

Abstract
Background
The balance of fibrinolytic mediators is crucial to the survival of the critically ill patient, with tissue plasminogen activator (t-PA) and plasminogen activator inhibitor-1 (PAI-1) playing significant roles. While elevated levels of PAI-1 are associated with increased morbidity and mortality, the source of this PAI-1 remains elusive. Platelets contain 90% of circulating plasma PAI-1, however, their ability to release active PAI-1 is controversial. We hypothesize platelets contain active PAI-1 in α-granules capable of immediate degranulation when exposed to high concentrations of thrombin.
Methods
In vitro apheresis platelets were stimulated with thrombin (1IU/ml, 5IU/ml) followed by the collection of supernatant (5-120 minutes). Supernatant and lysate PAI-1 was measured by ELISA. The experiment was repeated in the presence of t-PA followed by measurement of t-PA:PAI-1 complex measurement by ELISA. Finally, healthy whole blood underwent dilution with control and thrombin-treated platelet lysate followed by thrombelastography (TEG) in a t-PA-stimulated TEG.
Results
Thrombin provoked immediate near-complete degranulation of PAI-1 from α-granules (median 5m 5IU/ml thrombin 125.1ng/ml, 1IU/ml thrombin 114.9ng/ml, control 9.9ng/ml). The released PAI-1 rapidly complexed with t-PA, with a 4-fold increase in complex formation in the thrombin-treated supernatant. Conversely, PAI-1 in the control lysate demonstrated a 6-fold increase in complex formation compared to thrombin lysate. Lastly, control platelet lysate inhibited t-PA induced fibrinolysis by TEG (median LY30 control 15m 7.9%), while thrombin-treated platelet lysates, after PAI-1 degranulation, were unable to affect the fibrinolysis profile (median LY30 5IU/ml 28.5%, 1IU/ml 12.4%).
Conclusion
Thrombin provokes rapid α-degranulation of active PAI-1, capable of complexing with t-PA and neutralizing t-PA induced fibrinolysis by TEG.
Keywords: fibrinolysis, fibrinolysis shutdown, trauma, thrombelastography
Introduction
The balance of pro-fibrinolytic and anti-fibrinolytic contributors is crucial to the survival of the sick and injured patient. This balance is maintained through a multitude of proteins, with two of the most significant being tissue plasminogen activator (t-PA), a serine protease, and plasminogen activator inhibitor-1 (PAI-1).(1-3) PAI-1 is a serine protease inhibitor that rapidly binds to t-PA covalently and blocks its ability to convert plasminogen to plasmin. It is well accepted that t-PA is released from endothelial cells exposed to hypoxia, shock, and other inflammatory mediators;(4, 5) however, the source and release of PAI-1 is not fully understood.
While PAI-1 is crucial to controlling t-PA and the risk of bleeding, excess PAI-1 can be detrimental and has been shown to correlate with increased morbidity and mortality in ICU and trauma patients, with increased risk of acute respiratory distress syndrome and multi-organ failure.(6-8) In addition, PAI-1 levels are known to be elevated in arterial thrombi, inhibiting t-PA and the ability to breakdown fibrin via plasmin.(9)
Multiple cell types are capable of synthesizing, storing, and releasing PAI-1 including endothelial cells, hepatocytes, neutrophils, macrophages, and platelets.(10-13) Platelets have been identified as a major storage of plasma PAI-1, accounting for more than 90% of circulating PAI-1,(14) however, their contribution to circulating PAI-1 is the subject of much debate stemming from discrepancy in the literature regarding its activity.(15-17)
Thrombin generation has repeatedly been shown to be elevated in the plasma of trauma patients, and thrombin activity is known to be elevated at the site of injury and thrombus.(18, 19) We hypothesize that platelets contain active PAI-1 in α-granules capable of immediate degranulation when exposed to thrombin.
Materials and Methods
Materials
All reagents were purchased from Sigma Chemical Company (St. Louis, MO) unless otherwise stated. Thrombin was purchased from Enzo Life Sciences (Farmingdale, NY). Human single chain tissue plasminogen activator (t-PA), human total PAI-1 and t-PA-PAI-1 complex ELISA kits were purchased from Molecular Innovations (Novi, MI). t-PA was diluted in 5% bovine serum albumin in phosphate buffered solution, separated into aliquots, and stored at −80°C until use. Human VEGF quantikine ELISA kit was purchased from R&D Systems Inc. (Minneapolis, MN).
Methods
Platelets
Platelets were obtained from healthy apheresis platelet donors at Bonfils Blood Center at day 1 of storage, when all units are routinely cultured. The platelets were washed at 800g for 15 minutes at room temperature and were resuspended in normal saline at a concentration of 3 × 108 cells/ml. Thrombin (1IU/ml, 5IU/ml) was added to the platelets for 5-120 minutes at 37°C, centrifuged at 800g for 15 minutes, and the supernatant isolated and frozen immediately at −80°C. The platelets were resuspended in normal saline followed by three freeze-thaw cycles (−80°C) to lyse the platelets. A platelet count was done to ensure lysis and stored at −80°C for future use. The experiment was then repeated in the presence of t-PA (150ng/ml) followed by supernatant storage for t-PA-PAI-1 complex measurement by ELISA.
