

Abstract
Loss of cystic fibrosis transmembrane conductance regulator (CFTR) function causes cystic fibrosis (CF), predisposing the lungs to chronic infection and inflammation. In young infants with CF, structural airway defects are increasingly recognized before the onset of significant lung disease, which suggests a developmental origin and a possible role in lung disease pathogenesis. The role(s) of CFTR in lung development is unclear and developmental studies in humans with CF are not feasible. Young CF pigs have structural airway changes and develop spontaneous postnatal lung disease similar to humans, therefore, we studied lung development in the pig model (non-CF and CF). CF trachea and proximal airways had structural lesions detectable as early as pseudoglandular development. At this early developmental stage, budding CF airways had smaller, hypo-distended lumens compared to non-CF airways. Non-CF lung explants exhibited airway lumen distension in response to forskolin/IBMX as well as to fibroblast growth factor (FGF)-10, consistent with CFTR-dependent anion transport/secretion, but this was lacking in CF airways. We studied primary pig airway epithelial cell cultures and found that that FGF10 increased cellular proliferation (non-CF and CF) and CFTR expression/function (in non-CF only). In pseudoglandular stage lung tissue, CFTR protein was exclusively localized to the leading edges of budding airways in non-CF (but not CF) lungs. This discreet microanatomic localization of CFTR is consistent with the site, during branching morphogenesis, where airway epithelia are responsive to FGF10 regulation. In summary, our results suggest that the CF proximal airway defects originate during branching morphogenesis and that the lack of CFTR-dependent anion transport/liquid secretion likely contributes to these hypo-distended airways.
INTRODUCTION
Cystic fibrosis (CF) is a life-limiting disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Lung disease, the leading cause of CF morbidity and mortality, is characterized by recurrent airway infection, inflammation and remodeling (e.g. bronchiectasis).(1–5) Airways are principal sites for CF lung disease and understanding its early pathogenesis could clarify disease mechanisms, offer new insights for treatment modalities, and identify optimal windows for effective therapies.(6)
Structural defects in the proximal airways are increasingly recognized in young infants with CF, many times before clinical disease is apparent and before sufficient postnatal time has elapsed for remodeling to occur. For example, a retrospective study of pediatric bronchoscopy cases found tracheomalacia in ~15% of CF individuals (median age with tracheomalacia ~ 14 months) and tracheomalacia was diagnosed as early as one month of age.(7) Likewise, a study of young infants with CF (median age, 3.6 months) found airway dilatation, airway wall thickening or air trapping in greater than 80% of the subjects by computed tomography, yet only ~16% of subjects had clinical respiratory disease.(8) In a review of published autopsy data from infants less than two weeks of age, the older and generally larger infants with CF had significantly smaller trachea caliber than the non-CF controls.(9, 10) Structural airway defects in young children with CF prior to the onset of significant clinical disease could suggest that these changes have fetal origins.
Studies on human fetal tissue are limited by ethical standards, tissue availability, and acceptable controls. An alternative approach to overcome these limitations is to study fetal development in an animal model. CF pigs spontaneously develop postnatal lung disease including lesions similar to humans with CF.(11–13) Additionally, newborn CF pigs, reminiscent of human infants with CF, exhibit a number of lung (e.g. air trapping) and proximal airway abnormalities including anterior cartilage defects, altered smooth muscle bundles, and hypoplastic submucosal glands.(9, 14) These congenital abnormalities point towards a fetal origin. We hypothesized that the absence of CFTR would cause morphological lesions during early fetal airway development. We studied non-CF and CF fetal tissues in parallel, so as to identify the time and onset of early lesions and to clarify the functional role(s) of CFTR in normal development.
MATERIALS AND METHODS
Tissues
All animal experiments were approved by the University of Iowa Institutional Animal Care and Use Committee (IACUC). Experimental methods were carried out in accordance with relevant federal guidelines including the Public Health Service policy on humane care and use of laboratory animals, and in accordance with the Animal Welfare Act. Null (CFTR−/−) and ΔF508 (CFTRΔF508/ΔF508) litters were produced from heterozygous matings(11, 13) and TgFABP-pCFTR pigs were produced by cloning as previously described(12). Date-mated sows (Exemplar Genetics, Sioux City, Iowa) were euthanized on days 36, 41, 54, 60, 69, and 82 of gestation and fetal tissues were collected (Supplemental Table 1). When only a small number of time points were available for trachea and lung analyses, gestational time points were sometimes combined into one group as long as it did not change the context of the data. In these situations, the merged time points are labeled accordingly. Male and female pigs were used as available in litters, whereas the cloned pigs were males. To evaluate the differences in non-CF and CF phenotype, littermates (i.e. siblings) were used in the studies to compare CF and non-CF groups for all time points with the exception of the TgFABP-pCFTR pigs (used only at d36) in which each litter were exclusively clones. Trachea and lungs from archival blocks at d90 gestation and newborn pigs were additionally used for CFTR immunohistochemistry.
Histopathology
At tissue collection, trachea and lung tissues were placed into 10% neutral buffered formalin (~ 3–5 days) and submitted to the Comparative Pathology Laboratory (University of Iowa, Iowa City, IA USA) for tissue processing, paraffin embedding, sectioning (~4 μm), and histochemical staining with hematoxylin and eosin (HE) stain and amylase-pretreated Periodic acid-Schiff (dPAS). Histopathological examination was performed by a veterinary pathologist experienced with the model. At early time points, some tissues were excluded from study due to insufficient tissue or irregular plane of sectioning (e.g. trachea). Morphometric (quantitative) analysis and histopathologic (semi-quantitative) scoring was done in a manner masked to group treatments so as to avoid bias.(15)
Immunohistochemistry
CFTR: CFTR immunohistochemistry was performed as previously described.(16) Briefly, paraffin embedded tissues were sectioned (~4 μm) and hydrated through a series of xylene and alcohol baths. Antigen retrieval (citrate buffer, pH 6.0, 110°C for 15 minutes) was performed using the NxGen Decloaking Chamber™ (Biocare Medical, Concord, CA USA). Endogenous peroxidase activity was quenched with hydrogen peroxide (3%, 8 min), endogenous avidin/biotin was blocked (Avidin/Biotin Blocking Kit, Vector Laboratories, Inc., Burlingame, CA USA), and nonspecific background blocked with equine serum (5% in 1× Dako Buffer). Primary Ab was generously supplied by Dr. John Riordan, Ph.D., University of North Carolina –Chapel Hill and the Cystic Fibrosis Foundation Therapeutics. Primary Ab (1:1200 in Dako diluent, 60 minutes) was applied and followed by secondary (1:200, 30 minutes, Vector Biotinylated Anti-Mouse IgG) and then Vector ABC Reagent (30 minutes, Standard VECTASTAIN® Elite® ABC Kit, Vector Laboratories, Inc., Burlingame, CA USA). Next, chromogen (DAB Plus - 5 minutes and then DAB enhancer, 3 minutes, room temperature) was applied and then the tissues counterstained with Harris hematoxylin (1 min., Surgipath, Leica Microsystems, Inc., Buffalo Grove, IL USA). Slides were blue in Scott’s Tap water, dehydrated through a series off alcohols and xylene baths, then coverslipped.
