

Abstract
Aquaporins are membrane proteins that regulate cellular water flow. Recently, aquaporins have been proposed as mediators of cancer cell biology. A subset of aquaporins, referred to as aquaglyceroporins are known to facilitate the transport of glycerol. The present study describes the effect of gene knockdown of the aquaglyceroporin AQP3 on MDA-MB-231 breast cancer cell proliferation, migration, invasion, adherence and response to the chemotherapeutic agent 5-fluorouracil. shRNA mediated AQP3 gene knockdown induced a 28% reduction in cellular proliferation (P<0.01), a 39% decrease in migration (P<0.0001), a 24% reduction in invasion (P<0.05) and a 25% increase in cell death at 100 µM 5-FU (P<0.01). Analysis of cell permeability to water and glycerol revealed that MDA-MB-231 cells with knocked down AQP3 demonstrated a modest decrease in water permeability (17%; P<0.05) but a more marked decrease in glycerol permeability (77%; P<0.001). These results suggest that AQP3 has a role in multiple aspects of breast cancer cell pathophysiology and therefore represents a novel target for therapeutic intervention.
Keywords: aquaporin, AQP3, breast cancer, proliferation, migration, invasion, glycerol
Introduction
Breast cancer is the leading cause of female mortality in the Western world (1) and is the most common form of cancer in the UK. One million diagnoses were made worldwide in 2011 (2), of which over 40,000 new cases were diagnosed in the UK (3). Importantly, metastatic relapse is a key predictor of survival with fewer than 5% of women with metastatic breast cancer being disease free at five years (4). Although significant improvements in the management of primary and metastatic breast cancers have been made, the search for a successful and less toxic treatment of the metastatic breast cancer is on-going.
The aquaporin (AQP) family of membrane protein channels facilitates rapid water transport across all cell membranes, thereby maintaining body water homeostasis (5). Thirteen human AQPs have been identified to date (6), which form two distinct sub-groups: Classical AQPs (AQP0, 1, 2, 4, 5, 6 and 8) that solely transport water, and aquaglyceroporins (AQP3, 7, 9 and 10) that additionally transport small, uncharged molecules such as glycerol and urea (6).
The aquaglyceroporin AQP3 is expressed widely in the body and has been implicated in an increasing number of physiological and pathophysiological processes. These include metabolism, type 2 diabetes and skin elasticity (7). AQP3-null mice have also been demonstrated to have a reduced capacity for wound healing (8).
Recent evidence has suggested that several AQP family members are involved in carcinogenesis, with altered expression of AQPs being detected in several types of cancer (9). AQPs are now known to regulate cancer cell proliferation, migration, angiogenesis and metastasis (10–12). AQP3 has further been suggested to have a role in bladder (13), colorectal (14), gastric (15,16), lung (17,18) and skin (19) cancers. In breast cancer, an increased expression of AQP3 was reported compared to healthy border tissue, although the reasons for this observation are unclear (20,21). The role of AQP3 in breast cancer is however controversial, as recent evidence has suggested that AQP3 expression may be linked to disease survival in HER2 positive breast cancer (22) and a study recently reported that AQP3 gene silencing significantly reduced oestradiol-induced profilferation, invasion and migration in a cell model of breast cancer (23). It is possible that increased AQP3 expression in breast cancer cells may facilitate glycerol transport into the cell. This may then contribute to the generation of ATP (19), providing cancer cells with increased energy required for proliferation and tumourigenesis. Alternatively, these observations may be explained by as yet unassigned effects of water transport through the cell. Triple-negative breast cancers are associated with poor survival outcomes, and have been linked to a lower 5-year survival rate (24). This is due to lack of response to therapies such as tamoxifen and Herceptin as well as rapid tumour development and metastasis (25) and as such makes studying triple negative breast cancer a priority.
These findings suggest that AQP3 may be involved in several key processes that are important in breast cancer cell biology. In the present study AQP3 gene silencing was used to establish a role for this aquaglyceroporin in breast cancer cell pathophysiology. The effects on cellular proliferation, migration, invasion and response to a chemotherapeutic agent in the invasive human breast cancer cell model MDA-MB-231 were compared.
Materials and methods
Cell culture and reagents
Reagents were purchased from Sigma-Aldrich (Dorset, UK) unless stated otherwise. MDA-MB-231 human breast cancer cells were purchased from ATCC. Cells were routinely cultured in RPMI-1640 (PAA Laboratories Ltd., Somerset, UK) supplemented with 10% foetal bovine serum (FBS; PAA Laboratories Ltd., Somerset, UK), 50 IU/ml penicillin, 50 µg/ml streptomycin and 2 mM glutamine at 37°C in a 5% CO2 incubator. 5-FU was purchased from Tocris Bioscience (Bristol, UK).
