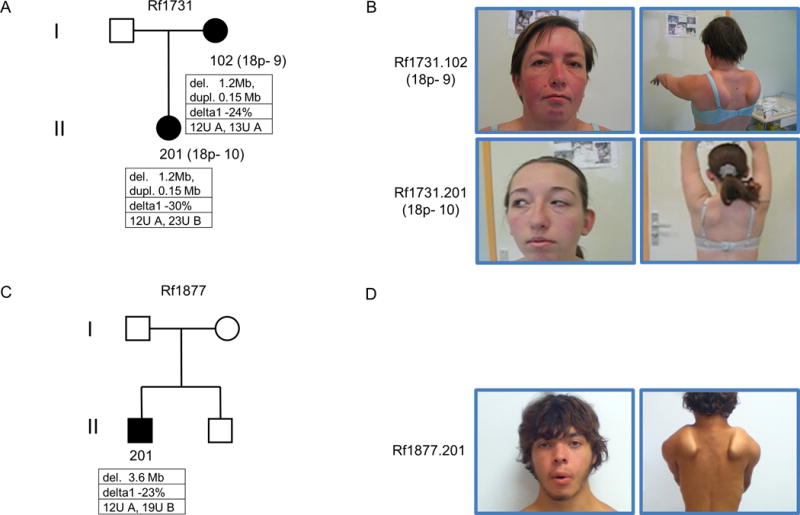
Pedigrees of families Rf1731 (A) and Rf1877 (C). Individuals with a phenotypic combination of 18p- syndrome and FSHD are marked with filled symbols. Below each individual, the size of 18p deletion or duplication is shown, followed by the delta1 values reflecting DNA methylation levels at D4Z4 and the number of D4Z4 units on both chromosomes 4 as well as chromosomal background (A: 4qA FSHD permissive; B: 4qB non-permissive) are depicted. Images of Rf1731.102 and Rf1731.201 (B) and Rf1877.201 (D) highlight facial characteristics compatible with 18p- syndrome, including ocular ptosis, strabismus, low-set ears. Right images show the presence of bilateral winging scapula which are characteristic of FSHD and is more pronounced on Rf1731.102 and Rf1877.201.
The daughter of the index case (Rf1731.201, 18p-10, Figure 1A, B) was examined first at the age of 16 at which age she did not present any sign of symptoms. She started to display signs at the age of 24 with mild scapular and abdominal weakness, as well as asymmetrical ocular ptosis and strabismus. No cardiac anomalies were detected by transthoracic echocardiography and by a 24-hour Holter monitoring, as well as by cardiorespiratory physical examination. Pulmonary function test indicated a moderate restrictive pattern. Physical examination also showed short stature, round fixed face, transverse smile, micrognathia, low-set ears with detached pinnae and short fingers. Intellectual disability was not evident but not formally measured. Muscle biopsy performed in vastus lateralis muscle showed mild non-specific dystrophic changes.
D4Z4 repeat sizing showed that she inherited the FSHD permissive D4Z4 repeat of 12 units from her mother and a 23 units long D4Z4 repeat on a 4qB non-permissive allele from her father. DNA methylation analysis was performed in PBMCs revealing a delta1 methylation value of −30% which is in the FSHD2 range (Figure 1A). Array CGH analysis showed the presence of the same heterozygous 1.2 Mb deletion and 151 Kb duplication on 18p as observed in the mother.
Based on the muscle phenotype, the presence of 12 and 13 units long D4Z4 repeats on permissive haplotypes, SMCHD1 haploinsufficiency and D4Z4 hypomethylation, the diagnosis of FSHD2 was confirmed in both individuals.
Family 2
The proband (Rf1877.201, Figure 1C, D) presented at age 13 with elevated levels of total blood CPK (up to 2727 UI/L), which dropped to slightly elevated levels at age 17 (262UI/L). Cardiac screening did not show abnormalities. He progressively developed difficulty in lifting his arms overhead, orbicularis oris and oculi muscle weakness with fixed face and transverse smile, moderate scapular and humeral muscle weakness with scapular winging and mild quadriceps femoris muscle weakness. Physical examination also revealed a short-broad neck, high-arched palate, hyperlordosis, as well as moderate calf hypertrophy and muscle wasting in pectoralis major and quadriceps femoris muscles. Muscle biopsy examination did not show any pathological changes, which is not uncommon for FSHD given the often focal nature of the disease. D4Z4 repeat sizing detected a repeat of 12 units on a FSHD permissive 4qA allele, which excluded FSHD1. DNA methylation analysis resulted in a delta 1 value of -23% which is in the FSHD2 range. Based on the presence of moderately sized permissive D4Z4 repeat at a permissive 4qA allele and low D4Z4 DNA methylation FSHD2 was diagnosed in this individual. Array CGH analysis showed the presence of a 3.7 Mb terminal deletion on 18p (arr[hg19] 18p11.32p11.31(136,226–3,709,404)×1) that besides SMCHD1 includes 19 additional protein coding genes. This 18p deletion was not detected in other members of the family.
18p hemizygosity leads to reduced SMCHD1 RNA and protein levels associated with DUX4 expression in muscle cells
To determine whether SMCHD1 hemizygosity leads to decreased SMCHD1 expression, RNA and protein from proliferating and differentiated muscle cell cultures was analyzed by qRT-PCR and western blotting, respectively. We assayed myotube cultures from individuals with 18p deletions who had been clinically diagnosed with FSHD2 but not with the clinical features of 18p- syndrome[12] and cultures from the 2 individuals in family Rf1731 presenting with features of 18p- syndrome and FSHD. SMCHD1 transcript levels were significantly reduced in 18p-myoblast (n=5, unpaired t-test test p<0.01) and myotube (n=5, unpaired t-test test p<0.05) cultures compared to control myoblast (n=7) and myotube (n=7) cultures (Figure 2A). Semiquantitative western blot analysis using HSP90 as loading control showed that control myotube cultures express significantly higher levels (unpaired t test p<0.05) of SMCHD1 than SMCHD1 hemizygous samples (Figure 2B and C).
Next, we asked if the lower SMCHD1 levels result in transcriptional derepression of DUX4 in the 18p deletion samples, as was shown in FSHD2 patient samples with SMCHD1 mutations[2, 4]. We detected DUX4 and DUX4 target gene expression by qRT-PCR in myoblast and myotube cultures of Rf1731.102 and Rf1731.201 (Figure 2D, Supplemental figure 2B, C) while myogenic differentiation was monitored by MYOG expression levels (Supplemental figure 2A). Although DUX4 target gene expression indicates that 18p deletion myotube cultures express DUX4 protein, we aimed to detect DUX4 by immunofluorescence microscopy to explore if individual 18p deletion myotubes exhibit the same pattern of sporadic nuclei expressing DUX4 as is found in FSHD. Sporadic expression of DUX4 was found in both myotube cultures, albeit with different frequencies (Figure 2E).
