

Abstract
Cysteamine dioxygenase (ADO) is a thiol dioxygenase whose study has been stagnated by ambiguity as to whether or not it possesses an anticipated protein-derived cofactor. Reported herein is the discovery and elucidation of a Cys-Tyr cofactor in human ADO, crosslinked between Cys220 and Tyr222 through a thioether (C–S) bond. By genetically incorporating an unnatural amino acid 3,5-difluoro-tyrosine (F2-Tyr) specifically into Tyr222 of human ADO, we identified an autocatalytic oxidative carbon–fluorine bond activation and fluoride release by mass spectrometry and 19F NMR spectroscopy. These results suggest that the cofactor biogenesis is executed by a powerful oxidant during an autocatalytic process. Unlike that of cysteine dioxygenase, the crosslinking results in a minimal structural change of the protein that is not detectable by routing techniques with low resolution. Finally, a new sequence motif C-X-Y-Y(F) is proposed for identifying Cys-Tyr crosslink.
Keywords: Aromatic C-H/C-F bond activation, cofactor biogenesis, thiol dioxygenase, unnatural amino acid, protein post-translational modification
COMMUNICATION
Cysteamine dioxygenase (ADO) has unique characters distinct from its sibling enzyme cysteine dioxygenase (CDO). Whether a Cys-Tyr crosslink is present in ADO is an unsolved question for a decade. By genetically incorporating difluorotyrosine into human ADO active site, the mysterious cofactor was uncovered by high-resolution MS/MS and 19F NMR. An autocatalytic oxidative C–F bond cleavage with a fluoride release was revealed, which lends credence to a Cys-Tyr cofactor with a new sequence motif for thiol dioxygenases.
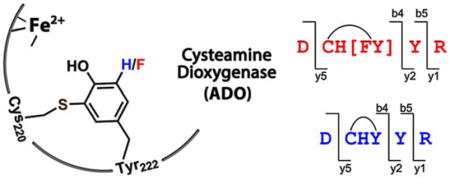
The non-heme iron-dependent enzymatic oxidation of 2-amino-ethanethiol (cysteamine) to hypotaurine has been known since 1963 (Scheme 1).[1] The responsible mammalian enzyme, cysteamine dioxygenase (ADO), was purified to homogeneity in 1971, and the characterization of the human enzyme was described in 2007.[2] Human ADO is one of two thiol dioxygenases, alongside cysteine dioxygenase (CDO), which plays critical roles in maintaining proper thiol level in cells, especially in managing metabolic cysteine and taurine concentrations. The dysfunction of ADO is associated with autoimmune and neurological conditions,[3] fat metabolism,[4] and oxidative stress.[5]
Scheme 1.

Thiol dioxygenases in cellular thiol metabolism.
The presence of a protein-derived cysteine-tyrosine (Cys-Tyr) cofactor crosslinked between two distant residues of the protein sequence, Cys93 and Tyr157, was revealed a decade ago from murine, rat, and human CDOs from independent studies by using X-ray crystallography.[6] The Cys-Tyr cofactor, formed through an autocatalytic process, increases the dioxygenase catalytic efficiency of CDO by an order of magnitude.[7]
ADO is a paralog of CDO.[2b] These two thiol dioxygenases belong to the Cupin superfamily with a conserved β-barrel fold[8] and share similar S atom-based dioxygenation with parallel substrate structures. Thus, it is sensible to anticipate that a corresponding Cys-Tyr cofactor is also formed through a similar self-processing reaction in human ADO, as previously found in CDO. The difficulty in crystallizing ADO hinders our further understanding towards this enzyme, especially if it possesses a Cys-Tyr cofactor. Notably, after nearly over a half-century since the discovery of ADO, its catalytic machinery remains elusive. The Cys-Tyr cofactor does not give rise to any discernible spectroscopic signature with which its presence can be exclusively identified and quantified. If the cofactor is present, its formation is likely to be an uncoupled, single-turnover reaction out of hundreds of cysteamine oxygenation (i.e., the coupled reaction) cycles. With a ferrous center, ADO is not amenable to characterization by optical spectroscopy since the absorbance of both its substrate and product overlap with the protein absorbance. Thus, the answer to the question whether or not a Cys-Tyr cofactor is present in ADO entails strenuous effort.
