

Abstract
A synthetic 8-mer amphipathic trans-acting poly-2′-O-methyluridylic thiophosphate triester RNA element (2′-OMeUtaPS) mediates an efficient delivery of uncharged polyA-tailed phosphorodiamidate morpholino sequences (PMO) to HeLa pLuc 705 cells or to myotube muscle cells. The transfected antisense PMO sequences induce alternate splicing of either the pre-mRNA encoding luciferase in HeLa pLuc 705 cells or exon 23 from the pre-mRNA encoding dystrophin in myotube muscle cells of the mdx mouse model of muscular dystrophy; the production of functional luciferase or dystrophin is restored without damaging cytotoxicity. The 2′-OMeUtaPS-mediated internalization of PMO sequences occurs through an energy-dependent mechanism; macropinocytosis has been determined to be the predominant endocytic pathway used in HeLa pLuc 705 cells.
Keywords: nucleic acid-based drug delivery; cellular uptake; antisense PNA, PMO and 2′-O-methyl RNA sequences; alternate splicing of luciferase pre-mRNA; exon 23 excision from mdx mouse dystrophin pre-mRNA
INTRODUCTION
Human genes undergo alternative splicing events, which are under the control of highly complex sequence-specific binding of various proteins to nuclear pre-mRNAs (Aartsma-Rus and van Ommen, 2007; Järver et al., 2014). Alternative splicing events are critically important to the treatment of Duchenne muscular dystrophy (DMD), which is a progressive deterioration of muscle function that is caused mainly by a frameshift deletion, nonsense or duplication mutation in the DMD gene encoding the protein dystrophin (Hoffman et al., 1987; Yokota et al., 2007; Aoki et al., 2013; Vila et al., 2015). Thus, skipping the exon(s) containing nonsense coding mutations or out-of-frame genomic deletions could permit restoration of the correct mRNA reading frame and production of a functional dystrophin protein (Muntoni and Wood, 2011; Shabanpoor et al., 2015). In this context, 2′-O-methyl phosphorothioate RNA and PMO sequences have been widely investigated in exon-skipping therapies for DMD; low levels of dystrophin production have however been reported (Shabanpoor et al., 2015). Although PMO sequences had demonstrated to be more efficient than 2′-O-methyl phosphorothioate RNA sequences at excising exon 23 from the dystrophin pre-mRNA in the mdx mouse model of muscular dystrophy (Fletcher et al., 2006), internalization of PMO sequences in muscle cells has been relatively poor. The chemical synthesis of 2′-OMeUtaPS, an octameric phosphorothioate 2′-O-methyluridylic acid functionalized with three N,N-dimethylaminopropyl and four octyl thiophosphate triester functions (Fig. 1) has recently been performed and reported to be efficient at delivering polyA-tailed PMO sequences in either HeLa pluc 705 cells or myotube muscle cells (Jain et al., 2017).
This unit describes the preparation of 2′-O-methylribonucleoside phosphoramidites (Basic Protocol 1) that will be used for solid-phase synthesis of the chimeric trans-acting polyuridylic acid thiophosphate triester element 2′-OMeUtaPS (Basic Protocol 2). The synthesis of the phosphordiamidites needed for the preparation of 2′-O-methylribonucleoside phosphoramidites is presented in Support Protocol 1. This unit also outlines: (i) the formation of PNA/PMO sequence:2′-OMeUtaPS complexes (Support Protocol 2); (ii) a flow cytometry analysis of the 2′-OMeUtaPS-mediated cellular internalization of PNA/PMO sequences in HeLa pLuc 705 cells (Basic Protocol 3); (iii) a luciferase assay for measuring luciferase activity production upon 2′-OMeUtaPS-mediated delivery of antisense PNA and PMO sequences in HeLa pLuc 705 cells (Basic Protocol 4); (iv) the 2′-O-MeUtaPS-mediated delivery of antisense PNA and PMO sequences in mdx mouse myorubes (Basic Protocol 5) and; (v) a nested PCR procedure for a qualitative agarose gel electrophoresis analysis of the extent of exon 23 skipping upon 2′-OMeUtaPS-mediated delivery of PNA and PMO sequences in mdx mouse myotubes (Basic Protocol 6).
With the intent of providing the novice experimenters with detailed information on the work recently published in the scientific literature (Jain et al., 2017), figures and textual materials have been taken from this article and converted into the stepwise procedures delineated herein to facilitate experimental reproduction of the original work; this resulted in unavoidable but necessary material similarities.
BASIC PROTOCOL 1. SYNTHESIS OF RIBONUCLEOSIDE PHOSPHORAMIDITES 3 AND 4
This protocol provides a general procedure for the preparation of ribonucleoside phosphoramidites 3 and 4 from commercial 5′-O-(4,4′-dimethoxytrityl)-2′-O-methyluridine, N,N,N′,N′-tetraisopropyl-O-[3-(N,N-dimethylamino)prop-1-yl]phosphordiamidite 1 or N,N,N′,N′-tetraisopropyl-O-(octan-1-yl)phosphordiamidite 2 (see Support Protocol 1) and 1H-tetrazole (Fig. 4.xx.1).
Figure 4.xx.1.