Enzyme-Linked Immunosorbent Assays (ELISAs)
Total PAI-1 and t-PA-PAI-1 complex were measured using commercial ELISAs according to manufacturer recommendations. PAI-1 release was measured in the supernatant as well as the lysate from platelets. To confirm α-degranulation in platelets, VEGF was measured in supernatant and platelet lysates via commercial ELISA, according to manufacturer recommendations.
Healthy volunteer blood collection
Informed consent was obtained and blood was collected from six healthy volunteers under the Colorado Multiple Institutional Board (COMIRB) protocol number 10-04777: 67% male, age 23–32, not taking medication, without chronic illness, not pregnant, non-smokers, and not obese.
t-PA-challenge thrombelastography
All blood samples were collected in 2.7mL buffered sodium citrate (3.2%) tubes (Vacutainer, Becton-Dickingson, Franklin Lakes, NJ). Whole blood then underwent a 20% dilution with platelet lysate in normal saline (400ul whole blood, 100ul platelet lysate in normal saline) or normal saline. Control platelet lysate at 15 and 30 minutes was contrasted to thrombin-treated (both 1IU/ml and 5IU/ml) platelet lysates at the same time point for the ability to influence fibrinolysis on a t-PA-stimulated TEG. Positive control samples underwent a 20% dilution with normal saline. Whole blood samples as well as whole blood/platelet lysate mixtures were assayed with TEG 5000 Thrombelastograph Hemostasis Analyzer (Haemonetics, Niles, IL). The methods for performing a t-PA-stimulated TEG to quantify fibrinolysis resistance/sensitivity were performed as previously described.(20) Briefly, blood samples were mixed with exogenous t-PA (75ng/ml) followed by clot evaluation with TEG. Sensitivity or resistance to t-PA was quantified using the lysis at 30 minute variable (LY30). R-time, angle, and maximum amplitude (MA) TEG values were also analyzed.
Statistics
Statistical analysis was completed using SPSS version 24 (IBM). As this was driven by a pre-specified hypothesis, control and platelet supernatant and lysates total PAI-1 and tPA-PAI-1 complex values at each time point were compared using a 2-tailed paired samples t-test with statistical significance set at p<0.05.(21, 22)
Mixed linear models for repeated measures with nested effect within donors were used. A Dunnett’s multiple comparison adjustment was applied. All tests were two-tailed and significance was established at p<0.05. LY30 was transformed via a Box Cox power transformation (lambda=0.5) to approximate normality. The other variables had their close to normal distribution assessed by inspecting histograms. These analyses were conducted with SAS 9.4 (SAS Institute Inc., Cary, NC, USA).
Results
Thrombin provokes immediate degranulation of PAI-1 from platelet α-granules
To determine the relative contribution of PAI-1 released, platelets (3×108cells/ml) were treated with thrombin (1IU/ml, 5IU/ml) and PAI-1 release was measured from 5-120 minutes. High dose thrombin (5IU/ml) elicited >95% release of PAI-1 and the lower dose thrombin (1IU/ml) elicited 85-95% release of PAI-1 (Figure 1). High PAI-1 levels were present in the supernatant at 5 minutes in both thrombin-treated platelet groups (5IU/ml median 125.1ng/ml, 1IU/ml median 119.6ng/ml) as compared to PAI-1 remaining in the cellular fraction (3.5ng/ml and 8.7ng/ml, respectively) and in control supernatants (9.9ng/ml, p=0.007). Further, PAI-1 release did not increase over the duration of the experiment (15-120 min) after either dose thrombin stimulation but remained significantly elevated compared to control (Figure 1). Release was likely from α-granules as 5IU/ml thrombin induced >95% release of cellular VEGF, a known α-granule constituent at 5 minutes (median 116.5pg/ml released vs, 5.7pg/ml remaining in the cellular fraction, n=2),(23) which did not change over the time course (Figure 2). As a further control, apheresis platelets were directly compared to platelets isolated from whole blood and demonstrated almost identical aggregation profiles to ADP (results not shown).
Figure 1. Thrombin provokes immediate PAI-1 release from platelets.