An EMBOSS pairwise alignment[9] suggested that ADO and CDO share 15.8% sequence identity and 26.5% similarity (Figure S1). Besides the preserved 3-His iron binding motif, there is a pair of the conserved tyrosine and cysteine residues (Tyr40 and Cys130 in CDO, Tyr91, and Cys183 in ADO). However, both Tyr40 and Cys130 are biologically irrelevant to CDO catalysis. Also, neither of the crosslinked residues of CDO, Cys93 and Tyr157, are conserved in the ADO sequences.
We cloned and overexpressed human ADO in Escherichia coli to determine if a Cys-Tyr cofactor is present. The as-isolated protein presented two significant bands by SDS-PAGE. However, these two bands appear ca. 40 kDa from each other, indicative of oligomerization changes rather than crosslinking. Further, the two bands coalesced after incubation with excess dithiothreitol (DTT) (Figure 1A), implicating disulfide bond(s). In contrast, the two bands from CDO are insensitive to DTT because its Cys-Tyr crosslink is irreversible and cannot be reduced by DTT. The higher molecular weight band in CDO corresponds to uncrosslinked polypeptide while the faster migrating band is mature CDO with crosslink.[10] Further processing of ADO with cysteamine and O2 did not lead to noticeable changes. Similarly, ADO eluted as at least three distinct fractions through size exclusion chromatography (SEC); however, the inclusion of DTT in the elution buffer substantially reduces other portions to one (Figure 1B). The different ADO conformations observed in solution and denatured forms are most likely due to the formation of reversible intermolecular disulfide bond(s). Thus, the experiments with low-resolution traditional approaches on crosslink are inconclusive.
Figure 1.

(A) SDS PAGE of ADO and CDO incubated with and without excess DTT. (B) SEC of ADO eluting from the buffer with and without 1 mM DTT.
We then attempted to solve the puzzle with high-resolution mass spectrometry. The expected 2-Da difference between Cys-Tyr crosslinked and uncrosslinked forms of WT protein is difficult to determine from intact protein MS.[11] Hence, we conducted a rigorous MS/MS analysis on human ADO. The native full-length protein was digested with trypsin and carbamidomethylated with all free cysteines. After examination of all the resulting peptides by high-resolution LC-MS/MS, a short hydrophilic peptide DCHYYR stood out with its early retention time (Figure S2).
At +2 charge state, both the crosslinked form and the uncrosslinked form with a carbamidomethyl cysteine were detected (Figure 2A). The crosslinked cysteine covalently bonds with another residue, and thus could not be carbamidomethylated. The two hydrogens loss (2 Da) in the crosslinked form and one additional carbamido-methylation (57 Da) in the uncrosslinked form resulted in a total 59-Da difference, indicating a crosslink present in the peptide and that Cys220, which is the only cysteine in this peptide, is involved.
Figure 2.

High-resolution mass spectra of WT (A–C) and F2-Tyr222 ADO (D–F). In WT ADO, both the crosslinked form at m/z 427.6680 and the uncrosslinked form at m/z 457.1865 were observed by LC-MS/MS (A). The predicted values are 427.6661 and 457.1847. CID spectra of the uncrosslinked (B) and crosslinked (C) forms identified the crosslink between Cys220 and Tyr222. In F2-Tyr222 ADO, both the crosslinked form at m/z 436.6638 and the uncrosslinked form at m/z 475.1778 were observed by LC-MS/MS (D). The predicted values are 436.6614 and 475.1753. CID spectra of uncrosslinked (E) and crosslinked (F) forms identified the crosslink was formed by Cys220 and F2-Tyr222 with only one fluorine. C* represents the carbamido-methylated cysteine. Arc represents crosslinking. The intact peptides (z = +2) are highlighted by red; specified fragments are shown in black while other observed values are in gray.
Since two tyrosine residues, Tyr222 and Tyr223, are present in the peptide we conducted further fragmentation with collision-induced dissociation (CID) to identify the exact crosslink position. The CID spectrum of uncrosslinked form indicated the presence of each amino acid in this peptide (Figure 2B). But the CID spectrum of crosslinked form was quite different (Figure 2C), which suggested that the fragmentation ions not containing Cys220, His221, Tyr222 residues (CHY) were conserved in the uncrosslinked peptide (e.g., y1, y2) while the ones containing CHY differed by 59 Da (e.g., y5, b4, b5). Fragments breaking CHY were not observable in the spectrum due to the sidechain crosslink. These data explicitly suggested that Cys220 and Tyr222 are crosslinked. Moreover, the electron-transfer dissociation (ETD) spectrum also confirmed the crosslink is located between these two residues (Figure S3A).
