

Abstract
Gene expression variation is an important mechanism underlying susceptibility to complex disease. In comparison with tobacco-related lung carcinogenesis, lung cancer in nonsmokers may involve important and etiologically distinct causal pathways. In this study, we conducted a genome-wide association study on spontaneous lung tumor incidence in inbred mice and identified a major susceptibility locus on mouse chromosome 2 (rs27328255, P = 6.68 × 10−7). We then evaluated the correlations of polymorphisms with the transcription of positional candidate genes in normal lungs. Single-nucleotide polymorphism rs27328255 was consistently and strongly associated (P = 7.42 × 10−9) in cis with transcript levels of Xrn2. We further showed that Xrn2 promotes proliferation and inhibits squamous differentiation in human lung epithelial cells and polymorphisms in human homolog XRN2 are associated with human lung cancer (rs2025811, P = 1.90 × 10−3, OR = 1.20). We conclude that genetic variants regulating Xrn2 expression in cis are determinants of spontaneous lung tumor susceptibility in mice and have genetic equivalents in lung cancer susceptibility in human beings. Identifying Xrn2 as a major candidate for spontaneous lung cancer has important implications for the diagnosis and treatment of lung cancer as well as delineation of the mechanisms underlying the genesis of lung cancer in nonsmokers.
Keywords: expression, association, lung cancer, mouse
Introduction
Lung cancer is the leading cause of cancer death for both men and women in the United States. In 2009, there was an estimated 219 440 cases of lung cancer diagnosed and only 15% of those patients are expected to survive for >5 years (Jemal et al., 2009). Genetic factors have important functions in lung cancer susceptibility (Matakidou et al., 2005), and several studies have recently been published on genetic factors that underlie susceptibility to lung cancer in smokers. Genome-wide association studies (GWASs) on lung cancer populations of European ancestry identified associations of common variants on chromosomal regions 15q24-25.1, 5p15.33, and 6p21.33 with lung cancer susceptibility (Amos et al., 2008; Hung et al., 2008; Liu et al., 2008; McKay et al., 2008; Thorgeirsson et al., 2008; Wang et al., 2008; Rafnar et al., 2009).
Mouse lung tumors have histological and molecular features similar to human pulmonary adenocarcinomas (Meuwissen and Berns, 2005). The majority of pulmonary tumors in inbred mice arise as hyperplasias, progress to adenomas, and ultimately result in carcinomas, paralleling the progression of lung tumors in human beings (Foley et al., 1991). Many genetic alterations identified in mouse models have a common genetic basis in human lung cancer and will identify pathways contributing to human lung cancer (You and Bergman, 1998). Mouse models have a number of advantages that favor detection of genetic associations that might remain elusive in human populations, including large variations in disease susceptibility among inbred strains, controlled environmental influences, generation of large cohorts, and a homozygous-inbred genome. These features are particularly useful when studying common diseases such as lung cancer in which environmental exposure represents an important factor contributing to the development and progression of disease.
Genetic studies of spontaneous lung tumor are merited for several reasons. Tobacco smoking is well established as the major risk factor for lung cancer, contributing to a 10-fold increase in risk in long-term smokers compared with nonsmokers (Doll and Peto, 1981). This is in contrast to small effects typically seen by common variants for most common diseases, which usually confer <50% increase in risk (corresponding to odd ratios <1.5) (Couzin and Kaiser, 2007). As a result, smoking behavior can be a major confounding variable in genetic association studies if smoking behavior of lung cancer cases and controls is even slightly unmatched. Owing to the potential confounding effect of smoking, genetic associations identified in lung cancer case–control studies could be primarily related to nicotine dependence and/or behavioral factors leading to a propensity to smoke and develop disease, rather than a direct effect of a genetic susceptibility factor per se (Amos et al., 2008; Liu et al., 2008; Wacholder et al., 2008). In addition, although most lung cancer deaths are caused by active cigarette smoking, 10–15% of patients diagnosed with lung cancer have never smoked, representing 17 000–26 000 deaths annually in the United States. This number of cancer deaths would rank among the top six to eight most common fatal cancers, if considered as a separate disease category (Thun et al., 2006). The mechanisms of nonsmoker lung carcinogenesis may represent important and etiologically distinct causal pathways, in comparison with tobacco-related lung carcinogenesis (Powell et al., 2003; Wong et al., 2003; Zeka et al., 2006).
To eliminate the confounding effects of cigarette smoking that may arise in human association studies and to identify genetic factors that are not directly related to carcinogen exposure, we performed an in silico GWAS of spontaneous lung tumors in inbred mice. Then, we systematically investigated the correlations of polymorphisms with the transcription of positional candidate genes in normal lung tissues. The singlenucleotide polymorphisms (SNPs) associated with spontaneous lung tumorigenesis were consistently and strongly associated in cis with transcript levels of Xrn2. Xrn2 is a member of 5′–>3′ exoribonucleases (XRNs) that comprise a large family of conserved enzymes in eukaryotes with crucial functions in RNA metabolism and RNA interference. We further showed that Xrn2 inhibited squamous differentiation in human lung epithelial cells and promotes proliferation and polymorphisms in human homolog XRN2 associated with lung cancer in human beings.
Results
Genome-wide association analysis of spontaneous lung tumors
To identify the genetic determinants of spontaneous lung tumors, we conducted a GWAS of spontaneous incidence of lung tumor in inbred mice. We first evaluated the cumulative distribution of P-values from our GWAS using the efficient mixed-model association (EMMA) approach. The distribution of observed P-values was similar to the expected uniform distribution [0, 1], indicating no inflation of test statistics from population structure or any other form of bias (Figure 1a). Therefore, any bias because of population structure and genetic relatedness among inbred strains has been largely eliminated by the EMMA approach. Using the permutation analysis, we established the genome-wide significance level of P = 8.24 × 10−7. The GWAS identified a cluster of SNPs on distal mouse chromosome 2 that were in strong linkage disequilibrium and strongly associated with spontaneous incidence of lung tumor in inbred mice (Figure 1b; Table 1). This major susceptibility locus spans ~1.65 megabases (145.70–147.35 Mb on chromosome 2) and was provisionally named Spontaneous Lung Tumor Susceptibility locus 1 (SLTS1). In contrast, there were no association signals on distal mouse chromosome 2 in a GWAS we earlier performed on carcinogen-induced lung tumor in inbred mice, suggesting that it is a distinct locus (Liu et al., 2006). To increase the SNP coverage, we chose additional 81 SNPs evenly distributed in the SLTS1 locus from the Perlegen SNP database and genotyped these SNPs in the inbred strains. The strongest evidence of association for the SLTS1 locus was at SNP rs27328255 (P = 6.68 × 10−7).
