

Abstract
Histone deacetylase 6 (HDAC6), a cytoplasmic enzyme that plays important roles in many biological processes, is one isoform of a family of HDAC enzymes that catalyse the removal of functional acetyl groups from proteins. HDAC6 stands out from the other members of this family because it almost exclusively deacetylates cytoplasmic proteins and exerts deacetylation-independent effects, which has led to the successful development of relatively isoform-specific inhibitors of its enzymatic action. Numerous studies have recently demonstrated that HDAC6 expression and activity are increased in kidney disease, such as autosomal dominant polycystic kidney disease (ADPKD), renal fibrosis, and acute kidney injury (AKI), among others. Moreover, HDAC6 inhibitors have been investigated for use in treating these diseases. In fact, HDAC6 inhibitors effectively limit the progression of kidney diseases, suggesting that targeting HDAC6 may provide a novel treatment approach. However, the primary challenge in developing HDAC6-targeted therapies is understanding how the renoprotective effect of NDAC6 inhibitors can be selectively harnessed. Here, we discuss the unique function of HDAC6 and recapitulate the alluring potential of its inhibitors in kidney diseases.
Keywords: Histone deacetylase 6, Autosomal dominant polycystic kidney disease, Renal fibrosis, Lupus nephritis, Acute kidney injury
Background
Histone deacetylase 6 (HDAC6) is a cytoplasmic protein that potentially regulates several cellular functions through deacetylase-dependent and/or deacetylase-independent mechanisms (Valenzuela-Fernandez et al. 2008). HDACs are a group of enzymes that play important roles in multiple cellular processes by removing the acetyl group from histone or nonhistone proteins (Li 2011). Based on their homology to yeast orthologous, mammalian HDACs are categorized into 4 groups: class I HDACs (HDACs1, 2, 3, and 8), class II HDACs (HDACs 4, 5, 6, 7, 9, and 10), class III HDACs (SIRT1–7), and class IV HDACs (HDAC11) (Batchu et al. 2016). HDAC6 was first discovered in 1999 because of its homology to HDAC1 (Grozinger et al. 1999; Verdel and Khochbin 1999). The gene encoding HDAC6 is localized to the sub-band border of chromosome Xp11.22–23 in humans (Voelter-Mahlknecht and Mahlknecht 2003). Unlike most HDAC isoforms, HDAC6 contains a Ser Glu-repeat domain (SE14) that acts as a cytoplasmic retention signal and mediates its stable anchorage in the cytoplasm (Bertos et al. 2004). At the N-terminus of the enzyme, there is a nuclear localization signal (NLS) that enables the deacetylase to shuttle between the nucleus and the cytoplasm (Liu et al. 2012a). HDAC6 is unique in that it has duplicated deacetylase domains as well as a C-terminal binder of the ubiquitin zinc (BUZ) finger domain, which binds ubiquitin (Seigneurin-Berny et al. 2001) (Fig. 1). Although this enzyme is named HDAC6, it does not exert detectable deacetylase activity towards histones in vivo (Zhang et al. 2003; Tran et al. 2007), while it deacetylates histones in vitro (Zhang et al. 2006). The most characterized substrates of HDAC6 include α-tubulin, heat shock protein 90 and cortactin (Zhang et al. 2003). As a key modulator, HDAC6 has been implicated in many cellular processes, including proliferation (Valenzuela-Fernandez et al. 2008), autophagy (Lin et al. 2017), apoptosis (Bitler et al. 2017), and DNA repair (Namdar et al. 2010) (Fig. 1). Furthermore, HDAC6 inhibitors have shown powerful potential therapeutic effects in multiple diseases, such as cancer, neurodegenerative diseases, cardiovascular diseases, and kidney disease, among others (Batchu et al. 2016).
Fig. 1.

Schematic diagram of the structure and pleiotropic functions of HDAC6 protein
HDAC6 has recently emerged as a vital cytokine in kidney diseases. Growing evidence has demonstrated that the expression and activity of HDAC6 are increased in various kidney diseases (Shi et al. 2017; Choi et al. 2015; Cebotaru et al. 2016), and inhibiting HDAC6 has mitigated the progression of kidney injury. In this review, we focus on the role of HDAC6 in the pathology of kidney diseases and recapitulate the alluring potential of its inhibitors, which may aid in the development of HDAC6-targeted therapies for kidney diseases.