To test if SMCHD1 hemizygosity results in reduced SMCHD1 levels in tissues other than muscle, we studied immortalized lymphoblast cell lines (LCLs) and fibroblasts from control, FSHD and individuals with 18p deletions who had not been diagnosed with FSHD2. SMCHD1 mRNA quantification by qRT-PCR in LCLs and primary fibroblasts with 1 copy or 2 copies of SMCHD1 (samples are listed in Supplemental table 1) showed that SMCHD1 mRNA levels were significantly reduced (fibroblast unpaired t-test test p<0.01, LCLs unpaired t-test p<0.0001) in samples with 1 copy of SMCHD1 (Figure 3A, B). Semiquantitative western blot analysis of primary fibroblasts with one (n=8) or 2 copies (n=8) of SMCHD1 using tubulin as control showed a significant (unpaired t-test p<0.05) reduction in SMCHD1 protein levels in 18p deletion primary fibroblasts compared to controls (Figure 3C, D).
Figure 3.
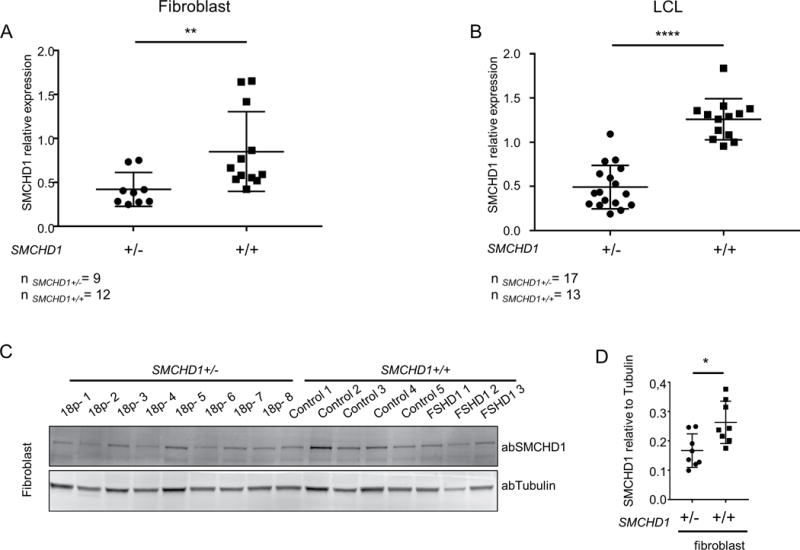
Hemizygosity of SMCHD1 is associated with reduced SMCHD1 transcript and protein levels in different tissues. SMCHD1 transcript levels determined by qRT-PCR and normalized to GUSB in primary fibroblasts (A) and LCLs (B). (C) SMCHD1 protein levels determined by western blot analysis in primary fibroblasts. (D) Intensity of SMCHD1 signal shown on panel C was normalized to Tubulin and statistical analysis of normalized values are shown. Scatter plots show mean and standard error of the mean. Statistical analysis was done by unpaired t-test followed Mann-Whitney test. * represents p<0.05 (D), ** represents p<0.01 (A) and ****represents p<0.0001 (B).
In summary, our results show that all 18p deletion samples have lower SMCHD1 RNA and protein content and that this situation is associated with DUX4 and DUX4 target expression in individuals with a relatively short D4Z4 repeat on a FSHD permissive chromosomal background, similar to FSHD.
D4Z4 chromatin in 18p- cell lines shows similar epigenetic characteristics as in FSHD2
Because of the reduced SMCHD1 levels, we asked if primary fibroblasts from individuals with 18p deletions who have not been diagnosed with FSHD2 also show D4Z4 chromatin changes characteristic of FSHD2. As previously reported in PBMCs from 18p- syndrome individuals[12], we found significantly reduced D4Z4 DNA methylation levels (n=6 control samples and n=8 18p-samples, unpaired t-test p<0.01)) in fibroblasts from these individuals, comparable to what was found in FSHD2 patients (Figure 4A). In concordance with previous observations in FSHD2 individuals, ChIP-qPCR on chromatin isolated from human primary fibroblasts (n=8 one copy and n=8 two copies of SMCHD1) showed a significant (unpaired t-test p<0.05) decrease in SMCHD1 binding at D4Z4 in samples hemizygous for SMCHD1 compared to controls (Figure 4B).
Figure 4. The D4Z4 chromatin landscape is similar in FSHD2 and 18p- syndrome fibroblasts.
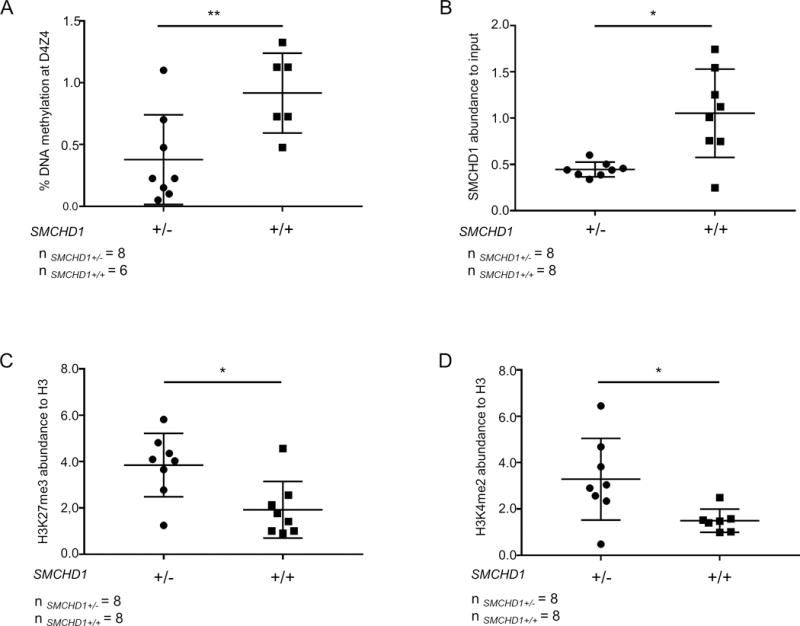
(A) DNA methylation at D4Z4 in control and 18p- fibroblasts (n=6 control and n=8 18p-) is shown as percentage methylation at the FseI site. (B) SMCHD1 levels at D4Z4 measured by ChIP-qPCR in control and 18p- primary fibroblasts. SMCHD1 enrichment values are shown as normalized to input and enrichment values of control 1 sample were set to 1. H3K27me3 (C) and H3K4me2 (D) levels at D4Z4 in the same samples shown in panel B. Histone modification levels are normalized to H3 and enrichment values of control 1 were set to 1. Statistical analysis was done by unpaired t-test followed Mann-Whitney test. * represents P<0.05 (B, C, D), ** represents P<0.01 (A).