Figure 1.
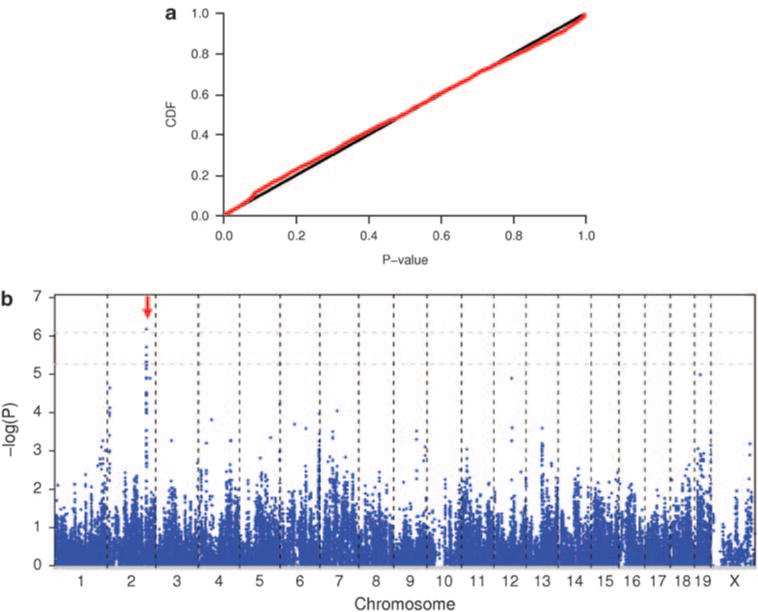
The results from genome-wide association analysis of spontaneous lung tumor in inbred mice. (a) Cumulative distribution of P-values from the EMMA approach in the GWAS. cdf refers to cumulative distribution function. (b) Scatter plot of P-values in −log scale for 84 010 SNPs. The two gray lines are genome-wide thresholds of P=0.05 and P=0.10, which correspond to point-wise P-values of 8.24 × 10−7 and P = 5.43 × 10−6, respectively.
Table 1.
SNPs associated with spontaneous incidence of lung tumor in inbred mice
| dbSNPa | Chr | Pos (bp) | Allelesb | MAF | Mean SI (%)c
|
P-values | |
|---|---|---|---|---|---|---|---|
| Mi/Mi | Ma/Ma | ||||||
| rs27328255 | 2 | 147 067 787 | T/C | 0.500 | 33.6 | 1.8 | 6.68E – 07 |
| rs13476822 | 2 | 147 204 386 | G/A | 0.478 | 32.9 | 1.8 | 1.94E – 06 |
| rs33240516 | 2 | 146 756 804 | T/C | 0.455 | 34.6 | 1.8 | 3.16E – 06 |
| rs27294933 | 2 | 146 900 320 | G/A | 0.455 | 33.1 | 1.8 | 4.79E – 06 |
Abbreviations: LD, linkage disequilibrium; MAF, minor allele of frequency; SNPs, single-nucleotide polymorphisms.
SNPs with P-value <P = 5.43 × 10−6 (that is, genome-wide threshold P = 0.10) were presented; for those SNPs that are in complete LD, only one of them was presented.
Alleles in bold are minor allele of SNPs.
SI indicates spontaneous incidence of lung tumor (%); Mi is a minor allele and Ma is a major allele.
Eleven annotated genes were identified on the SLTS1 locus, including Rin2 (Ras and Rab interactor 2), Nat5 (N-acetyltransferase 5), Crnkl1 (Crn, crooked neck-like 1), 4930529M08Rik, Insm1 (insulinoma-associated 1), A230067G21Rik, Ncrna00153, Xrn2 (5′-3′ exoribonuclease 2), Nkx2-4 (NK2 transcription factor related, locus 4), Nkx2-2 (NK2 transcription factor related, locus 2), and Pax1 (paired box gene 1). The marker rs27328255 showing the strongest evidence of association on the SLTS1 is located within 160 kb downstream of Xrn2 (Figures 2a and b). This disease-associated marker accounted for about 60% of variation in spontaneous incidence of lung tumor in mouse-inbred strains. Mice carrying the allele T of rs27328255 have on an average a 32% higher incidence of spontaneous lung tumor than noncarriers.
Figure 2.
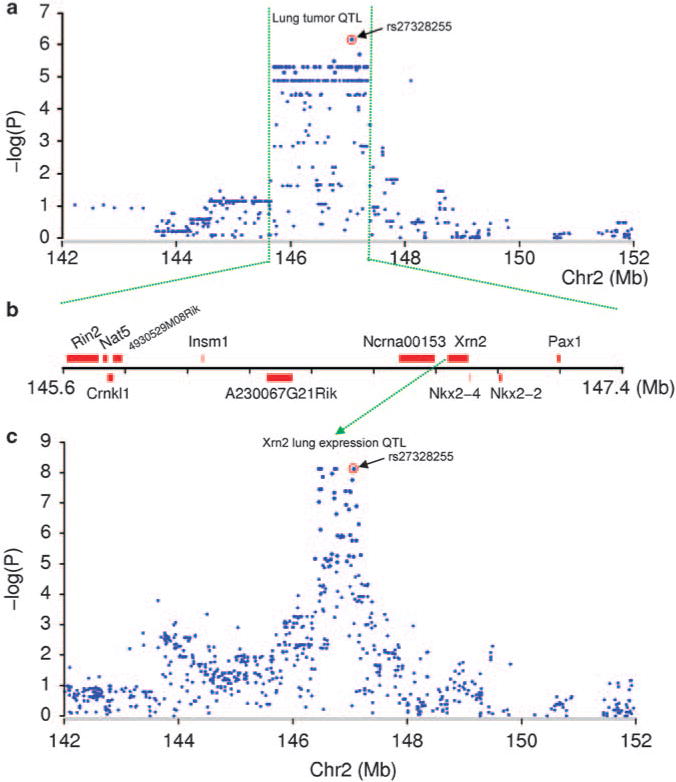
SNPs are associated with spontaneous lung tumor susceptibility and Xrn2 expression. (a) SNPs in the SLTS1 locus on mouse chromosome 2 are associated with spontaneous incidence of lung tumor. (b) Physical map of the SLTS1 locus. (c) SNPs in the SLTS1 locus are associated with Xrn2 expression.