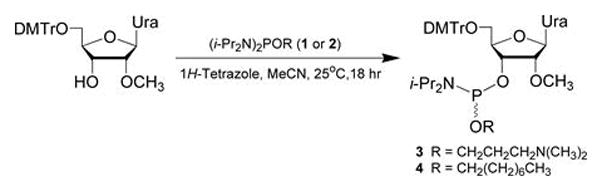
Preparation of ribonucleoside phosphoramidites 3 and 4. DMTr = 4,4′-dimethoxytrityl; Ura = uracil-1-yl; i-Pr = isopropyl.
Materials
5′-O-(4,4′-Dimethoxytrityl)-2′-O-methyluridine (ChemGenes Corporation)
N,N,N′,N′-Tetraisopropyl-O-[3-(N,N-dimethylamino)prop-1-yl]phosphordiamidite (1, see Support Protocol 1)
N,N,N′,N′-Tetraisopropyl-O-[octan-1-yl]phosphordiamidite (2, see Support Protocol 1)
0.45 M 1H-tetrazole in MeCN (Glen Research)
Anhydrous acetonitrile (MeCN, Glen Research)
Anhydrous benzene (Aldrich)
Argon or nitrogen source
Dichloromethane (CH2Cl2; Fisher Scientific)
Hexane (Fisher Scientific)
Triethylamine (Aldrich)
50-, 100-, and 250-mL round-bottom flasks (Kontes)
1-, 3- and 10-mL Luer-tipped glass syringes (Kimble)
5-mm NMR tubes (Wilmad)
Silica gel (60-Å, 230 to 400 mesh; EMD)
Dry ice/acetone bath
Magnetic stirrer and stir bars (VWR)
Rubber septa for 14/20- and 24/40-glass joints (Aldrich)
2.5 × 20-cm disposable Flex chromatography columns (Kontes)
2.5 × 7.5-cm EMD TLC plates pre-coated with a 250-μm layer of silica gel 60 F254
NMR spectrometer (Bruker)
Rotary evaporator equipped with dry ice condenser and connected to oil pump (Büchi)
Hand-held UV254 lamp (UVP)
Lyophilizer
Prepare the ribonucleoside phosphoramidites
-
1
Place N,N,N′,N′-tetraisopropyl-O-[3-(N,N-dimethylamino)prop-1-yl]phosphordiamidite (1, 2.0 mmol) or N,N,N′,N′-tetraisopropyl-O-[octan-1-yl]phosphordiamidite (2, 2.0 mmol) into a 50-mL round-bottom flask and add a magnetic stir bar. Seal the flask with a rubber septum.
-
2
Under an argon atmosphere, add 20 mL of anhydrous acetonitrile using a 10-mL glass syringe. Stir the solution with a magnetic stirrer.
-
3
While stirring the solution, add vacuum-dried 5′-O-(4,4′-dimethoxytrityl)-2′-O-methyluridine (1.0 mmol) dissolved in 2.2 mL of 0.45 M 1H-tetrazole (1.0 mmol) in MeCN using a 3-mL syringe.
Other activators including 5-ethylthio-1H-tetrazole or 5-benzylthio-1H-tetrazole could be used as a safer alternative, but have not been tested. -
4
Carefully transfer ~0.5 mL of the reaction mixture by syringe to a dry 5-mm NMR tube. Monitor the progress of the reaction in the NMR tube by 31P NMR spectroscopy. Upon completion of the reaction, as seen by the disappearance of 1 or 2, return the NMR sample to the main reaction mixture before proceeding to the next step.
The 31P NMR signal corresponding to 1 or 2 (δP 123 ppm) disappears within 6 or 18 hr at ambient temperature and thus indicates complete phosphitylation of the 3′-hydroxyl group in ribonucleoside. -
5
Add 1 mL of triethylamine and immediately concentrate the reaction mixture to a syrup using a rotary evaporator connected to a vacuum pump.
Purify and isolate the ribonucleoside phosphoramidites
-
6
Suspend the crude product in ~3 mL of 95:5 (v/v) hexane/triethylamine and spread the suspension on the top of a 2.5-cm × 20-cm disposable Flex chromatography column containing ~25 g silica gel that has been equilibrated in 95:5 (v/v) hexane/triethylamine (APPENDIX 3E).
-
7
Elute the column using a gradient of dichoromethane (0 → 95%) in 95:5 (v/v) hexane/triethylamine and collect 6 mL fractions.
-
8
Analyze the fractions by TLC (APPENDIX 3D) on a 2.5 × 7.5-cm EMD silica gel 60 F254 TLC plate using 9:1 (v/v) benzene/triethylamine as the eluent. Pool appropriate fractions in a 250-mL round-bottom flask and rotoevaporate under reduced pressure (~20 mmHg) until a white amorphous solid is obtained.
TLC analysis of diastereomeric 3 or 4 reveals two tight spots; the Rf of each pair of spots is ~0.6 or ~0.8, respectively, when9:1(v/v) benzene/triethylamine is used as the eluent. -
9
Dissolve the solid in ~5 mL anhydrous benzene and add the solution using a 10-mL pipette, at the rate of ~ 1 mL/min, to ~100 mL vigorously stirred hexane in a 250-mL round-bottom flask placed in a water bath kept at ~4°C.