Next we investigated if changes in histone modifications are present on D4Z4 in 18p- fibroblasts. Consistent with our previous report[4], ChIP-qPCR with antibodies against H3K27me3, H3K9me3 and H3K4me2 showed that H3K9me3 levels at D4Z4 do not differ (Supplemental figure 3) but that H3K27me3 levels, like in FSHD2, were significantly increased in 18p- samples (unpaired t-test p<0.05, Figure 4C). Furthermore, we detected significantly increased H3K4me2 levels (unpaired t-test p<0.05) at D4Z4 in fibroblasts hemizygous for SMCHD1 compared to controls, as reported in FSHD[16] (Figure 4D).
Altogether our study revealed striking similarities between the D4Z4 chromatin structure in FSHD2 and 18p- fibroblasts suggesting that indeed 18p- patients are at risk of expressing DUX4 in somatic tissue.
DUX4 is expressed in different in vitro systems originating from patients with 18p deletions
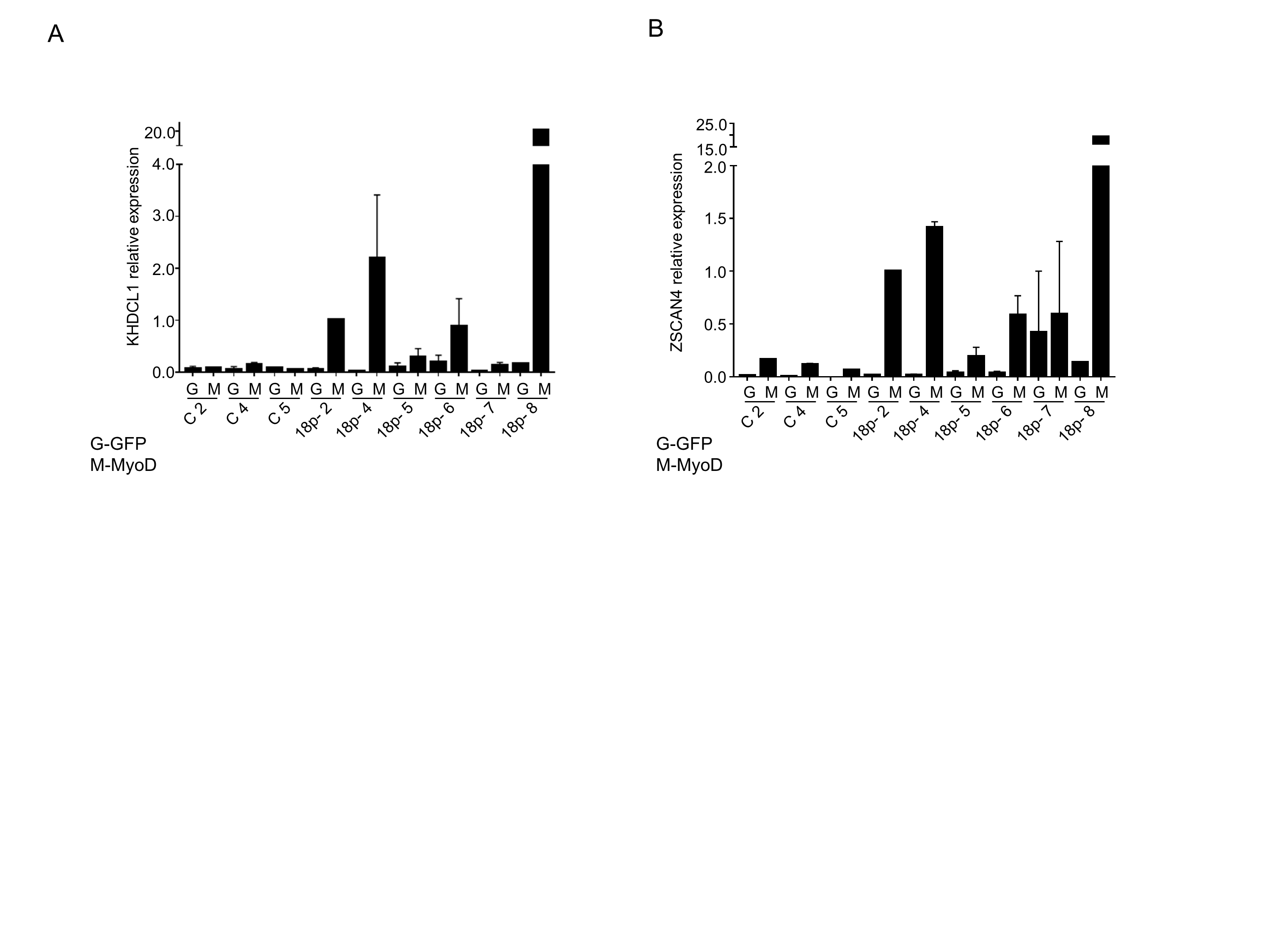
Besides in myogenic cells DUX4 expression has also been reported in FSHD LCLs[18] and in FSHD primary fibroblasts after transdifferentiation in myocytes[13]. To test for DUX4 expression in individuals with 18p- and a permissive 4q35 allele, we transdifferentiated control (n=3) and 18p- (n=6) primary fibroblasts by lentiviral MyoD transduction. Transdifferentiation was monitored by the presence of the late myogenic marker MYH3 (Figure 5A). DUX4 (Figure 5B) and DUX4 targets KHDCL1 and ZSCAN4[20] (Supplemental figure 4A, B) were detected in 18p-samples at similar levels as FSHD1 and FSHD2 fibroblasts after efficient transdifferentiation, while fibroblasts transduced with GFP as control expressed no or negligible levels of DUX4 and its targets.[13]
Because of the variegated nuclear expression of DUX4 in FSHD, we employed high content immunofluorescence microscopy allowing us to screen at least 10,000 nuclei per sample for the presence of DUX4. High content imaging of transdifferentiated control (n=3) and 18p- (n=7) fibroblasts allowed, despite the variability in transdifferentiation, for the detection of DUX4 only in 18p- cell cultures but not controls. We detected DUX4 positive nuclei in 5 out of 7 18p- cell cultures with the number of DUX4 positive nuclei varying between the cell lines (Figure 5C, Supplemental table 2).