Cis-regulation of Xrn2
Expression data were available for 7 of 11 annotated genes on the SLTS1 locus. The other four genes are either not expressed in lung or expressed at levels below the limit of detection by microarrays. We found that the expression level of one gene, Xrn2, was strongly and consistently associated with the same SNPs identified in the association analysis of spontaneous lung tumor (rs27328255, P = 7.42 × 10−9) (Figure 2c; Supplementary Figure S1). The allele T of rs27328255 is associated with a 16% increased expression in Xrn2 as compared with the allele C. Xrn2 expression accounted for 35% of the variation in spontaneous incidence of lung tumor in inbred strains (P = 6.82 × 10 13, adjusted R2 = 0.35) (Figure 3a). To confirm this finding, we measured mRNA levels of Xrn2 in normal lung in the three most resistant (AKR/J, C57/L, and CE/J) and susceptible (MA/MJ, SWR/J, and A/J) inbred strains using the quantitative RT–PCR analysis (Figure 3b). We found that Xrn2 expression in lung is significantly higher in susceptible than in resistant inbred strains (P = 4.71 × 10−5). None of the other transcripts on the SLTS1 locus or elsewhere in the genome showed a significant relationship to the disease-associated SNPs in our expression study.
Figure 3.
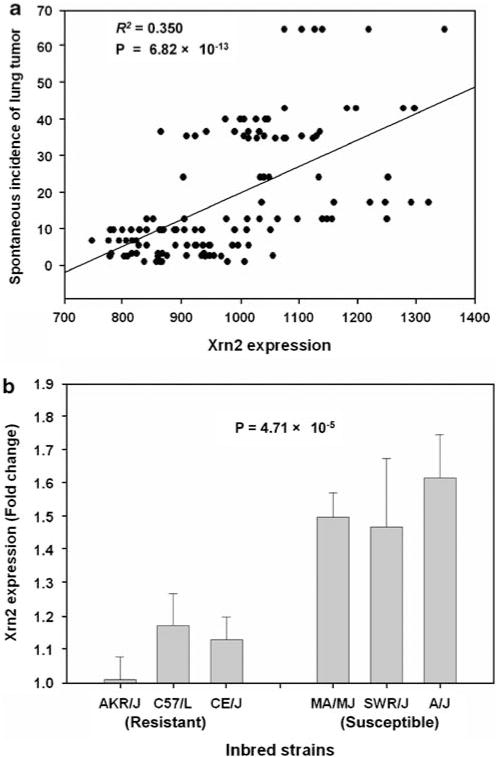
Association of Xrn2 expression with spontaneous lung tumor. (a) Correlation between Xrn2 expression with spontaneous incidence of lung tumor. Nineteen strains that have available data on both Xrn2 expression from microarrays and spontaneous incidence of lung tumor were analyzed. (b) Difference in Xrn2 expression from quantitative RT–PCR between the three most susceptible and resistant inbred strains, three male, and three female for each strain, P-value = 4.71 × 10−5.
Xrn2 modulates epithelial differentiation and promotes proliferation
We performed functional assays to examine the effects of Xrn2 expression on lung tumorigenesis. Xrn2 has been found to modulate differentiation in human lymphocytes (Park et al., 2007). We, therefore, hypothesized that Xrn2 may negatively regulate differentiation in human lung epithelial cells, thereby promoting proliferation and tumorigenesis. To determine the effect of Xrn2 on differentiation of a relevant lung epithelial cell type, we overexpressed Xrn2 in human Beas-2B cells. Beas-2B cells are of lung epithelial origin and retain the ability to be induced into squamous epithelial differentiation by 10% fetal calf serum (FCS) (Ke et al., 1988; Kuntz et al., 2006). To study differentiation, we used involucrin, a known marker of squamous epithelium and squamous differentiation in normal and tumor tissues including lung tumors (Said et al., 1983; Walts et al., 1985). Involucrin is a crosslinker of the keratin envelope in epithelial cells. Using this marker, we measured squamous differentiation in Beas-2B cells cultured with 10% FCS for 72 h. Immunoblot analysis shows transient expression and overexpression of HAtagged Xrn2 in Beas-2B cells (Figure 4a). Growth of Beas-2B cells is arrested under FCS; however, Xrn2 overexpression promotes proliferation under these differentiating conditions (Figure 4b). In cells transfected with vector control, FCS induced a 14-fold increase in involucrin mRNA (Figure 4c). Cells overexpressing Xrn2 exhibit only an eightfold increase in involucrin mRNA after FCS treatment. This translates into a 44% inhibition of involucrin transcript induction with Xrn2 overexpression (P<0.0002). Beas-2B cell morphology under differentiating conditions is drastically affected by Xrn2 overexpression. Cells with vector alone grown with 10% FCS have increased size; seem less migratory as they grow in small, tightly associated colonies; and take on a ‘fried egg’ appearance (Figure 4d). Cells overexpressing Xrn2 under the same conditions seem normal, compact, migratory, and proliferative. Although we found no observable effect of Xrn2 overexpression on the proliferation of one human and two mouse lung tumor cell lines in culture (data not shown), Xrn2 overexpression clearly inhibits differentiation and promotes proliferation in immortalized human lung epithelial cells under differentiating conditions.