-
10
Allow the suspension to settle and carefully decant off most (~95%) of the supernatant.
-
11
Rotoevaporate the wet material to dryness under reduced pressure and then dissolve in ~10 mL anhydrous benzene.
-
12
Pour the solution into a 50-mL round-bottom flask, freeze the solution in a dry ice/acetone bath and lyophilize under high vacuum to afford triethylamine-free 3 (0.85 mmol) or 4 (0.80 mmol) as a white powder in a yield of 85% or 80%, respectively.
5′-O-(4,4′-dimethoxytrityl)-3′-O-[(N,N-diisopropylamino)(3-[N,N-dimethylamino]prop-1-yl)oxy]phosphinyl-2′-O-methyluridine (3): 31P NMR (121 MHz, C6D6): δ 149.5, 149.1 ppm. +HRMS; calcd for C42ClH57N4O9P [M + Cl]− 827.355, found 827.351.5′-O-(4,4′-Dimethoxytrityl)-3′-O-[(N,N-diisopropylamino)(octan-1-yl)oxy]phosphinyl-2′-O-methyuridine (4). 31P NMR (121 MHz, C6D6): δ 148.6, 148.3 ppm. +HRMS; calcd for C45ClH62N3O9P [M + Cl]− 854.391, found 854.393.
Support Protocol 1. SYNTHESIS OF N,N,N′,N′-TETRAISOPROPYLPHOSPHORDIAMIDITES 1 AND 2
This protocol outlines the reaction of commercial bis(N,N-diisopropylamino)chlorophosphine with commercial 3-(N,N-dimethylamino)propan-1-ol or 1-octanol in the presence of triethylamine to provide the phosphordiamidites 1 or 2 (see Fig. 4.xx.2).
Figure 4.xx.2.
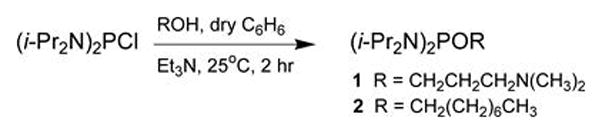
Preparation of the phosphordiamidites 1 and 2. i-Pr = isopropyl; Et3N = triethylamine.
Additional Materials (also see Basic Protocol 1)
bis(N,N-diisopropylamino)chlorophosphine (Aldrich)
3-(N,N-Dimethylamino)propan-1-ol (Aldrich)
1-Octanol (Aldrich)
1-mL glass syringes (Kimble)
Pasteur pipets (VWR)
Büchner funnel setup with vacuum source (Kontes)
Vacuum desiccator (Fisher Scientific)
High-vacuum oil pump (Edwards)
Prepare phosphordiamidites
-
1
Place 1.00 g (3.70 mmol) of bis(N,N-diisopropylamino)chlorophosphine and a magnetic stir bar in an oven-dried 50-mL round-bottom flask. Seal the flask with a rubber septum.
-
2
Using a 10-mL glass syringe, add 20 mL of anhydrous benzene under an argon atmosphere. While stirring the suspension with a magnetic stirrer add, consecutively, 436 μL (3.70 mmol) of 3-(N,N-dimethylamino)propan-1-ol or 583 μL (3.70 mmol) of 1-octanol and 1.00 mL (7.17 mmol) of triethylamine using 1-mL syringes. Stir the reaction mixture under argon at ~25°C until the reaction is complete.
Monitor the progress of the reaction by 31P NMR spectroscopy. The formation of 1 or 2 (singlet, 123.2 ppm, downfield relative to a phosphoric acid external standard) is complete within 2 hr at ~25°C.
Purify and isolate phosphordiamidites
-
3
Using a Pasteur pipette, the entire reaction mixture is loaded on the top of a 2.5-cm × 20-cm disposable Flex chromatography column containing ~15 g silica gel that has been equilibrated in 9:1 (v/v) benzene/triethylamine.
-
4
Elute the column with 100 mL 9:1 (v/v) benzene/triethylamine. Collect the eluate in a 250-mL round-bottom flask.
-
5
Rotoevaporate the volatiles under reduced pressure (~20 mmHg) to a viscous oil.
-
6
Dissolve the oily material in a 50-mL round bottom flask with 10-mL of anhydrous benzene.
-
7
While swirling the flask in a dry ice-acetone bath, freeze the solution. Lyophilize under high vacuum to give the phosphordiamidite 1 or 2 as a viscous oil.
