

Abstract
Corroborating evidence indicate that the downregulation of GABAA receptor subunit expression may underlie tolerance to the anticonvulsant and anxiolytic actions of benzodiazepine (BZ) ligands that act as full allosteric modulators (FAMs) of GABA actions at a variety of GABAA receptor subtypes. We and others have shown that 10‐14 days treatment with increasing doses of diazepam (a FAM) resulted in anticonvulsant tolerance and decreased the expression of the α1 GABAA receptor subunit mRNA and protein in frontal cortex. In addition, we have also shown that long‐term treatment with imidazenil, a partial allosteric modulator of GABA action at selective GABAA receptor subtypes, fail to change the expression of the α1 subunit mRNA or induce tolerance to its anticonvulsant or anxiolytic action. However, little is known regarding the potential role of epigenetic mechanisms on long‐term BZ‐induced downregulation of GABAA receptor subunit. Therefore, we examined the role of histone acetylation and DNA methylation mechanisms on long‐term diazepam‐induced downregulation of the α1 subunit mRNA expression in rat frontal cortex. We found that 10 days treatment with increasing doses of diazepam but not imidazenil decreased the expression of the α1 GABAA receptor subunit mRNA and promoter acetylation in frontal cortex. In addition, we also found that 10 days treatment with diazepam but not imidazenil increased the expression of histone deacetylase (HDAC) 1 and 2 in frontal cortex. Thus, the increased expression of HDAC1 and HDAC2 (class 1 HDACs) and consequently increased histone deacetylation mechanism of this class 1 HDACs, may underlie long‐term diazepam‐induced decreased expression of the α1 GABAA receptor subunit mRNA in frontal cortex.
Keywords: diazepam, histone deacetylation, imidazenil, α1 GABAA receptor subunit
Abbreviations
- GABA
g‐aminobutyric acid
- BZ
benzodiazepines
- FAM
full allosteric modulator
- PAM
partial allosteric modulator
- HDAC
histone deacetylase
- MeCP2
methyl CpG binding protein 2
- SAHA
Suberoylanilide hydroxamic acid
- TSA
Trichostatin A
- GAPDH
Glyceraldehyde‐3‐phosphate dehydrogenase
- H3K9me2
histone H‐3 dimethylated
- DNMT
DNA methyl transferase
- TET
ten‐eleven translocas0065
1. INTRODUCTION
Long‐term and repeated daily administration of benzodiazepines with full allosteric modulatory (FAM) (eg, diazepam, triazolam, and alprazolam) but not those with partial allosteric modulatory (PAM) actions (imidazenil and bretazenil) at a variety of GABAA receptor subtypes has been well established to lead to tolerance and dependence liabilities.1, 2, 3 Although considerable biochemical and electrophysiological evidence suggests that tolerance to the anxiolytic and anticonvulsant effects of FAM are associated with downregulation of receptor subunits expression and GABAA receptor function,4, 5, 6, 7, 8, 9 the precise molecular mechanism underlying the downregulation of GABAA receptor subunits function is still not understood.
It is now widely recognized that epigenetic mechanisms such as DNA promoter methylation, histone acetylation, and methylation play significant roles in the pathophysiology of brain disorders including addictive behaviors, tolerance, and dependence liabilities.10, 11, 12, 13, 14 Histone tail lysine acetylation which involves the addition of acetyl groups to lysine residues on histone tails leads to a relaxed (open) chromatin structure and consequently the activation of target gene expression. In addition, the acetylation of histone tails is also regulated by histone acetyltransferases and histone deacetylases.15, 16, 17 Conversely, DNA promoter methylation which involves the covalent coupling of a methyl group to the C5 position of cytosine residues of CpG dinucleotide upstream of the transcription start site, is catalyzed by DNA methyltransferases [DNA methyltransferase 1 (Dnmt1), DNA methyltransferase 3a (Dnmt13a) DNA methyltransferase 3b (Dnmt3b) DNA methyltransferase 3 l (Dnmt3 l)] and is generally associated with gene silencing.11, 12 Recent studies have also shown that DNA methylation is kept in a steady state by the action of a demethylase system which includes a first step 5‐hydroxy methylation driven by ten‐eleven translocase (TET) enzymes.18 The role of these enzymes is difficult to establish because of the lack of specific pharmacological agents targeting these enzymes.
Histone acetylation and DNA methylation are targeted by histone deacetylase (HDAC) inhibitors and DNA methyltransferase inhibitors, respectively.10, 11, 18 Thus, the epigenetic regulation of targeted gene expression with selective pharmacotherapeutic strategies is increasingly becoming a viable alternative for the design and development of new therapeutic strategies to treat specific types of malignancies and neuropsychiatric disorders such as Huntington disease, schizophrenia and depression.19, 20, 21, 22, 23, 24 Interestingly, it has also been reported that chronic antidepressant‐induced increase acetylation status of histone proteins in frontal cortex and hippocampus contributes to the therapeutic effects of antidepressant interventions.25 These findings raise the possibility that long‐term diazepam exposure may lead to alterations of epigenetic mechanisms regulating the expression of α1‐GABAA receptor subunit resulting to the downregulation of this receptor subunit, and a process that may underlie tolerance to the pharmacological actions of diazepam.
