

TO THE EDITOR
Point mutations in the transcription factor TP63 gene underlie a subset of ectodermal dysplasias, including ankyloblepharon-ectodermal defects-cleft lip/palate syndrome [AEC; OMIM #106260], a developmental disorder associated with severe skin erosions (McGrath et al., 2001) (Figure 1a). TP63-AEC mutations cluster in exons 13 and 14, exons that encode putative protein-protein interaction domains (Figure 1b). Molecular mechanisms causing skin erosions in AEC are poorly understood. Both mouse models (Ferone et al., 2013, Koster et al., 2009, Russo et al., 2018) and human keratinocyte models (Zarnegar et al., 2012) have been used to investigate aspects of AEC. Although both approaches yielded important results, they also suffered from significant drawbacks. The mouse models did not fully replicate AEC skin phenotypes, while the cell culture models used non-physiological overexpression of mutant TP63-AEC in primary human keratinocytes [see discussion in (Koch et al., 2014)]. In the present study, we utilized AEC patient-derived cells that express physiological levels of TP63-AEC alleles on a genetic background susceptible to the disease. The latter point is important as disease severity among patients carrying the same TP63 mutation can vary dramatically (e.g. Bertola et al., 2004).
Figure 1. Generating iPSC and iPSC-derived keratinocytes from AEC patient skin.
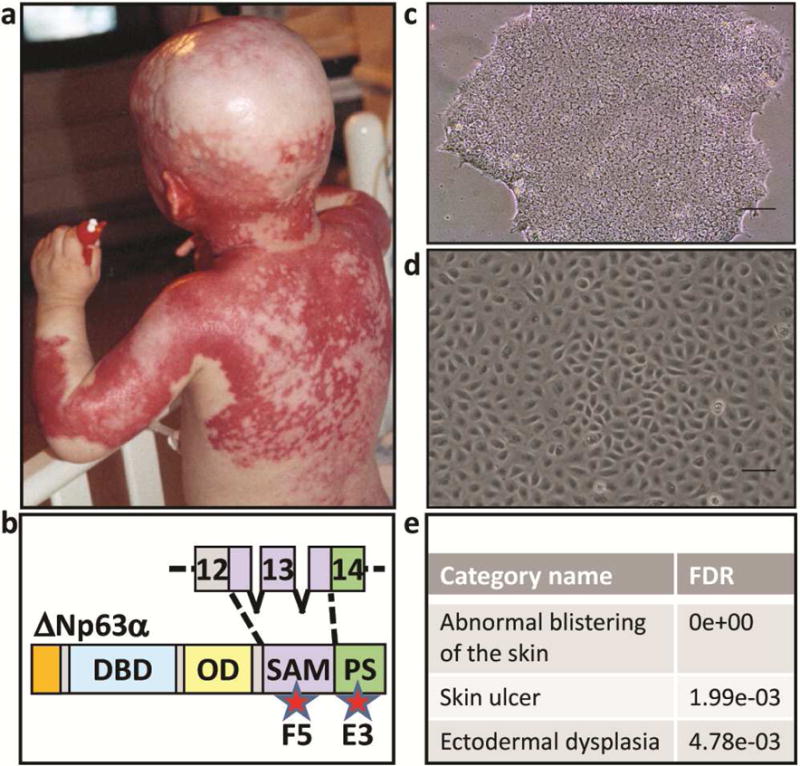
(a) Patient affected by AEC exhibiting severe skin erosions. The patient and his parents consented to the use of this image in this publication. (b) TP63 mutations in AEC patients occur mainly in exons 13-14, encoding putative protein-protein interaction domains. Approximate location of mutations in the AEC patient cells (F5 and E3) used in this manuscript are indicated by stars. The protein schematic shown is of ∆Np63α, the predominantly expressed TP63 isoform in human keratinocytes. DBD: DNA binding domain, OD: oligomerization domain, SAM: sterile alpha motif, PS: post-SAM domain. Phase contrast images of (c) human iPSC colony and (d) iPSC-derived keratinocytes. (e) Disease pathways associated with the TP63 mutations in E3 and F5 iPSC-K as determined by a Human Phenotype Ontology analysis of our RNAseq data (FDR; false discovery rate).