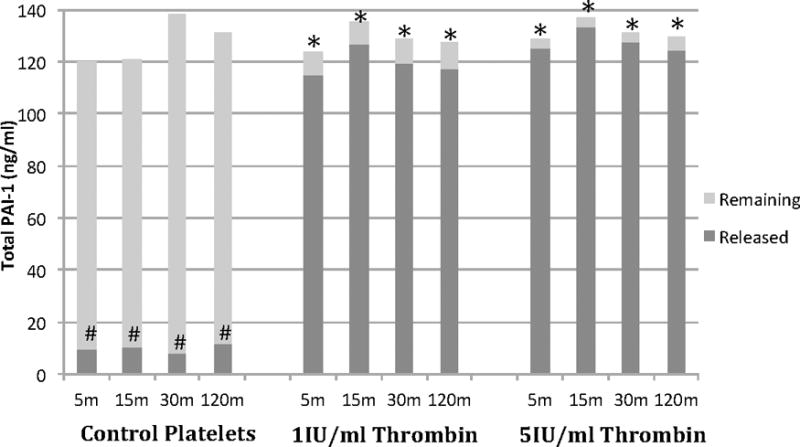
Platelets underwent near complete (>95%) degranulation of PAI-1 in response to thrombin stimulation as shown with released versus remaining. Control platelets did not degranulate over the duration of the experiment with a significant amount of PAI-1 remaining in the cells at the end of the experiment. n=4. *p<0.013 when compared to control lysate (same time point). #p<0.007 compared to thrombin supernatant.
Figure 2. Thrombin stimulates VEGF release from platelets, confirming near complete (>95%) α-degranulation.
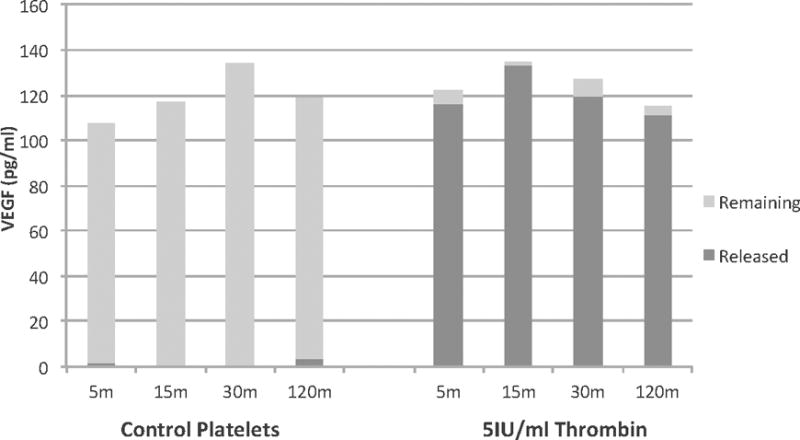
Thrombin stimulated platelets released nearly all stored VEGF at 5 minutes (116.5pg/ml released, 5.7pg/ml remaining (in lysate)) and this did not increase over the time course (120 minutes). Control platelets (in saline) did not release significant amounts of VEGF (1.8pg/ml at 5 minutes, 3.2pg/ml at 120 minutes). n=2.
PAI-1 is active, capable of complexing with t-PA
To evaluate the activity of this platelet released PAI-1, the experiment was repeated with the addition of t-PA to the supernatant of 5IU/ml thrombin and control platelets as well as lysate followed by t-PA-PAI-1 complex measurements (Figure 3). 5IU/ml thrombin provoked the release of active PAI-1 capable of complexing with t-PA. Thrombin-treated platelet supernatant experienced a significant increase in complex formation compared to control platelet supernatant, and this did not change significantly over the course of the experiment. Control platelet lysate, on the other hand, with preserved PAI-1, demonstrated about 5-6-fold increase in t-PA-PAI-1 complex formation as compared to thrombin platelet lysate at all time points (Figure 3).
Figure 3. Thrombin provokes the release of active PAI-1, capable of complexing with t-PA.
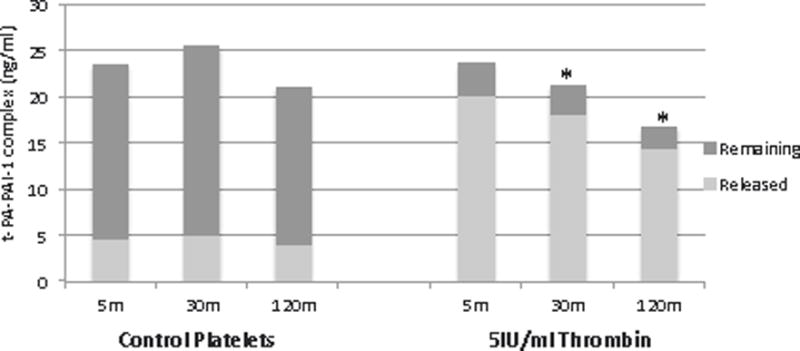
Supernatant and lysate were combined with t-PA (150ng/ml) followed by measurement of t-PA-PAI-1 complex by ELISA. Thrombin-treated supernatant demonstrate a 3-4 fold increase in complex formation compared to control demonstrating release of active PAI-1. Conversely, control lysate had a 5-6-fold increase in complex formation compared to thrombin-treated, confirmed storage of active PAI-1. n=3-4, values reported as median. *p<0.03 compared to control lysate (same time point). #p<0.002 compared to thrombin supernatant (same time point).