HDAC6 and ADPKD
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common hereditary disorders in humans, affecting 1/500 individuals in the United States (Gabow 1993). The hallmark of ADPKD is the development of multiple bilateral renal cysts that replace normal renal parenchyma, resulting in end-stage renal disease (ESRD) in approximately 50% of individuals with ADPKD by the age of 50 (Chebib et al. 2015). Most cases of ADPKD are caused by mutations in one of two genes: PKD1, accounting for 85–95% of cases, and PKD2, accounting for most of the remaining cases (Peters and Sandkuijl 1992). Aberrant Ca2+ signalling and cyclic adenosine monophosphate (cAMP) signalling are the leading causes of ADPKD (Chebib et al. 2015), and aberrant Ca2+ metabolism causes a switch to a proliferative cAMP-dependent phenotype in ADPKD (Chang and Ong 2013). The hypothesis is that Ca2+ restriction in ADPKD cells causes cAMP-dependent activation of the B-Raf/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway, resulting in increased cell growth (Yamaguchi et al. 2004). Similarly, increased Ca2+ influx into ADPKD cells restores normal cAMP signalling, thereby reducing cell growth (Yamaguchi et al. 2006). However, this hypothesis remains debated, and thus, the function of PC1 and PC2 in Ca2+ signalling is controversial (Chebib et al. 2015).
HDAC6 promotes cyst formation and disease by enhancing intracellular cAMP in ADPKD. Abnormal proliferation of the cyst-lined epithelium and increased intra-cystic fluid secretion via the cystic fibrosis transmembrane conductance regulator (CFTR) may contribute to cyst growth in ADPKD (Ramasubbu et al. 1998; Hanaoka et al. 1996). CFTR, a cAMP-activated chloride channel, is expressed in the apical epithelia in ADPKD cystic tissue, and its function can be measured by cAMP-mediated activation of chloride currents (Hanaoka et al. 1996). Cebotaru et al. found that the HDAC6-specific inhibitor tubacin inhibited cystic cell proliferation and reduced cAMP-triggered activation of CFTR in canine renal epithelial cells (Cebotaru et al. 2016). Consistently, Yanda and colleagues also found that treatment with ACY-1215, a specific HDAC6 inhibitor, slowed cyst growth in a mouse model of ADPKD by lowering cAMP levels (Yanda et al. 2017a). Interestingly, Yanda et al. found that HDAC inhibition decreased intracellular resting Ca2+ and increased ATP-simulated Ca2+ release in PC1 knockout (KO) cells. HDAC6 inhibition reduced the release of Ca2+ from the ER induced by thapsigargin, an inhibitor of the endoplasmic reticulum, Ca2 + -ATPase. HDAC6 inhibition and treatment of cells with the intracellular Ca2+ chelator BAPTA-AM reduced cAMP levels in PC1 KO cells (Yanda et al. 2017b). The poorly understood roles of PC1 and PC2 may responsible for this paradoxical phenomenon.