Transfection and selection of stably expressing AQP3 shRNA clones
shRNA plasmids for AQP3 were purchased from Origene, USA. Of the four plasmid sequences, the plasmid that caused the most significant downregulation was chosen for production of stably expressing cells (termed shAQP3 cells). Transfection of AQP3 and control shRNA plasmids [including a red fluorescent protein (RFP) sequence and a puromycin-N-acetyl transferase gene located downstream of the SV40 promoter] into MDA-MB 231 cells was performed with Genjet transfection reagent (GenJet™ DNA in vitro Transfection Reagent; SignaGen Laboratories, Rockville, MD, USA). Cells were subsequently treated with 2 µg/ml puromycin for 2 weeks before colonies of puromycin resistant cells were picked using glass colony selection cylinders. Cells then underwent a 5-fold serial dilution with the last dilution being assessed for RFP expression and sub-culturing.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and western blot analysis of AQP expression
RNA was isolated from all samples using a proprietary RNA isolation kit (E.Z.N.A. ® Total RNA kit I; Omega Bio-Tek, Inc., Norcross, GA, USA). Quantification of total RNA was performed using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Inc., Leicester, UK). 1 µg total RNA from each sample was reverse transcribed using a proprietary cDNA synthesis kit (Primerdesign Ltd., Cambridge, UK) for 20 min at 55°C. Resulting cDNA was diluted 1 in 10 and subjected to SYBR®-Green Real-Time PCR using pre-validated sequence specific primers for AQPs purchased from Primerdesign Ltd. Housekeeping genes that were used to normalise data were chosen from a pool of candidate normalising genes with the two most stably expressed genes being employed (β-actin and YWHAZ). Samples were analysed using Precision SYBR-Green PCR Mastermix (Primerdesign Ltd.) and a Stratagene MX3000P thermal cycler (Stratagene, Stockport, UK). Comparisons were made for each sample between the average crossing point (Cq) obtained from the genes of interest and the geometric mean of the Cq obtained from the housekeeping genes, β-actin and YWHAZ (giving ΔCq).
For protein analysis, cells were lysed using RIPA buffer (Merck Millipore, Watford, UK) and insoluble material removed by centrifugation at 13,000 RCF, with the insoluble pellet being discarded. The protein content of the lysate was quantified using a modified Lowry method (DC Protein Assay; Bio-Rad Laboratories Ltd., Watford, UK) and photometrically measured at 690 nm. All samples were compared to a standard curve using BSA of a known concentration. Briefly, for analysis of AQP3 protein expression, 60 µg protein per sample was denatured and separated using 12% SDS-PAGE. Following SDS-PAGE, protein was transferred to a pure nitrocellulose blotting membrane (VWR International Ltd., Leicestershire, UK) at 100 V for 1 h and subsequently blocked using 5% non-fat milk (Marvel) in 1X TBS buffer (Sigma-Aldrich) containing 0.1% Tween-20 (TBS-T). After blocking, the membrane was washed three times in TBS-T and once in TBS buffer and then incubated overnight at 4°C with rabbit anti-human AQP3 antibody (cat. no. sc-20811; Santa Cruz Biotechnology, Inc., Dallas, TX, USA). The membrane was again washed before incubation at room temperature for 1 h with a goat anti-mouse IgG-HRP conjugated secondary antibody (Abcam, Cambridge, UK). Following secondary antibody incubation, the membrane was washed and subsequently exposed to the EZ-ECL chemiluminescence western blotting detection system (Geneflow, Lichfield, UK) for 5 min. Bands were then visualised using the GBOX HR 16 imaging system and GeneSys software (Geneflow). Blots were then stripped and probed for tubulin (using an anti-tubulin antibody from Abcam; cat. no. ab6046) as a loading control. Band density was measured using ImageJ and AQP3 band density was calculated as a ratio of AQP3: Tubulin.
Cellular proliferation assay
All cell counts were performed using a Countess® Automated Cell Counter (Thermo Fisher Scientific, Inc.). To measure cellular proliferation, wild type MDA-MB-231 and shAQP3 cells were grown to confluence and serum-starved overnight to synchronise the cell cycle. 2×105 cells were seeded into 75 cm2 cell culture vessels and allowed to grow for 72 h under standard conditions. Cells were subsequently detached using Accutase (PAA Laboratories Ltd.) before being re-counted. For each count, both chambers of a Countess slide were used and an average figure recorded.
Cellular migration assays
Two separate methodologies were employed to assess cell migration. For the wound scratch assay (26) WT and shAQP3 cells were seeded into 6-well plates, grown to confluence then serum-starved overnight. A sterile 20 µl pipette tip was used to produce a cross shaped scratch in the middle of each well. Images were taken using a Leica DMI4000 B inverted microscope at time points 0, 24, 48 and 72 h. Wound scratch analysis software TScratch was used to calculate the percentage wound closure for the different cell types at all time points (27).
Cellular migration was also assessed using a Cell-IQ® automated image capture system (CM Technologies, Tampere, Finland). WT and shAQP3 cells were seeded into 6-well plates and allowed to become confluent prior to overnight serum starvation. Cells were then scratched with a 20 µl pipette tip, and washed twice with HBBS to remove floating cells. Cells were treated with 5 µg/ml mitomycin C to inhibit cellular proliferation. The plates were placed on the Cell-IQ® system and two regions of interest (ROI) were selected for each cell type. The images were obtained every 15 min continuously over a 24 h period. Cell migration was expressed as the percentage of the wound closure relative to the initial wound scratched area. Digitised images were then analysed by Cell-IQ Analyser™ software to calculate the percentage of wound closure. Fig. 3C represents the mean of three separate experiments.
Figure 3.

Reducing AQP3 expression inhibits cellular proliferation rate. Cell viability assay comparing stable AQP3 knock-down MDA-MB-231 cells (shAQP3) with WT at 24, 48 and 72 h (n=8). **P<0.01 vs. shAQP3. Data points represent mean ± SEM.