Figure 4.
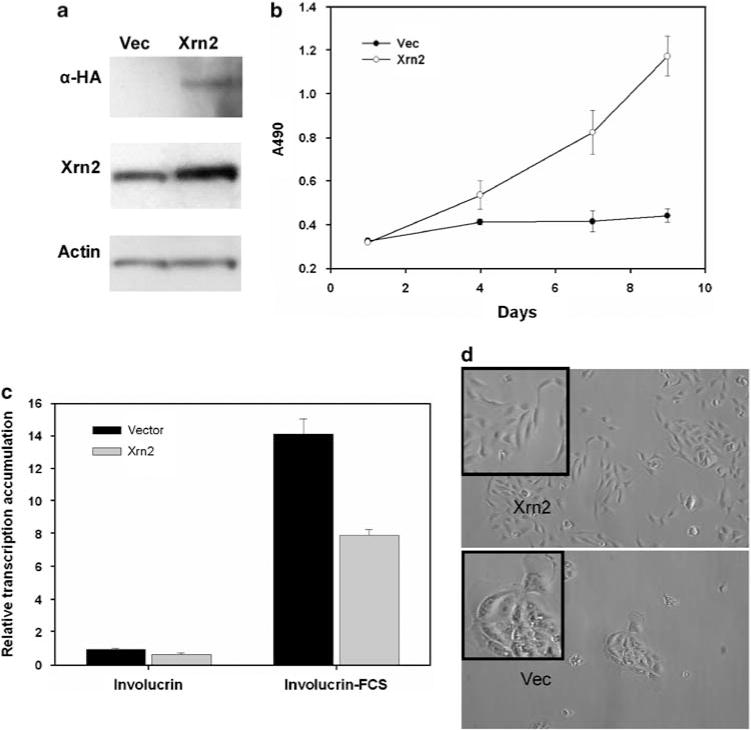
Xrn2 enhances proliferation and inhibits expression of the squamous differentiation marker involucrin in Beas2b cells. (a) Immunoblot for HA-tagged Xrn2 expression in Beas-2B cells. (b) MTT assay for proliferation of Beas-2B cells cultured under nondifferentiating conditions (SF) or differentiating conditions (FCS). (c) Quantitative real-time PCR to detect involucrin transcript accumulation in Beas-2B cells overexpressing Xrn2 cultured under nondifferentiating conditions with serum-free media (SF) or differentiating conditions with FCS. (d) Micrographs of Beas-2B cells cultured under differentiating conditions (FCS). Insets depict magnifications of representative areas of the micrographs to show differences in morphology.
Association of XRN2 with human lung cancer
Human XRN2 shares high homology with mouse Xrn2, maps to 20p11.2-p11.1 and is detected in several human tumor tissues (Li et al., 2005). We searched the ONCOMINE microarray database to study the expression of XRN2 in various human tumor tissues (http://www.oncomine.org). Twenty-three records in the database indicated differential expression of XRN2 between normal and tumor tissues (P<0.01). Except for one invasive breast cancer study, all the others observed that XRN2 was consistently up-regulated in tumor tissues, including brain, cervix uteri, colon, lung, head and neck, prostate, testis cancers, and myeloma (Supplementary Table S3). To evaluate the association of XRN2 in human beings, we analyzed the publicly available lung cancer GWAS datasets from the International Agency for Research on Cancer (IARC) (Lyon, France) (Hung et al., 2008). The IARC lung cancer study contained 1989 lung cancer cases and 2625 controls from six central European countries. The data from the IARC indicated associations of SNPs in XRN2 with human lung cancer (rs2025811, P = 1.90 × 10−3, OR = 1.20, 95% CI = 1.07–1.35) (Supplementary Table S4). The minor allele C of rs2025811 is associated with increased risk of lung cancer with its frequency of 17.16% in cases versus 14.73% in controls. In fact, SNPs in XRN2 are among the most significant hits for lung cancer on chromosome 20p11. These data suggest that Xrn2 in mice has a common genetic equivalent in lung cancer susceptibility in human beings.
Discussion
Variation in gene transcription is an important mechanism in mediating disease susceptibility, and the transcript abundances of genes may be directly modified by polymorphisms in regulatory elements (Schadt et al., 2003; Morley et al., 2004; Moffatt et al., 2007; Cookson et al., 2009). Here, we integrated a GWAS of spontaneous incidence of lung tumor and expression QTL analysis in normal lung tissue from inbred mice to identify Xrn2 as a major candidate gene for spontaneous lung tumor susceptibility. Xrn2 possesses 5′–>3′ exoribonuclease activity and promotes the termination of transcription by RNA polymerase II. It has been found to have an important function in cell differentiation in human lymphocytes (Park et al., 2007). Our study further showed that Xrn2 regulates squamous differentiation in human lung epithelial cells and promotes cell proliferation under differentiating conditions. Although there is no direct evidence to support the function of squamous differentiation in lung adenoma formation, a relationship between differentiation and tumorigenesis is evident. Several studies (Borczuk et al., 2003; Borczuk and Powell, 2007) have shown that histologic diversity in cancers of the lung is accompanied by various degrees of differentiation arrest of common progenitor cells and that the grade of lung tumors is inversely proportional to terminal differentiation. Lung adenocarcinomas have been shown to express involucrin focally indicating foci of adenosquamous differentiation (Said et al., 1983). This study used involucrin expression to show inhibition of a terminally differentiated phenotype in lung airway epithelial cells, and to hypothesize that such inhibition in lung tissues promotes the formation of these tumors of epithelial origin.