PAI-1 stored active, capable of inhibiting t-PA-induced fibrinolysis on TEG
To confirm platelet PAI-1 activity in a fibrinolysis model, whole blood underwent a 20% dilution with control and thrombin (5IU/ml, 1IU/ml) platelet lysates in normal saline (15 and 30 minute time points) followed by clot evaluation by t-PA-stimulated TEG. R-time, angle, MA, and LY30 t-PA-challenge TEG values exhibited significant differences across the groups at 15 and 30 minutes (Table 1). The rate of clot initiation (R-time) was significantly different across dilution groups at 15 (p<0.001) and 30 minutes (p<0.001). Thrombin lysates, with internalized thrombin, had a reduced R-time compared to the control lysates at the respective time points (Table 1). Rate of clot formation (angle) was significantly different across dilution groups (15m p=0.003, 30m p=0.005) with the platelet lysate dilution groups showing a faster rate of clot formation as compared to whole blood. There was no significant difference between either thrombin dose and control platelet lysate in rate of clot formation (angle) or maximum clot amplitude (MA).
Table 1.
t-PA-challenge TEG
| R-time (min) | Angle (degrees) | MA (mm) | LY30 % | |
|---|---|---|---|---|
| Whole blood | 11.2 (8.1-13.0) | 39.3 (35.3-47.4) | 35.4 (27.2-57.5) | 15.9 (11.7-19.3) |
| Normal saline | 9.6 (7.5-10.5) | 51.3 (42.4-58.8) | 38 (25.6-52.4) | 35.5 (27.0-50.7) |
| Control lysate- 15m | 6.1 (5.4-7.8) | 55.5 (46.6-60.4)# | 48.9 (35.4-57.8)§ | 7.9 (4.3-11.8)* |
| 1IU Thrombin lysate- 15m | 5.9 (3.7-6.9)† | 57.0 (47.4-63.6) | 59.0 (38.6-65.0) | 12.4 (11-21.1) |
| 5IU Thrombin lysate- 15m | 1.7 (1.4-2.7)† | 59.3 (51.8-64.3) | 42.2 (22.9-61.1) | 28.5 (17.0-34.1) |
| Control lysate- 30m | 6.4 (5.5-7.6) | 51.3 (42.9-53.8)# | 47.6 (37.7-58.8)§ | 6.1 (5.1-12.3)* |
| 1IU Thrombin lysate- 30m | 2.0 (1.8-4.4)† | 53.4 (42.9-61.1) | 58.0 (51.3-62.9) | 13.7 (8.7-16.9) |
| 5IU Thrombin lysate- 30m | 2.0 (1.8-4.4)† | 60.7 (48.2-64.1) | 40.9 (23.0-57.9) | 29.6 (19.2-41.1) |
Thrombelastography variables for whole blood, normal saline dilution (20% dilution of whole blood), control and thrombin (1IU, 5IU/ml) platelet lysate at 15 minutes, and control and thrombin (1IU, 5IU/ml) platelet lysate at 30 minutes (lysate in normal saline, 20% dilution of whole blood) in the presence of exogenous t-PA (75ng/ml). Thrombin lysate, with internalized thrombin, demonstrated reduced R-time compared to the control platelet lysate. In addition, control platelet lysate had an increased angle and MA compared to whole blood. Control platelet lysate, with preserved PAI-1, inhibited tPA-stimulated TEG fibrinolysis, with a reduced LY30 compared to normal saline and both thrombin platelet lysates (1IU, 5IU/ml) after degranulation of PAI-1. Control platelet lysate also demonstrated reduced LY30 compared to whole blood.
p<0.01 compared to normal saline, whole blood, and thrombin lysates (5IU, 1IU) LY30 at the same time point.
p<0.01 compared to control platelet lysates R-time at the same time point.
p<0.002 compared to whole blood angle.
p<0.02 compared to whole blood angle.
Fibrinolysis, as measured by LY30, differed significantly across dilution groups at both 15 and 30 minutes (p<0.0001 for both). The control lysate, with high levels of PAI-1 shown earlier, was able to inhibit the majority of t-PA-induced fibrinolysis, with a significant reduction in t-PA-induced fibrinolysis compared to both thrombin lysates and normal saline dilution at 15 minutes (5IU p=0.0005, 1IU p=0.03, NS p<0.0001) and 30 minutes (5IU p<0.0001, 1IU p=0.04, NS p<0.0001) (Table 1). Control lysate also had reduced fibrinolysis (15m median LY30 7.9%; 30m 6.1%) compared to whole blood (15.9%) although not significant at 15 minutes (15m p=0.05; 30m p=0.01). High dose thrombin (5IU/ml) lysate, after degranulation of PAI-1, was unable to influence t-PA-induced fibrinolysis, with a similar t-PA-TEG tracing to the normal saline dilution at both time points (Table 1). Low dose thrombin (1Iu/ml) also showed increase susceptibility to t-PA induced fibrinolysis compared to the control lysate. A sample t-PA-stimulated TEG tracing for whole blood, whole blood and normal saline, both thrombin lysates, and control lysate is shown in Figure 4.