FGF10: Paraffin embedded tissues were sectioned, hydrated, antigen retrieved and blocked with endogenous peroxidase, similar to the CFTR protocol above. General background was blocked with Background Buster (30 min., Innovex Biosciences, Inc., Richmond, CA USA). Primary antibody (rabbit polyclonal, 1:200 × 2hr, Santa Cruz #sc7917, Santa Cruz Biotechnology, Dallas, TX USA) was followed by washing, secondary antibody (biotinylated anti-rabbit 1:500 × 30 min.) and Vector ABC Reagent (30 minutes, Standard VECTASTAIN® Elite® ABC Kit, Vector Laboratories, Inc., Burlingame, CA USA). Application of chromogen, counterstain, and coverslipping was performed as stated above for CFTR.
Morphometry
Tracheas were examined from fetuses for d54/60, 69 and 82 days gestational age. In earlier time points (d36 and d41), some tissues were not consistently available for examination and interpretation, so were excluded. Tracheas were histopathologically examined (typically mean of ~3 samples per animal) and morphometrically assessed in cross section similar to that described for neonatal CF pigs.(9) Briefly, high resolution digital images were collected on a BX51 microscope with DP73 digital camera (Olympus, Center Valley, PA USA) and analyzed with CellSens Software (Olympus, Center Valley, PA USA). Trachea tissues were examined in cross section. Tracheal size (i.e. caliber) was evaluated by lumen circumference. The growth of submucosal glands and trachealis smooth muscle tissues was assessed through tissue area normalized by lumen circumference (i.e. area/circumference) for each tissue section. For quantitative measurements, the mean value for each animal was used in the analysis. The detection of discontinuous cartilage and for smooth muscle bundles was scored in a binary manner (0 – absent, 1 present) for all tissues sections/animal and the median score used for analysis.
Lungs were evaluated at d36–60 gestational age for pseudoglandular development. The pseudoglandular stage represents the development of large airways.(17) The number of airways sampled per lung ranged between 150–300 structures (mean ~230) except when limited by tissue sample size (e.g. d36 samples) in which all available airways were sampled. To examine lumen diameters, the length of the minor diameter was consistently selected for each airway in cross sections. So as to be consistent for each sample, all primitive airways were measured including those in which the lumen was not observed and was given a value of zero. To evaluate the incidence of unexpanded primitive airways, the percentage of the total airways with a diameter value of <6 μm were determined. The basophilic histochemical staining qualities of luminal material (”staining scores”) was studied using histochemical (HE) stained samples and ordinally scored based on its appearance as: 0 – none, 1 – eosinophilic, 2 – mixture of eosinophilic and basophilic material, and 3 – basophilic material. Airway filling of luminal material (“filling scores”) was ordinally scored by estimating the % of filling for each airway as: 0 – none, 1 – 1 to 25%, 2 – 26 to 50%, 3 – 51 to 75% and 4 – 76% to complete obstruction.
Lung explant studies
After tissue collection, lungs were transported in DMEM/F12 with penicillin/streptomycin, gentamicin, and amphotericin B. Explants were placed on collagen coated Costar Transwells (Corning, Corning, NY USA) and cultured in Ham’s F12 with 10% FCS, 10mM HEPES, and penicillin/streptomycin.
Explants were treated with 10 μM Forskolin and 100 μM IBMX, and either 50ng/ml FGF10 or DMSO control then imaged, depending on the study, every four hours for 24 hours, or daily for up to as much as 7 days. Images were taken on an Olympus IX-81 microscope, and lumen area was measured using ImageJ.(18)
Primary culture studies for FGF10 stimulation studies
Well-differentiated primary porcine cultured epithelia were grown on filters and incubated for 0, 24, or 72 hours with 100ng/mL of recombinant human FGF10 in growth media (cat # 345-FG-025, R&D Systems, Minneapolis MN, USA). CFTR protein expression was examined by enumerating the number of positive immunostained cells (apical brown color) standardized per length of filter evaluated in sections. Differences in epithelial height were determined by evaluating the total area of the cultured epithelia divided by the length of filter evaluated in sections (area/length) to obtain a mean height (this approach avoided sampling bias).
Electrophysiology
Cultures were mounted in modified Ussing chambers (Physiologic Instruments, Inc., San Diego, CA USA). Cultured epithelia were bathed on both surfaces with solution containing (mM): 135 NaCl, 2.4 K2HPO4, 0.6 KH2PO4, 1.2 CaCl2, 1.2 MgCl2, 10 dextrose, 5 HEPES (pH = 7.4) at 37 °C and gassed with compressed air.
Vt was maintained at 0 mV to measure short-circuit current (Isc). Transepithelial electrical conductance was measured by intermittently clamping Vt to +5 and/or −5 mV.
Parameters were measured under basal condition and after the following interventions: 100 μM apical amiloride (as previously performed in our lab),(12, 19, 20) 100 μM apical DIDS (4,4- diisothiocyano stilbene-2,2-disulfonic acid), 10μM forskolin and 100μM IBMX (3-isobutyl-2-methylxanthine), and100μM apical GlyH-101.
RNA extraction and Reverse Transcription
RNA was extracted using QIAzol reagent from RNeasy Lipid Tissue Mini Kit (QIAGEN, Valencia, CA, USA) and contaminating DNA was removed using DNase according to the manufacturer’s instructions. 500 ng of total RNA was reverse transcribed using SuperScript VILO MasterMix (Life Technologies, Foster City, CA, USA) according to the manufacturers’ specifications.
Real-time qPCR analysis and normalization
qPCR was performed using Takara Bio SYBR Green Master Mix with ROX (Clontech, Mountain View, CA, USA). Primer sequences were as follows CFTR forward: 5′-CTG GAG CCT TCA GAG GGT AAA AT -3′, CFTR reverse: 5′-AGT TGG CAC GCT TTG ATG ACA CTC C -3′; β-actin forward: 5′-CTG CGG CAT CCA CGA AAC T -3′, β-actin reverse: 5′-GTG ATC TCC TTC TGC ATC CTG TC -3′. The cDNA was amplified in the 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Relative mRNA expression was calculated using the ΔΔCt method. Individual data were normalized against the housekeeping gene β-actin. Results were given as fold change in expression over the non-CF control (no treatment at 0 hr time point) for each time point and donor.
FGF10 RNA isolation and sequencing
Total RNA was isolated from in vivo porcine fetal lungs on day 37 of gestation using the mirVanaTM miRNA isolation kit (Ambion, Grand Island, NY USA). Total RNA was tested on an Agilent Model 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA USA). Samples with an RNA integrity number (RIN) greater than 8.0 were selected for further processing.
Libraries were prepared using the TruSeq RNA Sample Prep (Illumina, San Diego, CA) and submitted to the University of Iowa DNA Facility for deep sequencing. 12 paired-end DNA libraries were sequenced to an average depth of 212 million read pairs (range of 179–241 million) with 100 base reads. The sequences were aligned to the Sus scrofa genome (release 10.2) using TopHat (v2.0.10) and known genes were annotated using Ensembl (release 74). Gene expression differences between CF (5 replicates) and non-CF (7 replicates) groups were analyzed using Cuffdiff (v2.1.1).(21)
Statistics
Comparison of morphometric and semi-quantitaive scoring data between non-CF and CF groups was performed with the nonparametric Mann Whitney or Bonferroni post-tests unless otherwise stated. Evaluation of anterior cartilage defects and smooth muscle bundles were statistically evaluated through a Chi-Square test. Statistical significance was placed at P<0.05.
RESULTS
CF tracheal abnormalities begin early in fetal development
We studied tissues from non-CF and CF fetal pigs (term = 114 days). Similar to newborn CF pigs,(9) fetal CF pigs had small caliber tracheas (Figure 1a, b) with cartilage (arrows, Figure 1a; Supplemental Table 2) and smooth muscle defects (Figure 1c,d, Supplemental Table 3) that were detected as early as d54/60 of gestation. In contrast, submucosal glands were first observed at d82 gestation and consisted only of primitive ducts that lacked serous acini or mucinous tubules, but no group differences were seen in submucosal gland growth (Figure 1e, f). The origin of submucosal glands after onset of other tracheal lesions indicates that submucosal glands were not causative in these developmental abnormalities.