Initially, we generated fibroblast-derived induced pluripotent stem cells (iPSC) from two AEC patients carrying mutations in either exon 13 [I537T (line F5)] or exon 14 [R598L (line E3)] of the TP63 gene (Figures 1b, c)(biopsies were obtained with institutional approval and written informed patient consent). Next, we used CRISPR/CAS- and TALEN-mediated gene editing to correct the TP63-AEC mutations in both lines (Supplementary Figure S1). This yielded conisogenic pairs of iPSC lines that were genetically identical except for the presence or absence of the TP63-AEC mutation [E3, E3GC (gene-corrected); F5, F5GC]. These cell pairs are ideally suited to investigate molecular pathways while minimizing genetic background effects. The iPSC were then differentiated into keratinocytes [Figure 1d and (Dinella et al., 2014)]. These iPSC-derived keratinocytes (iPSC-K) were similar to primary human keratinocytes in that they expressed keratinocyte markers [TP63 and KRT14] and assembled desmosomes containing DSG1/2, DSC3, DSG3, and JUP (Figure 2). As TP63, KRT14, DSG3, and DSC3 are only co-expressed in stratified epithelia, these findings demonstrate that our iPSC-K show basic properties of keratinocytes. Next, we subjected the conisogenic iPSC-K lines, grown under conditions that suppress differentiation (low calcium culture conditions), to an RNAseq analysis (accession number GSE109185). Upon analyzing the ranked gene lists using the Human Phenotype Ontology tool (Kohler et al., 2017), we identified several abnormalities in gene expression patterns of AEC iPSC-K which are relevant for the AEC skin phenotype (Figure 1E). Further, analysis of the transcriptome data using Gene Ontology revealed a significant effect of the AEC mutations on genes in the “Desmosome” category (FDR 1.49e-03). Specifically, we identified a downregulation of desmosomal cadherins, a finding that we independently verified by qRT-PCR (data not shown). Next, we induced iPSC-K differentiation and desmosome assembly by exposing the cells to media with elevated calcium concentrations and performed immunofluorescence analysis for various desmosomal markers. We observed striking differences between AEC iPSC-K and gene-corrected iPSC-K in the expression level and distribution of several desmosomal transmembrane receptors (Figures 2b-d; F5 and F5GC cells are shown). To determine whether the abnormal expression and distribution of desmosomal proteins affected cell adhesion, we subjected these cells to a dispase cell adhesion assay (Hartlieb et al., 2014). In sharp contrast to gene-corrected iPSC-K sheets, AEC iPSC-K sheets rapidly disintegrated upon exposure to mechanical stress (Figure 2e). Enzyme-release assays suggested acantholysis (loss of cell-cell adhesion) rather than cytolysis as the underlying cause (data not shown).
Figure 2. Characterization of desmosomal protein expression in AEC iPSC-K and AEC patient skin.
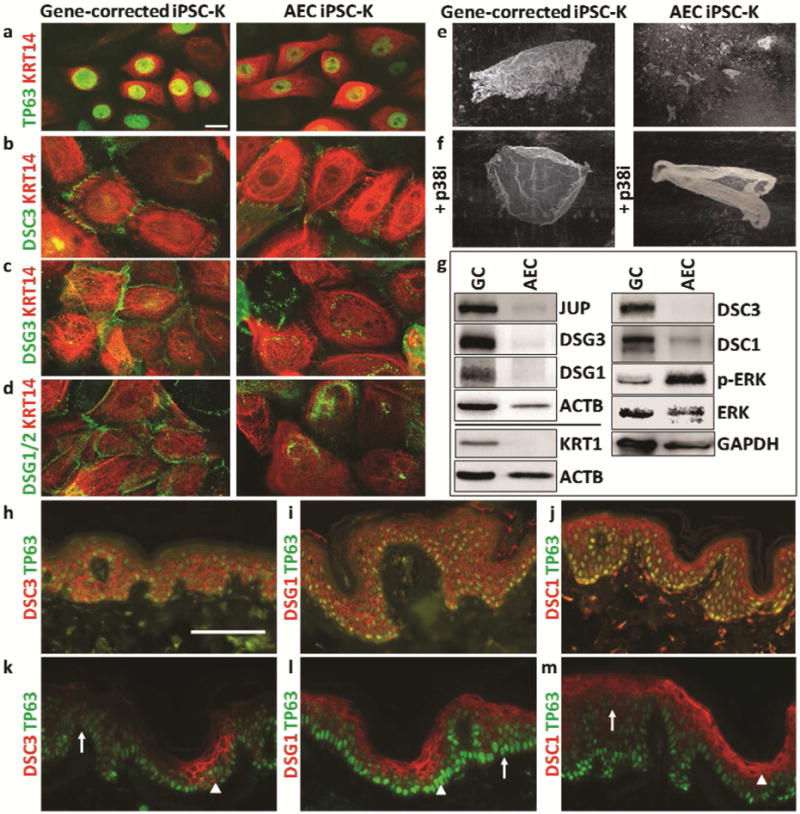
Immunofluorescence analysis of a conisogenic pair of AEC and gene-corrected iPSC-K (F5, F5GC) in low calcium (a) and high calcium media for 24 hours (b-d). (a) TP63 and KRT14, (b) DSC3 and KRT14, (c) DSG3 and KRT14, and (d) DSG1/2 and KRT14 expression and localization. (e) Dispase assay showing accelerated fragmentation of AEC iPSC-K sheets in response to mechanical stress. (f) Treatment with the p38MAPK inhibitor SB202190 prevents AEC iPSC-K sheet fragmentation. (g) Western blot analysis for the indicated proteins of F5 and F5GC iPSC-K after exposure to high calcium conditions for 24 hours. Similar data for E3 and E3GC are shown in Supplemental Figure 2. Note downregulation of several proteins in AEC iPSC-K (JUP, 0.24; DSG3, 0.18; DSG1, 0.41; KRT1, 0.063; DSC3, 0.061; DSC1, 0.49). The ratio of pErk/Erk was increased 4.2 fold in AEC iPSC-K. (h-j) Immunofluorescence analysis of normal human skin and (k-m) AEC skin for (h,k) DSC3, (i, l) DSG1, and (j, m) DSC1. Arrowheads point towards normal expression while arrows point towards abnormal expression in panels h-j. Size bars: 10μm (a-d), 100μm (h-m)