We and others have previously shown that long‐term treatment of rats with increasing doses of diazepam or triazolam (FAMs) for 10‐14 days induced anticonvulsant tolerance and decreased expression of mRNA and protein encoding for the α1 GABAA receptor subunit in prefrontal cortex.2, 7, 8, 26, 27 In contrast, long‐term treatment with equipotent anti‐bicuculline doses of partial allosteric modulators (PAM) of GABAA receptors (imidazenil or bretazenil) fail to elicit anticonvulsant tolerance or change the expression of mRNA and protein encoding for α1 GABAA receptor subunit.7, 8, 26, 27 Furthermore, Rudolph et al. 28 demonstrated that the protective action of diazepam against pentylenetetrazole‐induced tonic‐clonic seizure is significantly reduced in α1 GABAA receptor subunit mutant mice when compared to their wild‐type counterparts. It has also been suggested that α1‐containing GABAA receptors probably mediate the sedative, amnesic and anticonvulsant actions of classical benzodiazepines.29, 30 Taken together, these findings suggest that the downregulation of α1‐containing GABAA receptors by FAM of GABA action might underlie anticonvulsant tolerance and dependence liabilities.
Recent studies in cerebral cortical cultured neurons have shown that the decreased α1 GABAA receptor subunit expression observed following chronic alcohol exposure is mediated by histone modifications.31 To the best of our knowledge there are no published studies which have examined the role of epigenetic mechanisms on long‐term benzodiazepines‐induced downregulation GABAA receptor subunit expression. In light of these considerations, we examined the role of histone acetylation and DNA methylation on long‐term imidazenil or diazepam‐induced downregulation of α1‐GABAA subunit mRNA expression in rat frontal cortex. Our results show that 10 days of repeated oral treatment with increasing doses of diazepam but not imidazenil resulted in decreased expression of α1‐GABAA subunit mRNA and promoter acetylation in frontal cortex.
2. MATERIALS AND METHODS
2.1. Animals, drugs, and reagents
Adult male Sprague Dawley rats (Harlan, Indianapolis) were received and after 1 week of acclimatization they were housed individually, maintained on a 12/12‐h light‐dark cycle with standard food and water made available ad libitum. All experiments were carried out in accordance with the National Institute of Health Guidelines for the Care and Use Laboratory Animals and approved by the animal committee at the University of Illinois at Chicago.
Diazepam and imidazenil were obtained from Hoffman‐La Roche (Nutley, NJ); TRIzol reagent was obtained from Life Technologies (Life Technologies Corporation, USA); Qiagen RNeasy Kit from Qiagen (Valencia, CA, USA); Fermentas Maxima SYBR Green/ROX qPCR Master Mix obtained from Fermentas (Fermentas International Inc., Canada); Novex 4%‐12% Tris‐Glycine gels obtained from Invitrogen (Carlsbad, CA); HRP‐conjugated secondary anti‐mouse or anti‐rabbit antibodies from GE Healthcare (Arlington Heights, IL); anti‐GAPDH, Immobilon Western Chemiluminescent HRP Substrate, anti‐H3K9me2, anti‐MeCP2, anti‐H3, and anti‐acetyl‐H3 from Millipore (Billerica, MA).
2.2. Schedule for long‐term treatment
For long‐term diazepam, imidazenil or vehicle treatment, we used the same treatment schedule used in our previous studies in which we demonstrated anti‐bicuculline tolerance and downregulation of α1 GABAA receptor subunit expression following long‐term treatment with FAMs of GABA action at GABAA receptors.7, 8, 26, 27 In brief, diazepam or imidazenil was suspended in water containing 0.05% Tween‐20 and administered in 1 mL volumes by oral gavage 3 times daily (at approximately 9:00 am, 2:00 pm and 7:00 pm) for 10 days at increasing doses (diazepam: days 1‐3, 17.6 μmol/kg; days 4‐6, 35.2 μmol/kg and days 7‐10, 52.8 μmol/kg; imidazenil: days 1‐3, 2.85 μmol/kg; days 4‐6, 5.70 μmol/kg; and days 7‐10, 8.55 μmol/kg). Control rats received vehicle (0.05% Tween 20 in water) treatment. Rats were sacrificed at least 18 hours after the last diazepam or imidazenil dose and frontal cortex dissected and stored at −80°C until used for RT‐qPCR, ChIP assay or western blot.
2.3. RNA extraction and real‐time polymerase chain reaction (PCR) quantification
Total RNA was isolated from frontal cortices using TRIzol reagent and further purified using Qiagen RNeasy Kit. Total RNA was converted to cDNA using the Applied Biosystems (USA) High Capacity Archive Kit (4368813). Relative quantitative real‐time polymerase chain reaction (qPCR) was performed with the Applied Biosynthesis Real‐Time PCR system using Fermentas Maxima SYBR Green/ROX qPCR Master Mix (K0222; Fermentas International Inc., Canada). PCR mixtures were run on a Stratagene (USA) Mx3005P QPCR System. Primers were designed to cross over one intron to amplify cDNA and yield an amplicon of between 90 and 130 base pairs (primer sequences are provided in Table 1). Dissociation curves were conducted to establish the presence of a single amplicon at the predicted melting temperature and a lack of primer‐dimer formation. A comparative threshold cycle (C T) validation experiment was done to determine target and reference primer efficiency. For normalization of mRNAs expression, GAPDH was used as reference gene. C T value was used for relative quantification of target gene expression and normalized to GAPDH and the relative expression levels were calculated as C T.32, 33
Table 1.