As our results in mice indicated that Xrn2 affected lung tumor susceptibility, we examined polymorphisms of XRN2 in human beings in the publicly available lung cancer GWAS datasets from IARC. The IARC data indicated associations of multiple SNPs in XRN2 with human lung cancer. It should be noted that the association of XRN2 in human beings is highly significant (Supplementary Table S4) as it is a hypothesis-driven, candidate-gene association study. However, the IARC study contained mixed samples of smokers and nonsmokers, which may not be eligible for assessing the XRN2 association, because of smoking confounding effects. Future studies are needed to further investigate the validity of this association in large samples of nonsmoker lung cancer. In addition, we observed that the expression of XRN2 is consistently up-regulated in various tumor tissues. In inbred mice, higher expression in Xrn2 is associated with increased risk in spontaneous lung tumor (Figure 3a).
The SLTS1 locus accounted for about 60% of variation in spontaneous incidence of lung tumor in mouse-inbred strains; whereas the Xrn2 expression only accounted for 35% of the variation. Therefore, there may exist other genetic variations to be identified in the SLTS1 contributing to the variation in spontaneous lung tumor. Eleven genes were identified in the SLTS1 locus. Ncrna00153, Nkx2-4, and Nkx2-2 are near Xrn2 and are particularly interesting because of known functional relationship to cancer. Ncrna00153 is a nonprotein coding RNA gene and is homologous to Drosophila bam. Ncrna00153 was suggested to have an important function in mammalian germ line stem cell self-renewal and differentiation (Tang et al., 2008). The transcription factors Nkx2-2 and Nkx2-4 are members of the larger NK2 family of homeodomain containing proteins. Several studies have implicated NKX2 family members in cancer. The related gene Nkx2-1 (also called TITF1, thyroid transcription factor 1) is a key regulator of normal lung development and cell differentiation (Kimura et al., 1996). Recently, characterization of copy-number alterations in primary lung adenocarcinomas identified chromosome 14q13.3, a locus that includes NKX2-1, as the most frequent focal amplification (about 12%) in this tumor type (Weir et al., 2007). NKX2-1 is a lineage-specific oncogene in lung cancer and its amplification and overexpression contribute to lung cancer cell proliferation rates and survival (Kwei et al., 2008). Small-interfering RNA (siRNA)-mediated NKX2-1 knockdown reduced lung cancer cell proliferation specifically in cells with 14q13.3 amplification and expressing NKX2-1 (Kendall et al., 2007; Tanaka et al., 2007; Weir et al., 2007; Kwei et al., 2008). Interestingly, gain of 14q13.3 is significantly associated with EGFR activating mutations, which occur very frequently in nonsmokers with lung adenocarcinomas (Shigematsu and Gazdar, 2006), but is not associated with KRAS or TP53 mutations (Weir et al., 2007). More evidence implicating NKX family members in cancer includes evidence of NKX2-8 in liver cancer and evidence that genetic ablation of Nkx2-9, the mouse homolog of human NKX2-8, deregulates progenitor cells in the large airways and leads to bronchial dysplasia (Tian et al., 2006). In addition, NKX2-2 is an EWS/FLI-regulated gene that is necessary for oncogenic transformation in Ewing’s sarcoma (Smith et al., 2006).
In summary, we identified a major susceptibility locus SLTS1 on distal mouse chromosome 2 for spontaneous lung cancer through GWAS in inbred mice. Subsequent expression QTL analysis established Xrn2 as a primary candidate for the SLTS1 locus. The results indicated that genetic variants regulating Xrn2 expression in cis are determinants of susceptibility to spontaneous lung tumor. We further showed that Xrn2 regulates differentiation in human lung epithelial cells and promotes proliferation, and polymorphisms in XRN2 may also affect human lung cancer susceptibility. The identification of Xrn2 as a major candidate for spontaneous lung tumor has important implications for the diagnosis and treatment of lung cancer as well as delineation of the mechanisms underlying the genesis of lung cancer in nonsmokers.
Materials and methods
Lung tumor and SNP data
In our analysis, we used earlier published data on 5311 mice from 24 inbred strains, which were measured for spontaneous incidence of lung tumors at 18–24 months (Supplementary Table S1). Spontaneous incidence of lung tumors for a strain was defined as percentage of mice of that strain developing lung tumors that occurs naturally in pathogenand carcinogen-free conditions. We acknowledge that there may be a measurement bias in spontaneous incidence of lung tumor from earlier studies. However, this bias should be small as tumor incidence is a simple measurement without treatment of carcinogens, and large sample sizes (221 mice, on average, per strain) were used for phenotype measurement. The spontaneous incidence of lung tumor ranged from 0 to 82% with a mean of 17.7% (±21.4%), suggesting a wide range of variation in spontaneous lung tumorigenesis between inbred mouse strains. These strains were derived from different genealogies, including Castle’s mice (129S1/SvJmJ, A/J, AKR/J, BALB/cByJ, C3H/HeJ, CBA/J, DBA/2J, DBA/1J, LP/J, NZB/BlNJ, NZO/HILtJ, O20, P/J, and RF/J), C57related strains (C57BL/10J, C57BL/6J, C57BR/cdJ, C57L/J, and MA/MyJ), Swiss mice (FVB/NJ, SJL/J, and SWR/J), and other inbred strains (CE/J and RIIIS/J). The SNP data were obtained from the Mouse Phenome Database (http://phenome.jax.org/), which contained 190 903 SNPs on commonly used mouse-inbred strains. These SNP data were further filtered by removing SNPs with <18 strains typed or without genetic mapping information. To be included, each SNP allele also had to be present in five or more inbred strains. The resulting data consisted of 84 010 SNPs, spanning the mouse genome at an average density of ~30 kb per SNP.
Genome-wide SNP association analysis
To correct for population structure and genetic relatedness among inbred strains, we used a recently developed method EMMA to assess association of lung tumor susceptibility with SNPs (Kang et al., 2008). Specifically, the mixed model in the EMMA method can be represented by:
where y is an n × 1 vector of observed phenotypes (that is spontaneous incidence of lung tumor) and X is an n × q matrix of fixed effects including mean, SNPs, and other covariate variables. β is a q × 1 vector representing coefficients of the fixed effects. Z is an n × t incidence matrix mapping each observed phenotype to one of t inbred strains. u is the random effect (that is strain effects) of the mixed model with , where K is the t × t kinship matrix inferred from genotypes and e is an n 1 vector of residual effect such that . The overall phenotypic variance–covariance matrix can be represented as .