Figure 4. Sample t-PA-stimulated TEG tracings.
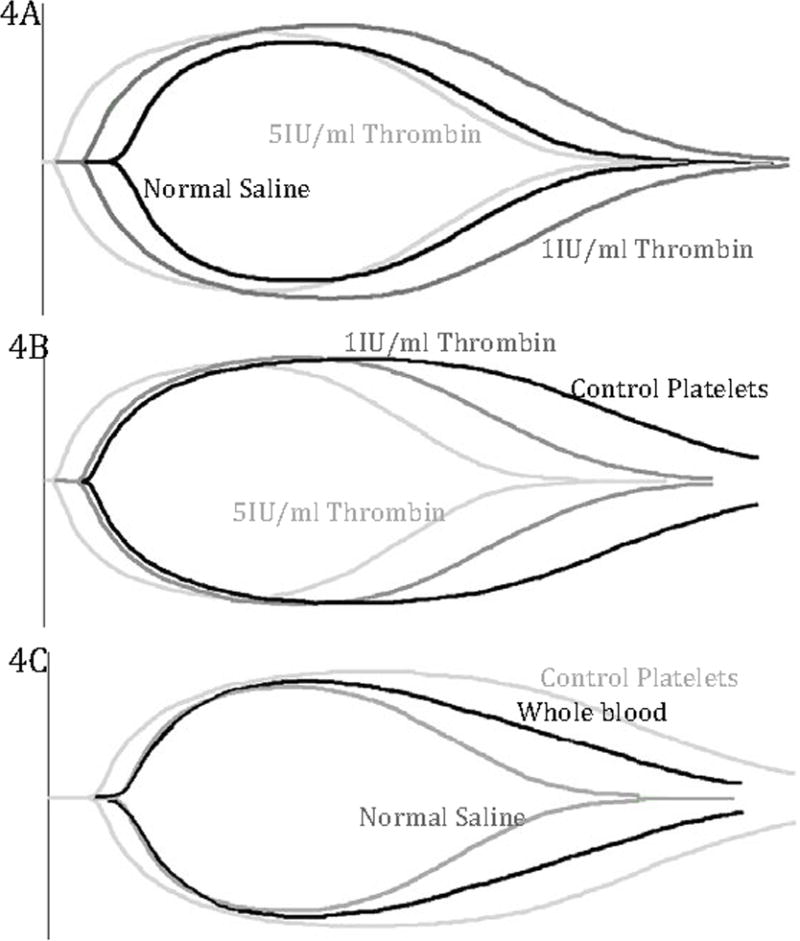
Sample TEG tracings of whole blood, normal saline dilution (20% dilution of whole blood), 1IU/ml and 5IUml thrombin platelet lysates and control platelet lysates (20% dilution of whole blood) in the presence of exogenous t-PA (75ng/ml). A) Normal and thrombin platelet lysates manifest rapid clot breakdown, or fibrinolysis, in the presence of t-PA and have similar TEG tracings. B) Control, thrombin (1IU/ml, 5IU/ml) platelet lysates. Control platelet lysate, with preserved PAI-1, manifest resistance to t-PA compared to both thrombin lysates. Thrombin platelet lysates (1IU/ml, 5IU/ml), after degranulation of PAI-1, lose the ability to resist increased lysis in the presence of t-PA. C) Control platelet lysate, whole blood, and normal saline dilution. Control platelet lysate, with PAI-1, resists changes to fibrinolysis compared to whole blood and normal saline dilution.
Discussion
Elevated plasma PAI-1 portends poor outcomes.(6, 8) While 90% of the circulating PAI-1 is contained in platelets, the ability to degranulate PAI-1 as well as the activity of this PAI-1 has been debated.(14-17) Thrombin directly causes α-degranulation of active PAI-1, capable of complexing with t-PA and inhibiting fibrinolysis on a t-PA-modified TEG.
Platelet dysfunction in trauma and ICU patients is common and associated with worse outcomes.(24, 25) Platelet lysate has previously been shown to attenuate t-PA-induced fibrinolysis, however the mechanism was unknown.(26) While the authors speculated the implication of PAI-1 as well as other proteins accounting for this resistance to fibrinolysis, our data show platelet PAI-1 is active, capable of complexing with t-PA (Figure 3). These complex values were less than expected in the presence a high concentration of t-PA (150ng/ml), but this demonstrates the short half-life of PAI-1, the instability of active PAI-1 without the necessary cofactors such as vitronectin, and the effect of freeze-thaw cycles.(27) Even with these limitations, thrombin-treated platelet supernatant had a 4-fold increase in t-PA-PAI-1 complex formation while the control lysates retained a 6-fold increase in complex formation compared to thrombin. In addition, control platelet lysate, with preserved PAI-1, is able to inhibit t-PA-induced fibrinolysis, whereas thrombin-treated platelet lysate, after degranulation of PAI-1, loses this ability and functions similar to the positive control (Figure 4, Table 1). While the definition of platelet dysfunction in critically-ill patients typically involves aggregation assays (adenosine diphosphate, thrombin receptor activating peptide, arachidonic acid),(24, 25) it likely involves degranulation as well, including the α-granule, leading to elevated plasma PAI-1.
