

Abstract
Histone deacetylases (HDACs) represent a family of enzymes that are targets for epigenetic modulation of genomic activity that may be beneficial in the treatment of many diseases, including cancer and CNS disorders. In animal models, HDAC inhibitors have neuroprotective, antiepileptogenic, and antidepressant effects. Assaying HDAC activity provides a robust method for identifying HDAC inhibitors and for assessing their effects under various physiological conditions or after pathological insults. In this unit, a simple and sensitive assay for measuring HDAC activity is described. HDAC activity in tissue lysates can be assessed fluorometrically using a Boc-Lys(Ac) HDAC activity kit. HDACs catalyze the deacetylation of the substrate, Boc-Lys(Ac)-AMC. Addition of a trypsin-containing developer converts the deacetylated product to a quantifiable fluorophore that can be used both as a screening method to identify putative HDAC inhibitors. and to assess the effects of these inhibitors on tissue and animal epigenetic-modulated phenotypes.
Keywords: HDAC, Epigenetic, HDAC substrate, Sodium Butyrate, Trichostatin-A, nuclear protein, fluorophore
Introduction
The epigenetic modification of histones plays a pivotal role in the regulation of cell-specific expression of genes required for development and maintenance of cellular function. Epigenetics refers to reversible or ‘plastic’ alterations in gene expression that are constantly reprogrammed by the environment and other factors (Younus and Reddy, 2017). This continuously changing landscape is maintained by enzymes that are termed ‘writers’ and ‘erasers’. These encode or remove epigenetic markers, respectively. Histone acetyltransferases (HATS) serve as “writer” enzymes that transfer acetyl groups to the lysine residues of histone proteins, transforming DNA to a more open conformation by reducing its affinity to the histone. The weakened interaction between DNA and histones allows for greater interaction of the DNA with transcription factors; therefore, increased acetylation of histones is associated with upregulated gene expression (De Ruijter et al., 2003). In contrast, histone deacetylases (HDACs) are a family of enzymes that “erase” these transient modifications to the N-terminal tails of histone proteins, restoring the DNA to a more tightly bound conformation that is associated with decreased gene expression (i.e. gene silencing). Together, HATs and HDACs serve as epigenetic regulators of gene expression (Verdone et al., 2005; Henshall and Kobow, 2015). Additional “readers” also play a key role in regulating chromatin access and gene expression (Andrews et al., 2016) with a prominent role for acetyl “reader” (bromodomain containing) proteins that bind to acetyl groups, recruit large protein complexes, and thereby alter gene expression (Fujisawa and Filippakopoulos, 2017).
There are 18 HDAC enzymes which can be categorized into four subclasses (I-IV) based on their sequence similarity and function (Table 1; Seto and Yoshida, 2014; Volmar and Wahlestedt, 2015). HDAC classes I, II, and IV can all be inhibited by Trichostatin A (TSA) and are reliant on zinc as a cofactor, whereas class III HDACs (Sirtuin family) are NAD+ dependent. Classes I and II have high expression in the brain and are therefore the most relevant to neurological conditions such as epilepsy, depression, brain injury, and Alzheimer’s disease (Gray and Ekström, 2001; Volmar and Wahlestedt, 2015; Descalzi et al., 2015).
Table 1.
HDAC classes and their inhibitors.
| HDAC Class | Member | Subcellular Localization | Tissue Distribution | Inhibitors |
|---|---|---|---|---|
| Class I | HDAC 1 | Nucleus | Ubiquitous | SAHA, TSA, Entinostat (MS-275), Crebinostat, Valproic acid, compound 60, Fingolimod, Mocetinostat |
| HDAC 2 | Nucleus | Ubiquitous | SAHA, TSA, Entinostat (MS-275), Crebinostat, Valproic acid, Compound 60, Fingolimod, Apicidin | |
| HDAC 3 | Nucleus | Ubiquitous | SAHA, TSA, Entinostat (MS-275), Crebinostat, Valproic acid, Fingolimod, Apicidin, | |
| HDAC 8 | Nucleus, Cytoplasm | Mostly Ubiquitous | Valproic acid, Fingolimod, NBM-T-L-BMX-OS01 | |
| Class IIa | HDAC 4 | Nucleus, Cytoplasm | Brain, Heart, Skeletal Muscle | SAHA, Trichostatin A, Sodium Butyrate, MC1568 |
| HDAC 5 | Nucleus, Cytoplasm | Brain, Heart, Skeletal Muscle | SAHA, 4-Phenylbutyrate | |
| HDAC 7 | Nucleus, Cytoplasm, Mitochondria | Heart, Skeletal Muscle, Placenta | SAHA | |
| HDAC 9 | Nucleus, Cytoplasm | Brain, Skeletal Muscle | SAHA | |
| Class IIb | HDAC 6 | Predominantly cytoplasm | Heart, Liver, Placenta, Kidney | SAHA, Crebinostat, Tubacin, Tubastatin A, ACY-738, ACY-775 |
| HDAC 10 | Predominantly cytoplasm | Liver, Kidney, Spleen | Tubastatin A, Bufexamac | |
| Class III | Mammalian sirtuins (SIRT1-7) | Nucleus, Cytoplasm, Mitochondria | Fetal brain>adult brain (except SIRT 2 &5). SIRT4 highly expressed in astrocytes (postnatal brain) & in radial glia (embryonic tissues) | Resveratrol, Suramin, Nicotinamide, SRT1720, SRT3657 |
| Class IV | HDAC 11 | Nucleus, Cytoplasm | Brain, Heart, Kidney, Skeletal Muscle | Trichostatin A |
SAHA: Vorinostat; Fingolimod: phosphorylated-FTY720; TSA: Trichostatin A
(Volmar and Wahlestedt, 2014; Dokmanovic et al., 2007; de Ruijter et al., 2003)
HDACs play a dynamic role in the brain and help regulate a wide range of normal cellular activities. Aberrant expression of epigenetic factors, such as an extreme increase or decrease in histone marks (such as acetyl groups) can result in increased or decreased gene expression respectively, lead to genetic syndromes and altered cellular function (Carey and La Thangue, 2006). The inhibition of different HDAC proteins has been reported to be beneficial in neurological conditions such as Alzheimer’s disease (Lee et al., 2018), Huntington’s disease (Bassi et al., 2017), traumatic brain injury (TBI) (Wong and Langley, 2016), depression (Covington et al., 2015), epilepsy (Reddy et al., 2018), cancer (Rajendran et al., 2015), and neuropathic pain (Descalzi et al., 2015). Due to the neuroprotective effects of some HDAC inhibitors, this evolving drug class has been increasingly evaluated as potential therapeutic agents for CNS diseases (Huang et al., 2002; Younus and Reddy, 2017). However, available HDAC inhibitors are associated with adverse effects. Thus, the discovery and development of new, brain-specific HDAC inhibitors will help elucidate the specific functional role of HDACs in neuronal physiology and disease. It has been challenging to design highly specific HDAC inhibitors due to the sequence similarity of the zinc-containing catalytic site of the different HDACs. Most of the currently available HDAC inhibitors affect the activity of several HDACs within a class and often affect more than one class of HDACs. Table 2 lists several compounds with their reported potencies in relation to various HDAC proteins.