By Western blotting, we determined that DSG3 and JUP were downregulated in AEC iPSC-K (Figure 2g and Supplemental Figure 2). Downregulation of these two proteins has been shown to activate p38MAPK signaling and ultimately cause acantholysis in keratinocyte models of the autoimmune disease pemphigus vulgaris (Hartlieb et al., 2014, Spindler et al., 2014). To test whether aberrant DSG3-JUP-p38MAPK signaling might contribute to acantholysis in AEC iPSC-K, we repeated the dispase assays in the presence of a p38MAPK inhibitor (SB202190). The inhibitor stabilized the AEC iPSC-K sheets (Figure 2f), suggesting that activation of p38MAPK signaling contributes to acantholysis. Interestingly, we have previously shown that JUP can act as a positive regulator of Dsc3 gene expression, and that loss of Dsc3 or Dsg3 leads to acantholysis in mouse epidermis (Chen et al., 2008, Koch et al., 1997, Tokonzaba et al., 2013). This suggests that simultaneous downregulation of all three proteins might exacerbate desmosomal adhesion defects in AEC iPSC-K.
We also observed reduced expression of cytoskeletal and desmosomal proteins associated with keratinocyte differentiation, including DSG1, DSC1, and KRT1 in AEC iPSC-K cultured in high calcium media (Figure 2g). A direct role for DSG1 in mediating keratinocyte differentiation through suppression of ERK signaling has been demonstrated (Getsios et al., 2009). These authors showed that knockdown of DSG1 in 3D keratinocyte cultures leads to increased ERK signaling and reduced expression of the differentiation markers DSC1 and KRT1. Our Western blot data suggest that this mechanism also operates in AEC iPSC-K (Figure 2g). Thus, two previously identified signaling functions of desmosomes, DSG3-JUP-p38MAPK-controlled cell adhesion and DSG1-ERK-controlled differentiation, are affected by the two different TP63-AEC mutations analyzed.
To determine whether our iPSC-based in vitro system phenocopies AEC epidermis, we next performed immunofluorescence staining of perilesional skin of AEC patients and demonstrated focal loss (or reduced expression) of DSC3, DSG1, DSC1, and KRT1 [Figure 2h-m; and (Koster et al., 2009)], thereby validating the results obtained with our iPSC-based system. It remains to be seen why these desmosomal defects occur focally given that there is no evidence of mosaicism in AEC.
In summary, we have developed a human iPSC-based in vitro system that enables us to identify molecular mechanisms underlying skin fragility in AEC patients. As iPSC-K from two patients with two different mutations demonstrated similar desmosomal abnormalities, our data suggest that these defects are of general relevance for AEC. Our in vitro system will be ideally suited to identify new disease pathways in AEC and other ectodermal dysplasias caused by TP63 mutations.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health (NIH) under Award Numbers R56 AR066713 and R01AR072621. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Further support was provided by research grants from the National Foundation for Ectodermal Dysplasias (NFED). MIK and PJK were the recipients of both grant mechanisms. iPSC line generation and characterization were performed with support from the Gates Bioengineering Core at the University of Colorado Anschutz Medical Campus. Bioinformatics support was provided by Drs. Michael Edwards and Tzu Phang (University of Colorado). We would like to thank the patients who supported this research by providing skin biopsies.