Cellular invasion assay
Cell invasion assays were carried out with WT and shAQP3 cells in 96-well transwell plates with 8 µm pores using the Cultrex® BME Cell Invasion Assay system (R&D Systems Abington, UK). Cells were starved in serum-free medium for 18 h before analysis. Basement membrane extract (BME) was used to coat transwell plates for 4 h at 37°C in a CO2 incubator before being aspirated and 50,000 cells were subsequently seeded per transwell. 10% FBS medium was added to the bottom chambers. Cells were incubated at 37°C in CO2 incubator for 48 h. After 24 h the top and bottom chambers of the invasion devices were carefully aspirated, washed and Calcein-AM (in a cell lysis buffer) was added to the bottom chambers and incubated at 37°C in CO2 incubator for 1 h. The top chambers were removed and plates were read on a SPECTRAmax® GEMINI-XS Spectrofluorometer (Molecular Devices Corporation, Sunnyvale, CA, USA) at 485 nm excitation, 520 nm emission.
Cellular adhesion assay
Wild type and shAQP3 cells were seeded into 96-well plates coated with fibronectin (R&D Systems), where adherent cells were captured. After 4 h unbound cells were washed away, and the adherent cells are were exposed to Calcein-AM solution before fluorescence was read at on a SPECTRAmax® GEMINI-XS Spectrofluorometer (Molecular Devices Corporation) at 485 nm excitation, 520 nm emission.
Cell viability assay
For analysis of 5-FU induced cell death, WT and shAQP3 cells were seeded into 96-well plates at a density of 10,000 cells per well. After 48 h cells were treated with 5-FU at a concentration of 100 µmol/l for 48 h. A proprietary resazurin-based cell viability assay was used to measure induced cell death following the manufacturer's protocol (PrestoBlue™; Thermo Fisher Scientific, Inc.) using a SPECTRAmax® GEMINI-XS Spectrofluorometer.
Fluorescent cell swelling assay
In order to assess transport of water and glycerol into AQP3 silenced cells, a Calcein AM-based cell swelling assay was employed. Briefly, 2.5×104 cells were seeded in triplicate in 96-well tissue culture treated plates (Falcon) 24 h before the experiment. Cells were subsequently incubated with 5 µM calcein-AM (Molecular Probes; Invitrogen Life Technologies) in complete cell culture media for 90 min. Excess calcein-AM was removed by washing. Calcein AM fluorescence time-series were measured in a BioTek Synergy HT plate reader using the 495 nm excitation and 515 nm emission filters, with the internal temperature held at 37°C.
For water permeability analysis fluorescence readings were taken every 50 ms. After 5 sec, 75 µl of hypotonic culture media (170 mOsm/kg H2O) was added to the 75 µl media (340 mOsm/kg H2O) already on the cells (to give a final extracellular osmolality of 255 mOsm/kg H2O; ΔOsm=85 mOsm/kg H2O) using the plate reader injector system, at a rate of 350 µl/sec. Fluorescence was then measured for 49.8 sec. For glycerol permeability analysis, fluorescence readings were taken every 100 ms due to the slower rate of swelling associated with glycerol transport. After 10 sec, 75 µl of an isoosmotic (340 mM in ddH2O) glycerol solution was added to the 75 µl media already on the cells using the plate reader injector system, at a rate of 350 µl/sec. Fluorescence was then measured for 99.9 sec. As the glycerol solution was isoosmotic, any changes in cell volume could be attributed to changes in intracellular osmolality and subsequent osmosis due to glycerol uptake. As a negative control, an isoosmotic mannitol solution (340 mM in ddH2O) was injected, and no change in fluorescence intensity was observed (data not shown). All solution osmolalities were measured with an Osmomat 3000 freezing point depression osmometer (Gonotec, Berlin, Germany). Fluorescence readings from cells not loaded with calcein AM were taken as background and subtracted from the raw data. Data were normalised to the average of the first five readings after injection. Exponentially decaying growth curves of the form C-Ae-kt were fitted subject to the constraint C-A=1, using the Solver tool in Microsoft Excel. Curves were fitted to 3 fluorescence traces per experimental repeat.
Statistical analysis
Statistical analysis of data was performed using GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla,. CA, USA). One-way analysis of variance (ANOVA) with Tukey's post hoc test for comparisons of ≥3 groups or Student's t-test for comparisons of <3 groups were performed for the statistical analysis between experimental conditions. P<0.05 were considered significant as indicated by *; P<0.01 are indicated by **; and P<0.001 are indicated by ***. Where n is stated, n refers to the number of separate biological replicates assessed.
Results
AQP expression in primary human breast tissue and MDA-MB-231 cells
Expression of all 13 human AQP family members was investigated in the invasive breast cancer cell line MDA-MB-231 using SYBR®-Green Real-Time PCR analysis. AQP3 was the most abundant transcript of all AQP family members, with an average Ct value of 24 (±0.89, Fig. 1A and B). Analysis of AQP3 gene expression in primary human breast tissue showed a threefold higher level of AQP3 expression (Fig. 1C; P=0.018, n=6) in cancerous breast tissue compared to healthy border tissue. After shRNA-mediated AQP3 gene silencing, both qPCR and western blotting (Fig. 2) were used to assess gene knock-down.