Finally, we analyzed a cohort of 18p- (n=17), control (n=13) and FSHD1 (n=10) LCL cell lines (the same cohort as included in the SMCHD1 expression analysis) for DUX4 and DUX4 target gene expression. All samples included in the study were sized for D4Z4 repeat at 4q35 and all cell lines had their shortest D4Z4 repeat on a FSHD permissive DUX4 PAS containing chromosome. Since D4Z4 array length also correlates with DUX4 expression[1] control LCLs were selected with similar D4Z4 repeat sizes at a permissive 4q35 with sizes ranging between 13 and 35 units (Supplemental table 1). We detected significantly higher DUX4 levels (FSHD1; one way ANOVA p<0.0001, 18p-; one-way ANOVA p<0.05) in FSHD1 and 18p- LCLs compared to controls (Figure 6A). The relative expression of three DUX4 target genes was also determined. MBD3L2 and TRIM43 expression was significantly increased in FSHD1 (Figure 6B, C, one way ANOVA, TRIM43 p<0.05, MBD3L2 p<0.001) and 18p- LCLs (one way ANOVA, TRIM43 p<0.001, MBD3L2 p<0.001). ZSCAN4 expression levels did not significantly differ between controls and FSHD1 (Figure 6D) while it was significantly increased in 18p- samples (one way ANOVA, p<0.01).
Figure 6.
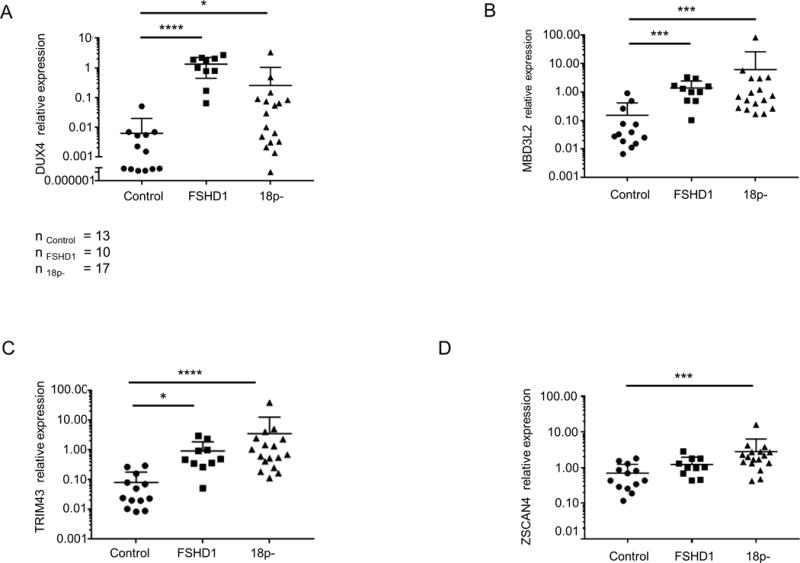
DUX4 and DUX4 target gene expression in control, 18p- and FSHD1 LCLs. Relative expression of DUX4 (A) and DUX4 targets TRIM43 (B), ZSCAN4 (C), and MBD3L2 (D) in a cohort of control, FSHD1 and 18p- LCLs. Scatter plots show expression levels normalized to GUSB and relative expression level of FSHD1–2 was set to 1. Statistical analysis was done by one way ANOVA followed by Kruskal-Wallis test. * represents p<0.05 (A, B), *** represents p<0.01 (C, D) and **** represents p<0.0001 (A, B).
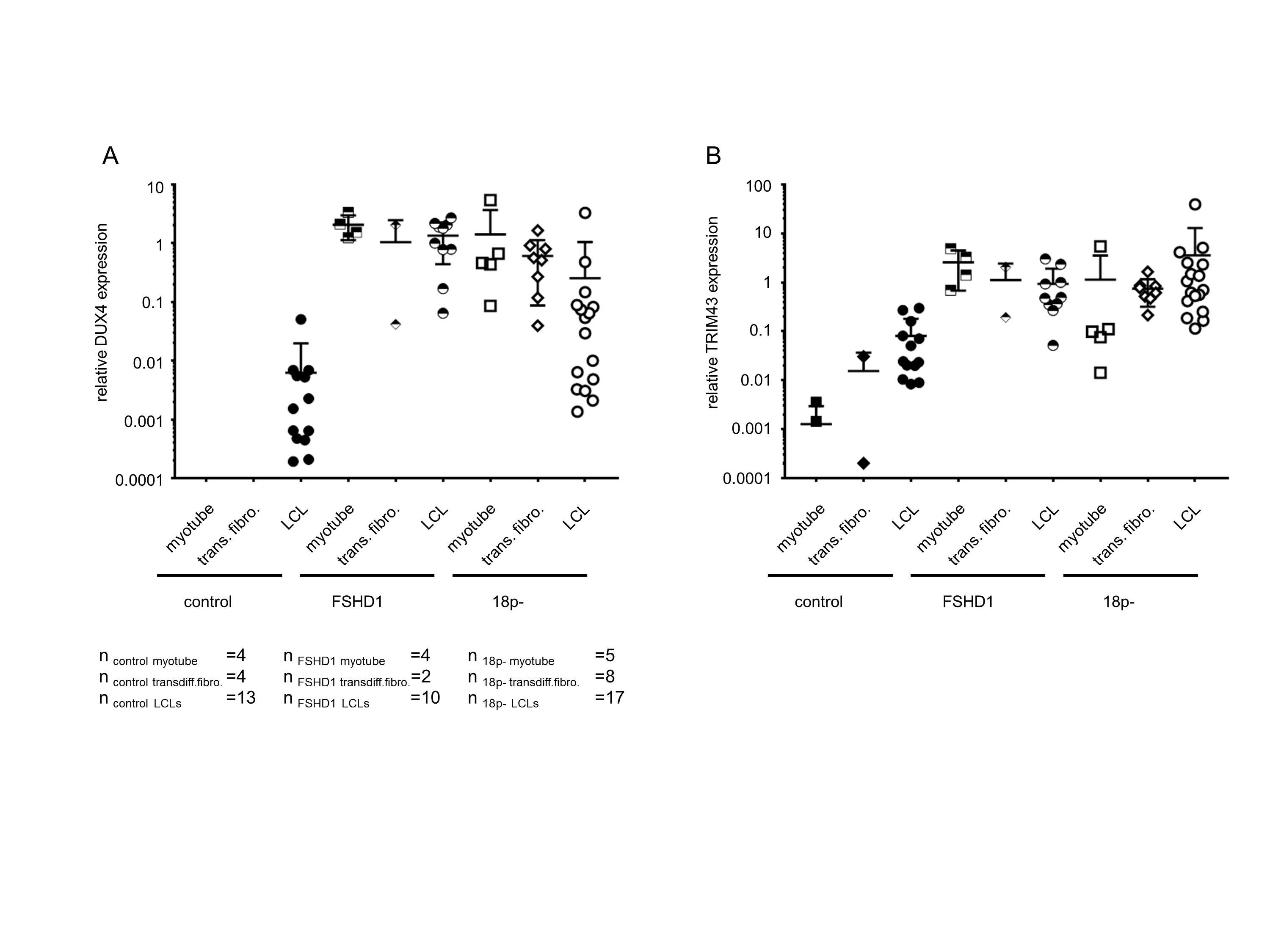
In general, DUX4 and TRIM43 expression levels were similar between myotubes, LCLs and transdifferentiated fibroblasts of FSHD1 and 18p- individuals, although quite some variability was observed in transdifferentiated fibroblasts possibly as a consequence of the transduction efficiency (Supplemental figure 5). Highest DUX4 expression was detected in 18p- LCL samples with the shortest D4Z4 repeat of 13 units (n=2, Supplemental figure 6) and 15 units (n=1). Interestingly DUX4 expression of 18p- samples with repeats of 25 D4Z4 units was still above control levels. Together these results show that different cell systems originating from 18p-individuals can express DUX4 and DUX4 targets similar to FSHD and that the level of DUX4 expression is dependent on the size of the permissive D4Z4 repeat.