Figure 1. Fetal trachea from non-CF and CF pigs at d54/60, d69 and d82 of gestation (term ~ 114d).
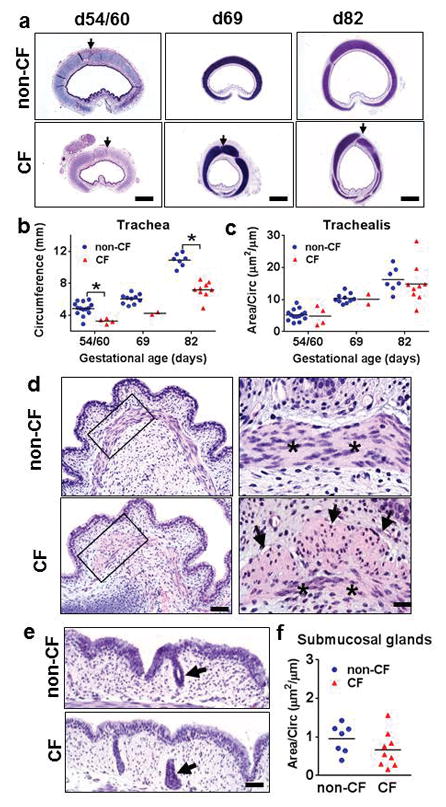
a) In tissue sections, CF tracheas had smaller caliber with anterior cartilage defects (arrows). HE stain, bars = 420μm, 840μm, and 1mm, respectively. b) Tracheal caliber was reduced in CF pigs as early as d54/60, (*P<0.01, Mann Whitney test). c) The trachealis muscle of non-CF and CF tracheas had similar amount of growth from d54 to d82, (NS, Mann Whitney test). d) The trachealis muscle (asterisks) had lesions, i.e. morphological evidence of smooth muscle bundles (arrows) in cross sections as early as d60 in CF pigs. HE stain, bars = 62 and 20μm, respectively. e) Submucosal glands were first detected as early as d82 in tracheas and at this time were composed of budding ducts (arrows) in both groups, HE stain, bar = 40μm. f) Submucosal glands had similar early growth in CF and non-CF at d82 (P=0.173, Mann Whitney test). Graph lines = mean.
CF airways have abnormalities during pseudoglandular development
Fetal CF tracheas exhibited among other things small caliber, similar to the smaller caliber trachea and bronchi we previously detected in newborn and postnatal CF pigs.(9, 14, 22) These proximal airway defects suggested a common etiology, and so we examined lungs at the pseudoglandular stage, when the proximal conducting airways are formed by branching morphogenesis.(17)
During early pseudoglandular development (d36 and d41), CF pig airways had smaller lumens compared to non-CF airways, but by d54/60 (Figure 2a, b) no differences were detected. To evaluate the smaller CF airways from another perspective, we evaluated the percentage of “hypo-distended” airways (i.e. <6 μm diameter) in fetal lung. We chose <6 μm as a cutoff because the smallest non-CF airways in both d36 and d41 lungs represented a nominal portion (~15%) of the distribution. CF airways had a significantly higher percentage of hypo-distended airways compared to non-CF in early pseudoglandular lung growth (Figure 2c). In fetal airways, wispy to amorphous material was sometimes observed within fetal airway lumens of both groups (Figure 2d). The lumen material appeared to have more basophilic staining in some CF airways using routine hematoxylin and eosin (HE) stains. To test this observation, the extent of basophilia in the lumen material was scored using an ordinal system.(15) CF lumen material had higher scores indicative of more basophilic staining than non-CF (Figure 2e) and the basophilic luminal material was reminiscent of mucus staining that is sometimes basophilic; however, it was negative for dPAS (Figure 2f) suggesting that it was not mucus. While the specific cause of the increased basophilic staining was not identified, many factors (e.g. pH, fixative type, etc.) can influence histochemical stain qualities including the extent of eosinophilia and basophilia in a routine HE.(23–25) The histochemical staining of the CF airway lumen material most likely reflects changes in the local environment that have been described in CF airways such as anion secretion,(26) liquid volume,(27) or pH.(28, 29)
Figure 2. Histopathology and morphometry of pseudoglandular fetal lung (d36–60).
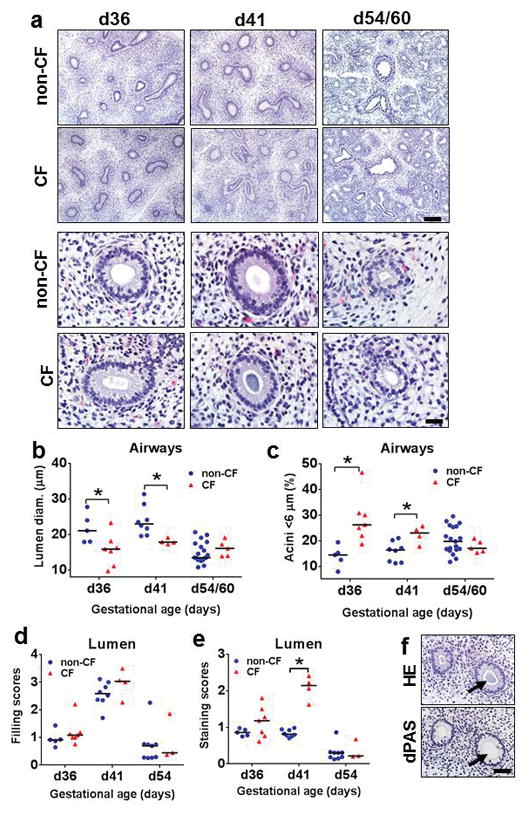
a) Tissue sections of fetal lung from CF and non-CF pigs at d36, d41, and d54/60, HE stain, bars = 161 and 27μm, respectively. b,c) CF airways had reduced lumen size (b) with increased percentage of hypo-expanded airways (c) compared to non-CF lungs at d36 and d41 in early pseudoglandular growth. d,e) Airways in both groups had similar filling scores for luminal material (d), but CF airways had increased basophilic staining scores noted at d41 (e). f) The altered composition of luminal material by HE stain (top, arrow) was negative for dPAS+ mucus (bottom, arrow) on serial sections. Bar = 54μm. Graph lines = mean.
cAMP- and growth factor-mediated liquid secretion validates fetal human lung studies
Early gestation fetal lung explants from humans are reported to show cAMP-mediated liquid secretion (manifested as luminal distension of fetal airways) in normal, but not CF, lungs; however, interpretation of these studies was limited by the small number of CF cases evaluated (n=2).(27, 30) To replicate and extend these limited observations in human tissues and investigate the utility of the pig model for developmental studies, we studied fetal lung explants from non-CF and CF pigs. Over the course of seven days, fetal pig lung explants lacked differences between non-CF and CF groups under basal (unstimulated) conditions (Figure 3a). In contrast, forskolin/IBMX stimulation of non-CF, but not CF, fetal pig lungs produced marked airway lumen distension consistent with cAMP-mediated secretion (Figure 3b, c).
Figure 3. Influence of cAMP and FGF10 on fetal pig lung epithelia.