Primer Sequences used for real‐time PCR
| Gene | Forward | Reverse |
|---|---|---|
| α1GABAR | 5′‐GTGCAAGTTAAATTGCGCTGCA | 5′‐GCTTCCCAATATCCAATCTGCAGC |
| DNMT1 | 5′‐AAGCCAGCTATGCGACTTGGAAAC | 5′‐ACA ACC GTTGGCTTTCTGAGTGAG |
| DNTM3b | 5′‐TGTGCAGAGTCCATTGCTGTAGGA | 5′‐GCT TCCGCCAATCACCAAGTCAAA |
| MeCP2 | 5′‐ACCAGCTCCAACAGGATTCCAT | 5′‐ TCTGCAGAATGGTGGGCTGA |
| HDAC1 | 5′‐GACATGCCAAGTGTGTGGAGT | 5′‐ CACAGCTGTCTCGTAAGTCCAG |
| HDAC2 | 5′‐GTGAAGCTGAACCGTCAACAGA | 5′‐CTCGAGGACAGCAAGCACAA |
| HDAC3 | 5′‐TGGTGGTCTACATCATGCCAA | 5′‐CGAGGGTGGTATTTGAGCAG |
| GAPDH | 5′‐ACCAGGGCTGCCTTCTCT | 5′‐ATCTCGCTCCTGGAAGATGGT |
2.4. Chromatin immunoprecipitation assay
In order to establish a possible role for epigenetic mechanisms in long‐term diazepam‐induced downregulation of α1 GABAA receptor subunit mRNA expression, we examined DNA methylation, histone acetylation, and methylation levels at α1 GABAA receptor subunit promoter in frontal cortex. We used specific antibodies against H3K9me2, MeCP2, and acetylated histone H3 for chromatin immunoprecipitation (ChIP) assay as previously described.34, 35 In brief, frontal cortices were fixed in methanol‐free 1% formaldehyde by incubating on a rocking platform at room temperature for 15‐20 minutes followed by homogenization in lysis buffer and DNA shearing by sonication. The resulting cross‐linked DNA‐chromatin complex was immunoprecipitated (IP) with antibodies directed at H3K9me2, MeCP2, or acetylated histone H3, and the precipitated DNA quantified by Real‐Time PCR system using Fermentas Maxima SYBR Green/ROX qPCR Master Mix. Input DNA was used as internal standard for normalization. After subtraction of input DNA Ct value from the Ct values of the respective samples, the ΔΔCt method 32 was used to determine the fold change in levels of H3K9me2, MeCP2, H3‐total, or H3‐acetylated at the α1 GABAA receptor promoter. The percentages of IP DNA were calculated using the following formula: % (IP/total input) = 2(Ct (10% input−Ct (IP) × 100%. The primer sequences for α1 GABAA subunit promoter were: Forward‐5′‐GTGCAAGTTAAATTGCGCTGCA‐3′, Reverse‐5′‐GCTTCCCAA TATCCAATCTGCAGC‐3′ (see Figure 1).
Figure 1.
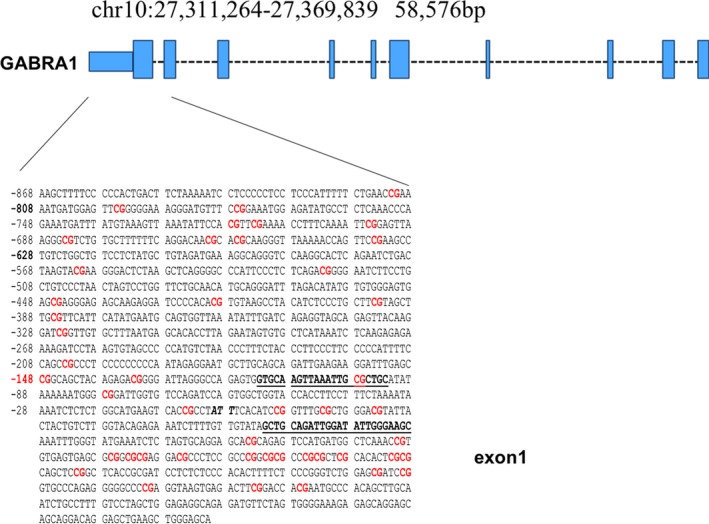
Sequence and genomic location for Gabra1 promoter region of interest including the first exon. ChIP assays were performed using the primer pair indicated in underlined bold letters and the transcription start site in italicized bold letters
2.5. Western blot
Frontal cortex of vehicle, imidazenil and diazepam‐treated rats were homogenized in HEPES‐buffered sucrose (320 mmol L−1 Sucrose, 4 mmol L−1 HEPES pH 7.4, 1% SDS). We used Pierce® Bicinchoninic acid (BCA) assay to determine protein concentration (Thermo Fisher Scientific, Waltham, MA). Lysates were resuspended in Laemmli reducing buffer and 30 μg of each sample were separated by electrophoresis on Novex 4%‐12% Tris‐Glycine gels. Proteins were then transferred to PVDF membrane in a buffer containing 35 mmol L−1 Tris, 192 mmol L−1 glycine, and 20% methanol at 4°C. Membranes were blocked using 5% milk and later incubated with the primary antibody, anti‐H3 (1:1000) or anti‐acetyl‐H3 (1:1500). Protein levels were normalized with GAPDH (1:5000). HRP‐conjugated secondary anti‐mouse or anti‐rabbit antibodies (1:10 000) were used. Membranes were developed with Immobilon Western Chemiluminescent HRP Substrate and densitometric analysis performed with Image StudioTM (LI‐COR, Bioscience). The expression levels for the respective proteins were normalized to GAPDH protein levels.
2.6. Statistical analysis
Data were analyzed by one‐way ANOVA followed by Tukey's post hoc comparison.