N,N,N′,N′-Tetraisopropyl-O-[3-(N,N-dimethylamino)prop-1-yl]phosphordiamidite (1): Yield (1.01 g, 3.03 mmol, 82%). 1H NMR (300 MHz, C6D6): δ 3.70 (dt, J = 7.3, 6.4 Hz, 2H), 3.55 (sept, J = 6.8 Hz, 2H), 3.52 (sept, J = 6.8 Hz, 2H), 2.36 (t, J = 7.1 Hz, 2H), 2.11 (s, 6H), 1.77 (quint, J = 6.8 Hz, 2H), 1.25 (d, J = 6.8 Hz, 12H), 1.20 (d, J = 6.8 Hz, 12H). 13C NMR (75 MHz, C6D6): δ 62.8 (d, 2JCP = 21.8 Hz), 56.9, 45.6, 44.6 (d, 2JCP = 12.6 Hz), 30.4 (d, JCP = 8.6 Hz), 24.8 (d, JCP = 8.6 Hz), 24.1 (d, JCP = 5.6 Hz). 31P NMR (121 MHz, C6D6): δ 123.3.N,N,N′,N′-Tetraisopropyl-O-[octan-1-yl]phosphordiamidite (2): Yield (1.17 g, 3.24 mmol, 88%). 1H NMR (300 MHz, C6D6): δ 3.65 (dt, J = 7.1, 6.4 Hz, 2H), 3.57 (sept, J = 6.8 Hz, 2H), 3.53 (sept, J = 6.8 Hz, 2H), 1.65 (dq, J = 6.6, 6.4 Hz, 2H), 1.43 (m, 2H), 1.34–1.18 (br m, 8H), 1.27 (d, J = 6.8 Hz, 12H), 1.22 (d, J = 6.8 Hz, 12H), 0.89 (dd, J = 6.8, 6.4 Hz, 3H). 13C NMR (75 MHz, C6D6): δ 64.7 (d, 2JCP = 21.8 Hz), 44.6 (d, 2JCP =12.6 Hz), 32.2, 32.1 (d, JCP = 8.6 Hz), 29.8, 29.7, 26.6, 24.8 (d, JCP = 8.5 Hz), 24.1 (d, JCP =5.6 Hz), 23.1, 14.3. 31P NMR (121 MHz, C6D6): δ 123.2.The phosphordiamidites 1 and 2 are of sufficient purity to be used without further purification in the preparation of deoxyribonucleoside phosphoramidites 3 and 4.
BASIC PROTOCOL 2. SOLID-PHASE SYNTHESIS OF THE CHIMERIC TRANS-ACTING POLY-2′-O-METHYLURIDYLIC THIOPHOSPHATE TRIESTER ELEMENT (2′-OMeUtaPS)
The chemical structure of 2′-OMeUtaPS is presented in Fig. 4.xx.3. This protocol delineates the solid-phase synthesis and characterization of this amphiphilic phosphorothioate RNA sequence.
Figure 4.xx.3.

Chemical structure of the chimeric trans-acting amphipathic poly-2′-O-methyl uridylic thiophosphate triester element (2′-OMeUtaPS) shown in its protonated state under physiological conditions.
Materials
5′-O-(4,4′-Dimethoxytrityl)-3′-O-[(N,N-diisopropylamino)(3-[N,N-dimethylamino] prop-1-yl)oxy]phosphinyl-2′-O-methyluridine (3, see Support Protocol 1)
5′-O-(4,4′-Dimethoxytrityl)-3′-O-[(N,N-diisopropylamino)(octan-1-yl)oxy]phosphinyl-2′-O-methyluridine (4, see Support Protocol 1)
-
Reagents for automated solid-phase oligonucleotide synthesis (Glen Research):
Activator solution: 1H-tetrazole in acetonitrile
Anhydrous acetonitrile
Cap A solution: acetic anhydride in THF/pyridine
Cap B solution: 1-methylimidazole in THF
Deblocking solution: trichloroacetic acid (TCA) in dichloromethane (CH2Cl2)
Succinylated long chain alkylamine controlled-pore glass (CPG) support loaded with 0.2 μmol 5′-O-(4,4′-dimethoxytrityl)-thymidine
Sulfuration solution: 0.05 M 3-(dimethylaminomethylidene)amino-3H-1,2,4-dithiazole-3-thione in (2:3 v/v) pyridine/MeCN
Methylamine gas cylinder (Aldrich)
Triethylamine (Aldrich)
1-mL glass syringe (Kimble)
1.5-mL microcentrifuge tubes (Thomas Scientific)
DNA/RNA synthesizer (e.g., 394 DNA/RNA synthesizer, Applied Biosystems)
Stainless-steel pressure vessel equipped with a valve system (Parr Instrument)
High-vacuum oil pump (Edwards)
UV/vis spectrophotometer (Agilent Technologies)
HPLC instrument (Agilent Technologies)
Synthesize the chimeric 2′-OMeUtaPS RNA sequence
-
1
Perform a 0.2-μmol scale solid-phase synthesis of the chimeric 2′-OMe RNA phosphorothioate sequence (Fig. 4.xx.3) using an Applied Biosystems 394 DNA/RNA synthesizer in “trityl-on” mode according to the manufacturer’s recommendations (see also APPENDIX 3C).
The 2′-O-methyl ribonucleoside phosphoramidites 3 and 4 are used as 0.1 M solutions in dry acetonitrile. All ancillary reagents required for the automated preparation of 2′-OMeUtaPS have been purchased from Glen Research and used as recommended by the manufacturer.The synthetic cycle recommended for the preparation of 2′-OMeUtaPS differs from the conventional cycle used for synthesis of unmodified oligonucleotides in that the “capping step” is performed after the oxidative sulfuration reaction (Iyer et al., 1990). The reaction time of each of the following synthesis cycle steps has been extended to ensure optimal production of 2′-OMeUtaPS: (i) 5′-deblocking reaction (3% TCA in CH2Cl2, 60 sec); (ii) 1H-tetrazole-assisted phosphoramidite coupling reaction (600 sec); (iii) oxidation reaction (sulfuration solution, 600 sec); (iv) capping reaction (Cap A and Cap B solutions, 120 sec). Upon complete assembly of 2′-OMeUtaPS, spectrophotometric (498 nm) measurements of the 4,4′-dimethoxytrityl cation, released from the final synthesis cycle, revealed an overall synthesis yield of 92 ± 5%.