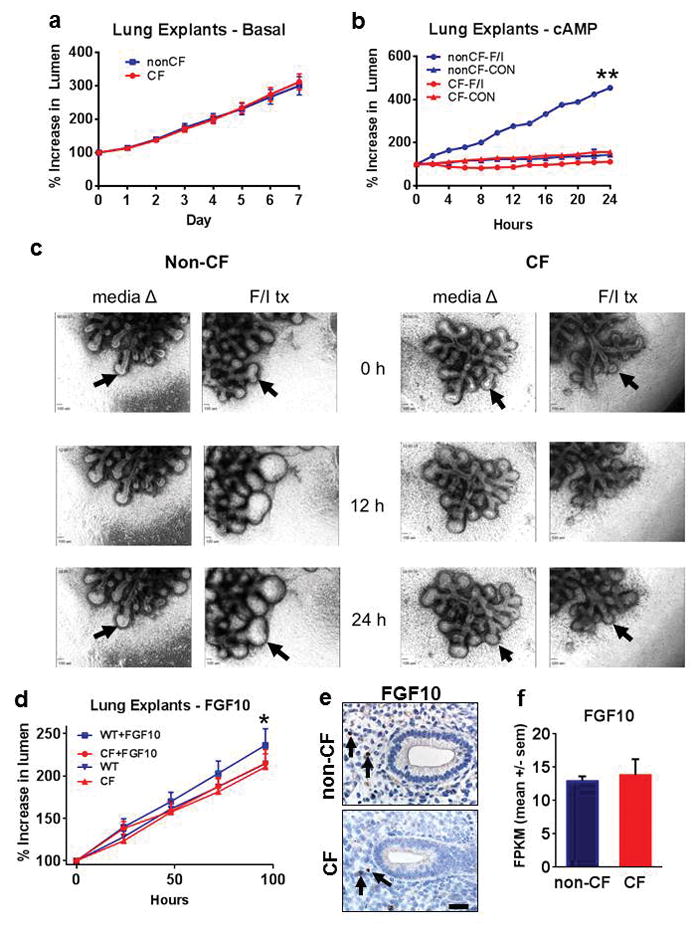
a) Fetal lung explants (d36) did not show differences in basal (unstimulated, media) lumen size between non-CF and CF groups. b) Fetal lung explants (d36) following forskolin/IBMX (FI) stimulation showed lumen expansion in non-CF airway lumens compared to CF airways (**P<0.01, nonCF-F/I vs CF-F/I groups at 24 hours) and unstimulated (media) controls. c) Morphology of non-CF and CF fetal lung explants using FI stimulation or media controls over 24 hours. Note the arrows (top and bottom) following airway lumen changes in forskolin/IBMX (F/I tx), but not in CF airways or unstimulated (media) controls. d) FGF10 stimulation of fetal lung explants (d36) caused expansion of airway lumens in non-CF (“WT + FGF10”, *P=0.013, linear regression), but not in CF airways or unstimulated (media) controls. e) FGF10 immunostaining of fixed fetal lung (d36) showing FGF10+ cells (arrows) similarly located in the mesenchymal tissues adjacent to the budding fetal airways, bar = 27μm. f) FGF10 mRNA abundance was similar in both non-CF and CF groups at d37 gestation (P=0.629, Cuffdiff test statistic, graph bars = mean +/− SEM).
Growth factors such as fibroblast growth factor (FGF)-10 have been reported to also stimulate liquid secretion in fetal lung explants from humans.(31) We evaluated fetal pig lung explants and found that FGF10 stimulated lumen expansion in non-CF, but not CF lung tissues (Figure 3d). While FGF10 significantly increased in lumen size in explants through CFTR-mediated secretion, this change was less robust than the forskolin/IBMX results. We speculate that the relative influence of FGF10 stimulation on CFTR-mediated secretion over the course (i.e. weeks) of early pseudoglandular growth could correlate biologically to the reduction in size of CF airways (<28% reduced compared to non-CF) during branching morphogenesis (see Figure 2 a–c). We localized FGF10 expression by immunostaining in fetal pig lungs (Figure 3e), and similar to studies in other species,(32, 33) we observed scattered FGF10 positive cells in mesenchymal tissues adjacent to airways. FGF10 mRNA abundance (Figure 3f) was studied in fetal lung, but there were no group-specific differences. These results demonstrate that loss of CFTR function did not alter FGF10 abundance and localization. Cumulatively, these data validate previous human fetal studies(27, 30, 31) and show that the fetal pig lung exhibits CFTR-mediated anion transport/liquid secretion in response cAMP agonists and FGF10.
FGF10 influence on primary airway epithelial cultures
To further understand how FGF10 might influence CFTR expression or function, we studied its effects on primary airway epithelial cultures from newborn pigs. Culture in the presence of FGF10 was associated with increased CFTR mRNA expression (Figure 4a) and increased immunostaining for CFTR protein (Figure 4b, c) in non-CF cells. In addition for both non-CF and CF cultures, FGF10 treated cells had increased epithelial height (Figure 4d) and Ki67 index (Figure 4e), both consistent with an increased cellular proliferation state. We also examined the effects of FGF10 on transepithelial short-circuit current. FGF10 increased cAMP-stimulated short-circuit current in non-CF cells (Figure 4f), but it did not affect conductance (Figure 4g) or amiloride-sensitive current (Supplemental Figure 1) in CF epithelia.
Figure 4. FGF10 affects CFTR-mediated secretion and growth of porcine epithelia.
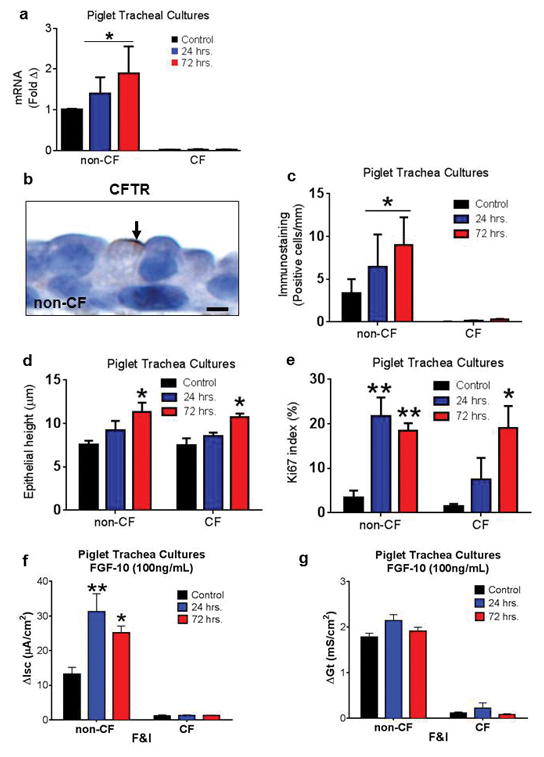
a–c) In primary pig epithelia cultures, FGF10 increased CFTR mRNA (a) and immunostaining (b,c, bar = 3μm) in non-CF cells (n=3/group) (Control versus combined FGF10 treated groups [24 hr + 72 hr], *P<0.05, Mann Whitney test). Note the example of staining for both groups in (b). d,e) In non-CF and CF primary pig epithelia cultures, FGF10 increased cell height (d) and increased Ki67 index (e) consistent with cellular proliferation (*P<0.05 and **P<0.01, Bonferroni post-test). f,g) Electrophysiology of non-CF and CF (n=3 & 4/group, respectively) primary pig epithelia cultured with FGF10. FGF10 treatment increased forskolin/IBMX (F&I) stimulated short-circuit current (f, 24 and 72 hrs, *P<0.001 and **P<0.01 versus control, Bonferroni post-test). FGF10 treatment did not significantly affect conductance (g, NS, Bonferroni post-test). Graph bars = mean +/− SEM.