Discussion
Based on the occurrence of a moderately sized D4Z4 repeat on a FSHD permissive chromosomal background we estimated that about 12% of 18p- patients are at risk of developing FSHD in the European population[12]. Our current study shows that individuals with 18p deletions, being hemizygous for SMCHD1 and few to 65 additional genes on 18p, can develop symptoms compatible with FSHD and display a molecular signature of FSHD in different cellular systems. We show in all in vitro models tested that SMCHD1 hemizygosity is associated with reduced SMCHD1 transcript and protein levels. At D4Z4, reduced SMCHD1 binding is associated with increased H3K27me3, H3K4me2 levels, and reduced DNA methylation levels, the latter corroborating our previous report of reduced D4Z4 DNA methylation levels in blood samples of people with 18 deletions[12]. The observed D4Z4 DNA hypomethylation, the significant increase in the transcriptionally permissive H3K4me2 histone modification and the increase in the repressive H3K27me3 modification, resembles the epigenetic landscape of D4Z4 in FSHD2, and suggests that the D4Z4 chromatin state is permissive for DUX4 transcription in somatic tissue of individuals with 18p-.
Indeed, we detected DUX4 expression in primary fibroblasts of individuals with variably sized deletions on chromosome 18p including SMCHD1 after transdifferentiation into myocytes. We extended our study to LCLs because a recent publication showed DUX4 and DUX4 target gene expression in FSHD LCLs[18]. We could confirm the presence of DUX4 and some DUX4 targets at equal levels in FSHD LCLs, the absence in control LCLs, and extended this observation by showing DUX4 and DUX4 target gene expression in LCLs of individuals with 18p-. Several studies have reported that the epigenetic structure of the disease locus is similar in different tissues[4, 21, 22], and DUX4 expression has also been reported in non-myogenic tissues of non-affected individuals[3, 23, 24]. However, given the significant transformation that LCLs have undergone, the clinical relevance of DUX4 expression in these cell lines is likely very limited.
Besides SMCHD1 activity, DUX4 expression is also dependent on the size of the D4Z4 repeat: longer D4Z4 repeats are less likely to express DUX4 than shorter repeats. We therefore measured D4Z4 repeat sizes in all samples included in the study and in agreement with earlier observations higher DUX4 expression was detected in 18p- samples with shorter D4Z4 repeats on a permissive chromosome 4qA than in those with longer repeats. Importantly, those 18p-samples with repeats of 13–15 units had the highest expression but 18p- samples harboring repeats of 15–25 D4Z4 units still expressed DUX4 above controls. Our initial estimation, predicting that about 12% individuals with 18p- might be at risk of developing FSHD based on SMCHD1 monosomy and a D4Z4 repeat ≤16 units on a permissive chromosome 4 may, in light of these findings, be an underestimation. When using a threshold of ≤25 units based on the observation of DUX4 expression in LCLs, almost one-third of individuals with 18p- might be at risk of developing FSHD. However, as we rarely find patients with >16 units in the FSHD2 patient population[14] it might suggest that DUX4 expression levels in tissue culture may not necessarily reflect the levels in vivo and be predictive for pathogenicity.
The clinical diagnosis of FSHD in individuals with 18p- may be challenging because of the presence of additional signs and symptoms related to 18p- syndrome. In particular, the presence of extra-ocular muscle involvement and dysmorphic features are confounding and may suggest other diagnosis. In family Rf1731 we identified a deletion on 18p that is almost identical to the deletions reported earlier in two classical FSHD2 families, affecting the same five genes on 18p. While clinically different, the main genetic difference between Rf1731 and the earlier reported FSHD2 families is the duplication of 150kb adjacent sequence in Rf1731 affecting exons 5, 6 and 7 of DLGAP1 and two antisense transcripts DLGAP1-AS1 and DLGAP1-AS2. The database of Genomic Variants lists 3 large duplications (nsv1059696[25], dgv184n21[26] and esv2758710[27], Supplemental figure 1B) that partially overlap with this region. These variants were found in studies performed in the control population and predicts that this duplication represents a variant without clinical significance. Although DLGAP1 is part of the postsynaptic scaffold and has been suggested to associate with obsessive-compulsive disorder[28], its role in the neurological disease aspects of this family remains enigmatic. Considering the clinical variability in FSHD and 18p- syndrome, each representing conditions in which the clinical variability may vary from non- or minimally affected to severe in individuals carrying the same genetic defect, it is perhaps not surprising that almost identical deletions at 18p may result in a pure FSHD phenotype or a combination of clinical features of FSHD and 18p-syndrome. Alternatively, since SNP array analysis cannot determine the genomic location of the 150kb duplication, it is possible that this duplication has integrated somewhere else in the genome and thereby disrupting or altering the expression of unknown loci.
In summary, our study highlights the complex and variable clinical outcome of SMCHD1 haploinsufficiency and supports the conclusion that people with 18pdeletions should routinely be genetically characterized for their D4Z4 repeat size and haplotype and monitored for clinical features of FSHD.
Supplementary Material
Supplemental figure 1. 18p chromosomal deletions and duplication detected by array CGH in families with combined 18p- and FSHD phenotype. (A) Top part of the figure shows ideogram of chromosome 18 and red rectangle specifies genomic region shown at higher magnification below. Participant study numbers are shown to the left of the bar that indicates the intact portion of the chromosome. The 1.2Mb interstitial deletion (represented by white gap) and 150kb duplication (represented by pink bar) detected in individuals of Rf1731 and 3.6Mb telomeric deletion in Rf1877 in UCSC genome browser. For reference, the 18p deletion detected in the families described in reference 12 are also shown. Genes localized in the shown genomic region are shown at the bottom. (B) The duplicated region ([hg19] 18p11.31(3,553,032–3,703,842) affects several exons of DLPAG1 and 2 antisense transcripts as shown in the UCSC genome browser screen shot. Copy number variations deposited in the Genomic Variant Database for the depicted region. Duplications are shown as solid blue bars and deletions are shown as solid red bars.