3. RESULTS
3.1. Long‐term diazepam treatment alters the expression of α1‐GABAA receptor subunit and Dnmt3b in frontal cortex
In previous studies, we demonstrated that 5‐10 days treatment with diazepam but not imidazenil resulted to anticonvulsant tolerance against bicuculline‐induced tonic convulsion 26 in Sprague Dawley rats. In subsequent studies we also demonstrated that this same diazepam treatment schedule consistently elicited downregulation of α1 GABAA receptor subunit mRNA and cognate protein expression in rat frontal cortex.7, 8, 27, 36 Here, we measured the expression of mRNAs encoding for α1‐GABAA receptor subunit, MeCP2, Dnmt1 and Dnmt3b in frontal cortex 18 hours after the last dose of 10 days diazepam, imidazenil, or vehicle treatment. As previously reported, we have confirmed that long‐term treatment with diazepam but not imidazenil in doses that induced anticonvulsant tolerance 27 resulted in a significantly decreased expression of mRNA encoding for the α1‐GABAA receptor subunit. In addition, 10 days of diazepam treatment also elicited a significantly increased the expression of mRNA encoding for Dnmt3b but failed to alter the expression of mRNAs encoding for Dnmt1 and MeCP2 in frontal cortex (Figure 2).
Figure 2.
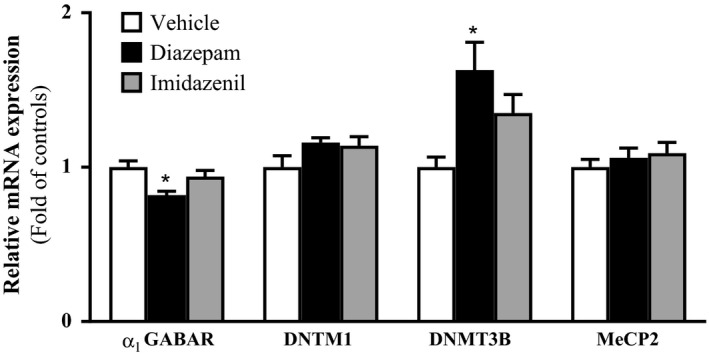
Effects of long‐term diazepam and imidazenil treatment on mRNA profiling in frontal cortex. Frontal cortices of rats treated with increasing doses of diazepam, imidazenil or vehicle for 10 days were sacrificed 18 hours after the last dose, brains collected and frontal cortices dissected for mRNA analysis. Values are expressed as mean ± SEM of the fold change in controls, n = 5 rats. *P < .05 significantly different from controls, one‐way ANOVA followed by post hoc Turkey's test
3.2. Decreased α1GABAA receptor subunit mRNA expression is associated with decreased acetylated histone H3 without altering the level of H3K9me2
To investigate a possible role for epigenetic mechanisms on long‐term diazepam‐induced downregulation α1GABAA receptor subunit expression, we examined whether the decreased expression of this subunit mRNA in frontal cortex is associated with increased DNA methylation (MeCP2 occupancy), increased occupancy of H3K9me2 or decreased histone H3 acetylation at the α1 GABAA receptor subunit promoter. In Figure 3, we show that long‐term treatment with diazepam but not imidazenil significantly decreased the occupancy of acetylated histone H3 at α1GABAA receptor subunit promoter. In addition, diazepam treatment also resulted in a significantly increased of the occupancy of MeCP2 in the absence of expression changes but failed to alter the level of H3K9me2 at this GABAA receptor subunit promoter while imidazenil treatment had no effect. These results indicate that only long‐term diazepam treatment resulted in a significant reduction of the level of acetylated histone H3 at α1GABAA receptor subunit promoter thereby leading to a decreased expression of this receptor subunit in frontal cortex. Thus long‐term diazepam exposure will most likely result to the induction of chromatin and synaptic remodeling in frontal cortex.
Figure 3.
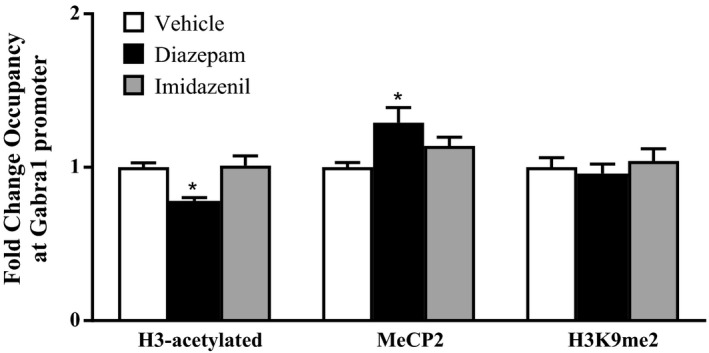
Effects of long‐term diazepam and imidazenil treatment on histone H3‐acetylated, histone H‐3 dimethylated (H3K9me2) and MeCP2 occupancy at Gabra1 gene promoter as shown in Figure 1. Frontal cortices of rats treated with increasing doses of diazepam, imidazenil or vehicle for 10 days were used to measure occupancy at the Gabra1 promoter by chromatin immunoprecipitation (ChIP) assay. Values are represented as the mean ± SEM of the fold change in controls, n = 5 rats. *P < .05 significantly different from controls, one‐way ANOVA followed by post hoc Turkey's test
3.3. Long‐term diazepam treatment alters the expression of HDAC1 and HDAC2 but not HDAC3
Since long‐term diazepam treatment decreased the levels of acetylated histone H3 at α1‐GABAA receptor subunit promoter, we also examined whether the decreased levels of acetylated histone H3 was mediated by increased expression of histone deacetylase (HDAC) enzymes. It has been suggested that HDAC2 plays a significant role in the development of alcohol dependence in rodents,10, 37, 38, 39 therefore we examined the expression of class 1 HDACs (HDAC 1, 2, and 3), which are localized in the nucleus.40 In Figure 4, we show that long‐term treatment with diazepam but not imidazenil significantly increased the expression of HDAC1 and HDAC2 without changing the expression of HDAC3. These results suggest that the decreased levels of acetylated histone H3 at the α1‐GABAA receptor subunit promoter and consequently the decreased α1‐GABAA receptor subunit expression may be due to increased expression and most likely, the enzymatic activity of HDAC1 and HDAC2.