Table 2.
HDAC inhibitor compounds and their reported IC50 (nM). Hull et al., 2016; Beckers et al., 2007; Selleckchem.com
| HDACi | HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC4 | HDAC6 | HDAC7 | HDAC9 | Broad Spectrum |
|---|---|---|---|---|---|---|---|---|---|
| TSA | 2±0 | 3±0 | 4±1 | 456±59 | 6±2 | 3±1 | 5±2 | 6±5 | 1.8 |
| Vorinostat (SAHA) | 68±14; 21±13; 10 | 164±45 | 48±17; 37±11; 20 | 1524±463; 1200±380 | 101±31 | 90±26; 25±6 | 104±35 | 107±21 | 10 |
| Entinostat (MS275) | 181±62; 180±70 | 1155±134 | 2311±803; 740±250; 1700 | 44900±18100 | >10,000 HDAC4, 6, 7, 8, 9 | ||||
| Romidepsin (FK228) | 1.6 ; 36 | 3.9 ; 47 | |||||||
| Mocetinostat (MGCD0103) | 34±17 ; 820±1.4 ; 150 | 34±8 ; 290 | 998±431 ; 620±160 ; 1660 | >10,000 HDAC4, 6, 7, 8, 9; 590 HDAC11; | |||||
| Panobinostat (LBH589) | 3±0 | 3±0 | 4±1 | 12±5 | 61±1 | 14±7 | 3±2 | 5-20 | |
| Belinostat | 41±6 | 125±21 | 30±0 | 216±43 | 115±16 | 82±19 | 67±22 | 128±46 | 27 |
| Apicidin | 0.299 | 120±28 | 43±7 | 575±111 | |||||
| Valproic acid | 171000 ; 400000 | ||||||||
| Dacinostat (LAQ824) | 1.8±0.6 | 3.7±3 | 140±34 | 15±7 | 32 | ||||
| Sodium Butyrate | 175000 ; 300000 | 400000 | 300000 |
Recently, we reported that HDAC inhibition is associated with effective disease-modification of epileptogenesis (Reddy et al., 2018; Clossen and Reddy, 2017). Epilepsy is a complex disease state that can be triggered by a precipitating injury, such as a traumatic brain injury, yet the extent of epigenetic modification following such incidents is still unknown. Thus, HDAC inhibitors may hold great potential as antiepileptogenic agents for curing epilepsy and to investigate the resulting changes in epigenetic regulation of gene expression. Sodium butyrate been identified as a one such compound that may retard the process of epileptogenesis (Reddy et al., 2018). Sodium butyrate inhibits a broad range of HDACs in the brain (Ferrante et al., 2003; Davie, 2003; Takuma et al., 2014). In vitro or ex vivo assays are the typical standard for determining the mode of action and the concentrations of HDAC inhibitors (HDACi) required to inhibit 50% of the deacetylase activity (IC50). The present Unit describes a simple protocol for measuring HDAC activity in brain and its inhibition by HDAC inhibitors, such as sodium butyrate.
STRATEGIC PLANNING
HDAC enzymes are present in both the nuclear and cytoplasmic compartments, and act on both histone and non-histone proteins to affect changes in protein acetylation status and function. It is imperative that tissue samples should be processed to capture HDACs in nuclear or cytosolic, or both, protein purifications. Moreover, the different HDAC classes have varying levels of expression throughout the body (Table 1). The experimental tissue lysate should have high levels of HDAC expression in order to yield reliable results from the HDAC activity assay. Generally, the higher the expression level, the higher the overall yield of HDAC in the tissue lysate and the reliability of the final results of the assay.