Cleave 2′-OMeUtaPS from the CPG solid support
-
2
Place the 2′-OMeUtaPS synthesis column into a stainless steel pressure vessel. Connect the valve system of the vessel to a high vacuum oil pump. Turn on the valve and evacuate the vessel over a period of 5 min. Turn off the valve system and disconnect it from the vacuum pump.
-
3
Connect the pressure vessel to a methylamine gas cylinder through the vessel’s valve system. Turn on the valves of both the gas cylinder and vessel. Fill the pressure vessel to maximum gas pressure (~2.5 bar, 25°C).
-
4
Expose the 2′-OMeUtaPS synthesis column to pressurized methylamine gas for 3 min. Turn off the valves of both the gas cylinder and vessel. Disconnect the gas cylinder from the vessel and open the valve system of the vessel to release excess methylamine gas. Connect the valve system of the vessel to a high vacuum oil pump. Evacuate the vessel over a period of 2 min and remove the vacuum pump.
Do not use an aqueous methylamine or ammonia solution to release2′-OMeUtaPSfrom the solid support as it may result in partial internucleotidic chain cleavage of the chimeric RNA sequence. -
5
Using a 1-mL glass syringe, elute 2′-OMeUtaPS from the synthesis column with 2 mL of 1:60:39 (v/v/v) triethylamine/acetonitrile/water. Collect the eluate in two 1.5-mL microcentrifuge tubes.
-
6
Evaporate the eluate of each tube to about half its volume at ambient temperature using a stream of air. Combine the eluate into one 1.5-mL micocentrifuge tube.
-
7
Measure the concentration of the 2′-OMeUtaPS solution using a UV/vis spectrophotometer at 260 nm. Evaporate the solution to dryness using a stream of air. Store the dried chimeric RNA sequence at −20°C.
2′-OMeUtaPS: +ESI-MS: calcd for C127H205N19O54P7S7 [M + 2H]+2 corresponding to a calculated mass of 3301.0, found 3300.9.
Support Protocol 2. GENERAL PROCEDURE FOR THE FORMATION OF PNA OR PMO SEQUENCE:2′-OMeUtaPS COMPLEXES
Mixing 2′-OMeUtaPS with commercial PNA or PMO sequences (Fig. 4.xx.4) results in the formation of complexes, which are needed to demonstrate cellular uptake and bioactivity of the PNA and PMO sequences in HeLa pLuc 705 cells or myotube muscle cells.
Figure 4.xx.4.

Commercial PNA and PMO sequences needed to demonstrate the 2′-OMeUtaPS-assisted delivery and bioactivity of these sequences in live mammalian cells.
Additional Materials (also see Basic Protocol 2)
PMO sequences 5–7 and 10–14 (GeneTools, LLC, Fig. 4.xx.4)
PNA sequences 8, 9 and 15 (PNA Bio, Inc., Fig. 4.xx.4)
2′-OMe RNA phosphorothioate sequence 16 (Donated by K. Nagaraju, Children’s National Health System, Washington, D.C.)
2′-OMeUtaPS (see Basic Protocol 2)
OptiMEM (Life Technologies)
Pipettor (Corning)
20- and 1000-μl pipet tips (Fisher Scientific)
Water bath set at 37°C (Thermo Scientific)
Prepare a 2×stock solution of PNA or PMO sequence:2′-OMeUtaPS complexes
-
To a 1.5-mL microcentrifuge tube, containing 20 μL OptiMEM, add 1 μL of 100 μM PNA or 100 μM PMO stock solution using a pipettor.
Each commercial PNA or PMO sequence is reconstituted in an appropriate volume of sterile water to provide a 100 μM PNA or PMO stock solution. -
Dissolve 2′-OMeUtaPS (Basic Protocol 2, step 7) in a solution of 1:60:39 (v/v/v) triethylamine/acetonitrile/water to provide a 100 μM 2′-OMeUtaPS stock solution.
This stock solution must be used immediately after preparation and be freshly prepared for any subsequent experiment. Pipet 2 μL 100 μM 2′-OMeUtaPS stock solution and add it to the oligomer solution of step 1 followed by 27 μL of OptiMEM to produce a 2× stock solution of PNA or PMO sequence:2′-OMeUtaPS complexes i.e., 2 μM PNA or PMO sequence and 4 μM 2′-OMeUtaPS in 50 μL OptiMEM.
-
Incubate the solution at 37°C for 30 min. Store the 2× stock solution of PNA or PMO sequence:2′-OMeUtaPS complexes at 4°C for 15 min prior to immediate use in cellular uptake experiments.
When used in cellular uptake experiments, this 2× stock solution of the complexes will provide an extracellular concentration of 1.0 μM in PNA or PMO sequence and 2.0 μM in 2′-OMeUtaPS (see Basic protocol 3).