Figure 4.
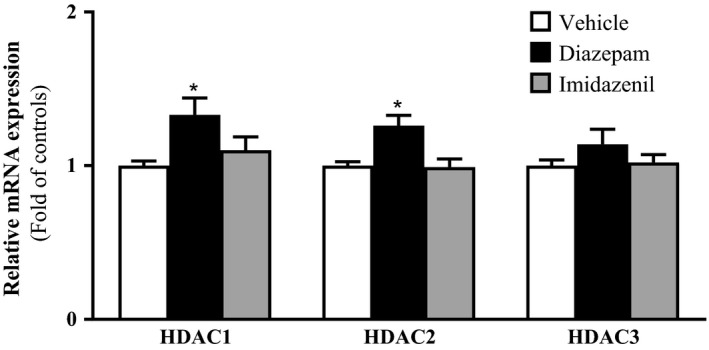
Effects of long‐term diazepam and imidazenil treatment on class 1 HDACs mRNA profiling in frontal cortex. Long‐term diazepam but not imidazenil increased HDAC1 and HDAC2 expression in frontal cortex. Frontal cortices of rats treated with increasing doses of diazepam, imidazenil or vehicle for 10 days were sacrificed 18 hr after the last dose, brains collected and frontal cortices dissected for mRNA analysis. Values are expressed as mean ± SEM of the fold change in controls, n = 5 rats. *P < .05 significantly different from controls, one‐way ANOVA followed by post hoc Turkey's test
3.4. Long‐term diazepam treatment does not alter the expression of total histone H3 or acetylated histone H3 in frontal cortex
Because 10 days diazepam treatment significantly decreased the occupancy of acetylated histone H3 at the promoter of the α1 GABAA receptor subunit, we examined whether long‐term treatment with diazepam or imidazenil led to changes in the expression of total or acetylated histone H3 protein in frontal cortex. The data expressed as the ratio of the optical density of the target protein (total histone H3 or acetylated histone H3) and GAPDH indicated that there were no statistical significant differences between the levels of total histone H3 (vehicle 0.818 ± 0.079, diazepam 0.557 ± 0.070, imidazenil 0.614 ± 0.082; F (2,11) = 3.491, P = .067) and acetylated histone H3 (vehicle 0.742 ± 0.051, diazepam 0.576 ± 0.049, imidazenil 0.564 ± 0.044; F (2,11) = 4.245, P = .063) in frontal cortex of long‐term diazepam or imidazenil‐treated groups when compared with the vehicle‐treated group. These results indicate that the decreased acetylated histone H3 bound to the α1 GABAA receptor subunit is not due to changes in the levels of histone H3 proteins resulting from long‐term diazepam treatment. Thus, the decreased levels of histone H3 acetylated at α1‐GABAA receptor subunit promoter resulting from long‐term diazepam treatment may underlie the decrease expression of this GABAA receptor subunit in frontal cortex.
4. DISCUSSION
The outcomes of in vivo and in vitro studies on the regulation of GABAA receptor subunit expression show that the magnitude and direction of long‐term benzodiazepine‐induced alterations of GABAA receptor subunit expression is subunit‐specific, brain region‐specific, and treatment duration dependent. In addition, overwhelming evidence show a consistent downregulation of α1 GABAA receptor subunit expression following long‐term treatment with FAMs of GABA action at a variety of GABAA receptor subtypes.2, 41 Moreover, the concurrent decreases in α1 subunit mRNA and protein expression which occurs following chronic diazepam treatment,8 suggests that transcriptional mechanisms might play a significant role in regulating the expression of GABAA receptor subunits during long‐term benzodiazepine exposure. This study focuses on the role of epigenetic mechanisms in long‐term diazepam‐induced downregulation of α1 GABAA receptor subunit expression. To the best of our knowledge, this is the first study reporting association between alterations of acetylated histone H3 and long‐term benzodiazepine‐induced downregulation of GABAA receptor subunit expression. Specifically, we report here that the resulting decrease in the expression of α1 GABAA receptor subunit in frontal cortex associated with long‐term treatment with diazepam (a FAM) but not imidazenil (a PAM) might be due to decreased levels of acetylated histone H3 at the promoter of α1‐containing GABAA receptors. Furthermore, the increased occupancy of MeCP2 may recruit HDAC1 and HDAC3 to this promoter complex and thus augment histone deacetylation mechanisms and indirectly DNA methylation mechanisms at this promoter.42 Taken together, the increased expression of HDAC1 and HDAC2 and consequently histone deacetylation mechanism in frontal cortex may underlie long‐term diazepam‐mediated decrease in the levels of acetylated histone H3 at this GABAA receptor subunit promoter.