Basic Protocol 1: HDAC Activity Assay
As previously noted, any tissue or cell line can be measured for HDAC activity levels, assuming adequate HDAC enzyme expression. In this protocol, we used mouse brain tissue. To determine HDAC activity and its inhibition by sodium butyrate, brain tissue samples were extracted from control and butyrate-treated (600 mg/kg, i.p.) mice (n=6 mice/group). HDAC activity was measured in nuclear lysates using the fluorometric HDAC substrate Boc-Lys(Ac)-AMC/Boc-Lys-AMC assay (I-1875 and I-1880. Bachem, Torrance, California), as reported previously (Nian et al., 2009; Rajendran et al., 2011; 2013; 2015). Briefly, incubation was performed at 37 °C for 30-min with cortex tissue lysates (15 μg protein/ well), and the HDAC reaction was initiated by the addition of Boc-Lys(Ac)-AMC substrate. HDAC enzymes convert the acetylated substrate, Boc-Lys(Ac)-AMC, to the deacetylated product. The addition of a trypsin-containing developer allows the product to be converted into a quantifiable fluorophore. HDAC inhibitors, such as Trichostatin A (TSA), can inhibit this reaction and serve as a positive control. All materials and equipment for preparing the fluorometric HDAC Activity Assay are commercially available. Unless otherwise stated, chemicals can be obtained from Sigma-Aldrich (SA). We have listed the equipment used in our laboratory, though there are many similar products available that will produce similar results.
Materials
Assay Buffer, pH 8.0 (see recipe)
Substrate (50mM, Boc-Lys(Ac)-AMC, Bachem #I-1875, see recipe)
Standard Inhibitor TSA Stock (1 mM TSA, MW: 302.37; see recipe)
Buffer for Developer, pH 8.0 (see recipe)
Developer (20×), pH 8.0 (see recipe)
Deacetylated Standard 10 mM (Boc-Lys-AMC, Bachem, #I-1880; see recipe)
Tris, Fisher Chemical, #T395-1
NaCl, SA#S5886
KCl, SA#P5405
MgCl2. 6H20, SA#M8266
TSA, VWR#TCT2477
DMSO, SA#D8418
Trypsin, VWR #97063-884
Distilled Water
HCl and NaOH for adjustment of pH
NUNC MaxiSorp* C-Bottom Framed Well Modules or other white microplate for fluorescent reading, Fisher Scientific #12-565-289A
Micropipets, Multi-channel pipets, and appropriate tips
Reagent Reservoirs for multi-channel pipets
Fluorescent Plate Reader—Biotek Synergy 2 or Spectra MAX Gemini XS (or other)
Benchtop Incubator or Heating block at 37°C.
- Prepare working solutions as described below.
-
1ATrichostatin A (0.1 μM): Combine 1 μl of 1 mM TSA Stock (solution 1C) with 10 ml of Assay buffer (solution 1 in REAGENTS AND SOLUTIONS section)
-
1BSubstrate (100 μM Boc-Lys(Ac)-AMC): Dilute substrate 500× by combining 10 μl Boc-Lys(Ac)-AMC stock (solution 2 REAGENTS AND SOLUTIONS section) + 4990 μl Assay buffer (solution 1 in REAGENTS AND SOLUTIONS section).
-
1CDeacetylated Standard (25 μM Boc-Lys-AMC): Dilute 2 μl Boc-Lys-AMC stock (solution 6 in REAGENTS and SOLUTIONS section) + 798 μl of Assay buffer (solution 1 in REAGENTS and SOLUTIONS) for a total of 800 μl.
-
1DHDAC Assay Developer: Dilute 600 μl of 20× developer (solution 5) in 11.4 ml Assay buffer (solution 1) for a total of 12 ml. The final molarities for trypsin and TSA should be 100 μM and 100 nM, respectively.
-
1ETissue Extract: Dilute S2 Tissue extract (see nuclear extraction protocol above) to 300 μg protein/ml in Assay buffer. Dilutions can be calculated using the results from the BCA protein concentration assay. Amount of assay buffer = (protein concentration from the BCA assay × stock amount/300) − stock amount.e.g. = ((1000 μg/ml × 15 μl)/300 μg/ml) − 15 μl) = 35 μl Assay buffer. Recommendation: 5 μg/15 μl protein per well.
-
1A
- Add the working solution reagents to the test well plate in the following order (Table 3), starting on well B1 of the 96-well plate: Assay buffer, Inhibitor (0.1 μM TSA solution, if needed), tissue extract. In the blank well(s) add 25 μl Assay buffer only. In tissue extract wells, add 10 μl assay buffer and 15 μl tissue extract. For the inhibitor control (TSA control), add 10 μl inhibitor (0.1 μM TSA) and 15 μl tissue extract to the wells. At this step, all wells should contain 25 μl total solution. Necessary controls include wells containing nuclear tissue extract from HCT116 cells extract (W09-001-GM5, tebu-bio) as a reference for standard HDAC activity, as can HeLa cell nuclear extract (Enzo Life Sciences, BML-KI140-0100) which can be used for inhibitor screening or immunoprecipitation of HDAC containing complexes (Rajendran et al., 2015). Table 3 shows the reagent scheme for the assay.
-
3Allow the well plate to equilibrate to assay buffer temperature for 5 min.
-
4aAdd 25 μl substrate (100 μM Boc-Lys(Ac)-AMC) to well plate and let the enzymatic reaction take place for 30 min at 37°C or at room temperature. Wells should now contain 50 μl solution.
-
4bDuring the 30 min wait time, add solutions to standard-curve wells. In row A of the well plate, skipping the blank (well A1), begin a standard-curve of deacetylated standard stock (solution 1C). The concentration of deacetylated standard stock should be 2500 nM. The final well plate standards should contain concentrations of 0 nM (blank), 50 nM, 250 nM, 500 nM, 1000 nM, 1500 nM, and 2000 nM. Table 4 describes plate location of the standard concentration curve wells.
-
4cAdd 50 μl HDAC Assay Developer (100 μM trypsin and 100 nM TSA) diluted developer to all wells. Wells should have a total solution of 100 μl/well. Wait at least 15 min before taking the fluorescent reading.