CFTR immunostaining is temporally and spatially regulated during development
CFTR mRNA and protein expression have been detected during fetal lung development in humans.(27, 34–37) In pseudoglandular lung, human CFTR protein has been localized immunohistochemically in the developing epithelium and adjacent mesenchyme; however, the extensive cellular immunostaining that has been historically reported does not clarify a role for CFTR in lung development.(34, 38) We localized CFTR protein in fetal tissues to better understand its normal developmental role and to study the potential consequences of its absence in CF. CFTR immunostaining was seen only in non-CF tracheas supporting the specificity of the immunohistochemical technique.
CFTR immunostaining was evaluated in trachea at d60 and d90 during fetal growth and these were compared to newborn trachea (Figure 5a–d). At d60, submucosal glands were not yet developed, but weak CFTR staining was detected in scattered cells of the airway surface epithelium (Figure 5a). By d90, CFTR had similar, but stronger immunostaining in scattered uncommon cells of the surface epithelium as well as serous cells of the submucosal glands (Figure 5b). Newborn trachea had more robust CFTR immunostaining than d90 in scattered cells of the surface epithelium and serous cells of submucosal glands (Figure 5c,d).
Figure 5. Localization of immunostaining in fetal and newborn (NB) non-CF pigs. CF tissues consistently lacked CFTR immunostaining.
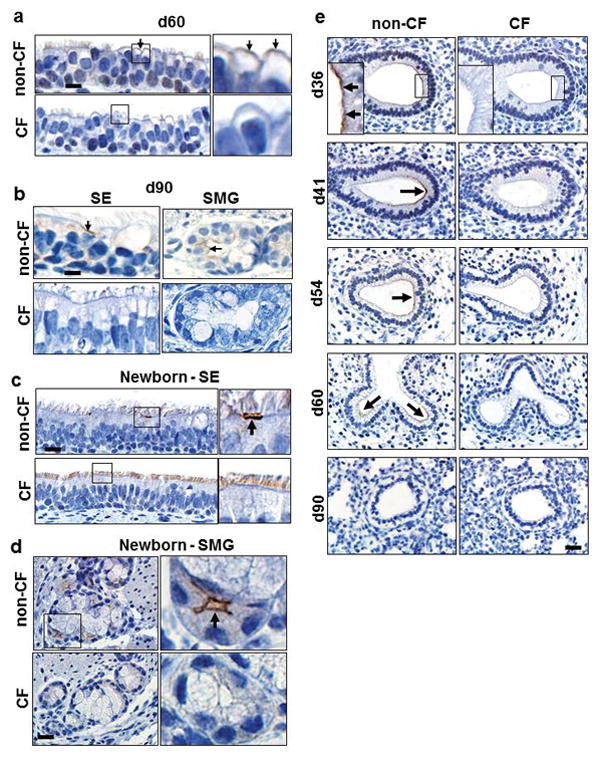
a) Fetal non-CF pig trachea (d60) with weak apical CFTR immunostaining (arrows, insets) of nonciliated epithelial cells, bar = 11 and 3μm, respectively. b) Fetal non-CF pig trachea (d90) with apical CFTR immunostaining (arrows) on solitary surface epithelial (SE) cells and serous cells of the submucosal glands (SMG), bar = 10 and 5 μm, respectively. c,d) Newborn non-CF trachea with apical CFTR immunostaining (arrows) in solitary cells of the SE (c, bar = 15 and 7μm, respectively) and in most serous cells of the SMG (d, bar = 22 and 5μm, respectively). e) CFTR immunostaining in non-CF pseudoglandular lung (d36–60) was typically located on the apical cell surface along leading edge of budding airways (arrows and top left inset panel), bar = 29 and 10μm, respectively. CFTR immunostaining intensity decreased with age and was absent in distal airspaces at d90 (canalicular lung).
In contrast, newly-formed airways undergoing branching morphogenesis had robust immunostaining at d36 (Figure 5e), but CFTR intensity gradually declined through the pseudoglandular period and was absent in distal lung by d90 (Figure 5e). Importantly, during branching morphogenesis CFTR protein was exclusively localized to the leading edge of budding airways (Figure 5e). This previously undocumented localization pattern is novel and consistent with the specific site of FGF10 responsive cells during early branching morphogenesis.(32, 39) These data suggest that CFTR expression has temporal and spatial changes during fetal airway development. CFTR expression is most robust in branching airways early in lung development, but later in gestation is robustly expressed in submucosal glands and scattered surface epithelial cells of trachea and bronchi.
DISCUSSION
From this current study, several conclusions emerged regarding CFTR during lung development.
First, the absence of CFTR during fetal growth produces airway defects that are detectable early in the pseudoglandular stage of lung development. Lesions in CF fetal airways during early branching morphogenesis are consistent with the developmental origins of proximal airways, the same airways that have structural abnormalities in newborn CF pigs.(9, 14) In contrast, CF and non-CF airways lacked differences in lumen distention at later time points during branching morphogenesis, which corresponds developmentally with the origins of more distal airways that lack known structural differences in newborn CF and non-CF pigs. The lack of group differences in later pseudoglandular stages might reflect one or more changes: 1) transition of CFTR protein (i.e. secretion) from distal lung to large airway submucosal glands and surface epithelia (see Figure 5); 2) an anatomic “dilution” effect from significantly more, variably sized airways and airspaces being formed as the lungs start to transition from pseudoglandular to canalicular stage of development; and/or 3) increased diversity of microanatomic structure during the transition from pseudoglandular to canalicular stages makes “apples to apples” comparison more challenging.
Structural defects in the proximal airways have been reported shortly after birth in CF infants (7–9) and similarly in animal models such as CF mice,(40) CF pigs,(9, 13, 14, 22, 41) and CF rats.(42) CFTR expression is reportedly higher during early fetal life compared to adult lung tissues in humans(27, 34, 36, 37) and many animal models(43–46). Cumulatively, these data corroborate the expected early fetal origin for proximal airway defects seen in newborn CF pigs.
Second, our data suggest that CFTR contributes to, but is not essential for, lung liquid secretion and airway distension during early fetal lung growth. In the developing fetal airways, liquid secretion is coupled to active Cl− transport(47) and cAMP-stimulated secretion contributes to this luminal volume.(27) In addition to these secretions, liquid volume in fetal lungs is maintained through the collective actions of low elastic recoil, diaphragmatic contractions (fetal breathing), and glottic closure.(48) The combined actions of secretion and maintenance of fetal lung liquid produces a slightly positive intra-luminal pressure to distend airways.(49) Some in the CF community had wondered why a lack of CFTR-dependent anion transport/secretion during early development of CF babies did not cause lung hypoplasia. Thus, questions concerning a role for CFTR during early fetal life have persisted and not been possible to study, until the advent of newer animal models such as the CF pig.
During development, airway distension provides a mechanical “stretch” signal (i.e. mechanotransduction) necessary for proper lung development and CFTR has been linked with this signaling pathway.(27, 50) Our data suggest that in CF, deficient CFTR-mediated liquid secretion reduces, but does not eliminate, airway distension (Figure 6a). We speculate that this reduced airway distension improperly “imprints” downstream signaling pathways in the affected fetal airways resulting in morphological differences in the proximal (red, Figure 6b,c), but not the distal (blue, Figure 6b,c) airways of the newborn non-CF and CF pigs.
Figure 6. A proposed model for CFTR in early lung development.