Chronic exposure to ethanol, a positive allosteric modulator of GABA action at GABAA receptors results in decreases inmRNA and protein expression for α1 GABAA receptor subunit both in vitro and in vivo.43, 44, 45 Most importantly, Arora et al. 37 reported that treatment with HDAC inhibitors attenuated the decrease in α1 protein expression induced by chronic ethanol exposure. In a different but related study, Di et al. 46 reported that α1 (A322D) gene mutation leads to the decrease expression of α1 subunit found in epilepsy. Together, the results of these studies suggest a possible involvement of histone acetylation and/or DNA methylation mechanisms in the regulation of α1 GABAA receptor subunit expression. The results of our studies indicate that long‐term diazepam‐induced downregulation of α1 subunit expression might in part be mediated by alterations of the histone acetylation mechanisms associated with the α1 GABAA receptor subunit promoter. It has been established that the deacetylation of histone core wrapped around promoters leads to a closed or condensed chromatin structure, which has been associated with reductions in gene transcription and in rare cases gene silencing precludes changes in histone acetylation.47
It is important to note that long‐term treatment with imidazenil, a PAM which does not elicit anticonvulsant tolerance or the downregulation of the α1 subunit following chronic treatment,2, 7, 8, 26, 27, 48, 49 fail to downregulate α1 subunit expression, change the levels of acetylated histone H3 at the α1 GABAA receptor subunit promoter or change expression of HDAC1 and HDAC2 in frontal cortex (Figures 2 and 4). Therefore one can surmise that HDAC1 and HDAC2 mediated histone deacetylation of the α1 GABAA receptor subunit promoter might regulate the downregulation of the α1 subunit expression during long‐term diazepam treatment. This inference is in agreement with the studies by Arora et al.,37 in which they found that blockade of the upregulation of HDAC2 with HDAC inhibitors [Suberoylanilide hydroxamic acid (SAHA) or Trichostatin A (TSA)] normalized the hyposensitivity of dopaminergic ventral tegmental area GABAergic neurons to GABA. In addition, it has been suggested that HDAC2 is involved in the development of alcohol dependence.38, 39 It is also interesting to note that HDAC1 and HDAC2 which are typically localized in the nucleus, are structurally identical and are often found in the same repressive complexes with MeCP2.40 MeCP2, which is often found in a complex together with class I HDACs acts as a transcriptional repressor by recruiting HDACs to active gene promoters 50, 51 and may also repress the transcription of the α1 receptor subunit by recruiting HDAC1 and HDAC2 to this GABAA receptor subunit promoter complex. Thus, HDAC1 and HDAC2‐mediated histone deactylation at the α1 GABAA receptor subunit promoter may trigger the signaling mechanisms that underlie the downregulation of the α1 subunit that is associated with long‐term benzodiazepine treatment. Accordingly, pretreatment or concurrent treatment with selective HDAC1 and HDAC2 inhibitors might prevent the decrease in α1 GABAA receptor subunit expression associated with chronic benzodiazepine treatment. Therefore future experiments will examine whether concurrent treatment with selective HDAC inhibitors will attenuated the decrease α1 subunit expression associated with anxiolytic and anticonvulsant benzodiazepines tolerance and/or dependence.
In conclusion, the outcome of our study points to a new and important role for histone deacetylation in chronic benzodiazepine‐induced downregulation of α1 GABAA receptor subunit expression. However, further studies will be needed to establish whether the decreased α1 subunit expression elicited by long‐term diazepam can be attenuated by pretreatment or concurrent treatment with class 1 HDAC inhibitors. Nonetheless, our study presents a central role for epigenetic mechanisms in the transcriptional regulation of GABAA receptor subunit expression during chronic benzodiazepine exposure. Furthermore, understanding the role of HDAC inhibitors in benzodiazepine tolerance and the downregulation of GABAA subunit expression may uncover new therapeutic targets to prevent benzodiazepine tolerance, dependence and/or withdrawal syndrome.
AUTHORS’ CONTRIBUTIONS
Participated in research design: J. Auta, A. Guidotti.
Conducted Experiments: J. Auta, E. Gatta.
Contributed reagents and analytical tools: A. Guidotti, S.C. Pandey, J. M. Davis.
Performed data analysis: J. Auta, E. Gatta, A. Guidotti.
Wrote or contributed to the writing of the manuscript: J. Auta, S.C. Pandey, A. Guidotti.
ACKNOWLEDGEMENT
This study was supported by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) grant P50AA022538 to Alessandro Guidotti and Subhash Pandey, Senior Research Career Scientist award from the Department of Veterans Affairs to Subhash Pandey, 1RO1MH101043 and 1RO1MH093348 to Alessandro Guidotti.