-
5Use a Biotek Synergy 2 or other fluorescent plate reader to read the plate at an excitation wavelength of 360 nm emission wavelength of 460 nm.
-
6Follow usual laboratory procedures for data acquisition and analysis. If the readings for the standard concentration curve do not follow a linear pattern, there may have been pipetting errors or air bubbles in the well plate. Stock standards should be prepared before starting any major project. In order to compare HDAC activity levels in the unknown samples, normalize unknown sample readings to the standard concentration curve containing known values of inhibition by Trichostatin A (row A of well plate).
-
3
Table 3. General reagent scheme for the fluorometric HDAC activity assay.
HCT116 nuclear extraction (high HDAC activity) was used as a positive tissue control. Volume of HCT116 needed depends on protein content calculated from BCA assay; keep in mind that protein content differs between tissue type. Abbreviation: Ac, acetylated substrate.
| Sample | Assay buffer | Inhibitor TSA (5×) | Tissue Extract (minimum 3 μg/15μl) | Substrate (Ac, 100 μM) | Diluted Developer | Total (μl) | |
|---|---|---|---|---|---|---|---|
| Blank No enzyme | 25 μl | — | — | 25 μl | Equilibrate buffer, HCT116 extract and inhibitors to assay temperature. Initiate HDAC reaction by adding substrate, Incubate 30 min at 37°C. Add developer (15 min) to stop reaction | 50 μl | 100 |
| Tissue sample | 10 μl | — | 15 μl | 25 μl | 50 μl | 100 | |
| Tissue + Trichostatin
A (0.1 μM stock) |
— | 10 μl | 15 μl | 25 μl | 50 μl | 100 | |
| Nuclear extract from HCT116 cells, if needed | 23 | 2 μl | 25 μl | 50 μl | 100 | ||
| Nuclear extract from HCT116 cells +TSA, if needed | 13 | 10 μl | 2 μl | 25 μl | 50 μl | 100 | |
| Deacetylated Standard (BL, if need. see below) | varies | Various amounts of standard needed to have the final concentrations of standard at 0 μM - 2.5 μM in 50 μl of assay buffer | 50 μl | 100 | |||
Concentration of test inhibitors depends on their potency (IC50 for HDACi).
Table 4.
Standard deacetylated concentration stock.
| Concentration (nM) | Assay buffer (μl) | Deacetylated standard (μl) | Plate location | Final volume (μl) |
|---|---|---|---|---|
| 0 | 100 | 0 | A2 | 100 |
| 50 | 98 | 2 | A3 | 100 |
| 250 | 90 | 10 | A4 | 100 |
| 500 | 80 | 20 | A5 | 100 |
| 1000 | 60 | 40 | A6 | 100 |
| 1500 | 40 | 60 | A7 | 100 |
| 2000 | 20 | 80 | A8 | 100 |
| 2500 | 360 | 40 | A9 | 100 |
Support Protocol 1: Isolation of nuclear protein from brain tissues
In the present support protocol, mouse brain cerebral cortex, following traumatic brain injury (TBI), are used as a nuclear protein source since Class I and Class II HDACs are highly expressed in this tissue. Cortex tissue samples were collected from mouse brain following the TBI procedure (see Reddy et al 2018 for additional information on inducing TBI injury in mice).
All materials and equipment for tissue preparation and Pierce BCA protein concentration assay are commercially available. The Pierce BCA assay is a concise, high-precision kit to measure total protein concentration compared to a standard curve using Bovine Serum Albumin as the protein standard. To reproduce this experiment, equipment and materials have been listed with the company source, comments, and catalog number if needed. There are many similar products available that will produce comparable results.
Materials
Nuclear Extraction Kit comprising 10× Pre-Extraction Buffer, Extraction Buffer, 1000× DTT Solution, 1000× Protease Inhibitor Cocktail (PIC) Stored at 4°C. Abcam #ab113474
1.5 ml – Microcentrifuge Tubes
15 ml – Conical Tubes
Pipettes and Pipette Tips
Pierce BCA Protein Assay Reagent A, Thermo Scientific #23223, Room Temperature
Pierce BCA Protein Assay Reagent B, Thermo Scientific #23224, Room Temperature
Pierce Bovine Serum Albumin Standard, 2 mg/ml, Thermo Scientific #23210, Room Temp
Thermo Scientific Pierce 96-Well Plates, Thermo Scientific #15041
Distilled Water, Room Temperature
Benchtop Centrifuge, up to 14,000 RPM, refrigerated 4°C
Branson Sonifier or any Homogenizer
Vortex Mixer
Rad BioTek Synergy2 Plate Reader, BioTek
Nuclear Extraction
Rapidly dissect the whole brain from the mouse, and micro-dissect specific regions if necessary (e.g. hippocampus, cortex, amygdala etc.). Weigh the tissue and flash-freeze tissue in isopentane and dry ice to inhibit proteases. Store at –80 °C until needed.
- Prepare working reagents.
- 1× Pre-Extraction Buffer: Dilute 10× Pre-Extraction Buffer with cold distilled water at a ratio of 1:10 (e.g. 1 ml 10× Pre-Extraction Buffer + 9 ml distilled water). Add 10 μl of DTT and 10 μl PIC to 1× Pre-Extraction Buffer (1:1000 ratio). If excess solution is made, store at 4 °C.
- Extraction Buffer: Add 10 μl DTT to 10 ml Extraction Buffer (1:1000 ratio). No PIC (Protease Inhibitor Cocktail) is needed when the samples are used for a nuclear HDAC activity/inhibition assay.
Add 10 ml 1× Pre-Extraction Buffer containing 10 μl DTT and 10 μl PIC per gram of tissue per sample (as 10% w/v). If weight of tissue is 100 mg, add 1ml 1× Pre-Extraction Buffer.