{kind=link}
Supplemental figure 2. Expression analysis of myogenic cells originating from 18p- individuals. Relative expression of MYOG (A) and DUX4 targets ZSCAN4 (B) and TRIM43 (C) in control and 18p- myoblasts and myotubes (samples are also shown in Figure 2). Expression levels were normalized to GUSB.
{kind=link}
Supplemental figure 3. Relative abundance of H3K9me3 at D4Z4 in control and 18p- primary fibroblasts. Enrichment was calculated relative to H3 and enrichment of control 1 was set to 1.
{kind=link}
Supplemental figure 4. DUX4 target gene quantification in transdifferentiated 18p- and control fibroblasts. Relative expression levels of DUX4 target KHDCL1 (A) and ZSCAN4 (B) in MyoD (M) or GFP (G) transduced primary fibroblasts (presented also in Figure 4A, B). Expression levels were normalized to GUSB with 18p-2 being set to one. Bar diagrams show the mean and SD of 2 independent experiments.
{kind=link}
Supplemental figure 5. Relative expression levels of DUX4 (A) and DUX4 target gene TRIM43 (B) in myotubes, transdifferentiated fibroblasts and LCLs of control, FSHD1 and 18p- samples. The number of samples included in the study is depicted in the figure. Expression levels were normalized to GUSB.
{kind=link}
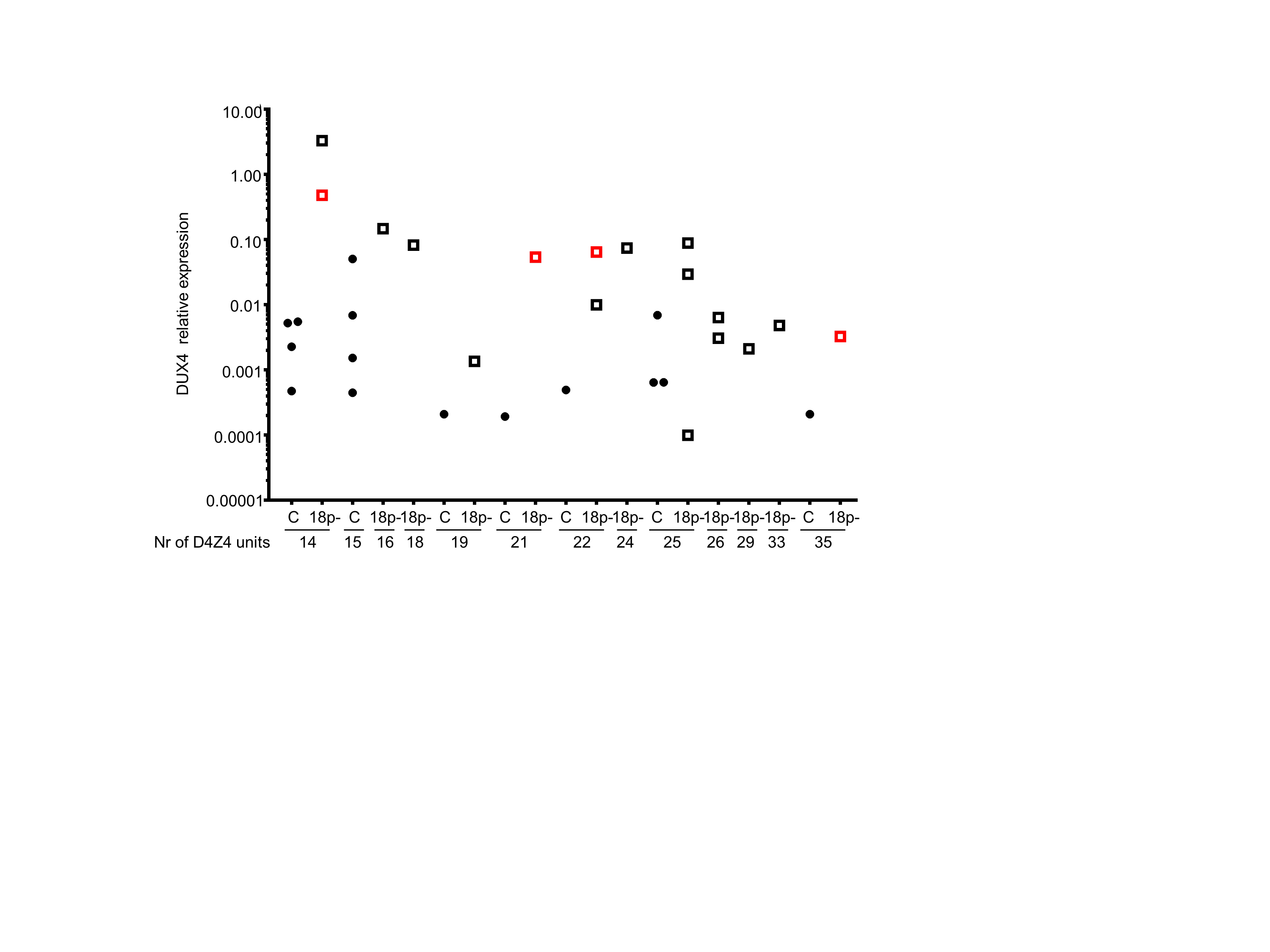
Supplemental figure 6. Relative expression levels of DUX4 (also presented in Figure 5A) in control and 18p- LCLs with different D4Z4 repeat sizes. The repeat size in units (U) of shortest FSHD permissive D4Z4 repeat is depicted below the sample identifier. Control (black dots) and 18p- (open squares) samples are ordered according to their D4Z4 repeat size. Red open squares depict LCL samples originating from people with 18p deletion whose primary fibroblast samples were also included in our study. DUX4 expression values were normalized to GUSB and are shown on a logarithmic scale.
{kind=link}
Supplemental table 1. List of samples included in the study.
Supplemental table 2. Quantification of DUX4 expressing nuclei after immunofluorescence staining of transdifferentiated primary fibroblasts.
Acknowledgments
We thank all patients and family members for their participation in our studies. We also would like to thank Fatiha Amghar-el Bouazzaoui for technical assistance.
Funding
This study was supported with grants from the US National Institutes of Health (NIH) (National Institute of Neurological Disorders and Stroke (NINDS) P01NS069539, and National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) R01AR045203 and R01AR066248, the Prinses Beatrix Spierfonds (W.OP14-01 and W.OR14-04), and Spieren voor Spieren.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributions
JB, SM, conceived the study, JB designed the experiments, analyzed the data, JB, RG, KRS, RJFL, PV, CR and AH performed the experiments. LC, NC, LF, VM, JC, MK, CDS, PH, JDC, RT, SS, SJT contributed with clinical data and patient material. JB, SM, SS and CDS wrote the manuscript.
Conflicts of interest
All authors declare to have no conflicts of interest.