In the EMMA approach, a kinship matrix based on genetic similarity was inferred from SNP genotype data. However, most Mouse Phenome Database SNPs were initially discovered by comparison of the genomes of several classical inbred laboratory mouse strains (such as C57BL/6J and 129S1/SvImJ). Although this SNP ascertainment will not likely introduce false positives in association mapping, it certainly affects any inference about the population on the basis of these SNPs (Clark et al., 2005). Recently, Perlegen Science reported 8.27 million high-quality SNPs by resequencing the genomes of 4 wild derived and 11 classical strains (Frazer et al., 2007), which is a more representative set of SNPs. Therefore, to correctly infer a kinship among inbred strains, we randomly chose 624 SNPs from the Perlegen SNP database (http://mouse.perlegen.com/mouse/download.html) and typed these SNPs in these inbred strains to infer kinship matrix. SNP genotyping was performed by the Sequenom mass array spectrometry system at the Human Genetics Division Genotyping Core of Washington University (St Louis, MO, USA).
An R package implementation of the EMMA method is publicly available (http://mouse.cs.ucla.edu/emma/). A twosided P-value from the EMMA for each SNP was obtained for testing hypothesis of no association between the SNP and spontaneous incidence of lung tumor. As the spontaneous incidence of lung tumor is frequency data, we performed arcsine transformation on the data before statistical analysis. We also applied logit transformation on the data: ln (y/(1 y)), where y is tumor incidence and y is reset as 0.25/n (n is sample size) when tumor incidence is zero. Similar results were obtained from the association analysis for two different transformations and only the association results from arcsine-transformed data are presented.
One thousand permutations were used to establish a genome-wide threshold (a global P-value of 0.05) for declaring significant associations in the association analysis. Specifically, lung tumor phenotype was randomly reshuffled among subjects while fixing the genotypes. For each permutation, the EMMA approach described above was implemented, and the most significant −log(P) was recorded. Sorting the maximum −log(P) from large to small, the 5% quantile of the empirical distribution was taken as the genome-wide threshold (a global P-value of 0.05).
Expression QTL analysis
Variation in gene expression is an important mechanism underlying susceptibility to complex disease (Cookson et al., 2009). We, therefore, measured global gene expression in normal lung tissues from 40 laboratory inbred mouse strains using Affymetrix Mouse Exon 1.0 ST microarrays. These strains included 129S1/SvImJ, 129X1/SvJ, A/J, AKR/J, BALB/cByJ, BTBR_T + _tf/J, BUB/BnJ, C3H/HeJ, C57BL/6J, C57BR/cdJ, C57 L/J, C58/J, CAST/EiJ, CBA/J, CE/J, CZECHII/EiJ, DBA/1J, DBA/2J, FVB/NJ, JF1/Ms, KK/HlJ, LG/J, LP/J, MA/MyJ, MOLF/EiJ, MSM/Ms, NOD/LtJ, NON/LtJ, NZB/BlNJ, NZW/LacJ, PERA/EiJ, PL/J, PWD/PhJ, RIIIS/J, SEA/GnJ, SJL/J, SM/J, SPRET/EiJ, SWR/J, and WSB/EiJ. Nineteen strains (bold in Supplementary Table S1) overlapped between lung tumor GWAS and expression QTL experiments. Seven-week old mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and housed for 1 week in the Washington University Animal Facility. At the age of 8 weeks, all mice were killed by cervical dislocation; lung tissues were removed and frozen at −80 1C for subsequent molecular assays. Totally, 240 chips with three female and three male mice per strain were analyzed. Gene-level signal estimates were derived from the Affymetrix CEL files by quantile sketch normalization using Iterplier algorithm, as implemented with Expression Console v1.1.1 (http://www.affymetrix.com/products_services/software/specific/expression_console_software.affx).
Transcription of positional candidate genes from the lung tumor GWAS was analyzed. Similar to spontaneous incidence of lung tumor, transcription abundance was treated as a quantitative phenotype, and genome-wide SNP association of transcription abundance was performed by the EMMA approach as described above. The same set of Mouse Phenome Database SNP data and filtering criteria were used in the expression association analysis. A complete description of the global results will be presented in another paper.
Xrn2 overexpression in lung epithelial cells
Beas-2B human cell line was obtained from ATCC and cultured on dishes coated with 0.01 mg/ml fibronectin, 0.01% BSA, and 0.03% Bovine Collagen Type I in complete BEGM media with supplements (Lonza/Clonetics, San Diego, CA, USA). Cells were grown at 37 celsius under 5% CO2. For overexpression of mouse Xrn2, an N-terminal three hemagglutinin-tagged full-length Xrn2 cDNA or the 3HA tag alone was cloned into pCDNA3 for expression of HA-tagged Xrn2 protein. Cells at ~60% confluence were transfected with 8 mg of HA-Xrn2 or empty HA-vector using Lipofectamine 2000 reagents per manufacturer’s protocol (Invitrogen) in serum-free media with supplements. Cells were incubated overnight, media was replaced, and cells were split 1:4 into BEGM containing 200 mg/ml Geneticin (Gibco). Cells were grown in selective media until mock-transfected cells were dead.
Quantitative real-time PCR
RNA was isolated using Trizol reagent as per manufacturer’s instructions (Invitrogen). Two micrograms of total RNA per sample were converted to cDNA using the SuperScript First-Strand Synthesis system for RT–PCR (Invitrogen). Primers for quantitative RT–PCR analysis were designed using Primer Express software version 2.0 (Applied Biosystems) (Supplementary Table S2). Quantitative RT–PCR assay was carried out using the SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). One microliter of cDNA was added to a 25 ml total volume reaction mixture containing water, SYBR Green PCR Master Mix, and primers. Each real-time assay was carried out in triplicate on a Bio-Rad MyIQ machine. Data were collected and analyzed with Stratagene Mx3000 software. Gene b-actin was used as an internal control to compute the relative expression level for each sample, ΔCT = (CT,target–CT,b-actin,) where CT is cycle number at which the fluorescence signal exceeds background. The average Xrn2 expression of strain AKR/J (one of the most resistant strain) was treated as the baseline. Then, the fold change of gene expression in inbred mice was calculated as where ΔCT* is the baseline mRNA level. The differences in expression between two groups were determined by two-tailed Student’s t-test.