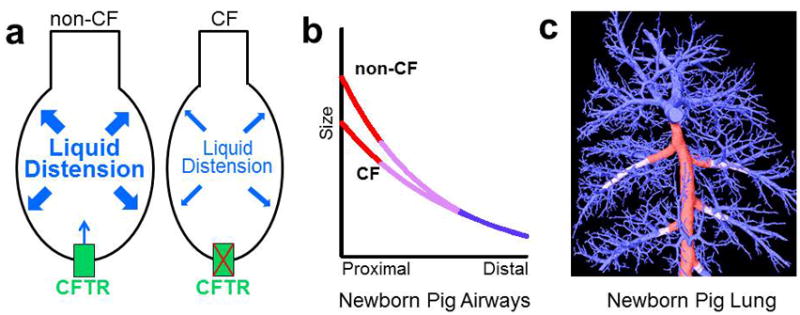
a) During early branching morphogenesis, CFTR transport/liquid secretion contributes to, but is not essential for, distension of fetal airways (a). Fetal airway “stretch” (i.e. mechanical distension) directs signaling pathways needed for normal airway development. In CF, we speculate that diminished airway distension dysregulates downstream stretch-mediated signaling pathways resulting in proximal airway lesions at birth. b,c) Newborn CF pig airways have structural differences compared to non-CF airways. This difference is preferentially seen in the proximal airways (red, b,c) with a transition zone (lilac, b,c) to similar sized distal airways (blue, b,c). Proximal airways originate during early pseudoglandular growth and this time period corresponds to when differences in fetal airways were observed (see Figure 2a–c). c) A computed tomography reconstruction of the newborn pig airway tree shows the location where size differences are noted between non-CF and CF in the proximal airways (red line) with a transition zone (lilac line) to the distal airways (blue line), which are similar in size. b and c adapted from Adam et al.(14)
Lastly, we were able to validate previous human studies(27, 30) regarding CFTR-dependent liquid secretion in fetal lung and demonstrate that the pig model is useful for the study of lung development. In fetal pig lung explants, CFTR transport/liquid secretion, stimulated by forskolin/IBMX or FGF10, distended airway lumens in nonCF lungs. This distension was absent in CF lungs. Lumen distension in explants may be useful to study CFTR function and therapies and parallels endpoints described for epithelium-derived organoids.(51–53) While organoids offer several opportunities for study of CF, fetal lung explants from models such as the pig have advantages in that these represent native tissue. Accordingly, fetal explants retain epithelial and mesenchymal tissues that are increasingly recognized to cross-talk in normal lung development.(54, 55) Our data suggest insufficient liquid secretion as a contributing mechanism for reduced lumen size in CF fetal airways, but we cannot completely exclude the contributions of other mechanisms (e.g. contracted airway smooth muscle)..(56) Additionally, our work does not exclude the participation of other factors that can influence liquid secretion including keratinocyte growth factor(31) or extracellular calcium-sensing receptors.(38)
We were also able to study FGF10 and its influence on fetal airway secretion and CFTR expression in lung epithelia. FGF10 is an essential signaling factor regulating early lung development(32, 55) and is secreted by lung mesenchymal cells to bind its receptor (FGFR2 IIIb) on fetal airway endoderm. (32, 57–59) Previous studies have suggested that secretion of FGF10 by lung mesenchymal cells is a factor influencing branching morphogenesis of the nascent fetal airways. (32, 39) Interestingly, our data show that CFTR is localized to FGF10-regulated regions undergoing branching morphogenesis. Previous work by Graeff et al. demonstrated that FGF10 could stimulate liquid secretion in non-CF fetal human lung explants; however, a relationship to CFTR expression or function was not evaluated.(31) Wong et al. reported that FGF10 could transform and differentiate human embryonic stem cells into airway epithelia that was identified, in part, by increased CFTR expression.(60) Mouse models have helped elucidate the effects of dysregulated FGF10 in lung development. For example, mice that are homozygous null for FGF10 or its receptor FGFR2 IIIb both have pulmonary agenesis.(58, 61) In contrast, hypomorphic mutants developed lungs with altered airway size and defective development of smooth muscle and cartilage,(33, 62, 63) features that are similar in scope to that observed in newborn CF pigs.(9, 14) Salivary glands also undergo branching morphogenesis similar to lung during fetal development and recent work suggests cAMP-mediated CFTR secretion participates in lumen expansion in fetal salivary glands.(64) These data along with our other results suggest a compelling and novel association between FGF10 and CFTR during early branching morphogenesis – the time when proximal airways develop.
This study has limitations. 1) We do not know with certainty that early fetal lung development is similar between pigs and humans. But the similarity of the porcine and human lung anatomy and physiology, morphologic stages of lung growth, and the phenotypes CF pigs and CF humans suggest they have significantly overlapping developmental patterns.(41, 65) 2) Our investigation did not focus on CFTR interaction(s) with cell signaling pathways (e.g. Wnt/beta-catenin)(66) that might influence specific signaling pathways in development. However, our demonstration of hypo-distension of the proximal CF airways provides compelling evidence for mechanotransduction pathways as the target of future studies. 3) Lastly, we did not evaluate the initial stage (e.g. embryonic stage) of fetal lung growth. However, the overlapping abnormalities in fetal and newborn CF airways and trachea (a structure formed during embryonic development) would predict that embryonic lung growth may have similar roles for CFTR function.
Our study also has several advantages. 1) Study of the fetal pig lung model allowed us to examine early developmental time points using age-matched controls (not possible in human studies). 2) Caesarian-section derived pigs (non-CF and CF) allowed us to collect “pristine” lung tissues (i.e. lacking autolytic or degenerative changes often present in fetal lungs from humans). While CFTR protein localization has been examined in adult and fetal lungs from humans,(34, 38, 67, 68) nonspecific background has been a concern for CFTR immunohistochemistry techniques. For example, harvest to fixation times in human tissues can varying depending on circumstances and the source (e.g. autopsy, biopsy etc.), thus influencing tissue autolysis.(16) 3) Fetal lung tissues from pig models are readily accessible. These features make the pig model attractive for translational studies that are largely impossible in humans.
In conclusion, our data suggest several novel and compelling findings. 1) Hypo-distension of CF airways is the principal lesion seen at the early stage of branching morphogenesis, a time point when the proximal airways (i.e. those prone to structural defects seen at birth) are formed. 2) Fetal lungs from non-CF, but not CF, pigs exhibit CFTR-dependent anion transport/liquid secretion, corroborating previous human studies and validating the fetal lung pig model. 3) FGF10 stimulates airway proliferation (non-CF and CF) and increased CFTR expression and secretion (non-CF only). 4) CFTR protein is exclusively localized in airway epithelial sites that are prone to FGF10 regulation during branching morphogenesis, a novel finding. Our data suggest that in normal lung development, CFTR-dependent anion transport/liquid secretion is associated with FGF10 regulation of branching morphogenesis and that in CF, hypo-distended airways may lead to “downstream” signaling changes during development resulting in airway defects.
Supplementary Material
Acknowledgments
This work was supported by the National Heart Lung and Blood Institute (grant HL51670 and HL091842), the National Institute of Diabetes and Digestive and Kidney Diseases (grant DK54759), and the Cystic Fibrosis Foundation. GlyH-101 was a generous gift from the Cystic Fibrosis Foundation Therapeutics and Robert Bridges, Ph.D. CFTR antibody was a generous gift from John Riordan, Ph.D., University of North Carolina – Chapel Hill, and Cystic Fibrosis Foundation Therapeutics.
Footnotes
The authors declare no conflict of interest.