HDAC6 exaggerates ADPKD by upregulating epidermal growth factor receptor (EGFR) activity (Liu et al. 2012b). EGFR, which is also known as ErbB receptor tyrosine kinase, plays important roles in renal development, renal electrolyte homeostasis and tubule repair following injury (Melenhorst et al. 2008). Studies have shown that inhibition of EGFR tyrosine kinase activity, either genetically or pharmacologically, significantly reduces renal cyst formation and improves renal function in rodent models of PKD (Melenhorst et al. 2008; Sweeney et al. 2000; Torres et al. 2003), indicating that persistent EGF signalling is a primary factor of disease progression in PKD. HDAC6, a microtubule-associated α-tubulin deacetylase, exhibits increased expression and activity in Pkd1 mutant mouse embryonic kidney cells (Liu et al. 2012b). Liu et al. (Liu et al. 2012b) found that targeting HDAC6 with a general HDAC inhibitor, trichostatin, or a specific HDAC6 inhibitor, tubacin, increased the acetylation of α-tubulin and downregulated the expression of EGFR in Pkd1 mutant renal epithelial cells. In addition, inhibition of HDAC activity decreased the phosphorylation of ERK1/2, a downstream target of the EGFR axis, and normalized EGFR localization from apical to basolateral in Pkd1 KO mouse kidneys (Liu et al. 2012b). Thus, these authors suggested that Pkd1 mutation-induced upregulation of HDAC6 might slow the trafficking of EGFR from early endosomes to late endosomes along microtubules for degradation through deacetylation of α-tubulin, leading to phosphorylation of ERK1/2 to facilitate cyst formation (Liu et al. 2012b). Additionally, EGF contributes to ADPKD by inducing abnormal activation of the Wnt/β-catenin-dependent pathway and nuclear translocation of β-catenin (Li et al. 2008). It has been shown that HDAC6 regulates EGF-induced nuclear translocation of β-catenin (Deribe et al. 2009). EGF receptor activity is increased, and the receptor is mis-localized to the apical membrane in Pkd1 KO mice, whereas inhibition of HCAC6 activity in Pkd1 KO mice restores EGF localization to the basolateral cell membrane (Liu et al. 2012b).
Collectively, HDAC6 contributes to cyst growth by promoting cell proliferation and fluid secretion induced by aberrant Ca2+ signalling, abnormal cAMP signalling, and prolonged EGFR signalling (Fig. 2).
Fig. 2.
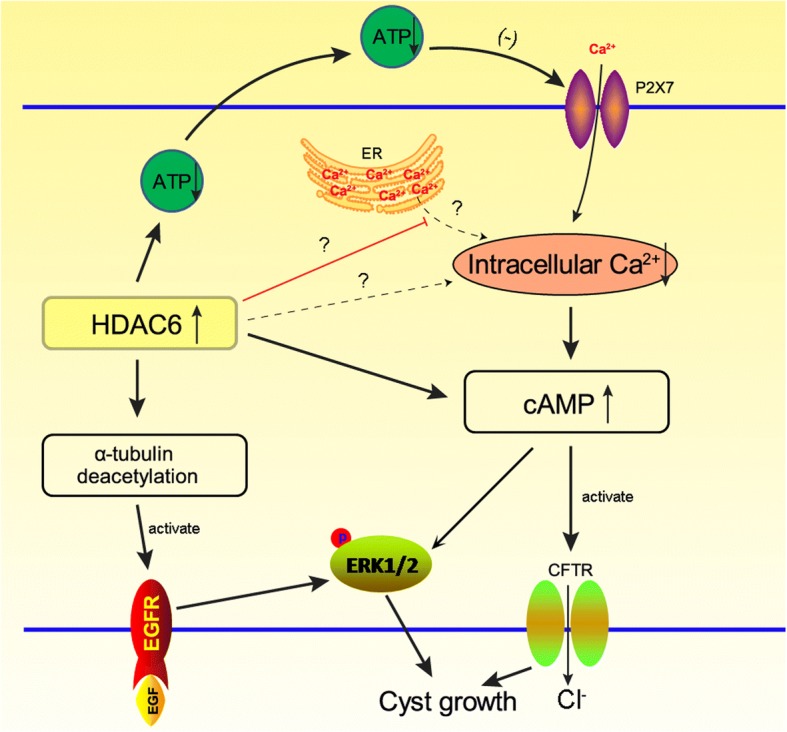
Schematic presentation of the signalling pathways by which HDAC6 contributes to cyst growth by promoting cell proliferation and fluid secretion induced by aberrant Ca2+ signalling, abnormal cAMP signalling, and prolonged EGFR signalling
HDAC6 and renal fibrosis
Renal fibrosis, characterized by fibroblast proliferation and the accumulation of extracellular matrix (ECM), is the endpoint of chronic kidney disease. Connective tissue growth factor (CTGF), implicated in the formation of ECM, including the ECM proteins fibronectin and collagen, is considered to be a molecular hallmark of renal fibrosis (Gao et al. 2008). Renal fibrosis is a complicated process, and multiple cellular cytokines are involved in the onset of renal fibrosis. As a participant in the process of fibrosis through activation of Smad2 and Smad3 phosphorylation, TGF-β/Smad signalling is considered to be the key regulator in renal fibrosis (Wang et al. 2014).