BASIC PROTOCOL 3. 2′-OMeUtaPS-MEDIATED CELLULAR INTERNALIZATION OF PNA OR PMO SEQUENCES IN HeLa pLuc 705 CELLS
This protocol details the 2′-OMeUtaPS-mediated cellular internalization of fluorescently-labelled PMO sequence 6 or PNA sequence 8 in live HeLa pLuc 705 cells. Fig. 4.xx.5 shows the histogram of the cellular uptake of the PMO sequence 6:2′-OMeUtaPS and PNA sequence 8:2′-OMeUtaPS complexes in the above-mentioned cell line, as analysed by fluorescence-activated cell sorting (FACS) flow cytometry. Fig. 4.xx.6 provide FACS evidence that the extension of PMO sequences with a short PMO-polyA stretch is necessary and sufficient for 2′-OMeUtaPS-mediated internalization in HeLa pLuc 705 cells.
Figure 4.xx.5.
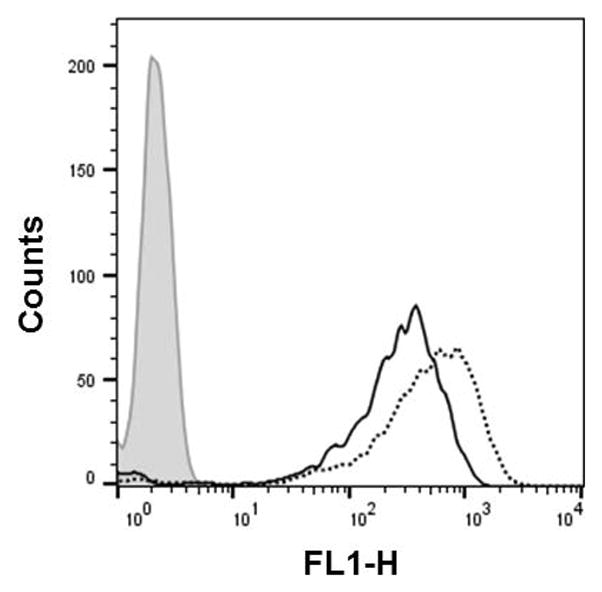
Flow cytometry analysis of the 2′-OMeUtaPS-assisted cellular uptake of the control PMO sequence 6 (peak area with solid black border) and PNA sequence 8 (peak area with a dotted black border) in HeLa pLuc 705 cells. Pale gray peak area, untreated HeLa pLuc 705 cells.
Figure 4.xx.6.
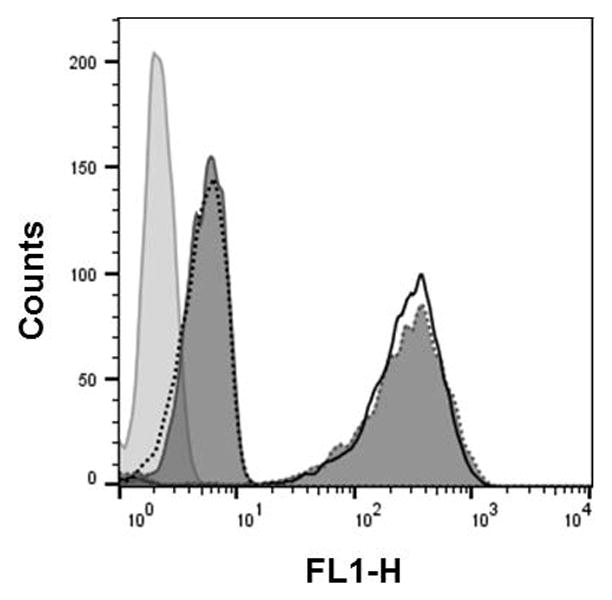
Flow cytometry analysis of the 2′-OMeUtaPS-assisted cellular uptake of the PMO sequence 5 (peak area with a solid black border), control PMO sequence 6 (dark gray peak area with a dotted border), PMO sequence 12 (peak area with a black dotted border) and PMO sequence 13 (dark gray peak area with solid border) in HeLa pLuc 705 cells. Pale gray peak area accounts for untreated HeLa pLuc 705 cells.
Materials
HeLa pLuc 705 cells (donated by Professor Rudolph Juliano,
University of North Carolina, Chapel Hill School of Medicine, arjay@med.unc.edu)
Dulbecco’s minimum essential medium (DMEM) supplemented with 10% or 20% heat-inactivated fetal bovine serum (10% FBS-DMEM and 20% FBS-DMEM) (see Reagents and Solutions UNIT 4.69)
Dulbecco’s Phosphate-buffered saline, pH 7.4, supplemented with 2% fetal bovine serum (Life Technologies)
1× Phosphate Buffered Saline pH 7.4 (PBS, ThermoFisher)
0.25% Trypsin (MediaTech)
Ice-cold 0.4% Trypan blue (MediaTech)
Monensin (Sigma)
100× L-glutamine (Life Technologies)
100× penicillin-streptomycin (Life Technologies)
100 mM sodium pyruvate (Life Technologies)
50 mg/mL hygromycin B (Life Technologies)
75-cm2 flask (Corning)
100- and 1000-μl pipet tips (Fisher Scientific)
Flat-bottom, 96-well tissue-culture plates (BD-Falcon)
FACS tubes (Falcon)
1.5-mL microcentrifuge tubes (Thomas scientific)
Pipettor (Corning)
Cellometer Auto T4 Cell Viability Counter (Nexcelom)
37°C, 5% CO2 humidified incubator (Thermo Scientific)
Vacuum aspirator (VWR)
FACScan flow cytometer (Becton Dickinson)
Culture cells
-
1
HeLa pLuc 705 (1×106 cells) are allowed to grow at 37°C in a 75-cm2 flask containing 15 mL of 10% FBS-DMEM in a 37°C, 5% CO2 humidified incubator until cells are 70% confluent.