Abbreviations
- AEC
Ankyloblepharon-ectodermal defects-cleft lip/palate syndrome
- DSG
Desmoglein
- DSC
Desmocollin
- JUP
Plakoglobin
- KRT
Keratin
- iPSC
Induced Pluripotent Stem Cells
- iPSC-K
iPSC-Derived Keratinocytes
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors state no conflicts of interest
References
- Bertola DR, Kim CA, Albano LM, Scheffer H, Meijer R, van Bokhoven H. Molecular evidence that AEC syndrome and Rapp-Hodgkin syndrome are variable expression of a single genetic disorder. Clinical genetics. 2004;66(1):79–80. doi: 10.1111/j.0009-9163.2004.00278.x. [DOI] [PubMed] [Google Scholar]
- Chen J, Den Z, Koch PJ. Loss of desmocollin 3 in mice leads to epidermal blistering. J Cell Sci. 2008;121(Pt 17):2844–9. doi: 10.1242/jcs.031518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinella J, Koster MI, Koch PJ. Use of induced pluripotent stem cells in dermatological research. The Journal of investigative dermatology. 2014;134(8):e23. doi: 10.1038/jid.2014.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferone G, Mollo MR, Thomason HA, Antonini D, Zhou H, Ambrosio R, et al. p63 control of desmosome gene expression and adhesion is compromised in AEC syndrome. Human molecular genetics. 2013;22(3):531–43. doi: 10.1093/hmg/dds464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getsios S, Simpson CL, Kojima S, Harmon R, Sheu LJ, Dusek RL, et al. Desmoglein 1-dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. The Journal of cell biology. 2009;185(7):1243–58. doi: 10.1083/jcb.200809044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlieb E, Rotzer V, Radeva M, Spindler V, Waschke J. Desmoglein 2 compensates for desmoglein 3 but does not control cell adhesion via regulation of p38 mitogen-activated protein kinase in keratinocytes. The Journal of biological chemistry. 2014;289(24):17043–53. doi: 10.1074/jbc.M113.489336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch PJ, Dinella J, Fete M, Siegfried EC, Koster MI. Modeling AEC-New approaches to study rare genetic disorders. American journal of medical genetics Part A. 2014;164A(10):2443–54. doi: 10.1002/ajmg.a.36455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, et al. Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol. 1997;137(5):1091–102. doi: 10.1083/jcb.137.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler S, Vasilevsky NA, Engelstad M, Foster E, McMurry J, Ayme S, et al. The Human Phenotype Ontology in 2017. Nucleic acids research. 2017;45(D1):D865–D76. doi: 10.1093/nar/gkw1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster MI, Marinari B, Payne AS, Kantaputra PN, Costanzo A, Roop DR. DeltaNp63 knockdown mice: A mouse model for AEC syndrome. Am J Med Genet A. 2009;149A(9):1942–7. doi: 10.1002/ajmg.a.32794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JA, Duijf PH, Doetsch V, Irvine AD, de Waal R, Vanmolkot KR, et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Human molecular genetics. 2001;10(3):221–9. doi: 10.1093/hmg/10.3.221. [DOI] [PubMed] [Google Scholar]
- Russo C, Osterburg C, Sirico A, Antonini D, Ambrosio R, Wurz JM, et al. Proceedings of the National Academy of Sciences of the United States of America. 2018. Protein aggregation of the p63 transcription factor underlies severe skin fragility in AEC syndrome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindler V, Dehner C, Hubner S, Waschke J. Plakoglobin but not desmoplakin regulates keratinocyte cohesion via modulation of p38MAPK signaling. The Journal of investigative dermatology. 2014;134(6):1655–64. doi: 10.1038/jid.2014.21. [DOI] [PubMed] [Google Scholar]
- Tokonzaba E, Chen J, Cheng X, Den Z, Ganeshan R, Muller EJ, et al. Plakoglobin as a regulator of desmocollin gene expression. The Journal of investigative dermatology. 2013;133(12):2732–40. doi: 10.1038/jid.2013.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnegar BJ, Webster DE, Lopez-Pajares V, Vander Stoep Hunt B, Qu K, Yan KJ, et al. Genomic profiling of a human organotypic model of AEC syndrome reveals ZNF750 as an essential downstream target of mutant TP63. American journal of human genetics. 2012;91(3):435–43. doi: 10.1016/j.ajhg.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.