Figure 1.

AQP gene expression in breast cancer biopsies and MDA-MB-231 cells. (A) qPCR analysis of AQP family mRNA expression in breast cancer biopsy samples. Data expressed as mean Cq value ± SEM (n=8). (B) Graphical representation of qPCR expression in MDA-MB-231 cells with all AQPs normalised to the expression level of AQP0. Bars represent mean ± SEM. (C) qPCR analysis of AQP3 mRNA expression in primary healthy breast tissue (HBT) and matched cancerous tissue (CT). Bars represent mean expression levels normalised to HBT (n=6). *P<0.05 vs. HBT.
Figure 2.

AQP expression after shRNA-mediated gene-silencing in MDA-MB-231 cells. (A) qPCR analysis of AQP3 mRNA expression levels in stably shRNA transfected MDA-MB-231 cells (shAQP3) compared to wild type (WT) cells (n=8). (B) AQP3 protein expression in shAQP3 cells measured by semi-quantitative western blotting, (n=6). *P<0.05. Bars represent mean ± SEM.
Semi-quantitative western blotting band density analysis demonstrated that protein expression was reduced by 60% (±12%) in the AQP3 shRNA stable clone (shAQP3) compared to WT cells (P=0.012).
shRNA-mediated AQP3 gene silencing significantly reduces MDA-MB-231 cellular proliferation
AQP3 has been identified as a regulator of cellular proliferation in several cell types (8,28–30). The effect of silencing AQP3 in MDA-MB-231 cells on cellular proliferation rate was measured. Analysis of total cell count 72 h post-seeding showed that AQP3-silenced cells (shAQP3) had a 28% reduction (±9%) in cell numbers compared to WT cells (P=0.01, Fig. 3). From repeated counting at 24, 48 and 72 h, the doubling time of cells was calculated as follows: WT=30.98 h, shAQP3=39.27 h (P<0.05 for shAQP3 vs. WT; n=8). AQP3 silencing therefore significantly decreased the rate of cellular proliferation in MDA-MB231 cells resulting in an increased doubling time. This suggests that AQP3 is involved in MDA-MB-231 cell proliferation, and may therefore represent a novel target for reducing levels of breast cancer cell growth.
Silencing AQP3 mRNA causes a significant reduction in cellular migration
The role of AQP3 in cell migration and wound healing is well established (8,14,28). The present study therefore assessed the impact of AQP3 gene silencing of MDA-MB-231 cells, a cell line known to have migratory potential (31). In order to assess cell migration, WT and shAQP3 cells were subjected to two assays; a wound scratch assay, an established measure of MDA-MB-231 cell migration (31,32) and analysis using the Cell IQ system. For the wound scratch assay, after 72 h, WT cells showed a wound closure of 58% (±6%), n=5 (Fig. 4). Wound closure was significantly reduced in the shAQP3 cells to just 25% (±6%; P=0.001). Using the Cell IQ system, in conjunction with mytomycin C treatment to inhibit cellular proliferation, shAQP3 cells showed a 39% reduction in cellular migration after 24 h compared to WT cells. These data demonstrate that AQP3 is an important regulator of MDA-MB-231 cell migration, and that cell migration can be potently inhibited in vitro by targeting AQP3 expression. As mytomycin C, a mitotic inhibitor, was used for the CellIQ assay these results suggest that the difference in wound closure observed was not due to the effects of AQP3 silencing on proliferation.
Figure 4.

AQP3 silenced cells exhibit reduced migration rate. Analysis of cellular migration using the CellIQ imaging and automated analysis system after 24 h (n=3). Data represents mean ± SD. ***P<0.0001.
Stable AQP3-silenced cells show decreased levels of cellular invasion but unchanged cellular adhesion
The process of metastasis, the spread of cancer cells from a primary site to distant parts of the body, is initiated by cellular invasion and is considered a pivotal aspect of cancer mortality (33). In order to assess the levels of invasiveness of cells in which AQP3 had been silenced, proprietary BME- and collagen-based cellular invasion assays was used, whereby cells were challenged using BME or collagen coated transwell dishes through which they were required to invade in order to reach a bottom chamber. After a 72 h incubation period, shAQP3 cells showed a significantly reduced level of invasion through BME- and collagen coated plates of 76% (±3%) and 77% (±4%) respectively compared to WT cells (Fig. 5A and B, n=6).
Figure 5.

AQP3 mediates cellular invasion but not adhesion. Graphical representation of the relative level of shAQP3 and wild type (WT) invasion through (A) BME substrate and (B) collagen substrate after 24 h. n=8. **P<0.01. (C) Cellular adhesion of shAQP3 and WT cells as measured by calcein AM fluorescence after 4 h incubation in fibronectin coated plate. n=8. Bars represents mean ± SEM.
Cellular adhesion plays an important part in cancer progression and metastasis (34) with circulating cancer cells ultimately interacting with endothelial cells, leading to extravasation (35). WT and shAQP3 cells were seeded into fibronectin coated plates for 4 h to assess levels of cellular adhesiveness. Analysis of Calcein-AM staining in each well after this period showed that there was no significant difference in the levels of cellular adhesion between the two cell types (P=0.56, Fig. 5C; n=8) suggesting that AQP3 does not have a role in MDA-MB-231 cell adhesion.