-
2
Vacuum-aspirate the medium. Wash the cells with 5 mL PBS using a pipettor. Vacuum-aspirate the PBS.
-
3
Detach cells from the flask by adding 2 mL of 0.25% Trypsin using a pipettor. Incubate at 37°C for 3 to 5 min in a 5% CO2 humidified incubator. Pipet 3 mL of 10% FBS-DMEM to the cell suspension and transfer 100 μL of the suspension to a 1.5-mL microcentrifuge tube using a pipettor.
-
4
Count cells using a Cellometer T4 cell viability counter according to the manufacturer’s instructions.
Plate cells
-
5
In a flat-bottom tissue culture 96-well plates, add 2 × 104 cells/well of HeLa pLuc 705 using a pipettor and allow the cells to grow in 100 μL of 10% FBS-DMEM for 24 hr in a 37°C, 5% CO2 humidified incubator.
Internalize the PNA or PMO sequence:2′-OMeUtaPS complexes in the cells
-
6
Remove the culture medium from all wells using a vacuum aspirator. Pipet into each well 50 μL of 20% FBS-DMEM followed by 50 μL of 2× stock solution of PMO sequence 6:2′-OMeUtaPS or PNA sequence 8:2′-OMeUtaPS or PMO sequence 5, 12 or 13:2′-OMeUtaPS complexes. Incubate the cell cultures at 37°C for 18 hr in a 37°C, 5% CO2 humidified incubator.
The final concentration of PMO sequence:2′-OMeUtaPS or PNA sequence:2′-OMeUtaPS complexes in each well should be 2 μM in 2′-OMeUtaPS and 1.0 μM in PMO or PNA sequences. -
7
Remove the medium from each well by vacuum-assisted suction. Pipet 50 μL of 0.25% Trypsin into each well using a pipettor.
Complete cell detachment occurs within 5–10 min at37°C. -
8
Pipet into each well 100 μL of ice-cold Dulbecco’s Phosphate Buffered Saline (pH 7.4) supplemented with 2% Fetal Bovine Serum.
Perform FACS analyses
-
9
Pipet the cells of each well into individual FACS tubes followed by 50 μL of 0.4% Trypan blue and 20 μL of 200 μM monensin using a pipettor. Place the tubes in an ice-bath and then perform analysis of each tube by flow cytometry using a FACScan™ flow cytometer according to the manufacturer’s recommendations.
Cell analyses should be performed within 1 h in order to prevent production of misleading results caused by cell death. A total of 5,000 events are counted; the results of the FACS analysis are reported as a percentage of the cells that have internalized the fluorescently-labelled sequences. The extent of 2′-OMeUtaPS-mediated cellular uptake of PMO or PNA sequences in HeLa pLuc 705 cells is presented in Figs 4.xx.5 and 4.xx.6, respectively.
BASIC PROTOCOL 4. LUCIFERASE ASSAY FOR DETERMINING THE BIOACTIVITY OF PNA OR PMO SEQUENCES IN HeLa pLUC 705 CELLS
HeLa cells, that are stably transfected with the recombinant plasmid pLuc 705 carrying a luciferase gene interrupted by a mutated β-globin intron, exhibit aberrant splicing of luciferase pre-mRNA, which prevents translation of functional luciferase (Kang et al., 1998). However, treatment of HeLa pLuc 705 cells with PNA sequence 9:2′-OMeUtaPS or PMO sequence 10:2′-OMeUtaPS complexes, which is complementary to the mutated splice site, induces correct splicing and restoration of luciferase activity (Fig. 4.xx.7). Thus, HeLa pLuc 705 cells provide a functional assay for evaluating the efficiency and functionality of 2′-OMeUtaPS-mediated internalization of PNA and PMO sequences through quantitative measurements of luciferase production.
Figure 4.xx.7.