HDAC6 contributes to renal fibrosis through regulation of epigenetic histone modification and Smad3-dependent fibrotic genes (Choi et al. 2015). A previous study showed that the expression of HDAC6 was increased in a hypertensive kidney damage mouse model (Choi et al. 2015), and inhibition of HDAC6 or small interfering RNA against HDAC6 attenuated hypertensive stimuli-induced renal fibrosis and inflammation (Choi et al. 2015). Choi et al. investigated the fibrotic mechanism of HDAC6 and found that HDAC6 may participate in the regulation of epigenetic histone modification and facilitate phospho-Smad2/3 to Smad3 binding elements in fibrosis-associated gene promoters (Choi et al. 2015). Additionally, Shan et al. revealed a novel function of HDAC6 in epithelial-mesenchymal transition (EMT) by intercepting the TGF-β-SMAD3 signalling cascade (Shan et al. 2008). Aberrant EMT has been well documented in renal fibrosis. Shan and colleagues found that TGF-β1-induced EMT is accompanied by HDAC6-dependent deacetylation of α-tubulin. Importantly, inhibition of HDAC6 attenuated TGF-β1-induced EMT markers, such as aberrant expression of epithelial and mesenchymal peptides, as well as the formation of stress fibres. Reduced expression of HDAC6 also impaired SMAD3 activation in response to TGF-β1. Conversely, inhibition of SMAD3 activation substantially impaired HDAC6-dependent deacetylation of α-tubulin as well as the expression of EMT markers (Shan et al. 2008).
Finally, a recent study reported that unilateral ureteral obstruction disrupted microtubules, accompanied by less reduction of HDAC6 and α-tubulin acetyl transferase, which acetylates tubulin, a component of the microtubule; this finding means that HDAC6 may lead to renal fibrosis by inducing deacetylation of α-tubulin (Noh et al. 2018). Hence, HDAC6 may be a valuable therapeutic target for the treatment of renal fibrosis.
HDAC6 and lupus nephritis
Systemic lupus erythematosus (SLE) is an autoimmune disease in which the immune system produces autoantibodies against normal healthy tissue or cellular components to form immune complexes that are deposited in various tissues and subsequently induce inflammation, leading to tissue damage (Apostolidis et al. 2011). Lupus nephritis (LN) occurs in approximately 50% of SLE patients and is a major cause of morbidity and mortality due to this disease (Almaani et al. 2017). The pathogenesis of LN is complicated. Studies using lupus-prone mice have demonstrated that ablation of plasmacytoid dendritic cells (pDCs), a major source of interferon-α (IFN-α), prevented LN progression, reduced autoreactive T and B cell activation, and decreased autoantibodies in circulation and renal deposition (Sisirak et al. 2014; Rowland et al. 2014). Several studies have also shown a critical role of IFN-α in LN (Abdel Galil et al. 2017; Ji et al. 2015). Nevertheless, the role of IFN-α in LN is still unknown.
HDAC6 may play an active role in LN by increasing IFN-α levels. Several studies have recently provided evidence that epigenetic factors, including acetylation of histones and non-histone proteins, play crucial roles in the initiation and development of SLE, including LN (Hedrich 2017). Ren et al. found that HDAC6 expression is increased in animal models of SLE and that inhibition of HDAC6 decreased the disease. Furthermore, these authors examined the function of the HDAC6 inhibitor ACY-738 in LN and revealed that the IFN-α-producing ability of pDCs was decreased along with immunoglobulin isotype switching and the generation of pathogenic autoantibodies. Additionally, renal tissue showed decreased immunoglobulin deposition and reduced inflammation, as determined by glomerular and interstitial inflammation (Ren et al. 2017). This study indicates that HDAC6 is indeed involved in the development of LN, although further studies are needed to clarify the function of HDAC6 in the immune response.