AQP3 silencing increases 5-FU induced cell death
5-FU is a fluoropyrimidine pro-drug commonly given to treat bowel, breast or stomach cancer (36). A regulatory role for AQP3 in chemotherapy-induced breast cancer cell death has previously been suggested (20). We therefore investigated the role of AQP3 in cell death by exposing WT and shAQP3 cells to a range of 5-FU concentrations for 48 h and subsequently measuring cell viability. shAQP3 cells showed significantly lower (maximal 25% reduction, P<0.05; n=5) levels of cell viability at all 5-FU concentrations tested when compared to WT cells (Fig. 6), suggesting that AQP3 has a cytoprotective effect in breast cancer cells.
Figure 6.
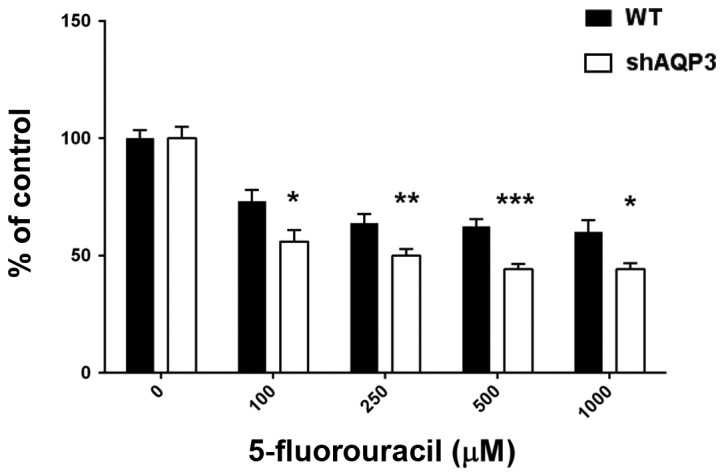
AQP3 gene silencing sensitises MDA-MB-231 cells to 5-fluouracil-induced cell death. Cell viability assay comparing shAQP3 cells and wild type (WT) exposed to 5FU after 48 h (n=6). Black bars=WT, open bars=shAQP3. *P<0.05, **P<0.01, ***P<0.001 vs. WT. Bars represent mean ± SEM.
AQP3 silencing markedly decreases glycerol permeability in MDA-MB-231 cells
As AQP3 is known to be permeable to both water and glycerol, analysis of the transport of both of these molecules was performed in WT and shAQP3 cells to provide information regarding the likely role of AQP3 in these cells. Analysis of cellular fluorescence using cells pre-exposed to calcein AM (adapted from (37)) showed that shAQP3 cells displayed a significant decrease of 17% in cellular permeability to water (P=0.016, n=3) compared to control cells (Fig. 7). There was however a more marked decrease in glycerol permeability in shAQP3 cells (Fig. 8), with a 77% decrease in glycerol permeability being observed (P=0.0018, n=3) compared to control cells, suggesting that AQP3 in MDA-MB-231 is preferentially a glycerol channel and that the effects of AQP3 silencing are likely due to a reduction in glycerol transport.
Figure 7.

AQP3 gene silencing induces a small decrease in water transport. Representative calcein AM fluorescence intensity time-series of (A) WT and (B) shAQP3 cells subjected to a 85 mOsm inwardly directed osmotic gradient (black), and fitted exponential decay function of the form C-Ae-kt (red). (C) Osmotic water permeability of WT and shAQP3 cells normalised to the average wild type permeability. n=3, with each n an average over 3 fluorescence time-series. *P<0.05. Bars represent mean ± SEM.
Figure 8.

AQP3 gene silencing induces a marked decrease in glycerol transport. Representative calcein fluorescence intensity time-series of (A) WT and (B) shAQP3 cells subjected to a 170 mM inwardly directed glycerol gradient (black), and fitted exponential decay function of the form C-Ae-kt (red). (C) Glycerol permeability of WT and shAQP3 cells normalised to the average wild type permeability. n=3, with each N an average over 3 fluorescence time-series. ***P<0.001. Bars represent mean ± SEM.
Discussion
The rates of breast cancer remain high in most societies, as do the mortality-incidence ratios (38). With this in mind, identification of novel cellular targets for therapeutic interventions is of key importance for developing the repertoire available to clinicians. It is known from previous work that AQP3 is expressed in most epithelia (39) and that expression levels are significantly upregulated in cancerous breast epithelia (40). In addition, previous studies have suggested that AQP3 is involved in oestrogen-induced breast cancer cell migration, invasion and proliferation (23). This study reports for the first time that downregulation of AQP3 expression in breast cancer cells significantly modulates several clinically relevant aspects of breast cancer cell biology.
A key finding presented here is that silencing AQP3 expression produced a marked reduction in cellular proliferation rate (leading to an increase in the doubling time) in MDA-MB-231 cells. Previous evidence suggests that AQP3 is involved in epithelial cell proliferation (29), although no evidence of this effect in breast epithelial cells has previously been reported. Targeting the proliferation rate of cancerous breast epithelial cells is of great importance as cellular proliferation (measured by Ki67 expression or mitosis counting) has been significantly correlated with disease prognosis in numerous studies (41,42). These data, in combination with the findings presented here, suggest that targeting cellular proliferation by pharmaceutical downregulation or inhibition of AQP3 expression or function is clinically relevant and might be associated with a better prognosis.