References
- 1.Wang LH, Tawil R. Facioscapulohumeral Dystrophy. Current Neurology and Neuroscience Reports. 2016;16(7):66. doi: 10.1007/s11910-016-0667-0. [DOI] [PubMed] [Google Scholar]
- 2.Lemmers RJ, Tawil R, Petek LM, Balog J, Block GJ, Santen GW, Amell AM, van der Vliet PJ, Almomani R, Straasheijm KR, Krom YD, Klooster R, Sun Y, den Dunnen JT, Helmer Q, Donlin-Smith CM, Padberg GW, van Engelen BG, de Greef JC, Aartsma-Rus AM, Frants RR, de Visser M, Desnuelle C, Sacconi S, Filippova GN, Bakker B, Bamshad MJ, Tapscott SJ, Miller DG, van der Maarel SM. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nature genetics. 2012;44(12):1370–1374. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Das S, Chadwick BP. Influence of Repressive Histone and DNA Methylation upon D4Z4 Transcription in Non-Myogenic Cells. PloS one. 2016;11(7):e0160022. doi: 10.1371/journal.pone.0160022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balog J, Thijssen PE, Shadle S, Straasheijm KR, van der Vliet PJ, Krom YD, van den Boogaard ML, de Jong A, RJ FL, Tawil R, Tapscott SJ, van der Maarel SM. Increased DUX4 expression during muscle differentiation correlates with decreased SMCHD1 protein levels at D4Z4. Epigenetics. 2015;10(12):1133–1142. doi: 10.1080/15592294.2015.1113798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Iaco A, Planet E, Coluccio A, Verp S, Duc J, Trono D. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nature genetics. 2017 doi: 10.1038/ng.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whiddon JL, Langford AT, Wong CJ, Zhong JW, Tapscott SJ. Conservation and innovation in the DUX4-family gene network. Nature genetics. 2017 doi: 10.1038/ng.3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hendrickson PG, Dorais JA, Grow EJ, Whiddon JL, Lim JW, Wike CL, Weaver BD, Pflueger C, Emery BR, Wilcox AL, Nix DA, Peterson CM, Tapscott SJ, Carrell DT, Cairns BR. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nature genetics. 2017 doi: 10.1038/ng.3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kowaljow V, Marcowycz A, Ansseau E, Conde CB, Sauvage S, Matteotti C, Arias C, Corona ED, Nunez NG, Leo O, Wattiez R, Figlewicz D, Laoudj-Chenivesse D, Belayew A, Coppee F, Rosa AL. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscular disorders : NMD. 2007;17(8):611–623. doi: 10.1016/j.nmd.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 9.Bosnakovski D, Lamb S, Simsek T, Xu Z, Belayew A, Perlingeiro R, Kyba M. DUX4c, an FSHD candidate gene, interferes with myogenic regulators and abolishes myoblast differentiation. Experimental neurology. 2008;214(1):87–96. doi: 10.1016/j.expneurol.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 10.Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camano P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, Miller DG, Tapscott SJ, Tawil R, Frants RR, van der Maarel SM. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science (New York, NY) 2010;329(5999):1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemmers RJ, van der Vliet PJ, van der Gaag KJ, Zuniga S, Frants RR, de Knijff P, van der Maarel SM. Worldwide population analysis of the 4q and 10q subtelomeres identifies only four discrete interchromosomal sequence transfers in human evolution. American journal of human genetics. 2010;86(3):364–377. doi: 10.1016/j.ajhg.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemmers RJ, van den Boogaard ML, van der Vliet PJ, Donlin-Smith CM, Nations SP, Ruivenkamp CA, Heard P, Bakker B, Tapscott S, Cody JD, Tawil R, van der Maarel SM. Hemizygosity for SMCHD1 in Facioscapulohumeral Muscular Dystrophy Type 2: Consequences for 18p Deletion Syndrome. Human mutation. 2015;36(7):679–683. doi: 10.1002/humu.22792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van den Boogaard ML, Lemmers RJ, Balog J, Wohlgemuth M, Auranen M, Mitsuhashi S, van der Vliet PJ, Straasheijm KR, van den Akker RF, Kriek M, Laurense-Bik ME, Raz V, van Ostaijen-Ten Dam MM, Hansson KB, van der Kooi EL, Kiuru-Enari S, Udd B, van Tol MJ, Nishino I, Tawil R, Tapscott SJ, van Engelen BG, van der Maarel SM. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. American journal of human genetics. 2016;98(5):1020–1029. doi: 10.1016/j.ajhg.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lemmers RJ, Goeman JJ, van der Vliet PJ, van Nieuwenhuizen MP, Balog J, Vos-Versteeg M, Camano P, Ramos Arroyo MA, Jerico I, Rogers MT, Miller DG, Upadhyaya M, Verschuuren JJ, Lopez de Munain Arregui A, van Engelen BG, Padberg GW, Sacconi S, Tawil R, Tapscott SJ, Bakker B, van der Maarel SM. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Human molecular genetics. 2015;24(3):659–669. doi: 10.1093/hmg/ddu486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeng W, de Greef JC, Chen YY, Chien R, Kong X, Gregson HC, Winokur ST, Pyle A, Robertson KD, Schmiesing JA, Kimonis VE, Balog J, Frants RR, Ball AR, Jr, Lock LF, Donovan PJ, van der Maarel SM, Yokomori K. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD) PLoS genetics. 2009;5(7):e1000559. doi: 10.1371/journal.pgen.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balog J, Thijssen PE, de Greef JC, Shah B, van Engelen BG, Yokomori K, Tapscott SJ, Tawil R, van der Maarel SM. Correlation analysis of clinical parameters with epigenetic modifications in the DUX4 promoter in FSHD. Epigenetics. 2012;7(6):579–584. doi: 10.4161/epi.20001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasi-Zogaj M, Sebold C, Heard P, Carter E, Soileau B, Hill A, Rupert D, Perry B, Atkinson S, O’Donnell L, Gelfond J, Lancaster J, Fox PT, Hale DE, Cody JD. A review of 18p deletions. American journal of medical genetics Part C, Seminars in medical genetics. 2015;169(3):251–264. doi: 10.1002/ajmg.c.31445. [DOI] [PubMed] [Google Scholar]