Notably, Ren et al. concluded that HDAC6 inhibition may suppress B cell development and responses (Ren et al. 2017), which plays an essential role in the pathogenesis of LN (Suso et al. 2018). Similarly, primary membranous nephropathy (MN) is one of the most common types of autoimmune nephritis, in which phospholipase A2 receptor (PLA2R) and thrombospondin type-1 domain-containing 7A (THSD7A) are the two major autoantigens (Beck Jr et al. 2009; Tomas et al. 2014). Autoreactive B-cell clones can produce anti-PLA2R and anti-THSD7A antibodies, which interact with their corresponding antigens, resulting in the deposition of immune complexes in primary MN (Ruggenenti et al. 2017). Taken together, HDAC6 inhibitors may exert a renoprotective role in both LN and primary MN; however, no study has reported this effect in primary MN.
HDAC6 and AKI
Acute kidney injury (AKI), which is characterized by a rapid decline in the glomerular filtration rate, is a serious clinical problem. AKI is not only associated with high rates of mortality but also an increased risk of chronic kidney disease (Geng et al. 2015). Rhabdomyolysis, one of the major causes of community-acquired AKI, accounting for 5–15% of cases, is induced by different conditions, including crush injuries, severe trauma, intense physical exercise and some medications and illicit drugs, such as cocaine (Komada et al. 2015; Panizo et al. 2015). Rhabdomyolysis induces multiple deleterious effects on the kidney, including apoptosis, inflammation, oxidative stress, vasoconstriction, and tubular obstruction (Panizo et al. 2015; Humphreys et al. 2008). During these pathological processes, multiple signalling pathways and numerous genes involved in cell death and inflammatory responses are activated and/or upregulated (Geng et al. 2015). However, the detailed mechanism responsible for the pathogenesis of rhabdomyolysis-induced AKI is poorly understood. Understanding the molecular basis of these processes will aid in devising therapeutic strategies to treat AKI.
Inhibition of HDAC6 protects against ischaemic stroke and prolongs survival after sepsis in animal models (Wang et al. 2016; Li et al. 2015). Consistently, increased HDAC6 expression was observed in the cytoplasm of renal tubular cells, which commonly results in rhabdomyolysis-induced AKI, in a sepsis animal model (Shi et al. 2017). Administration of tubastatin A, significantly reduced the serum creatinine and blood urea nitrogen levels and attenuated renal tubular damage in the injured kidneys (Shi et al. 2017). Moreover, HDAC6 inhibition also resulted in decreased expression of NGAL, an injury marker of renal tubules, reduced apoptotic cells and decreased the expression of proinflammatory cytokines in the kidney after acute injury (Shi et al. 2017; Tang et al. 2018). Furthermore, HDAC6 inhibition reduced the level of oxidative stress by suppressing malondialdehyde (MDA) and preserved the expression of superoxide dismutase (SOD) in the injured kidneys (Shi et al. 2017). Recently, Yuying Feng et al. found that HDAC6 inhibitor shows renal protection via the reduction of endoplasmic reticulum (ER) stress-mediated apoptosis in tubular epithelial cells of rhabdomyolysis-induced AKI (Feng et al. 2018). Collectively, HDAC6 may accelerate AKI, mainly in rhabdomyolysis-induced AKI, by inducing oxidative stress, inflammation and ER stress.
Conclusion
HDAC6 plays a aggravating role in kidney diseases, and up-regulation of HDAC6 could exacerbate kidney diseases. In ADPKD, a role of HDAC6 in cytogenesis has been identified, as HDAC6 facilitates the progression of ADPKD by regulating Ca2+ signalling, cAMP signalling and EGFR endocytic trafficking and degradation. Moreover, treatment of ADPKD with HDAC6 inhibitors is effective, suggesting that these molecules are promising drug candidates for alleviating ADPKD. Furthermore, HDAC6 is closely related to other kidney diseases, including LN and AKI. Given its function in immune responses, HADC6 may participate in the onset of primary MN; however, additional studies are needed to confirm this idea.
Acknowledgments
Funding
This work was supported by grants from the National Natural Science Foundation of China (General Program 81760130).
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Authors’ contributions
XF and LY: Substantial contributions to the conception of the work, revising the article critically for important intellectual content, final approval of the submitted version, both agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. BK and YC: Drafting the article, final approval of the version to be published, agrees to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. WT and TY: Searching literatures and collecting some related information. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Ben Ke and Yanxia Chen contributed equally to this work.