References
- 1.Andersen DH. Pathology of cystic fibrosis. Ann NY Acad Sci. 1962;93:500–17. [Google Scholar]
- 2.Oppenheimer EH, Esterly JR. Pathology of cystic fibrosis review of the literature and comparison with 146 autopsied cases. Perspect Pediatr Pathol. 1975;2:241–78. [PubMed] [Google Scholar]
- 3.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 4.Welsh MJ, Ramsey BW, Accurso F, Cutting GR. Cystic Fibrosis. In: Scriver CR, ALB, Sly WS, Valle D, Childs B, Vogelstein B, editors. The Metabolic and Molecular Basis of Inherited Disease. New York: McGraw-Hill; 2001. pp. 5121–89. [Google Scholar]
- 5.VanDevanter DR, Kahle JS, O’Sullivan AK, Sikirica S, Hodgkins PS. Cystic fibrosis in young children: A review of disease manifestation, progression, and response to early treatment. J Cyst Fibros. 2015 doi: 10.1016/j.jcf.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. 2015;372:351–62. doi: 10.1056/NEJMra1300109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fischer AJ, Singh SB, Adam RJ, et al. Tracheomalacia is associated with lower FEV and Pseudomonas acquisition in children with CF. Pediatr Pulmonol. 2013 doi: 10.1002/ppul.22922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sly PD, Brennan S, Gangell C, et al. Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am J Respir Crit Care Med. 2009;180:146–52. doi: 10.1164/rccm.200901-0069OC. [DOI] [PubMed] [Google Scholar]
- 9.Meyerholz DK, Stoltz DA, Namati E, et al. Loss of cystic fibrosis transmembrane conductance regulator function produces abnormalities in tracheal development in neonatal pigs and young children. Am J Respir Crit Care Med. 2010;182:1251–61. doi: 10.1164/rccm.201004-0643OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sturgess J, Imrie J. Quantitative evaluation of the development of tracheal submucosal glands in infants with cystic fibrosis and control infants. Am J Pathol. 1982;106:303–11. [PMC free article] [PubMed] [Google Scholar]
- 11.Rogers CS, Stoltz DA, Meyerholz DK, et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. 2008;321:1837–41. doi: 10.1126/science.1163600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stoltz DA, Rokhlina T, Ernst SE, et al. Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J Clin Invest. 2013;123:2685–93. doi: 10.1172/JCI68867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ostedgaard LS, Meyerholz DK, Chen JH, et al. The DeltaF508 mutation causes CFTR misprocessing and cystic fibrosis-like disease in pigs. Sci Transl Med. 2011;3:74ra24. doi: 10.1126/scitranslmed.3001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adam RJ, Michalski AS, Bauer C, et al. Air trapping and airflow obstruction in newborn cystic fibrosis piglets. Am J Respir Crit Care Med. 2013;188:1434–41. doi: 10.1164/rccm.201307-1268OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibson-Corley KN, Olivier AK, Meyerholz DK. Principles for valid histopathologic scoring in research. Vet Pathol. 2013;50:1007–15. doi: 10.1177/0300985813485099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyerholz DK, Lambertz AM, Reznikov LR, et al. Immunohistochemical Detection of Markers for Translational Studies of Lung Disease in Pigs and Humans. Toxicol Pathol. 2015 doi: 10.1177/0192623315609691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burri PH. Fetal and postnatal development of the lung. Annu Rev Physiol. 1984;46:617–28. doi: 10.1146/annurev.ph.46.030184.003153. [DOI] [PubMed] [Google Scholar]
- 18.Michoud MC, Robert R, Hassan M, et al. Role of the cystic fibrosis transmembrane conductance channel in human airway smooth muscle. Am J Respir Cell Mol Biol. 2009;40:217–22. doi: 10.1165/rcmb.2006-0444OC. [DOI] [PubMed] [Google Scholar]
- 19.Li X, Tang XX, Vargas Buonfiglio LG, et al. Electrolyte transport properties in distal small airways from cystic fibrosis pigs with implications for host defense. Am J Physiol Lung Cell Mol Physiol. 2016;310:L670–9. doi: 10.1152/ajplung.00422.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang EH, Pezzulo AA, Meyerholz DK, et al. Sinus hypoplasia precedes sinus infection in a porcine model of cystic fibrosis. Laryngoscope. 2012;122:1898–905. doi: 10.1002/lary.23392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adam RJ, Abou Alaiwa MH, Bouzek DC, et al. Postnatal Airway Growth in Cystic Fibrosis Piglets. J Appl Physiol. 1985;2017 doi: 10.1152/japplphysiol.00263.2017. jap 00263 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiernan JA. Dyes and other colorants in microtechnique and biomedical research. Coloration Technology. 2006;122:1–21. [Google Scholar]
- 24.Sanjai K, Kumarswamy J, Patil A, Papaiah L, Jayaram S, Krishnan L. Evaluation and comparison of decalcification agents on the human teeth. J Oral Maxillofac Pathol. 2012;16:222–7. doi: 10.4103/0973-029X.99070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singer M, Morrison PR. The influence of pH, dye, and salt concentration on the dye binding of modified and unmodified fibrin. J Biol Chem. 1948;175:133–45. [PubMed] [Google Scholar]
- 26.Chen JH, Stoltz DA, Karp PH, et al. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell. 2010;143:911–23. doi: 10.1016/j.cell.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCray PB, Jr, Reenstra WW, Louie E, Johnson J, Bettencourt JD, Bastacky J. Expression of CFTR and presence of cAMP-mediated fluid secretion in human fetal lung. Am J Physiol. 1992;262:L472–81. doi: 10.1152/ajplung.1992.262.4.L472. [DOI] [PubMed] [Google Scholar]
- 28.Pezzulo AA, Tang XX, Hoegger MJ, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487:109–13. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shah VS, Meyerholz DK, Tang XX, et al. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science. 2016;351:503–7. doi: 10.1126/science.aad5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCray PB, Jr, Bettencourt JD, Bastacky J. Developing bronchopulmonary epithelium of the human fetus secretes fluid. Am J Physiol. 1992;262:L270–9. doi: 10.1152/ajplung.1992.262.3.L270. [DOI] [PubMed] [Google Scholar]
- 31.Graeff RW, Wang G, McCray PB., Jr KGF and FGF-10 stimulate liquid secretion in human fetal lung. Pediatr Res. 1999;46:523–9. doi: 10.1203/00006450-199911000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development. 1997;124:4867–78. doi: 10.1242/dev.124.23.4867. [DOI] [PubMed] [Google Scholar]
- 33.Mailleux AA, Kelly R, Veltmaat JM, et al. Fgf10 expression identifies parabronchial smooth muscle cell progenitors and is required for their entry into the smooth muscle cell lineage. Development. 2005;132:2157–66. doi: 10.1242/dev.01795. [DOI] [PubMed] [Google Scholar]
- 34.Marcorelles P, Montier T, Gillet D, Lagarde N, Ferec C. Evolution of CFTR protein distribution in lung tissue from normal and CF human fetuses. Pediatr Pulmonol. 2007;42:1032–40. doi: 10.1002/ppul.20690. [DOI] [PubMed] [Google Scholar]
- 35.Tiddens HA, Donaldson SH, Rosenfeld M, Pare PD. Cystic fibrosis lung disease starts in the small airways: can we treat it more effectively? Pediatr Pulmonol. 2010;45:107–17. doi: 10.1002/ppul.21154. [DOI] [PubMed] [Google Scholar]
- 36.Trezise AE, Chambers JA, Wardle CJ, Gould S, Harris A. Expression of the cystic fibrosis gene in human foetal tissues. Hum Mol Genet. 1993;2:213–8. doi: 10.1093/hmg/2.3.213. [DOI] [PubMed] [Google Scholar]