To facilitate a definitive mass spectrometry analyses and explore the oxidizing power of the oxidant for C-H bond activation during cofactor biogenesis we incorporated unnatural tyrosine into ADO through a genetic method. Using a tRNA(MjtRNATyrCUA)/F2-TyrRS system to target the TAG codon,[12] we generated an expression system that efficiently synthesizes human ADO with a specific substitution of Tyr222 to 3,5-difluoro-tyrosine (F2-Tyr). The presence of two fluorine atoms will potentially facilitate the mass spectrometry analysis if the same crosslink is formed because the mass difference would be 20 instead of 2 Da after a C-F bond and an S-H bond cleavage, forming a new C-S bond. After trypsin digestion on F2-Tyr222 ADO, we observed similar elution profile by LC-MS/MS as in WT ADO (Figure S2B). Interestingly, we still found two forms of the targeting peptide. At z = +2, a mono-fluoro-substituted crosslinked form and a difluoro-substituted uncrosslinked form with a carbamidomethylated cysteine were observed (Figure 2D). The 77-Da difference was caused by S-H and C-F bonds cleavage (20 Da for loss of an H and F) in crosslinked form and one additional carbamidomethylation (57 Da difference) in uncrosslinked form. The mass increase of total 36 Da in F2-Tyr222 ADO uncrosslinked form compared to WT ADO was expected for the successful incorporation of F2-Tyr222, the ortho positions of which are substituted by two fluorine atoms instead of two hydrogens.
Next, we performed CID fragmentation on both forms to further confirm the displacement of a fluorine atom upon crosslinking. Compared to the uncrosslinked form of WT ADO (Figure 2B), the uncrosslinked form of F2-Tyr222 ADO (Figure 2E) has an overall 36-Da increase only in fragments containing F2-Tyr222 (e.g., y3, y4, y5, b4, b5) and no increase in fragments without it (e.g., y1, b2). As for the crosslinked form, however, F2-Tyr222 ADO (Figure 2F) shows only an 18-Da increase on the fragments containing crosslink (e.g., y5, b4, b5) when compared to the WT (Figure 2C). The same results were obtained in the ETD spectrum (Figure S3B). Thus, there is only single fluorine in the crosslinked peptide. Together with the WT results, the presence of crosslink between Cys220 and Tyr222 is unambiguously established.
To reconcile the mass spectrometry results, we employed 19F NMR spectroscopy to attempt to detect the fate of the fluorine during cofactor biogenesis in F2-Tyr222 ADO. Under anaerobic condition, concentrated F2-Tyr222 ADO was mixed with substrate cysteamine in a sealed NMR tube. The resulting 19F NMR spectrum was completely silent; the absence of F2-Tyr222 signal may result from the coupling with the adjacent paramagnetic iron center. Then the NMR tube was opened and exposed to air for overnight in-tube reaction. The oxidized F2-Tyr222 ADO gave rise to a new signal at −121.14 ppm (Figure 3). Spiking in aqueous potassium fluoride overlaid with the observed new signal, which confirms the fluorine departed in the fluoride form during autocatalytic Cys-Tyr cofactor formation, which was promoted in the presence of the enzyme substrates (i.e., cysteamine and O2).
Figure 3.

19F NMR spectra of the F2-Tyr222 enzyme-substrate complex under anaerobic conditions (A), then exposed to air for an overnight reaction (B), and reaction mixture spiked with aqueous KF (C).
The C-F bond is one of the most durable single bonds in organic chemistry. The oxidative cleavage of one of the C-F bonds in F2-Tyr222 ADO is an intriguing observation. To estimate the bond strength of the C-F bond with an aromatic carbon, we performed density functional theory calculations on L-Tyr and F2-Tyr. The results show that the dissociation energy of the C-F bond (132.66 kcal/mol) is 10.12 kcal/mol greater than that of the corresponding C-H bond (122.54 kcal/mol) (Table S1), which is more significant than a phosphoanhydride bond in ATP (7.3 kcal/mol), yet the non-heme iron-bound oxidant possesses sufficient power to cleave a C-F bond.
We obtained a predicted model structure by using the Phyre2 server[13] to estimate the locations and relative distances of the cysteine and tyrosine residues. The model suggests that Tyr87, Tyr222, Tyr223, and Cys220 are within the second coordination sphere of the iron center while others such as Tyr91 and Cys183 conserved in CDO are likely located on the protein surface and remote from the iron center (Figure 4A). Notably, the side chains of Cys220 and Tyr222 are close to each other (~3 Å) but located on a different side of the iron as compared to CDO (Figure 4B). Moreover, our multiple sequence alignment revealed that Cys220 and Tyr222 are both conserved in the eukaryotic ADOs and a new Cys-Tyr motif C-X-Y-Y(F) is identified (Figure S1B).