Contributor Information
Xiangdong Fang, Email: xiangdongfang818@sina.com.
Liping Yang, Email: yangliping2018@sina.com.
References
- Abdel Galil SM, El-Shafey AM, Abdul-Maksoud RS, El-Boshy M. Interferon alpha gene expression and serum level association with low vitamin D levels in Egyptian female patients with systemic lupus erythematosus. Lupus. 2017; 10.1177/0961203317716321. [DOI] [PubMed]
- Almaani S, Meara A, Rovin BH. Update on lupus nephritis. Clin J Am Soc Nephrol : CJASN. 2017;12:825–835. doi: 10.2215/CJN.05780616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolidis SA, Crispin JC, Tsokos GC. IL-17-producing T cells in lupus nephritis. Lupus. 2011;20:120–124. doi: 10.1177/0961203310389100. [DOI] [PubMed] [Google Scholar]
- Batchu SN, Brijmohan AS, Advani A. The therapeutic hope for HDAC6 inhibitors in malignancy and chronic disease. Clin Sci. 2016;130:987–1003. doi: 10.1042/CS20160084. [DOI] [PubMed] [Google Scholar]
- Beck LH, Jr, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361:11–21. doi: 10.1056/NEJMoa0810457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertos NR, et al. Role of the tetradecapeptide repeat domain of human histone deacetylase 6 in cytoplasmic retention. J Biol Chem. 2004;279:48246–48254. doi: 10.1074/jbc.M408583200. [DOI] [PubMed] [Google Scholar]
- Bitler BG, et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat Cell Biol. 2017;19:962–973. doi: 10.1038/ncb3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebotaru L, et al. Inhibition of histone deacetylase 6 activity reduces cyst growth in polycystic kidney disease. Kidney Int. 2016;90:90–99. doi: 10.1016/j.kint.2016.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MY, Ong AC. New treatments for autosomal dominant polycystic kidney disease. Br J Clin Pharmacol. 2013;76:524–535. doi: 10.1111/bcp.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chebib FT, Sussman CR, Wang X, Harris PC, Torres VE. Vasopressin and disruption of calcium signalling in polycystic kidney disease. Nat Rev Nephrol. 2015;11:451–464. doi: 10.1038/nrneph.2015.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, et al. Tubastatin a suppresses renal fibrosis via regulation of epigenetic histone modification and Smad3-dependent fibrotic genes. Vasc Pharmacol. 2015;72:130–140. doi: 10.1016/j.vph.2015.04.006. [DOI] [PubMed] [Google Scholar]
- Deribe YL, et al. Regulation of epidermal growth factor receptor trafficking by lysine deacetylase HDAC6. Sci Signal. 2009;2:ra84. doi: 10.1126/scisignal.2000576. [DOI] [PubMed] [Google Scholar]
- Feng Y, et al. Selective histone deacetylase 6 inhibitor 23BB alleviated rhabdomyolysis-induced acute kidney injury by regulating endoplasmic reticulum stress and apoptosis. Front Pharmacol. 2018;9:274. doi: 10.3389/fphar.2018.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabow PA. Autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1993;22:511–512. doi: 10.1016/S0272-6386(12)80921-8. [DOI] [PubMed] [Google Scholar]
- Gao X, Li J, Huang H, Li X. Connective tissue growth factor stimulates renal cortical myofibroblast-like cell proliferation and matrix protein production. Wound Repair Regen. 2008;16:408–415. doi: 10.1111/j.1524-475X.2008.00380.x. [DOI] [PubMed] [Google Scholar]
- Geng X, et al. Differences in gene expression profiles and signaling pathways in rhabdomyolysis-induced acute kidney injury. Int J Clin Exp Pathol. 2015;8:14087–14098. [PMC free article] [PubMed] [Google Scholar]
- Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci U S A. 1999;96:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanaoka K, Devuyst O, Schwiebert EM, Wilson PD, Guggino WB. A role for CFTR in human autosomal dominant polycystic kidney disease. Am J Phys. 1996;270:C389–C399. doi: 10.1152/ajpcell.1996.270.1.C389. [DOI] [PubMed] [Google Scholar]