Homogenize samples with Branson Sonifier. Place sonifier tip in liquid and sonicate in 5 second increments at 20-30% max power until completely homogenized (no visible tissue pieces). Wash the probe before and after each sample with distilled water to prevent cross contamination. Cool the sample and probe in ice to avoid denaturing the protein, as the sonication produces considerable heat. Avoid foaming the homogenate by making sure the probe tip remains completely immersed in the homogenate solution during sonication. Keep homogenates on ice until all samples have been sonicated. Total homogenate can be stored at –80°C or used immediately.
Incubate samples on ice for 15 minutes and centrifuge for 10 minutes at 12,000 rpm at 4 °C.
Remove the supernatant (S1) and aliquot into 2 microcentrifuge tubes. Store at –80 °C.
Add 100 μl Extraction Buffer working solution (with DTT) for each 100 mg tissue to the nuclear pellet (P1). Vortex sample, then sonicate three times for 10 seconds to increase nuclear protein extraction.
Incubate the extract on ice for 15 minutes. Vortex samples every 3 minutes.
Centrifuge the sample for 15 minutes at 14,000 rpm at 4°C and transfer the nuclear protein supernatant (S2) into new microcentrifuge tubes. Keep the pellets (P2) in storage at –80 °C.
Transfer 6 μl of S2 with 54 μl 1× Extraction Buffer working solution to a clean microcentrifuge tube for protein concentration calculation (10× dilution). The final S2 protein concentration will be 10× the concentration of the reading from the BioTek Synergy 2. This dilution conserves protein for future assays. Use clean pipet tips for each sample.
Aliquot the remaining S2 into 2 microcentrifuge tubes and store at –80 °C until needed.
Measure protein concentration of nuclear extract using Pierce BCA Assay.
Pierce Bicinchoninic Acid (BCA) Assay
Prepare the tissue extracts as described above, then follow the kit instructions provided in the Assay Kit Manual or at the URL cited below.
Assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0011430_Pierce_BCA_Protein_Asy_UG.pdf Measure absorbance at 562 nm.
Notes
-
Increasing the incubation time or ratio of sample volume to working reagent increases the net 562 nm measurement for each well and lowers both the minimum detection level of the reagent and the working range of the assay. If all standards and unknowns are treated identically, such modifications may be useful.
Subtract the average 562 nm absorbance measurement of the Blank standard replicates from the 562 nm measurements of all other individual standard and unknown sample replicates.
Prepare a standard curve by plotting the average Blank-corrected 562 nm measurement for each BSA standard vs. its concentration in μg/ml. Use the standard curve to determine the protein concentration of each unknown sample.
REAGENTS AND SOLUTIONS
Prepare the following chemicals and reagents using the methods below. These stock solutions can be made ahead of time and stored until needed.
-
Assay Buffer, pH 8.0, 500 ml
50 mM Tris 3.03 g 137 mM NaCl 3.80 g 2.7 mM KCl 100.7 mg 1 mM MgCl2.6H2O 101.6 mg Adjust pH with HCl to 8.0, adjust volume with distilled water to 500 ml, aliquot to 25 ml per 50 ml centrifuge tubes for storing. Store at −20°C for up to 1 month.
-
Substrate (50 mM, Boc-Lys(Ac)-AMC, 100 μl)
Boc-Lys(Ac)-AMC 2.23 mg DMSO 100 μl Dissolve the substrate in DMSO and aliquot to 10 μl/micro tube for storing. Store at −80°C for up to 4 months.
-
Standard Inhibitor TSA Stock (1 mM, 5 ml)
Trichostatin A 1.52 mg DMSO 5 ml Dissolve TSA in DMSO and aliquot to 1 ml for 3 vials and the remaining 2 ml into 100 μl/microtubes for storing. Store at −80°C for up to 4 months.
-
Buffer for Developer, pH 8.0, 500 ml
Tris 3.03 g NaCl 5.54 g Dissolve the salts in 500 ml distilled water and adjust pH to 8.0 using NaOH. Store at 4 °C for up to 4 months.
-
Developer (20×), pH 8.0, 10 ml
Trypsin (2.14 mM) 500 mg Trichostatin A (2 μM) 20 μl of 1 mM Buffer for Developer 10 ml Dissolve Trypsin and Trichostatin A in developer buffer (Buffer for Developer from above) and adjust pH to 8.0 using HCl or NaOH. Aliquot 1 ml/microcentrifuge tube (1.7 ml tubes) and store at −20°C for up to 1 month.
-
Deacetylated Standard (Boc-Lys-AMC, 10 mM, 1 ml)
Boc-Lys-AMC 4.04 mg DMSO 1000 μl Dissolve the substrate in DMSO and aliquot to 20 μl/microtube for storing. Store at −80°C for up to 4 months.