Immunoblotting
Beas-2B cells transiently transfected with pCDNA3-3HA or pCDNA3-3HA-Xrn2 were plated in coated six-well tissue culture plates at ~50% confluence and cultured for 72 h in either complete BEGM with supplements or media without supplements and 10% FCS. Cells in each well of six-well plates were lysed with 250 ml of 1 sample buffer containing cocktailed proteinase inhibitors, sonicated, and then boiled for 10 min; 20 ml was resolved on SDS–PAGE and immunoblotting analyzed with indicated antibodies.
Proliferation assay
Beas-2B cells stably transfected with pCDNA3-3HA or pCDNA3-3HA-Xrn2 were harvested, counted on a hemacytometer, and seeded onto six-well tissue culture dishes at a density of 5000 cells per well. Cells were cultured in either complete BEGM with supplements or media without supplements and 10% FCS. Cells were assayed for viable cell numbers in triplicate using the MTT-based CellTiter 96 AQueous One Solution Cell Proliferation Assay kits (Promega), and absorbance at 490 nm measured on a plate reader periodically over 9 days in culture. Media was replaced every 2 days. P-values were determined by two-tailed Student’s t-test.
Supplementary Material
Acknowledgments
We thank the Broad Institute of Harvard/Massachusetts General Hospital and Massachusetts Institute of Technology, the Wellcome Trust Center for Human Genetics at Oxford University, and Perlegen Sciences for releasing inbred laboratory mouse SNP data. We thank the Mouse Phenome Project for collecting mouse SNP data. We thank the International Agency for Research on Cancer (Lyon, France) for releasing lung cancer GWAS data. We also thank investigators for generating spontaneous lung tumor data in inbred mice. The mouse SNP and phenotype data were crucial for the key findings described in this paper. We thank the staff of The Vanderbilt Microarray Shared Resource for microarray processing. MY was supported by grants from the US National Institutes of Health (CA099187, CA099147, ES012063, and ES013340). We thank Dr Jay W Tichelaar for his critical comments on the manuscript.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–622. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borczuk AC, Gorenstein L, Walter KL, Assaad AA, Wang L, Powell CA. Non-small-cell lung cancer molecular signatures recapitulate lung developmental pathways. Am J Pathol. 2003;163:1949–1960. doi: 10.1016/S0002-9440(10)63553-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borczuk AC, Powell CA. Expression profiling and lung cancer development. Proc Am Thorac Soc. 2007;4:127–132. doi: 10.1513/pats.200607-143JG. [DOI] [PubMed] [Google Scholar]
- Clark AG, Hubisz MJ, Bustamante CD, Williamson SH, Nielsen R. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 2005;15:1496–1502. doi: 10.1101/gr.4107905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10:184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couzin J, Kaiser J. Genome-wide association. Closing the net on common disease genes. Science. 2007;316:820–822. doi: 10.1126/science.316.5826.820. [DOI] [PubMed] [Google Scholar]
- Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Cancer Inst. 1981;66:1191–1308. [PubMed] [Google Scholar]
- Foley JF, Anderson MW, Stoner GD, Gaul BW, Hardisty JF, Maronpot RR. Proliferative lesions of the mouse lung: progression studies in strain A mice. Exp Lung Res. 1991;17:157–168. doi: 10.3109/01902149109064408. [DOI] [PubMed] [Google Scholar]
- Frazer KA, Eskin E, Kang HM, Bogue MA, Hinds DA, Beilharz EJ, et al. A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature. 2007;448:1050–1053. doi: 10.1038/nature06067. [DOI] [PubMed] [Google Scholar]
- Hung RJ, McKay JD, Gaborieau V, Boffetta P, Hashibe M, Zaridze D, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. caac.20006. [DOI] [PubMed] [Google Scholar]
- Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, et al. Efficient control of population structure in model organism association mapping. Genetics. 2008;178:1709–1723. doi: 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y, Reddel RR, Gerwin BI, Miyashita M, McMenamin M, Lechner JF, et al. Human bronchial epithelial cells with integrated SV40 virus T antigen genes retain the ability to undergo squamous differentiation. Differentiation. 1988;38:60–66. doi: 10.1111/j.1432-0436.1988.tb00592.x. [DOI] [PubMed] [Google Scholar]
- Kendall J, Liu Q, Bakleh A, Krasnitz A, Nguyen KC, Lakshmi B, et al. Oncogenic cooperation and coamplification of developmental transcription factor genes in lung cancer. Proc Natl Acad Sci USA. 2007;104:16663–16668. doi: 10.1073/pnas.0708286104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, et al. The T/ebp null mouse: thyroid-specific enhancerbinding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 1996;10:60–69. doi: 10.1101/gad.10.1.60. [DOI] [PubMed] [Google Scholar]
- Kuntz E, Hoeller U, Greatrix B, Lankin C, Seifert N, Acharya S, et al. Beta-carotene and apocarotenals promote retinoid signaling in BEAS-2B human bronchioepithelial cells. Arch Biochem Biophys. 2006;455:48–60. doi: 10.1016/j.abb.2006.08.023. [DOI] [PubMed] [Google Scholar]
- Kwei KA, Kim YH, Girard L, Kao J, Pacyna-Gengelbach M, Salari K, et al. Genomic profiling identifies TITF1 as a lineage-specific oncogene amplified in lung cancer. Oncogene. 2008;27:3635–3640. doi: 10.1038/sj.onc.1211012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zheng H, Ji C, Fei X, Zheng M, Gao Y, et al. A novel splice variant of human XRN2 gene is mainly expressed in blood leukocyte. DNA Seq. 2005;16:143–146. doi: 10.1080/10425170500066771. [DOI] [PubMed] [Google Scholar]