- Hedrich CM. Epigenetics in SLE. Curr Rheumatol Rep. 2017;19:58. doi: 10.1007/s11926-017-0685-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys BD, et al. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell. 2008;2:284–291. doi: 10.1016/j.stem.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Ji J, et al. A benzenediamine derivative fc-99 attenuates lupus-like syndrome in MRL/lpr mice related to suppression of pDC activation. Immunol Lett. 2015;168:355–365. doi: 10.1016/j.imlet.2015.10.017. [DOI] [PubMed] [Google Scholar]
- Komada T, et al. Role of NLRP3 Inflammasomes for rhabdomyolysis-induced acute kidney injury. Sci Rep. 2015;5:10901. doi: 10.1038/srep10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Epigenetics and autosomal dominant polycystic kidney disease. Biochim Biophys Acta. 2011;1812:1213–1218. doi: 10.1016/j.bbadis.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang X, Polakiewicz RD, Yao TP, Comb MJ. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J Biol Chem. 2008;283:12686–12690. doi: 10.1074/jbc.C700185200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. Inhibition of histone deacetylase 6 improves long-term survival in a lethal septic model. J Trauma Acute Care Surg. 2015;78:378–385. doi: 10.1097/TA.0000000000000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TW, et al. TDP-43/HDAC6 axis promoted tumor progression and regulated nutrient deprivation-induced autophagy in glioblastoma. Oncotarget. 2017;8:56612–56625. doi: 10.18632/oncotarget.17979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, et al. HDAC6 regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation in renal epithelial cells. PLoS One. 2012;7:e49418. doi: 10.1371/journal.pone.0049418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Peng L, Seto E, Huang S, Qiu Y. Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. J Biol Chem. 2012;287:29168–29174. doi: 10.1074/jbc.M112.371120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melenhorst WB, et al. Epidermal growth factor receptor signaling in the kidney: key roles in physiology and disease. Hypertension. 2008;52:987–993. doi: 10.1161/HYPERTENSIONAHA.108.113860. [DOI] [PubMed] [Google Scholar]
- Namdar M, Perez G, Ngo L, Marks PA. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc Natl Acad Sci U S A. 2010;107:20003–20008. doi: 10.1073/pnas.1013754107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh MR, Woo CH, Park MJ, In Kim J, Park KM. Ablation of C/EBP homologous protein attenuates renal fibrosis after ureteral obstruction by reducing autophagy and microtubule disruption. Biochim Biophys Acta. 2018;1864:1634–1641. doi: 10.1016/j.bbadis.2018.02.001. [DOI] [PubMed] [Google Scholar]
- Panizo N, Rubio-Navarro A, Amaro-Villalobos JM, Egido J, Moreno JA. Molecular mechanisms and novel therapeutic approaches to rhabdomyolysis-induced acute kidney injury. Kidney Blood Press Res. 2015;40:520–532. doi: 10.1159/000368528. [DOI] [PubMed] [Google Scholar]
- Peters DJ, Sandkuijl LA. Genetic heterogeneity of polycystic kidney disease in Europe. Contrib Nephrol. 1992;97:128–139. doi: 10.1159/000421651. [DOI] [PubMed] [Google Scholar]
- Ramasubbu K, Gretz N, Bachmann S. Increased epithelial cell proliferation and abnormal extracellular matrix in rat polycystic kidney disease. J Am Soc Nephrol : JASN. 1998;9:937–945. doi: 10.1681/ASN.V96937. [DOI] [PubMed] [Google Scholar]
- Ren J, et al. Selective HDAC6 inhibition decreases early stage of lupus nephritis by downregulating both innate and adaptive immune responses. Clin Exp Immunol. 2018;191(1):19–31. 10.1111/cei.13046 [DOI] [PMC free article] [PubMed]
- Rowland SL, et al. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med. 2014;211:1977–1991. doi: 10.1084/jem.20132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggenenti P, Fervenza FC, Remuzzi G. Treatment of membranous nephropathy: time for a paradigm shift. Nat Rev Nephrol. 2017;13:563–579. doi: 10.1038/nrneph.2017.92. [DOI] [PubMed] [Google Scholar]