COMMENTARY
Background Information
Epigenetic mechanisms include histone modifications, DNA methylation, and mRNA-based transcriptional control. A primary histone modification associated with many neurological disorders is lysine acetylation of histone tails (Kobow and Blumcke, 2014). Chromatin is often characterized as existing in two higher-order structures: euchromatin and heterochromatin. Euchromatin refers to the loosely packaged and open formation that is transcriptionally active, whereas heterochromatin is highly compact and transcriptionally inactive (Kouzarides, 2007). HDACs, and by extension HDAC inhibitors, can play a crucial role in regulating these changes in gene transcription via condensing or expanding the chromatin structure. Removal of the acetyl group by HDACs increases the positive charge of the histone and confers a more compact state, characteristic of gene repression. HDACs can also remove acetyl moieties from transcription factors, thereby further suppressing gene activity (Morris et al., 2010). Of the four HDAC superfamilies, classes 1 and 2 are the most relevant in neurological conditions as they are highly expressed in the brain (Gray and Ekstrom, 2001; Carey and La Thangue, 2006; Table 1). HDAC inhibitors are important tools for unraveling the pathology and mechanistic basis of brain diseases. There are many HDAC inhibtors such as TSA, sodium butyrate, vorinostat (SAHA), valproic acid, and entinostat (MS-275) that are used in animals (Ververis et al., 2013; Reddy et al., 2018). Sodium butyrate is widely used for broad-spectrum HDAC inhibition and consequent augmentation and acetylation of H3 and H4 histone proteins in the brain (Ferrante et al., 2003; Deutsch et al. 2008; Takuma et al., 2014). HDAC inhibitors can produce antidepressant-like and anxiolytic behavioral response in rodent models, which provides additional evidence for the brain bioavailability of these agents as well as the critical role of epigenetic modifications in CNS disorders (Covington et al., 2009; Golden et al., 2013). For additional detail on epigenetics and brain function, see Volmar and Wahlestedt, 2015.
Critical parameters
Selection of an optimal HDAC inhibitor and its dose are critical for this protocol. The dose of butyrate used in our study (600 mg/kg) is known to inhibit multiple HDACs and reliably increases the acetylation of H3 and H4 histones in the brain and other tissues (Ferrante et al., 2003; Takuma et al., 2014). Sodium butyrate effectively inhibits downstream H4 and H4 histone signaling in cells in vitro and after administration in animals leading to a wide range of molecular and behavioral responses (Davie, 2003; Minamiyama et al., 2004).
The most critical parameter for the fluorescent expression of HDAC activity is the purification of the nuclear protein lysate and optimal estimation of concentration by the BCA assay since incorrect plate readings will skew the final results. Care should be taken during the homogenizing stage of nuclear protein extraction and when pipetting small amounts of solution. If the sonifier tip is not completely immersed in the liquid, foaming of the tissue lysate can occur, causing protein to spray out or adhere to the walls of the tube. Furthermore, gently mixing the 96-well plate before BioTek Synergy2 reading will help to clear any protein or substrate that may adhere to the well walls. Once the protein has been extracted, store the extracts in vials at –80°C until needed. Heating the samples unnecessarily can denature the protein, which ultimately will affect the validity of the results. Frequent thawing and re-freezing can also damage the protein.
Trouble shooting
Low protein expression is the most common problem with this procedure. Brain tissue is high in both water and lipid content; therefore, it can sometimes be difficult to extract sufficient amounts of protein from the tissue samples. Moreover, structures in the brain contain varying levels of protein. Selecting large brain regions or highly defined structures will help to yield higher protein levels in the nuclear extract. In contrast, if the readings are too high across all the wells such that the data cannot be accurately interpreted, the cell/protein density may be too high. In this case, the cells/protein should be reduced. Once adequate protein content has been established, it is important to compare the unknown samples to positive tissue controls and to samples with a standard HDAC inhibitor (e.g. TCA).
Erratic duplicate or triplicate values may be caused by poor pipetting techniques or by the presence of air bubbles in the well. Improper pipetting techniques can also lead to unequal numbers of cells/protein content in the well. Be careful to release the liquid from the pipette gently, so that the contents do not splash onto the well walls or outside of the plate. Air bubbles can be removed by gently tapping the side of the well plate with a finger.
ANTICIPATED RESULTS
The described fluorometric HDAC assay can be used to determine the ability of a compound to inhibit HDAC activity. We performed this assay on cortical samples obtained from control and sodium butyrate-treated (600 mg/kg/day, i.p.) adult C57BL6 mice after sham or TBI. TBI was induced using a controlled-cortical impact that was induced at a velocity of 4.0 m/s and depth of 2-mm, using a 3-mm diameter rounded tip. Mice in the treatment cohort received 600 mg/kg sodium butyrate (i.p.) at 1 hr post-injury and 1 hr before dissection. Sodium butyrate is a short chain fatty acid Class I and II HDAC inhibitor. This dose of sodium butyrate is neuroprotective in a mouse model of temporal lobe epileptogenesis (Reddy et al., 2018); therefore, we selected the same regimen for the present study to determine the extent of HDAC inhibition. At 4 h and 24 hr post-TBI, brain tissues were flash frozen and dissected for the cerebral cortex. Tissues were stored at –80°C until needed for nuclear protein extraction.
To collect and transform the raw fluorescence emission units (FEU) data output into a form that could be used in publication, first export the two forms of data into a spreadsheet program (i.e. Excel): raw absorbance at 562 nm and “Blank” 562 nm absorbance. The “blank” data (well A1) compensates for the baseline absorption/background reading of the solutions. If your plate-reader software does not provide “Blank” 562 absorbance data, simply subtract the value of the “blank” well (A1) from all of the other well readings. If duplicates or triplicates were analyzed for the unknown samples, average those values so that there is now a single value of FEU for each unknown sample. Use the standard concentration curve (well rows A) to determine the estimated values of HDAC inhibition. Normalize the estimated values of the unknown samples (sham or TBI groups with/without butyrate administration in vivo) to the wells containing TSA as a ratio. The ratio of HDAC activity can be used to compare results in different groups. Nuclear extract from HCT116 cells can be used as a positive reference for HDAC activity as well as a way of limiting potential errors in routine experiments.