Auta J, Gatta E, Davis JM, Pandey SC, Guidotti A. Potential role for histone deacetylation in chronic diazepam‐induced downregulation of α1‐GABAA receptor subunit expression. Pharmacol Res Perspect. 2018;e00416 https://doi.org/10.1002/prp2.416
REFERENCES
- 1. Costa E, Auta J, Guidotti A. Tolerance and dependence to ligands of benzodiazepine recognition sites expressed by GABAA receptors In: Mohler H, ed. Handbook of Experimental Pharmacology, vol. 50 Berlin: Springer; 2001:227‐250. [Google Scholar]
- 2. Uusi‐Oukari M, Korpi ER. Regulation of GABAA receptor subunit expression by pharmacological agents. Pharmacological Rev. 2010;62:97‐135. [DOI] [PubMed] [Google Scholar]
- 3. Vinker CH, Olivier B. Mechanisms underlying tolerance after long‐term benzodiazepine use: a future for subtype‐selective GABAA receptors modulators? Adv Pharmacol Sci. 2012;2012:416864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gallager DW, Lakowski JM, Gonslaves SF, Rauch SL. Chronic benzodiazepine treatment decreases postsynaptic GABA sensitivity. Nature. 1984;308:74‐77. [DOI] [PubMed] [Google Scholar]
- 5. Gallager DW, Malcolm AB, Anderson SA, Gonslaves SF. Continuous releases of diazepam: electrophysiological, biochemical and behavioral consequences. Brain Res. 1985;342:26‐36. [DOI] [PubMed] [Google Scholar]
- 6. Hu X, Ticku MK. Chronic benzodiazepine treatment produces functional uncoupling of the GABA‐benzodiazepine receptor ionophore complex in cortical neurons. Mol Pharmacol. 1994;45:618‐625. [PubMed] [Google Scholar]
- 7. Impagnatiello F, Pesold C, Longone P, et al. Modifications of GABAA receptor subunit expression in rat neocortex during tolerance to diazepam. Mol Pharmacol. 1996;49:832‐841. [PubMed] [Google Scholar]
- 8. Pesold C, Caruncho HJ, Impagnatiello F, et al. Tolerance to diazepam and changes in GABAA receptor subunit expression in rat neocortical areas. Neuroscience. 1997;79:477‐487. [DOI] [PubMed] [Google Scholar]
- 9. Roca DJ, Schiller GD, Friedman L, Rozenberg I, Gibbs TT, Frab DH. γ‐aminobutyric acidA receptor regulation in culture: altered interactions following prolonged exposure to benzodiazepines, barbiturates, and methylxanthines. Mol Pharmacol. 1990;37:710‐719. [PubMed] [Google Scholar]
- 10. Berkel TDM, Pandey SC. Emerging role of epigenetic mechanisms in alcohol addiction. Alcohol Clin Exp Res. 2017;41:666‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Feng J, Fan G. The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int Rev Neurobiol. 2009;89:67‐84. [DOI] [PubMed] [Google Scholar]
- 12. Grayson DR, Kundakovic M, Sharma RP. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol Pharmacol. 2010;77:126‐135. [DOI] [PubMed] [Google Scholar]
- 13. Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorder. Nat Rev Drug Discov. 2008;7:854‐868. [DOI] [PubMed] [Google Scholar]
- 14. Renthal W, Nestler EJ. Epigenetic mechanisms in drug addiction. Trends in Mol Med. 2008;14:341‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kalkhoven E. CBP and p300: HATs for different occasions. Biochem Pharmacol. 2004;68:1145‐1155. [DOI] [PubMed] [Google Scholar]
- 16. Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferases activity is a critical component of memory consolidation. Neuron. 2004;42:961‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Strahl BD, Allis CD. The analogy of covalent histone modifications. Nature. 2000;403:41‐45. [DOI] [PubMed] [Google Scholar]
- 18. Grayson DR, Guidotti A. The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology. 2013;38:138‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deutsch SI, Rosse RB, Mastropaolo J, Long KD, Gaskins BL. Epigenetic therapeutic strategies for treatment of neuropsychiatric disorders: ready for prime time? Clin Neuropharmacol. 2008;31:104‐119. [DOI] [PubMed] [Google Scholar]
- 20. Ferrante RJ, Kubilus JK, Lee J, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorate the neurodegenerative phenotypic in Huntington's mice. J Neurosci. 2003;23:9418‐9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gardian G, Browne SE, Choi DK, et al. Neuroprotective effects of phenylbutyrate in N171‐82Q transgenic model of Huntington's disease. J Biol Chem. 2005;280:556‐563. [DOI] [PubMed] [Google Scholar]
- 22. Ryu H, Smith K, Camelo SI, et al. Sodium butyrate prolonged survival and regulates expression of anti‐apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 2005;93:1087‐1098. [DOI] [PubMed] [Google Scholar]
- 23. Steffan JS, Bodai L, Pallos J, et al. Histone deacetylase inhibitors arrest polyglutmate‐dependent neurodegeneration in Drosophila. Nature. 2001;413:739‐743. [DOI] [PubMed] [Google Scholar]
- 24. Ying M, Xu R, Wu X, et al. Sodium butyrate ameliorates histone hypoacetylation and neurodegenerative phenotypes in a mouse model for DRPLA. J Biol Chem. 2006;281:12580‐12586. [DOI] [PubMed] [Google Scholar]
- 25. Li J, Guo Y, Schroeder FA, et al. Dopamine D2‐like antagonists induce chromatin remodeling in striatal neurons through cyclic AMP‐protein kinase A and NMDA receptor signaling. J Neurochem. 2004;90:1117‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Auta J, Giusti P, Guidotti A, Costa E. Imidazenil, a positive allosteric modulator of GABAA receptors exhibit low tolerance and dependence liabilities. J Pharmacol Exp Ther. 1994;270:1262‐1269. [PubMed] [Google Scholar]