We can also report here that knockdown of AQP3 in MDA-MB-231 cells resulted in significant decreases in cellular migration and invasion and that these effects are in addition to the effects on proliferation. Recent data have shown that AQP3 is necessary for fibroblast growth factor-2 induced cell migration in MDA-MB-231 cells (43), suggesting that AQP3 is a key regulator of migration in these cells. These observations are of key importance as they suggest that targeted reduction in AQP3 levels may not only inhibit the ability of tumour cells to proliferate, but also reduce the ability of breast cancer cells to break through a basement membrane and spread to distant sites. Axillary lymph node metastases remains an important prognostic factor for breast cancer, and therefore any intervention that can reduce the capacity of primary breast cancer cells to spread to distant sites is of significant clinical interest.
The mechanism by which AQP3 may influence cellular migration is not known. It has been previously demonstrated that AQP3 mediated H2O2 transport, and that this has a potential role in the regulation of breast cancer cell migration (44). Our data show that glycerol permeability is significantly reduced in shAQP3 cells and this reduction is markedly more than that seen for water permeability (Figs. 7 and 8). Whilst this is not mechanistic proof, glycerol transport and possibly its role in cell metabolism may be the mechanism by which reducing AQP3 expression leads to changes in cellular behaviour.
AQP3 represents an exciting target for breast cancer therapy as it fulfils several criteria. First, it is a membrane protein and therefore relatively accessible using traditional pharmaceutical means (45). Secondly, research using AQP3 knockout mice has shown that the only major phenotypic consequences of global AQP3 knockdown are limited to dry skin and reduced wound healing (46–48); although a delay in wound healing is potentially an issue post-operatively, this can hypothetically be overcome by delaying any AQP3-based therapy after wound closure is complete. These observations suggest that there may be only limited consequences from using a global approach to reducing either AQP3 expression or function in breast epithelial cells. The lack of potential side effects that might be associated with global AQP3 knockdown is therefore a key strength of using AQP3 as a target for novel therapeutic interventions. This study provides a platform for further work; it would be beneficial to compare the effects of AQP3 in primary breast cancer epithelial cells and a range of other breast cancer cell models (including hormone-sensitive cell lines) as part of a wider programme for the translational impact of this study.
In conclusion, this study reports that AQP3 downregulation reduces breast cancer cell proliferation, migration and invasion in MDA-MB-231 cells whilst increasing sensitivity to the antimetabolite chemotherapy drug, 5-FU. This is the first study to identify AQP3 as a target for breast cancer therapy.
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Justo N, Wilking N, Jönsson B, Luciani S, Cazap E. A review of breast cancer care and outcomes in Latin America. Oncologist. 2013;18:248–256. doi: 10.1634/theoncologist.2012-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 4.Martino M, Ballestrero A, Zambelli A, Secondino S, Aieta M, Bengala C, Liberati AM, Zamagni C, Musso M, Aglietta M, et al. Long-term survival in patients with metastatic breast cancer receiving intensified chemotherapy and stem cell rescue: Data from the Italian registry. Bone Marrow Transplant. 2013;48:414–418. doi: 10.1038/bmt.2012.149. [DOI] [PubMed] [Google Scholar]
- 5.Conner AC, Bill RM, Conner MT. An emerging consensus on aquaporin translocation as a regulatory mechanism. Mol Membr Biol. 2013;30:1–12. doi: 10.3109/09687688.2012.743194. [DOI] [PubMed] [Google Scholar]
- 6.Ishibashi K, Kondo S, Hara S, Morishita Y. The evolutionary aspects of aquaporin family. Am J Physiol Regul Integr Comp Physiol. 2011;300:R566–R576. doi: 10.1152/ajpregu.90464.2008. [DOI] [PubMed] [Google Scholar]
- 7.Rojek A, Praetorius J, Frøkiaer J, Nielsen S, Fenton RA. A current view of the mammalian aquaglyceroporins. Annu Rev Physiol. 2008;70:301–327. doi: 10.1146/annurev.physiol.70.113006.100452. [DOI] [PubMed] [Google Scholar]
- 8.Hara-Chikuma M, Verkman AS. Aquaporin-3 facilitates epidermal cell migration and proliferation during wound healing. J Mol Med (Berl) 2008;86:221–231. doi: 10.1007/s00109-007-0272-4. [DOI] [PubMed] [Google Scholar]