Results from this protocol show sodium butyrate reduced HDAC activity in the butyrate-treated sham cortex at 4 hours post-sham (1.08 ± 0.01, n=6 mice) compared to the control sham group (n = 6 mice, 1.48 ± 0.17) (Fig. 1A). These results confirm inhibition of HDAC activity in mice following sodium butyrate treatment, consistent with earlier reports (Ferrante et al., 2003; Takuma et al., 2014). In addition, we determined the HDAC activity following TBI. Our results show HDAC activity in the cortex of TBI mice was significantly increased compared to those of sham mice at 4 hours (Fig. 1A) and 24 hours post-injury (Fig. 1B). Thus, these results demonstrate that brain trauma such as TBI can trigger HDAC enzyme activation in the cerebral cortex, possibly as a homeostatic compensation due to injury.
Fig. 1.
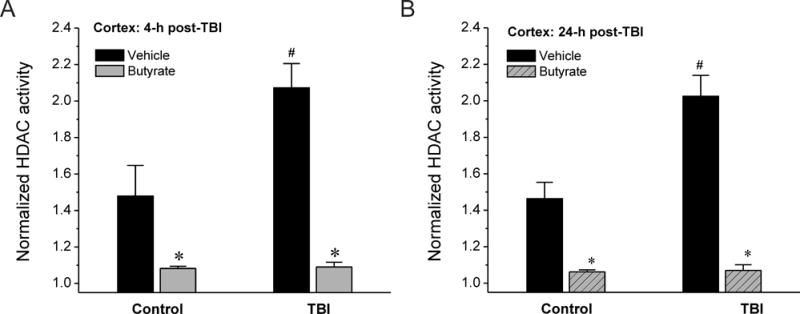
Changes in histone deacetylase (HDAC) activity in the cerebral cortex at 4 hours (A) and 24 hours (B) after traumatic brain injury (TBI) in adult mice. The HDAC inhibitor sodium butyrate significantly attenuated the HDAC activity in the cortex. The data were normalized to samples with trichostatin A and presented as the mean ± SEM. *p< 0.05 vs control or TBI without sodium butyrate. #p< 0.05 vs control without sodium butyrate.
Time considerations
There are several points in this procedure where the protocol can be paused a resumed at a later point, given that the products are stored at the proper temperatures. For example, dissected tissues can be stored at –80°C either directly after dissection or after homogenization with Pre-Extraction Buffer, the BCA assay can be performed any time after nuclear extraction has been completed, and the HDAC activity assay can be performed on a day separate from that of either the nuclear extraction or BCA assay following those procedures. The amount of time required to complete the nuclear extraction protocol depends on the number of samples being run. Our laboratory concurrently processes 12–24 tissue samples, which takes 3–4 hr. The BCA assay requires approximately 1 hour and thus can be run on the same day as nuclear extraction, so to reduce sample freezing/thawing. The HDAC activity assay requires approximately 2 hr if the working reagents are prepared ahead of time.
CONCLUSIONS
Despite the obvious correlation of HDAC activity with long-term changes in brain function, the signaling mechanisms and pathways underlying the HDAC regulation of neuronal function remain poorly understood. Most experimental studies rely on the outcomes of HDAC measurements, with the outcomes expressed as either increased or decreased expression of various HDACs. It is unclear whether such expression is associated with HDACs functional activity, either upstream or downstream in the cell signaling process. The present protocol describes the measurement of HDAC activity and its inhibitors in brain tissues including cerebral cortex.
Acknowledgments
This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs through the FY 2015 Epilepsy Research Program under Award #W81XWH-16-1-0660 (to D.S.R.). R.H.D. is supported by NCI R01 CA122959, NCI PREVENT grant HHSN26100004, the John S. Dunn Foundation, and a Chancellor’s Research Initiative from Texas A&M University.
ABBREVIATIONS
- HDAC
histone deacetylase
- mRNA
messenger RNA
- CNS
central nervous system
Footnotes
Conflict of Interest. The authors declare no competing financial interests.
Literature Cited
- Andrews FH, Strahl BD, Kutateladze TG. Insights into newly discovered marks and readers of epigenetic information. Nat Chem Biol. 2016;12(9):662–668. doi: 10.1038/nchembio.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassi S, Tripathi T, Monziani A, Di Leva F, Biagioli M. Epigenetics of Huntington’s Disease. Advances in Experimental Medicine and Biology. 2017;978 doi: 10.1007/978-3-319-53889-1_15. https://doi.org/10.1007/978-3-319-53889-1_15. [DOI] [PubMed] [Google Scholar]
- Carey N, La Thangue NB. Histone deacetylase inhibitors: Gathering pace. Current Opinion in Pharmacology. 2006;6(4):369–375. doi: 10.1016/j.coph.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Clossen BL, Reddy DS. Novel therapeutic approaches for disease-modification of epileptogenesis for curing epilepsy. Biochim Biophys Acta. 2017;1863(6):1519–1538. doi: 10.1016/j.bbadis.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Nestler EJ. Antidepressant actions of histone deacetylase inhibitors. J Neuroscience. 2009;29(37):11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, Maze I, Vialou V, Nestler EJ. Antidepressant action of HDAC inhibition in the prefrontal cortex. The Journal of Neuroscience. 2015;298:329–35. doi: 10.1016/j.neuroscience.2015.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie J. Inhibition of histone deacetylase activity by butyrate. The Journal of Nutrition. 2003;133(7 Suppl):2485S–2493S. doi: 10.1093/jn/133.7.2485S. [DOI] [PubMed] [Google Scholar]
- De Ruijter AJM, van Gennip AH, Caron HN, Kemp S, van Kuilenburg ABP. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochemical Journal. 2003;370(Pt 3):737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descalzi G, Ikegami D, Ushijima T, Nestler EJ, Zachariou V, Narita M. Epigenetic mechanisms of chronic pain. Trends in Neurosciences. 2015;38(4):237–246. doi: 10.1016/j.tins.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch SI, Rosse RB, Long KD, Gaskins BL, Burket JA, Mastropaolo J. Sodium butyrate, an epigenetic interventional strategy, attenuates a stress-induced alteration of MK-801’s pharmacologic action. European Neuropsychopharmacology. 2008;18(8):565–568. doi: 10.1016/j.euroneuro.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. Journal of Neuroscience. 2003;23(28):9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa T, Filippakopoulos P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol. 2017;18(4):246–262. doi: 10.1038/nrm.2016.143. [DOI] [PubMed] [Google Scholar]
- Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Golden SA, Christoffel D, Heshmati M, Hodes GE, Magida J, Davis K, Russo SJ. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nature Medicine. 2013;19(3):337–344. doi: 10.1038/nm.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SG, Ekstrom TJ. The Human Histone Deacetylase Family. Experimental Cell Research. 2001;262(2):75–83. doi: 10.1006/excr.2000.5080. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Kobow K. Epigenetics and Epilepsy. Cold Spring Harb Perspect Med. 2015;5(12):a022731. doi: 10.1101/cshperspect.a022731. Pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Doherty JJ, Dingledine R. Altered Histone Acetylation at Glutamate Receptor 2 and Brain-Derived Neurotrophic Factor Genes Is an Early Event Triggered by Status Epilepticus. Journal of Neuroscience. 2002;22(19):8422–8428. doi: 10.1523/JNEUROSCI.22-19-08422.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull EE, Montgomery MR, Leyva KJ. HDAC inhibitors as epigenetic regulators of the immune system: impacts on cancer therapy and Inflammatory Diseases. Biomed Res Int. 2016 doi: 10.1155/2016/8797206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger S, Nakashima K, Clemenson GD, Mejia E, Mathews E, Ure K, Hsieh J. Epigenetic Modulation of Seizure-Induced Neurogenesis and Cognitive Decline. Journal of Neuroscience. 2007;27(22):5967–5975. doi: 10.1523/JNEUROSCI.0110-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobow K, Bkumcke I. Epigenetic Mechanisms in Epilepsy. Prog Brain Res. 2014;213:279–316. doi: 10.1016/B978-0-444-63326-2.00014-4. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Lee J, Kim Y, Liu T, Hwang YJ, Hyeon SJ, Im H, Ryu H. SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer’s disease. Aging Cell. 2018;17(1) doi: 10.1111/acel.12679. https://doi.org/10.1111/acel.12679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamiyama M, Katsuno M, Adachi H, Waza M, Sang C, Kobayashi Y, Sobue G. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Human Molecular Genetics. 2004;13(11):1183–1192. doi: 10.1093/hmg/ddh131. [DOI] [PubMed] [Google Scholar]
- Morris MJ, Karra AS, Monteggia LM. Histone deacetylases govern cellular mechanisms underlying behavioral and synaptic plasticity in the developing and adult brain. Behavioural Pharmacology. 2010;21(5–6):409–419. doi: 10.1097/FBP.0b013e32833c20c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nian H, Bisson WH, Dashwood WM, Pinto JT, Dashwood RH. Alpha-keto acid metabolites of organoselenium compounds inhibit histone deacetylase activity in human colon cancer cells. Carcinogenesis. 2009;30(8):1416–1423. doi: 10.1093/carcin/bgp147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran P, Dashwood WM, Li L, Kang Y, Kim E, Johnson G, Dashwood RH. Nrf2 status affects tumor growth, HDAC3 gene promoter associations, and the response to sulforaphane in the colon. Clinical Epigenetics. 2015;7(1):102. doi: 10.1186/s13148-015-0132-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran P, Delage B, Dashwood WM, Yu TW, Wuth B, Williams DE, Dashwood RH. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Molecular Cancer. 2011;10(68):1–18. doi: 10.1186/1476-4598-10-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran P, Kidane AI, Yu TW, Dashwood WM, Bisson WH, Lohr CV, Dashwood RH. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics. 2013;8(6):612–613. doi: 10.4161/epi.24710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy SD, Clossen BC, Reddy DS. Epigenetic Histone Deacetylation Inhibition Prevents the Development and Persistence of Temporal Lobe Epilepsy. Journal of Pharmacology and Experimental Therapeutics. 2018;364(1):87–109. doi: 10.1124/jpet.117.244939. [DOI] [PubMed] [Google Scholar]
- Seto E, Toshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014;6(4):a018713. doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma K, Hara Y, Kataoka S, Kawanai T, Maeda Y, Watanabe R, Matsuda T. Chronic treatment with valproic acid or sodium butyrate attenuates novel object recognition deficits and hippocampal dendritic spine loss in a mouse model of autism. Pharmacology Biochemistry and Behavior. 2014;126:43–49. doi: 10.1016/j.pbb.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Verdone L, Caserta M, Di Mauro E. Role of histone acetylation in the control of gene expression. Biochemistry and Cell Biology. 2005;83(3):344–353. doi: 10.1139/o05-041. [DOI] [PubMed] [Google Scholar]
- Ververis K, Hiong A, Karagiannis TC, Licciardi PV. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics. 2013;7:47–60. doi: 10.2147/BTT.S29965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volmar CH, Wahlestedt C. Histone deacetylases (HDACs) and brain function. Neuroepigenetics. 2015:20–27. [Google Scholar]
- Wong VS, Langley BC. Epigenetic changes following traumatic brain injury and their implications for outcome, recovery and therapy. Neuroscience Letters. 2016;625:26–33. doi: 10.1016/j.neulet.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younus I, Reddy DS. Epigenetic interventions for epileptogenesis: A new frontier for curing epilepsy. Pharmacology & Therapeutics. 2017;177:108–122. doi: 10.1016/j.pharmthera.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]