Figure 4.

ADO structural model. (A) The cysteine and tyrosine residues are highlighted. Tyr87, Tyr222, Tyr223, and Cys220 are within the second coordination sphere of the iron center while others are likely located in protein surface and remote from the iron center. (B) Superposition of the active site of ADO model (green) and CDO crystal structure (PDB ID: 2IC1, gray).
We produced site-directed mutates of Tyr87, Tyr222, and Tyr223, altering them to alanine. The resulting variants were subjected to activity assay using an oxygen electrode. We quantitated the oxygen consumption and plotted Michaelis-Menten curves (Figure S4). The catalytic activity was most significantly reduced in Y222A ADO (ca. 23% of WT), with a modest reduction in Y87A (ca. 51% of WT), and no significant reduction in the activity of Y223A was observed (Table S2). To exclude the possibility of decreased activity is due to backbone distortions, we constructed a Y222F variant and again obtained similar assay results that showed mutation of Tyr222 led to a significant reduction of the ADO activity. Alternatively, F2-Tyr222 ADO only showed a modest decrease in activity (ca. 66% of WT). Thus, the structural modeling and site-directed mutagenesis analysis further corroborate the presence of Cys220-Tyr222 cofactor in human ADO and its enhancement to catalysis of the cysteamine deoxygenation is proven.
In the past decade, thiol dioxygenases including ADO and CDO have attracted a great deal of attention because of the potential of novel sulfur-based chemistry. The structure and function study of ADO has been hindered by ambiguity on its catalytic cofactor in the mature protein, and the enzymatic mechanism remains unknown even after nearly half a century of the discovery of its enzymatic activity. The ADO cofactor is less accessible relative to that of CDO because it is not formed from two remote residues and little impact made to the overall structure, so that there is seemingly only one form of the protein at the reduced state. Of note, the protein-derived Cys-Tyr redox cofactor is present in a growing number of iron- and copper-containing proteins. Its presence either enables or significantly enhances the capacity of the host metalloprotein to mediate a specific redox process, which may be achieved through correctly positioning substrate(s) or stabilizing the catalytic intermediates. It was reported that there are nearly three hundred candidate proteins identified for probable Cys-Tyr crosslinking based on an examination of published protein structures,[11] whereas few have been validated to possess such a crosslinking. The major hurdle for such limited number of the reported protein-derived Cys-Tyr cofactor is because of the dependence on crystallography as the identification utility, which is not always accessible to a specific system. A recent study has provided an alternative method that utilizes the native fluorescence, however, there is limited choice for the studied proteins since it requires small size and low tryptophan composition.[14] Herein, a high-resolution mass spectrometry-centered method coupled with the genetic incorporation of unnatural amino acid is described,[12, 15] which may be potentially developed into an efficient method to detect the presence of a Cys-Tyr cofactor.
In summary, the Cys-Tyr cofactor is firmly established as part of the catalytic machinery in human ADO by high-resolution mass spectrometry, mutagenesis analysis, activity assays, and 19F NMR data. ADO is the second human enzyme that is shown to contain a Cys-Tyr cofactor, and unlike the first one found in CDO, it has its own crosslink motif. The identification of the C-X-Y-Y(F) motif provides one more template for Cys-Tyr crosslink discovery in thiol dioxygenases. Additionally, we determined the metal-bound oxidant in ADO is potent enough to activate either an aromatic C-H or a more durable C-F bond when a genetically substituted fluorotyrosine probe is incorporated into the protein. These findings will facilitate the structure and mechanism studies of ADO.