- 37.Tizzano EF, Chitayat D, Buchwald M. Cell-specific localization of CFTR mRNA shows developmentally regulated expression in human fetal tissues. Hum Mol Genet. 1993;2:219–24. doi: 10.1093/hmg/2.3.219. [DOI] [PubMed] [Google Scholar]
- 38.Brennan SC, Wilkinson WJ, Tseng HE, et al. The extracellular calcium-sensing receptor regulates human fetal lung development via CFTR. Sci Rep. 2016;6:21975. doi: 10.1038/srep21975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clement R, Blanc P, Mauroy B, Sapin V, Douady S. Shape self-regulation in early lung morphogenesis. PLoS One. 2012;7:e36925. doi: 10.1371/journal.pone.0036925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonvin E, Le Rouzic P, Bernaudin JF, et al. Congenital tracheal malformation in cystic fibrosis transmembrane conductance regulator-deficient mice. J Physiol. 2008;586:3231–43. doi: 10.1113/jphysiol.2008.150763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stoltz DA, Meyerholz DK, Pezzulo AA, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. 2010;2:29ra31. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tuggle KL, Birket SE, Cui X, et al. Characterization of defects in ion transport and tissue development in cystic fibrosis transmembrane conductance regulator (CFTR)-knockout rats. PLoS One. 2014;9:e91253. doi: 10.1371/journal.pone.0091253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Broackes-Carter FC, Mouchel N, Gill D, Hyde S, Bassett J, Harris A. Temporal regulation of CFTR expression during ovine lung development: implications for CF gene therapy. Hum Mol Genet. 2002;11:125–31. doi: 10.1093/hmg/11.2.125. [DOI] [PubMed] [Google Scholar]
- 44.Jesse NM, McCartney J, Feng X, Richards EM, Wood CE, Keller-Wood M. Expression of ENaC subunits, chloride channels, and aquaporins in ovine fetal lung: ontogeny of expression and effects of altered fetal cortisol concentrations. Am J Physiol Regul Integr Comp Physiol. 2009;297:R453–61. doi: 10.1152/ajpregu.00127.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGrath SA, Basu A, Zeitlin PL. Cystic fibrosis gene and protein expression during fetal lung development. Am J Respir Cell Mol Biol. 1993;8:201–8. doi: 10.1165/ajrcmb/8.2.201. [DOI] [PubMed] [Google Scholar]
- 46.Tebbutt SJ, Wardle CJ, Hill DF, Harris A. Molecular analysis of the ovine cystic fibrosis transmembrane conductance regulator gene. Proc Natl Acad Sci U S A. 1995;92:2293–7. doi: 10.1073/pnas.92.6.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olver RE, Strang LB. Ion fluxes across the pulmonary epithelium and the secretion of lung liquid in the foetal lamb. J Physiol. 1974;241:327–57. doi: 10.1113/jphysiol.1974.sp010659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harding RHS. Lung growth and maturation. In: Rodeck CH, WM, editors. Fetal Medicine: Basic Science and Clinical Practice. 2. New York, NY, USA: Elsevier; 2009. pp. 133–46. [Google Scholar]
- 49.Vilos GA, Liggins GC. Intrathoracic pressures in fetal sheep. J Dev Physiol. 1982;4:247–56. [PubMed] [Google Scholar]
- 50.Liu M, Tanswell AK, Post M. Mechanical force-induced signal transduction in lung cells. Am J Physiol. 1999;277:L667–83. doi: 10.1152/ajplung.1999.277.4.L667. [DOI] [PubMed] [Google Scholar]
- 51.Okiyoneda T, Veit G, Dekkers JF, et al. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat Chem Biol. 2013;9:444–54. doi: 10.1038/nchembio.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dekkers JF, Wiegerinck CL, de Jonge HR, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19:939–45. doi: 10.1038/nm.3201. [DOI] [PubMed] [Google Scholar]
- 53.Boj SF, Vonk AM, Statia M, et al. Forskolin-induced Swelling in Intestinal Organoids: An In Vitro Assay for Assessing Drug Response in Cystic Fibrosis Patients. J Vis Exp. 2017 doi: 10.3791/55159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang H, Sweezey NB, Kaplan F. LGL1 modulates proliferation, apoptosis, and migration of human fetal lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2015;308:L391–402. doi: 10.1152/ajplung.00119.2014. [DOI] [PubMed] [Google Scholar]
- 55.Shannon JM, Hyatt BA. Epithelial-mesenchymal interactions in the developing lung. Annu Rev Physiol. 2004;66:625–45. doi: 10.1146/annurev.physiol.66.032102.135749. [DOI] [PubMed] [Google Scholar]
- 56.Cook DP, Rector MV, Bouzek DC, et al. CFTR in Sarcoplasmic Reticulum of Airway Smooth Muscle: Implications for Airway Contractility. Am J Respir Crit Care Med. 2015 doi: 10.1164/rccm.201508-1562OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Colvin JS, White AC, Pratt SJ, Ornitz DM. Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development. 2001;128:2095–106. doi: 10.1242/dev.128.11.2095. [DOI] [PubMed] [Google Scholar]
- 58.De Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rosewell I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127:483–92. doi: 10.1242/dev.127.3.483. [DOI] [PubMed] [Google Scholar]
- 59.Abler LL, Mansour SL, Sun X. Conditional gene inactivation reveals roles for Fgf10 and Fgfr2 in establishing a normal pattern of epithelial branching in the mouse lung. Dev Dyn. 2009;238:1999–2013. doi: 10.1002/dvdy.22032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wong AP, Bear CE, Chin S, et al. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat Biotechnol. 2012;30:876–82. doi: 10.1038/nbt.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sekine K, Ohuchi H, Fujiwara M, et al. Fgf10 is essential for limb and lung formation. Nat Genet. 1999;21:138–41. doi: 10.1038/5096. [DOI] [PubMed] [Google Scholar]
- 62.Ramasamy SK, Mailleux AA, Gupte VV, et al. Fgf10 dosage is critical for the amplification of epithelial cell progenitors and for the formation of multiple mesenchymal lineages during lung development. Dev Biol. 2007;307:237–47. doi: 10.1016/j.ydbio.2007.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sala FG, Del Moral PM, Tiozzo C, et al. FGF10 controls the patterning of the tracheal cartilage rings via Shh. Development. 2011;138:273–82. doi: 10.1242/dev.051680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nedvetsky PI, Emmerson E, Finley JK, et al. Parasympathetic innervation regulates tubulogenesis in the developing salivary gland. Dev Cell. 2014;30:449–62. doi: 10.1016/j.devcel.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rogers CS, Abraham WM, Brogden KA, et al. The porcine lung as a potential model for cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;295:L240–63. doi: 10.1152/ajplung.90203.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cohen JC, Larson JE, Killeen E, Love D, Takemaru K. CFTR and Wnt/beta-catenin signaling in lung development. BMC Dev Biol. 2008;8:70. doi: 10.1186/1471-213X-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gaillard D, Ruocco S, Lallemand A, Dalemans W, Hinnrasky J, Puchelle E. Immunohistochemical localization of cystic fibrosis transmembrane conductance regulator in human fetal airway and digestive mucosa. Pediatr Res. 1994;36:137–43. doi: 10.1203/00006450-199408000-00002. [DOI] [PubMed] [Google Scholar]
- 68.Crawford I, Maloney PC, Zeitlin PL, et al. Immunocytochemical localization of the cystic fibrosis gene product CFTR. Proc Natl Acad Sci U S A. 1991;88:9262–6. doi: 10.1073/pnas.88.20.9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.