- 9.Verkman AS, Hara-Chikuma M, Papadopoulos MC. Aquaporins-new players in cancer biology. J Mol Med (Berl) 2008;86:523–529. doi: 10.1007/s00109-008-0303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie Y, Wen X, Jiang Z, Fu HQ, Han H, Dai L. Aquaporin 1 and aquaporin 4 are involved in invasion of lung cancer cells. Clin Lab. 2012;58:75–80. [PubMed] [Google Scholar]
- 11.Zhang Z, Chen Z, Song Y, Zhang P, Hu J, Bai C. Expression of aquaporin 5 increases proliferation and metastasis potential of lung cancer. J Pathol. 2010;221:210–220. doi: 10.1002/path.2702. [DOI] [PubMed] [Google Scholar]
- 12.Tie L, Lu N, Pan XY, Pan Y, An Y, Gao JW, Lin YH, Yu HM, Li XJ. Hypoxia-induced up-regulation of aquaporin-1 protein in prostate cancer cells in a p38-dependent manner. Cell Physiol Biochem. 2012;29:269–280. doi: 10.1159/000337608. [DOI] [PubMed] [Google Scholar]
- 13.Otto W, Rubenwolf PC, Burger M, Fritsche HM, Rößler W, May M, Hartmann A, Hofstädter F, Wieland WF, Denzinger S. Loss of aquaporin 3 protein expression constitutes an independent prognostic factor for progression-free survival: An immunohistochemical study on stage pT1 urothelial bladder cancer. BMC Cancer. 2012;12:459. doi: 10.1186/1471-2407-12-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li A, Lu D, Zhang Y, Li J, Fang Y, Li F, Sun J. Critical role of aquaporin-3 in epidermal growth factor-induced migration of colorectal carcinoma cells and its clinical significance. Oncol Rep. 2013;29:535–540. doi: 10.3892/or.2012.2144. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Gui Z, Deng L, Sun M, Guo R, Zhang W, Shen L. c-Met upregulates aquaporin 3 expression in human gastric carcinoma cells via the ERK signalling pathway. Cancer Lett. 2012;319:109–117. doi: 10.1016/j.canlet.2011.12.040. [DOI] [PubMed] [Google Scholar]
- 16.Wang G, Gao F, Zhang W, Chen J, Wang T, Zhang G, Shen L. Involvement of Aquaporin 3 in Helicobacter pylori-related gastric diseases. PLoS One. 2012;7:e49104. doi: 10.1371/journal.pone.0049104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li B, Jin L, Zhong K, Du D. Correlation of aquaporin 3 expression with the clinicopathologic characteristics of non-small cell lung cancer. Zhongguo Fei Ai Za Zhi. 2012;15:404–408. doi: 10.3779/j.issn.1009-3419.2012.07.03. (In Chinese) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu YL, Matsuzaki T, Nakazawa T, Murata S, Nakamura N, Kondo T, Iwashina M, Mochizuki K, Yamane T, Takata K, Katoh R. Expression of aquaporin 3 (AQP3) in normal and neoplastic lung tissues. Hum Pathol. 2007;38:171–178. doi: 10.1016/j.humpath.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 19.Hara-Chikuma M, Verkman AS. Prevention of skin tumorigenesis and impairment of epidermal cell proliferation by targeted aquaporin-3 gene disruption. Mol Cell Biol. 2008;28:326–332. doi: 10.1128/MCB.01482-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trigueros-Motos L, Pérez-Torras S, Casado FJ, Molina-Arcas M, Pastor-Anglada M. Aquaporin 3 (AQP3) participates in the cytotoxic response to nucleoside-derived drugs. BMC Cancer. 2012;12:434. doi: 10.1186/1471-2407-12-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi Z, Zhang T, Luo L, Zhao H, Cheng J, Xiang J, Zhao C. Aquaporins in human breast cancer: Identification and involvement in carcinogenesis of breast cancer. J Surg Oncol. 2012;106:267–272. doi: 10.1002/jso.22155. [DOI] [PubMed] [Google Scholar]
- 22.Kang S, Chae YS, Lee SJ, Kang BW, Kim JG, Kim WW, Jung JH, Park HY, Jeong JH, Jeong JY, Park JY. Aquaporin 3 expression predicts survival in patients with HER2-positive early breast cancer. Anticancer Res. 2015;35:2775–2782. [PubMed] [Google Scholar]
- 23.Huang YT, Zhou J, Shi S, Xu HY, Qu F, Zhang D, Chen YD, Yang J, Huang HF, Sheng JZ. Identification of estrogen response element in aquaporin-3 gene that mediates estrogen-induced cell migration and invasion in estrogen receptor-positive breast cancer. Sci Rep. 2015;5:12484. doi: 10.1038/srep12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vici P, Pizzuti L, Natoli C, Gamucci T, Di Lauro L, Barba M, Sergi D, Botti C, Michelotti A, Moscetti L, et al. Triple positive breast cancer: A distinct subtype? Cancer Treat Rev. 2015;41:69–76. doi: 10.1016/j.ctrv.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 25.Lorusso G, Rüegg C. New insights into the mechanisms of organ-specific breast cancer metastasis. Semin Cancer Biol. 2012;22:226–233. doi: 10.1016/j.semcancer.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 26.Liang CC, Park AY, Guan JL. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 27.Gebäck T, Schulz MM, Koumoutsakos P, Detmar M. TScratch: A novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques. 2009;46:265–274. doi: 10.2144/000113083. [DOI] [PubMed] [Google Scholar]