- 18.Jones TI, Himeda CL, Perez DP, Jones PL. Large family cohorts of lymphoblastoid cells provide a new cellular model for investigating facioscapulohumeral muscular dystrophy. Neuromuscular disorders : NMD. 2017;27(3):221–238. doi: 10.1016/j.nmd.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ricci E, Galluzzi G, Deidda G, Cacurri S, Colantoni L, Merico B, Piazzo N, Servidei S, Vigneti E, Pasceri V, Silvestri G, Mirabella M, Mangiola F, Tonali P, Felicetti L. Progress in the molecular diagnosis of facioscapulohumeral muscular dystrophy and correlation between the number of KpnI repeats at the 4q35 locus and clinical phenotype. Annals of neurology. 1999;45(6):751–757. doi: 10.1002/1531-8249(199906)45:6<751::aid-ana9>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 20.Yao Z, Snider L, Balog J, Lemmers RJ, Van Der Maarel SM, Tawil R, Tapscott SJ. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Human molecular genetics. 2014;23(20):5342–5352. doi: 10.1093/hmg/ddu251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Overveld PG, Lemmers RJ, Sandkuijl LA, Enthoven L, Winokur ST, Bakels F, Padberg GW, van Ommen GJ, Frants RR, van der Maarel SM. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nature genetics. 2003;35(4):315–317. doi: 10.1038/ng1262. [DOI] [PubMed] [Google Scholar]
- 22.Jones TI, Yan C, Sapp PC, McKenna-Yasek D, Kang PB, Quinn C, Salameh JS, King OD, Jones PL. Identifying diagnostic DNA methylation profiles for facioscapulohumeral muscular dystrophy in blood and saliva using bisulfite sequencing. Clinical epigenetics. 2014;6(1):23. doi: 10.1186/1868-7083-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snider L, Geng LN, Lemmers RJ, Kyba M, Ware CB, Nelson AM, Tawil R, Filippova GN, van der Maarel SM, Tapscott SJ, Miller DG. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS genetics. 2010;6(10):e1001181. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gannon OM, Merida de Long L, Saunders NA. DUX4 Is Derepressed in Late-Differentiating Keratinocytes in Conjunction with Loss of H3K9me3 Epigenetic Repression. The Journal of investigative dermatology. 2016;136(6):1299–1302. doi: 10.1016/j.jid.2016.01.027. [DOI] [PubMed] [Google Scholar]
- 25.Coe BP, Witherspoon K, Rosenfeld JA, van Bon BW, Vulto-van Silfhout AT, Bosco P, Friend KL, Baker C, Buono S, Vissers LE, Schuurs-Hoeijmakers JH, Hoischen A, Pfundt R, Krumm N, Carvill GL, Li D, Amaral D, Brown N, Lockhart PJ, Scheffer IE, Alberti A, Shaw M, Pettinato R, Tervo R, de Leeuw N, Reijnders MR, Torchia BS, Peeters H, O’Roak BJ, Fichera M, Hehir-Kwa JY, Shendure J, Mefford HC, Haan E, Gecz J, de Vries BB, Romano C, Eichler EE. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nature genetics. 2014;46(10):1063–1071. doi: 10.1038/ng.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaikh TH, Gai X, Perin JC, Glessner JT, Xie H, Murphy K, O’Hara R, Casalunovo T, Conlin LK, D’Arcy M, Frackelton EC, Geiger EA, Haldeman-Englert C, Imielinski M, Kim CE, Medne L, Annaiah K, Bradfield JP, Dabaghyan E, Eckert A, Onyiah CC, Ostapenko S, Otieno FG, Santa E, Shaner JL, Skraban R, Smith RM, Elia J, Goldmuntz E, Spinner NB, Zackai EH, Chiavacci RM, Grundmeier R, Rappaport EF, Grant SF, White PS, Hakonarson H. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome research. 2009;19(9):1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mattheisen M, Samuels JF, Wang Y, Greenberg BD, Fyer AJ, McCracken JT, Geller DA, Murphy DL, Knowles JA, Grados MA, Riddle MA, Rasmussen SA, McLaughlin NC, Nurmi EL, Askland KD, Qin HD, Cullen BA, Piacentini J, Pauls DL, Bienvenu OJ, Stewart SE, Liang KY, Goes FS, Maher B, Pulver AE, Shugart YY, Valle D, Lange C, Nestadt G. Genome-wide association study in obsessive-compulsive disorder: results from the OCGAS. Molecular psychiatry. 2015;20(3):337–344. doi: 10.1038/mp.2014.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1. 18p chromosomal deletions and duplication detected by array CGH in families with combined 18p- and FSHD phenotype. (A) Top part of the figure shows ideogram of chromosome 18 and red rectangle specifies genomic region shown at higher magnification below. Participant study numbers are shown to the left of the bar that indicates the intact portion of the chromosome. The 1.2Mb interstitial deletion (represented by white gap) and 150kb duplication (represented by pink bar) detected in individuals of Rf1731 and 3.6Mb telomeric deletion in Rf1877 in UCSC genome browser. For reference, the 18p deletion detected in the families described in reference 12 are also shown. Genes localized in the shown genomic region are shown at the bottom. (B) The duplicated region ([hg19] 18p11.31(3,553,032–3,703,842) affects several exons of DLPAG1 and 2 antisense transcripts as shown in the UCSC genome browser screen shot. Copy number variations deposited in the Genomic Variant Database for the depicted region. Duplications are shown as solid blue bars and deletions are shown as solid red bars.
Supplemental figure 2. Expression analysis of myogenic cells originating from 18p- individuals. Relative expression of MYOG (A) and DUX4 targets ZSCAN4 (B) and TRIM43 (C) in control and 18p- myoblasts and myotubes (samples are also shown in Figure 2). Expression levels were normalized to GUSB.
Supplemental figure 3. Relative abundance of H3K9me3 at D4Z4 in control and 18p- primary fibroblasts. Enrichment was calculated relative to H3 and enrichment of control 1 was set to 1.
Supplemental figure 4. DUX4 target gene quantification in transdifferentiated 18p- and control fibroblasts. Relative expression levels of DUX4 target KHDCL1 (A) and ZSCAN4 (B) in MyoD (M) or GFP (G) transduced primary fibroblasts (presented also in Figure 4A, B). Expression levels were normalized to GUSB with 18p-2 being set to one. Bar diagrams show the mean and SD of 2 independent experiments.
Supplemental figure 5. Relative expression levels of DUX4 (A) and DUX4 target gene TRIM43 (B) in myotubes, transdifferentiated fibroblasts and LCLs of control, FSHD1 and 18p- samples. The number of samples included in the study is depicted in the figure. Expression levels were normalized to GUSB.
Supplemental figure 6. Relative expression levels of DUX4 (also presented in Figure 5A) in control and 18p- LCLs with different D4Z4 repeat sizes. The repeat size in units (U) of shortest FSHD permissive D4Z4 repeat is depicted below the sample identifier. Control (black dots) and 18p- (open squares) samples are ordered according to their D4Z4 repeat size. Red open squares depict LCL samples originating from people with 18p deletion whose primary fibroblast samples were also included in our study. DUX4 expression values were normalized to GUSB and are shown on a logarithmic scale.
Supplemental table 1. List of samples included in the study.
Supplemental table 2. Quantification of DUX4 expressing nuclei after immunofluorescence staining of transdifferentiated primary fibroblasts.