Supplementary Material
Acknowledgments
This work was supported in whole or part by the National Institutes of Health (NIH) grants GM107529 and GM108988, the National Science Foundation grant CHE-1623856, and the Lutcher Brown Distinguished Chair Endowment fund (to A.L.). The mass spectrometry facility was sponsored by National Institutes of Health Grant G12MD007591. The NMR spectrometer used is a shared instrument sponsored by the National Science Foundation (NSF) under award no. 1625963. We thank Dr. Jiangyun Wang for the generous gift of pEVOL-F2-TyrRS plasmid and Mr. Ian Davis for constructive discussions.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Cavallini D, Scandurra R, Demarco C. J Biol Chem. 1963;238:2999–3005. [PubMed] [Google Scholar]
- 2.a) Cavallini D, Scandurra R, Duprè S. Methods Enzymol. 1971;17(Part B):479–483. [Google Scholar]; b) Dominy JE, Jr, Simmons CR, Hirschberger LL, Hwang J, Coloso RM, Stipanuk MH. J Biol Chem. 2007;282:25189–25198. doi: 10.1074/jbc.M703089200. [DOI] [PubMed] [Google Scholar]
- 3.a) Bousquet M, Gibrat C, Ouellet M, Rouillard C, Calon F, Cicchetti F. J Neurochem. 2010;114:1651–1658. doi: 10.1111/j.1471-4159.2010.06874.x. [DOI] [PubMed] [Google Scholar]; b) Cisbani G, Drouin-Ouellet J, Gibrat C, Saint-Pierre M, Lagace M, Badrinarayanan S, Lavallee-Bourget MH, Charest J, Chabrat A, Boivin L, Lebel M, Bousquet M, Levesque M, Cicchetti F. Neurobiol Dis. 2015;82:430–444. doi: 10.1016/j.nbd.2015.07.012. [DOI] [PubMed] [Google Scholar]; c) Hara K, Nakamura M, Haranishi Y, Terada T, Kataoka K, Sata T. Amino Acids. 2012;43:397–404. doi: 10.1007/s00726-011-1094-9. [DOI] [PubMed] [Google Scholar]; d Coloso RM, Hirschberger LL, Dominy JE, Lee JI, Stipanuk MH. Adv Exp Med Biol. 2006;583:25–36. doi: 10.1007/978-0-387-33504-9_3. [DOI] [PubMed] [Google Scholar]
- 4.Ueki I, Stipanuk MH. J Nutr. 2009;139:207–214. doi: 10.3945/jn.108.099085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Nishimura T, Duereh M, Sugita Y, Yoshida Y, Higuchi K, Tomi M, Nakashima E. Placenta. 2015;36:693–698. doi: 10.1016/j.placenta.2015.02.014. [DOI] [PubMed] [Google Scholar]; b) Donnelly ET, McClure N, Lewis SE. Mutagenesis. 2000;15:61–68. doi: 10.1093/mutage/15.1.61. [DOI] [PubMed] [Google Scholar]
- 6.a) McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, Phillips GN., Jr Proc Natl Acad Sci U S A. 2006;103:3084–3089. doi: 10.1073/pnas.0509262103. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Simmons CR, Liu Q, Huang QQ, Hao Q, Begley TP, Karplus PA, Stipanuk MH. J Biol Chem, Vol. 2006;281:18723–18733. doi: 10.1074/jbc.M601555200. [DOI] [PubMed] [Google Scholar]; c) Ye S, Wu Xa, Wei L, Tang D, Sun P, Bartlam M, Rao Z. J Biol Chem. 2007;282:3391–3402. doi: 10.1074/jbc.M609337200. [DOI] [PubMed] [Google Scholar]
- 7.Dominy JE, Jr, Hwang J, Guo S, Hirschberger LL, Zhang S, Stipanuk MH. J Biol Chem. 2008;283:12188–12201. doi: 10.1074/jbc.M800044200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stipanuk MH, Simmons CR, Karplus PA, Dominy JE., Jr Amino Acids. 2011;41:91–102. doi: 10.1007/s00726-010-0518-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rice P, Longden I, Bleasby A. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 10.Stipanuk MH, Londono M, Hirschberger LL, Hickey C, Thiel DJ, Wang L. Amino Acids. 2004;26:99–106. doi: 10.1007/s00726-003-0001-4. [DOI] [PubMed] [Google Scholar]
- 11.Martinie RJ, Godakumbura PI, Porter EG, Divakaran A, Burkhart BJ, Wertz JT, Benson DE. Metallomics. 2012;4:1037–1042. doi: 10.1039/c2mt20093g. [DOI] [PubMed] [Google Scholar]
- 12.Li F, Shi P, Li J, Yang F, Wang T, Zhang W, Gao F, Ding W, Li D, Li J, Xiong Y, Sun J, Gong W, Tian C, Wang J. Angew Chem Int Ed. 2013;52:3958–3962. doi: 10.1002/anie.201300463. [DOI] [PubMed] [Google Scholar]
- 13.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hromada SE, Hilbrands AM, Wolf EM, Ross JL, Hegg TR, Roth AG, Hollowell MT, Anderson CE, Benson DE. J Inorg Biochem. 2017;176:168–174. doi: 10.1016/j.jinorgbio.2017.08.028. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Brock A, Herberich B, Schultz PG. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.