- Seigneurin-Berny D, et al. Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol Cell Biol. 2001;21:8035–8044. doi: 10.1128/MCB.21.23.8035-8044.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B, et al. Requirement of HDAC6 for transforming growth factor-beta1-induced epithelial-mesenchymal transition. J Biol Chem. 2008;283:21065–21073. doi: 10.1074/jbc.M802786200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, et al. Inhibition of HDAC6 protects against rhabdomyolysis-induced acute kidney injury. Am J Physiol Renal Physiol. 2017;312:F502–F515. doi: 10.1152/ajprenal.00546.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisirak V, et al. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med. 2014;211:1969–1976. doi: 10.1084/jem.20132522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suso JP, et al. Profile of BAFF and its receptors’ expression in lupus nephritis is associated with pathological classes. Lupus. 2018;27:708–715. doi: 10.1177/0961203317739132. [DOI] [PubMed] [Google Scholar]
- Sweeney WE, Chen Y, Nakanishi K, Frost P, Avner ED. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int. 2000;57:33–40. doi: 10.1046/j.1523-1755.2000.00829.x. [DOI] [PubMed] [Google Scholar]
- Tang J, et al. Blockade of histone deacetylase 6 protects against cisplatin-induced acute kidney injury. Clin Sci. 2018; 10.1042/CS20171417. [DOI] [PubMed]
- Tomas NM, et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med. 2014;371:2277–2287. doi: 10.1056/NEJMoa1409354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres VE, et al. EGF receptor tyrosine kinase inhibition attenuates the development of PKD in Han:SPRD rats. Kidney Int. 2003;64:1573–1579. doi: 10.1046/j.1523-1755.2003.00256.x. [DOI] [PubMed] [Google Scholar]
- Tran AD, et al. HDAC6 deacetylation of tubulin modulates dynamics of cellular adhesions. J Cell Sci. 2007;120:1469–1479. doi: 10.1242/jcs.03431. [DOI] [PubMed] [Google Scholar]
- Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18:291–297. doi: 10.1016/j.tcb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Verdel A, Khochbin S. Identification of a new family of higher eukaryotic histone deacetylases. Coordinate expression of differentiation-dependent chromatin modifiers. J Biol Chem. 1999;274:2440–2445. doi: 10.1074/jbc.274.4.2440. [DOI] [PubMed] [Google Scholar]
- Voelter-Mahlknecht S, Mahlknecht U. Cloning and structural characterization of the human histone deacetylase 6 gene. Int J Mol Med. 2003;12:87–93. [PubMed] [Google Scholar]
- Wang B, et al. Transforming growth factor-beta1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. 2014;85:352–361. doi: 10.1038/ki.2013.372. [DOI] [PubMed] [Google Scholar]
- Wang Z, et al. Tubastatin a, an HDAC6 inhibitor, alleviates stroke-induced brain infarction and functional deficits: potential roles of alpha-tubulin acetylation and FGF-21 up-regulation. Sci Rep. 2016;6:19626. doi: 10.1038/srep19626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol : JASN. 2006;17:178–187. doi: 10.1681/ASN.2005060645. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, et al. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem. 2004;279:40419–40430. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- Yanda MK, Liu Q, Cebotaru L. An inhibitor of histone deacetylase 6 activity, ACY-1215, reduces cAMP and cyst growth in polycystic kidney disease. Am J Physiol Renal Physiol. 2017;313:F997–1004. [DOI] [PMC free article] [PubMed]
- Yanda MK, Liu Q, Cebotaru V, Guggino WB, Cebotaru L. Histone deacetylase 6 inhibition reduces cysts by decreasing via cAMP and Ca2+ in knockout mouse models of polycystic kidney disease. J Biol Chem. 2017b;292:17897–908. [DOI] [PMC free article] [PubMed]
- Zhang Y, Gilquin B, Khochbin S, Matthias P. Two catalytic domains are required for protein deacetylation. J Biol Chem. 2006;281:2401–2404. doi: 10.1074/jbc.C500241200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.