- 27. Auta J, Impagnatiello F, Kadriu B, Guidotti A, Costa E. Imidazenil: a low efficacy agonist at α1‐ but high efficacy at α5‐GABAA receptors fail to show anticonvulsant cross tolerance to diazepam or zolpidem. Neuropharmacology. 2008;55:148‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rudolph U, Crestani F, Benke D, et al. Benzodiazepine actions mediated by specific gamma‐aminobutyric acid (A) receptor subtypes. Nature. 1999;401:796‐800. [DOI] [PubMed] [Google Scholar]
- 29. Mckernan RM, Tm Rosahl, Reynolds DS, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor α1 subtype. Nature Neurosci. 2000;3:587‐592. [DOI] [PubMed] [Google Scholar]
- 30. Rudolph U, Mohler H. GABA‐based therapeutic approaches: GABAA receptor subtype functions. Curr Opin Pharmacol. 2006;6:18‐23. [DOI] [PubMed] [Google Scholar]
- 31. Bohnsack JP, Patel VK, Morrow AL. Ethanol exposure regulates Gabra1 expression via histone deacetylation at the promoter in cultured cortical neurons. J Pharmacol Exp Ther. 2017;363:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Livak KJ, Schmittgen T. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta C(T)) method. Methods. 2001;25:402‐408. [DOI] [PubMed] [Google Scholar]
- 33. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative C(T) method. Nat. Protocol. 2008;3:1101‐1108. [DOI] [PubMed] [Google Scholar]
- 34. Dong E, Gavin DP, Chen Y, Davis J. Upregulation of TET1 and downregulation of APOBEC3A and APOBEC3C in the parietal cortex of psychotic patients. Transl Psychiat. 2012;2:e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pandey SC, Sakharkar AJ, Tang L, Zhang H. Potential role of adolescent alcohol exposure‐induced amygdaloid histone modification in anxiety and alcohol intake during adulthood. Neurobiol Dis. 2015;82:607‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Costa E, Auta J, Grayson DR, et al. GABAA receptors and benzodiazepines: a role for dendritic resident subunit mRNAs. Neuropharmacology. 2002;43:925‐937. [DOI] [PubMed] [Google Scholar]
- 37. Arora DS, Nimitvilai S, Teppen TL, et al. Hyposensitivity to gamma‐aminobutyric acid in the ventral tegmental area during alcohol withdrawal: reversal by histone deacetylase inhibitors. Neuropsychopharmacology. 2013;38:1674‐1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lopez‐Moreno JA, Marcos M, Calleja‐Conde J, et al. Histone deacetylase gene expression following binge alcohol consumption in rats and humans. Alcohol Clin Exp Res. 2015;39:1939‐1950. [DOI] [PubMed] [Google Scholar]
- 39. Moonat S, Sakkharkar AJ, Zhang H, Pandey SC. Aberrant histone deacetylase2‐mediated histone modifications and synaptic plasticity in the amygdala predisposes to anxiety and alcoholism. Biol Psychiatry. 2013;73:763‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Haberland M, Montgomery RL, Olsen EN. The many role of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Follesa P, Mancuso L, Biggio F, et al. Changes in GABAA receptor gene expression induced withdrawal of, but not by long‐term exposure to zaleplon or zolpidem. Neuropharmacology. 2002;42:191‐198. [DOI] [PubMed] [Google Scholar]
- 42. Detich N, Bovenzi V, Szyf M. Valproate induced replication‐independent active DNA methylation. J Biol Chem. 2003;278:27586‐27592. [DOI] [PubMed] [Google Scholar]
- 43. Devaud LL, Morrow AL. Interactions between neuroactive steroids and ethanol at GABAA receptors: effects of ethanol withdrawal. Stress, Gender and Alcohol‐Seeking Behavior, NIAAA Research Monograph. 1995:219‐240.
- 44. Devaud LL, Fritschy J‐M, Sieghart W, Morrow AL. Bidirectional alterations of GABAA receptors subunit peptide levels in rat cortex during chronic ethanol consumption and withdrawal. J Neurochem. 1997;69:126‐130. [DOI] [PubMed] [Google Scholar]
- 45. Kumar S, Sieghart W, Morrow AL. Association of protein kinase C with GABAA receptor containing a1 and a4 subunits in cerebral cortex: selective effects of chronic ethanol consumption. J Neurosci. 2002;82:110‐117. [DOI] [PubMed] [Google Scholar]
- 46. Di X‐J, Han D‐Y, Wang Y‐J, Chance MR, Mu T‐W. SAHA enhances proteostasis of epilepsy‐associated a1(A322D)/b2 g2 GABAA receptors. Chem Biol. 2013;20:1456‐1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang Z, Zang C, Rosenfeld JA, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genetic. 2008;40:897‐903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ghiani CA, Serra M, Motzo C, et al. Chronic administration of an anticonvuslsant dose of imidazenil fails to induce tolerance of GABAA receptor function in mice. Eur J Pharmacol. 1994;254:299‐302. [DOI] [PubMed] [Google Scholar]
- 49. Kadriu B, Larson J, Guidotti A, Davis JM, Nambiar MP, Auta J. Absence of tolerance to the anticonvulsant and neuroprotective effects of imidazenil against DFP‐induced seizure and neuronal damage. Neuropharmacology. 2011;61:1463‐1469. [DOI] [PubMed] [Google Scholar]
- 50. Nott A, Cheng J, Gao F, et al. Histone deacetylase 3 associates with MeCP2 to regulate FOXO and social behavior. Nat Neurosci. 2016;19:1497‐1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tesone‐Coelho C, Varela P, Escosteguy‐Neto JC, Cavarsan CF, Mello LE, Santos‐Junior JG. Effects of Ethanol on hippocampal neurogenesis depends on conditioned appetitive response. Addict Biol. 2013;18:774‐785. [DOI] [PubMed] [Google Scholar]