Platelets store PAI-1 in α-granules, which are capable of degranulating in response to trauma.(28, 29) The ability of thrombin to provoke degranulation of platelets is well-established,(15, 23, 30) and a high rate of degranulation occurs in response to thrombin stimulation. Thrombin levels in trauma patients are known to be elevated through the positive feedback loop with tissue factor,(19, 31, 32) with local areas of tissue injury may be capable of achieving high thrombin concentrations provoking immediate degranulation of α-granules and release of PAI-1.(15, 28) Near complete, rapid α-degranulation (>95%) was confirmed with subsequent measurement of VEGF levels in supernatant and lysate as VEGF is known to co-localize with PAI-1 in α-granules.(23) Following degranulation, platelets have the ability to synthesize more PAI-1 from mRNA,(15) but such synthesis occurs over a long period of time, rendering these platelets dysfunctional.
Interestingly, there is a stepwise progression of fibrinolysis from the control platelet lysate (LY30 15m 7.9%; 30m 6.1%), with near complete inhibition of t-PA induced fibrinolysis (due to retained PAI-1), to low-dose thrombin lysate (1IU/ml) (LY30 15m 12.4%; 30m 13.7%) and to high-dose thrombin lysate (5IU/ml) (LY30 28.5%; 30m 29.6%). Both of these thrombin doses released significant amounts of PAI-1 (1IU: 119.6ng/ml, 5IU: 125.1ng/ml) and the residual remaining in the lysate does not seem significant enough to account for the differences in their fibrinolysis profiles (lysate PAI-1: 1IU 8.7ng/ml; 5IU 3.5ng/ml). However, this can be accounted for in the methods section. When performing the whole blood dilution with platelet lysate, concentrated platelet lysate (3×109cells/ml), which is 10 times more concentrated then our initial PAI-1 measurements, was employed. This concentrated lysate would account for ~87ng/ml in the 1IU/ml thrombin lysate group versus ~35ng/ml in the high dose thrombin group, which likely accounts for the difference in the fibrinolysis profiles seen in the t-PA-stimulated TEG. PAI-1 concentration in the lysate (control lysate > low dose thrombin lysate > high dose thrombin lysate) correlates well with decreased fibrinolysis (LY30) by TEG (high dose thrombin lysate > low dose thrombin lysate > control platelet lysate).
There are limitations to this in vitro experiment. While thrombin generation is elevated in critically ill patients, thrombin is not the only stimulant acting on platelets in critically ill patients, with hypoxia, tissue factor, and TNFα also playing roles. Other cell types also undoubtedly affect the fibrinolytic balance, producing t-PA and PAI-1 and clearing t-PA-PAI-1 complexes. We also acknowledge the impact of multiple additional proteins in the fibrinolytic balance of the trauma patient, including antithrombin, activated protein C, and the coagulation factors. We were unable to measure the clotting properties by TEG of supernatant PAI-1 as the addition of thrombin at the start of the experiment produces a procoagulant environment with fast clot formation. Lastly, while a complete inhibition of t-PA-induced fibrinolysis with control lysate based on the PAI-1 values in the measured lysates was anticipated (Figure 1), the effect of 3 freeze thaw cycles on PAI-1 activity without cofactors present likely accounts for this discrepancy.
In conclusion, thrombin provokes rapid near-complete α-degranulation from platelets leading to the release of active PAI-1 capable of complexing with t-PA and inhibiting t-PA-induced fibrinolysis.
Acknowledgments
Conflict of Interest Statement
This study was supported in part by National Institutes of Health grants T32-GM008315 (National Institute of General Medical Sciences), P50-GM0492221 (National Institute of General Medical Sciences). Additional research support was provided by Haemonetics Corporation (Niles, IL).