- Liu P, Vikis HG, Wang D, Lu Y, Wang Y, Schwartz AG, et al. Familial aggregation of common sequence variants on 15q24-25.1 in lung cancer. J Natl Cancer Inst. 2008;100:1326–1330. doi: 10.1093/jnci/djn268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Wang Y, Vikis H, Maciag A, Wang D, Lu Y, et al. Candidate lung tumor susceptibility genes identified through wholegenome association analyses in inbred mice. Nat Genet. 2006;38:888–895. doi: 10.1038/ng1849. [DOI] [PubMed] [Google Scholar]
- Matakidou A, Eisen T, Houlston RS. Systematic review of the relationship between family history and lung cancer risk. Br J Cancer. 2005;93:825–833. doi: 10.1038/sj.bjc.6602769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay JD, Hung RJ, Gaborieau V, Boffetta P, Chabrier A, Byrnes G, et al. Lung cancer susceptibility locus at 5p15.33. Nat Genet. 2008;40:1404–1406. doi: 10.1038/ng.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen R, Berns A. Mouse models for human lung cancer. Genes Dev. 2005;19:643–664. doi: 10.1101/gad.1284505. [DOI] [PubMed] [Google Scholar]
- Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, Spielman RS, et al. Genetic analysis of genome-wide variation in human gene expression. Nature. 2004;430:743–747. doi: 10.1038/nature02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MH, Cho SA, Yoo KH, Yang MH, Ahn JY, Lee HS, et al. Gene expression profile related to prognosis of acute myeloid leukemia. Oncol Rep. 2007;18:1395–1402. [PubMed] [Google Scholar]
- Powell CA, Bueno R, Borczuk AC, Caracta CF, Richards WG, Sugarbaker DJ, et al. Patterns of allelic loss differ in lung adenocarcinomas of smokers and nonsmokers. Lung Cancer. 2003;39:23–29. doi: 10.1016/s0169-5002(02)00384-7. [DOI] [PubMed] [Google Scholar]
- Rafnar T, Sulem P, Stacey SN, Geller F, Gudmundsson J, Sigurdsson A, et al. Sequence variants at the TERT-CLPTM1 L locus associate with many cancer types. Nat Genet. 2009;41:221–227. doi: 10.1038/ng.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said JW, Nash G, Sassoon AF, Shintaku IP, Banks-Schlegel S. Involucrin in lung tumors. A specific marker for squamous differentiation. Lab Invest. 1983;49:563–568. [PubMed] [Google Scholar]
- Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, Colinayo V, et al. Genetics of gene expression surveyed in maize, mouse and man. Nature. 2003;422:297–302. doi: 10.1038/nature01434. [DOI] [PubMed] [Google Scholar]
- Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer. 2006;118:257–262. doi: 10.1002/ijc.21496. [DOI] [PubMed] [Google Scholar]
- Smith R, Owen LA, Trem DJ, Wong JS, Whangbo JS, Golub TR, et al. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell. 2006;9:405–416. doi: 10.1016/j.ccr.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Yanagisawa K, Shinjo K, Taguchi A, Maeno K, Tomida S, et al. Lineage-specific dependency of lung adenocarcinomas on the lung development regulator TTF-1. Cancer Res. 2007;67:6007–6011. doi: 10.1158/0008-5472.CAN-06-4774. [DOI] [PubMed] [Google Scholar]
- Tang H, Ross A, Capel B. Expression and functional analysis of Gm114, a putative mammalian ortholog of Drosophila bam. Dev Biol. 2008;318:73–81. doi: 10.1016/j.ydbio.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thun MJ, Henley SJ, Burns D, Jemal A, Shanks TG, Calle EE. Lung cancer death rates in lifelong nonsmokers. J Natl Cancer Inst. 2006;98:691–699. doi: 10.1093/jnci/djj187. [DOI] [PubMed] [Google Scholar]
- Tian J, Mahmood R, Hnasko R, Locker J. Loss of Nkx2.8 deregulates progenitor cells in the large airways and leads to dysplasia. Cancer Res. 2006;66:10399–10407. doi: 10.1158/0008-5472.CAN-06-1564. [DOI] [PubMed] [Google Scholar]
- Wacholder S, Chatterjee N, Caporaso N. Intermediacy and gene-environment interaction: the example of CHRNA5-A3 region, smoking, nicotine dependence, and lung cancer. J Natl Cancer Inst. 2008;100:1488–1491. doi: 10.1093/jnci/djn380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walts AE, Said JW, Siegel MB, Banks-Schlegel S. Involucrin, a marker of squamous and urothelial differentiation. An immunohistochemical study on its distribution in normal and neoplastic tissues. J Pathol. 1985;145:329–340. doi: 10.1002/path.1711450406. [DOI] [PubMed] [Google Scholar]
- Wang Y, Broderick P, Webb E, Wu X, Vijayakrishnan J, Matakidou A, et al. Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat Genet. 2008;40:1407–1409. doi: 10.1038/ng.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MP, Fung LF, Wang E, Chow WS, Chiu SW, Lam WK, et al. Chromosomal aberrations of primary lung adenocarcinomas in nonsmokers. Cancer. 2003;97:1263–1270. doi: 10.1002/cncr.11183. [DOI] [PubMed] [Google Scholar]
- You M, Bergman G. Preclinical and clinical models of lung cancer chemoprevention. Hematol Oncol Clin North Am. 1998;12:1037–1053. doi: 10.1016/s0889-8588(05)70040-x. [DOI] [PubMed] [Google Scholar]
- Zeka A, Mannetje A, Zaridze D, Szeszenia-Dabrowska N, Rudnai P, Lissowska J, et al. Lung cancer and occupation in nonsmokers: a multicenter case-control study in Europe. Epidemiology. 2006;17:615–623. doi: 10.1097/01.ede.0000239582.92495.b5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.