- 28.Ji C, Cao C, Lu S, Kivlin R, Amaral A, Kouttab N, Yang H, Chu W, Bi Z, Di W, Wan Y. Curcumin attenuates EGF-induced AQP3 up-regulation and cell migration in human ovarian cancer cells. Cancer Chemother Pharmacol. 2008;62:857–865. doi: 10.1007/s00280-007-0674-6. [DOI] [PubMed] [Google Scholar]
- 29.Levin MH, Verkman AS. Aquaporin-3-dependent cell migration and proliferation during corneal re-epithelialization. Invest Ophthalmol Vis Sci. 2006;47:4365–4372. doi: 10.1167/iovs.06-0335. [DOI] [PubMed] [Google Scholar]
- 30.Nakahigashi K, Kabashima K, Ikoma A, Verkman AS, Miyachi Y, Hara-Chikuma M. Upregulation of aquaporin-3 is involved in keratinocyte proliferation and epidermal hyperplasia. J Invest Dermatol. 2011;131:865–873. doi: 10.1038/jid.2010.395. [DOI] [PubMed] [Google Scholar]
- 31.Du J, Sun C, Hu Z, Yang Y, Zhu Y, Zheng D, Gu L, Lu X. Lysophosphatidic acid induces MDA-MB-231 breast cancer cells migration through activation of PI3K/PAK1/ERK signaling. PLoS One. 2010;5:e15940. doi: 10.1371/journal.pone.0015940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cazet A, Groux-Degroote S, Teylaert B, Kwon KM, Lehoux S, Slomianny C, Kim CH, Le Bourhis X, Delannoy P. GD3 synthase overexpression enhances proliferation and migration of MDA-MB-231 breast cancer cells. Biol Chem. 2009;390:601–609. doi: 10.1515/BC.2009.054. [DOI] [PubMed] [Google Scholar]
- 33.Liu L, Duclos G, Sun B, Lee J, Wu A, Kam Y, Sontag ED, Stone HA, Sturm JC, Gatenby RA, Austin RH. Minimization of thermodynamic costs in cancer cell invasion. Proc Natl Acad Sci USA. 2013;110:1686–1691. doi: 10.1073/pnas.1221147110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bendas G, Borsig L. Cancer cell adhesion and metastasis: Selectins, integrins, and the inhibitory potential of heparins. Int J Cell Biol. 2012;2012:676731. doi: 10.1155/2012/676731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Earley S, Plopper GE. Phosphorylation of focal adhesion kinase promotes extravasation of breast cancer cells. Biochem Biophys Res Commun. 2008;366:476–482. doi: 10.1016/j.bbrc.2007.11.181. [DOI] [PubMed] [Google Scholar]
- 36.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 37.Fenton RA, Moeller HB, Nielsen S, de Groot BL, Rützler M. A plate reader-based method for cell water permeability measurement. Am J Physiol Renal Physiol. 2010;298:F224–F230. doi: 10.1152/ajprenal.00463.2009. [DOI] [PubMed] [Google Scholar]
- 38.Forouzanfar MH, Foreman KJ, Delossantos AM, Lozano R, Lopez AD, Murray CJ, Naghavi M. Breast and cervical cancer in 187 countries between 1980 and 2010: A systematic analysis. Lancet. 2011;378:1461–1484. doi: 10.1016/S0140-6736(11)61351-2. [DOI] [PubMed] [Google Scholar]
- 39.Mobasheri A, Wray S, Marples D. Distribution of AQP2 and AQP3 water channels in human tissue microarrays. J Mol Histol. 2005;36:1–14. doi: 10.1007/s10735-004-2633-4. [DOI] [PubMed] [Google Scholar]
- 40.Niu D, Kondo T, Nakazawa T, Yamane T, Mochizuki K, Kawasaki T, Matsuzaki T, Takata K, Katoh R. Expression of aquaporin3 in human neoplastic tissues. Histopathology. 2012;61:543–551. doi: 10.1111/j.1365-2559.2011.04165.x. [DOI] [PubMed] [Google Scholar]
- 41.Urruticoechea A, Smith IE, Dowsett M. Proliferation marker Ki-67 in early breast cancer. J Clin Oncol. 2005;23:7212–7220. doi: 10.1200/JCO.2005.07.501. [DOI] [PubMed] [Google Scholar]
- 42.van Diest PJ, van der Wall E, Baak JP. Prognostic value of proliferation in invasive breast cancer: A review. J Clin Pathol. 2004;57:675–681. doi: 10.1136/jcp.2003.010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cao XC, Zhang WR, Cao WF, Liu BW, Zhang F, Zhao HM, Meng R, Zhang L, Niu RF, Hao XS, Zhang B. Aquaporin3 is required for FGF-2-induced migration of human breast cancers. PLoS One. 2013;8:e56735. doi: 10.1371/journal.pone.0056735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Satooka H, Hara-Chikuma M. Aquaporin-3 controls breast cancer cell migration by regulating hydrogen peroxide transport and its downstream cell signaling. Mol Cell Biol. 2016;36:1206–1218. doi: 10.1128/MCB.00971-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rucevic M, Hixson D, Josic D. Mammalian plasma membrane proteins as potential biomarkers and drug targets. Electrophoresis. 2011;32:1549–1564. doi: 10.1002/elps.201100212. [DOI] [PubMed] [Google Scholar]
- 46.Hara-Chikuma M, Verkman AS. Roles of aquaporin-3 in the epidermis. J Invest Dermatol. 2008;128:2145–2151. doi: 10.1038/jid.2008.70. [DOI] [PubMed] [Google Scholar]
- 47.Verkman AS. Knock-out models reveal new aquaporin functions. Handb Exp Pharmacol. 2009. pp. 359–381. [DOI] [PMC free article] [PubMed]
- 48.Hara M, Ma T, Verkman AS. Selectively reduced glycerol in skin of aquaporin-3-deficient mice may account for impaired skin hydration, elasticity, and barrier recovery. J Biol Chem. 2002;277:46616–46621. doi: 10.1074/jbc.M209003200. [DOI] [PubMed] [Google Scholar]