References
- 1.Cardenas JC, Matijevic N, Baer LA, Holcomb JB, Cotton BA, Wade CE. Elevated tissue plasminogen activator and reduced plasminogen activator inhibitor promote hyperfibrinolysis in trauma patients. Shock. 2014;41(6):514–21. doi: 10.1097/SHK.0000000000000161. [DOI] [PubMed] [Google Scholar]
- 2.Declerck PJ, De Mol M, Alessi MC, Baudner S, Paques EP, Preissner KT, Muller-Berghaus G, Collen D. Purification and characterization of a plasminogen activator inhibitor 1 binding protein from human plasma. Identification as a multimeric form of S protein (vitronectin) J Biol Chem. 1988;263(30):15454–61. [PubMed] [Google Scholar]
- 3.Binder BR, Christ G, Gruber F, Grubic N, Hufnagl P, Krebs M, Mihaly J, Prager GW. Plasminogen activator inhibitor 1: physiological and pathophysiological roles. News Physiol Sci. 2002;17:56–61. doi: 10.1152/nips.01369.2001. [DOI] [PubMed] [Google Scholar]
- 4.Emeis JJ, van den Eijnden-Schrauwen Y, van den Hoogen CM, de Priester W, Westmuckett A, Lupu F. An endothelial storage granule for tissue-type plasminogen activator. J Cell Biol. 1997;139(1):245–56. doi: 10.1083/jcb.139.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huber D, Cramer EM, Kaufmann JE, Meda P, Masse JM, Kruithof EK, Vischer UM. Tissue-type plasminogen activator (t-PA) is stored in Weibel-Palade bodies in human endothelial cells both in vitro and in vivo. Blood. 2002;99(10):3637–45. doi: 10.1182/blood.v99.10.3637. [DOI] [PubMed] [Google Scholar]
- 6.Gando S, Nakanishi Y, Tedo I. Cytokines and plasminogen activator inhibitor-1 in posttrauma disseminated intravascular coagulation: relationship to multiple organ dysfunction syndrome. Crit Care Med. 1995;23(11):1835–42. doi: 10.1097/00003246-199511000-00009. [DOI] [PubMed] [Google Scholar]
- 7.Semeraro N, Ammollo CT, Semeraro F, Colucci M. Sepsis, thrombosis and organ dysfunction. Thromb Res. 2012;129(3):290–5. doi: 10.1016/j.thromres.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 8.Madoiwa S, Nunomiya S, Ono T, Shintani Y, Ohmori T, Mimuro J, Sakata Y. Plasminogen activator inhibitor 1 promotes a poor prognosis in sepsis-induced disseminated intravascular coagulation. Int J Hematol. 2006;84(5):398–405. doi: 10.1532/IJH97.05190. [DOI] [PubMed] [Google Scholar]
- 9.Matsuura Y, Yamashita A, Iwakiri T, Sugita C, Okuyama N, Kitamura K, Asada Y. Vascular wall hypoxia promotes arterial thrombus formation via augmentation of vascular thrombogenicity. Thromb Haemost. 2015;114(1):158–72. doi: 10.1160/TH14-09-0794. [DOI] [PubMed] [Google Scholar]
- 10.Simpson AJ, Booth NA, Moore NR, Bennett B. Distribution of plasminogen activator inhibitor (PAI-1) in tissues. J Clin Pathol. 1991;44(2):139–43. doi: 10.1136/jcp.44.2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Booth NA, Anderson JA, Bennett B. Platelet release protein which inhibits plasminogen activators. J Clin Pathol. 1985;38(7):825–30. doi: 10.1136/jcp.38.7.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Booth NA, Simpson AJ, Croll A, Bennett B, MacGregor IR. Plasminogen activator inhibitor (PAI-1) in plasma and platelets. Br J Haematol. 1988;70(3):327–33. doi: 10.1111/j.1365-2141.1988.tb02490.x. [DOI] [PubMed] [Google Scholar]
- 13.Fearns C, Loskutoff DJ. Induction of plasminogen activator inhibitor 1 gene expression in murine liver by lipopolysaccharide. Cellular localization and role of endogenous tumor necrosis factor-alpha. Am J Pathol. 1997;150(2):579–90. [PMC free article] [PubMed] [Google Scholar]
- 14.Booth NA, Robbie LA, Croll AM, Bennett B. Lysis of platelet-rich thrombi: the role of PAI-1. Ann N Y Acad Sci. 1992;667:70–80. doi: 10.1111/j.1749-6632.1992.tb51599.x. [DOI] [PubMed] [Google Scholar]
- 15.Brogren H, Karlsson L, Andersson M, Wang L, Erlinge D, Jern S. Platelets synthesize large amounts of active plasminogen activator inhibitor 1. Blood. 2004;104(13):3943–8. doi: 10.1182/blood-2004-04-1439. [DOI] [PubMed] [Google Scholar]
- 16.Schleef RR, Sinha M, Loskutoff DJ. Immunoradiometric assay to measure the binding of a specific inhibitor to tissue-type plasminogen activator. J Lab Clin Med. 1985;106(4):408–15. [PubMed] [Google Scholar]
- 17.Fay WP, Eitzman DT, Shapiro AD, Madison EL, Ginsburg D. Platelets inhibit fibrinolysis in vitro by both plasminogen activator inhibitor-1-dependent and -independent mechanisms. Blood. 1994;83(2):351–6. [PubMed] [Google Scholar]
- 18.Matijevic N, Wang YW, Wade CE, Holcomb JB, Cotton BA, Schreiber MA, Muskat P, Fox EE, Del Junco DJ, Cardenas JC, Rahbar MH, Cohen MJ, P.S. Group Cellular microparticle and thrombogram phenotypes in the Prospective Observational Multicenter Major Trauma Transfusion (PROMMTT) study: correlation with coagulopathy. Thromb Res. 2014;134(3):652–8. doi: 10.1016/j.thromres.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dunbar NM, Chandler WL. Thrombin generation in trauma patients. Transfusion. 2009;49(12):2652–60. doi: 10.1111/j.1537-2995.2009.02335.x. [DOI] [PubMed] [Google Scholar]
- 20.Moore HB, Moore EE, Gonzalez E, Wiener G, Chapman MP, Dzieciatkowska M, Sauaia A, Banerjee A, Hansen KC, Silliman C. Plasma is the physiologic buffer of tissue plasminogen activator-mediated fibrinolysis: rationale for plasma-first resuscitation after life-threatening hemorrhage. J Am Coll Surg. 2015;220(5):872–9. doi: 10.1016/j.jamcollsurg.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothman KJ. No adjustments are needed for multiple comparisons. Epidemiology. 1990;1(1):43–6. [PubMed] [Google Scholar]
- 22.Green J, Britten N. Qualitative research and evidence based medicine. BMJ. 1998;316(7139):1230–2. doi: 10.1136/bmj.316.7139.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nylander M, Osman A, Ramstrom S, Aklint E, Larsson A, Lindahl TL. The role of thrombin receptors PAR1 and PAR4 for PAI-1 storage, synthesis and secretion by human platelets. Thromb Res. 2012;129(4):e51–8. doi: 10.1016/j.thromres.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 24.Ramsey MT, Fabian TC, Shahan CP, Sharpe JP, Mabry SE, Weinberg JA, Croce MA, Jennings LK. A prospective study of platelet function in trauma patients. J Trauma Acute Care Surg. 2016;80(5):726–32. doi: 10.1097/TA.0000000000001017. discussion 732-3. [DOI] [PubMed] [Google Scholar]
- 25.Kutcher ME, Redick BJ, McCreery RC, Crane IM, Greenberg MD, Cachola LM, Nelson MF, Cohen MJ. Characterization of platelet dysfunction after trauma. J Trauma Acute Care Surg. 2012;73(1):13–9. doi: 10.1097/TA.0b013e318256deab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore HB, Moore EE, Gonzalez E, Hansen KC, Dzieciatkowska M, Chapman MP, Sauaia A, West B, Banerjee A, Silliman CC. Hemolysis exacerbates hyperfibrinolysis, whereas platelolysis shuts down fibrinolysis: evolving concepts of the spectrum of fibrinolysis in response to severe injury. Shock. 2015;43(1):39–46. doi: 10.1097/SHK.0000000000000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Declerck PJ, Gils A. Three decades of research on plasminogen activator inhibitor-1: a multifaceted serpin. Semin Thromb Hemost. 2013;39(4):356–64. doi: 10.1055/s-0033-1334487. [DOI] [PubMed] [Google Scholar]
- 28.Levi M, Biemond BJ, van Zonneveld AJ, ten Cate JW, Pannekoek H. Inhibition of plasminogen activator inhibitor-1 activity results in promotion of endogenous thrombolysis and inhibition of thrombus extension in models of experimental thrombosis. Circulation. 1992;85(1):305–12. doi: 10.1161/01.cir.85.1.305. [DOI] [PubMed] [Google Scholar]
- 29.Lang IM, Schleef RR. Calcium-dependent stabilization of type I plasminogen activator inhibitor within platelet alpha-granules. J Biol Chem. 1996;271(5):2754–61. doi: 10.1074/jbc.271.5.2754. [DOI] [PubMed] [Google Scholar]
- 30.Kanter J, Khan SY, Kelher M, Gore L, Silliman CC. Oncogenic and angiogenic growth factors accumulate during routine storage of apheresis platelet concentrates. Clin Cancer Res. 2008;14(12):3942–7. doi: 10.1158/1078-0432.CCR-07-4824. [DOI] [PubMed] [Google Scholar]
- 31.Gando S, Nanzaki S, Sasaki S, Kemmotsu O. Significant correlations between tissue factor and thrombin markers in trauma and septic patients with disseminated intravascular coagulation. Thromb Haemost. 1998;79(6):1111–5. [PubMed] [Google Scholar]
- 32.Park MS, Owen BA, Ballinger BA, Sarr MG, Schiller HJ, Zietlow SP, Jenkins DH, Ereth MH, Owen WG, Heit JA. Quantification of hypercoagulable state after blunt trauma: microparticle and thrombin generation are increased relative to injury severity, while standard markers are not. Surgery. 2012;151(6):831–6. doi: 10.1016/j.surg.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]