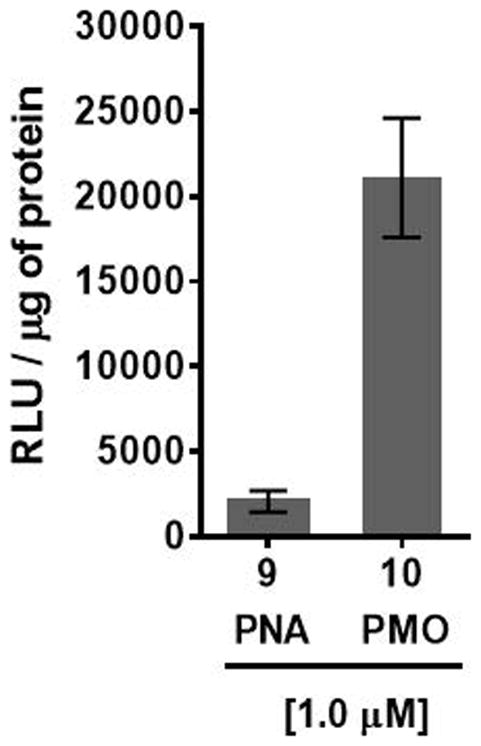
2′-OMeUtaPS-mediated delivery of polyA-tailed PNA and PMO sequences 9 and 10, respectively, in HeLa pLuc 705 cells cultured in serum-containing medium. The concentration of each sequence is kept at 1.0 μM whereas the concentration of 2′-OMeUtaPS is kept at 2.0 μM. Error bars represent the mean ± SD of three independent experiments. RLU = relative light units.
Materials
HeLa pLuc 705 cells (donated by Professor Rudolph Juliano, University of North Carolina, Chapel Hill School of Medicine, arjay@med.unc.edu)
Fetal bovine serum (FBS, Life Technologies)
DMEM (Life Technologies)
Serum-free medium (OptiMEM, Life Technologies)
2× stock solution of the 2′-OMeUtaPS:PNA sequence 9 and PMO sequence 10:2′-OMeUtaPS complexes (see Support Protocol 2)
2×Pierce luciferase cell lysis buffer (ThermoFisher Scientific)
Bright-Glow reagent (Promega)
Pierce Coomassie (Bradford) protein assay kit (ThermoFisher Scientific)
Black 96-well plate (Corning)
1.5-mL microcentrifuge tubes (Thomas Scientific)
White 96-well plate (Corning)
Vacuum aspirator (VWR)
37°C, 5% CO2 humidified incubator (Thermo Scientific)
Mechanical shaker (ThermoFisher Scientific)
Luminescence microplate reader (FilterMax F3 Multi-Mode microplate reader, Molecular Devices)
Pipettor (Corning)
Plate HeLa pLuc 705 cells and perform cellular internalization of PNA and PMO oligomers
-
1
In a black 96-well plate, allow 2 × 104 cells/well of HeLa pLuc 705 to grow in 100 μL of 10% FBS-DMEM at 37°C for 18 hr in a 37°C, 5% CO2 humidified incubator.
-
2
Remove the culture medium of each well by vacuum-assisted suction. Pipet 50 μL of 20% FBS-OptiMEM into each well.
Experiments are performed in triplicates. -
3
Using a pipettor, add 50 μL of 2× stock solution of PNA sequence 9:2′-OMeUtaPS or PMO sequence 10:2′-OMeUtaPS complexes in OptiMEM to the cells of each well. Incubate the plate at 37°C for 4 hr.
The extracellular concentration of the PNA or PMO sequence and 2′-OMeUtaPS is 1.0 μM and 2.0 μM, respectively. -
4
Remove the culture medium from each well using a vacuum aspirator.
Lyse the cells and perform the luciferase assay
-
5
Pipet 50 μL of Pierce luciferase cell lysis buffer to the cells of each well using a pipettor. Using a mechanical shaker, agitate the 96-well plate at 5°C for 10 min.
-
6
Transfer 30 μL of cell lysate from each well to individual wells of a white 96-well plate using a pipettor. Pipet 20 μL of Bright-Glow reagent into each well. Measure immediately luciferase activity using a luminescence microplate reader.
For each well, luminescence is integrated over a period of 1 sec and recorded as relative light units (RLU).Bright Glow reagent, that has been stored frozen at −20°C for less than one week or −80°C, should be used to ensure consistent luminescence measurements.Luminescence measurements are reported based on the amount of total protein present in the test sample. -
7
Measure protein concentrations from 5 μL of cell lysates obtained in step 6 using the Pierce Coomassie (Bradford) protein assay kit according to the manufacturer’s instructions.
The luciferase assay results supporting the bioactivity of PNA sequence 9 and PMO sequence 10 in HeLa pLuc 705 cells are shown in Figs 4.xx.7.
BASIC PROTOCOL 5. 2′-O-MeUtaPS-MEDIATED DELIVERY OF ANTISENSE PNA AND PMO SEQUENCES IN mdx MOUSE MYOTUBES
This protocol outlines the procedures for the culture and differentiation of H-2Kb-tsA58 (H2K) mdx mouse myoblasts and 2′-OMeUtaPS-assisted delivery of the PMO sequence 14 and PNA sequence 15 in mdx mouse myotubes.
Materials
H-2Kb-tsA58 (H2K) mdx myoblast cells (Donated by T.A Partridge, Children’s National Health System, Washington, D.C.)
PMO sequence 14 (GeneTools LLC)
PNA sequence 15 (PNA Bio, Inc.)
dTtaPS (UNIT 4.69, Basic Protocol 2)
2′-OMeUtaPS (Basic Protocol 2)
2′-OMe RNA sequence 16 (Donated by K. Nagaraju, Children’s National Health System, Washington, D.C.)
DMEM, high glucose, pyruvate (4500 mg/L glucose, 4 mM Glutamax, 110 mg/L Pyruvate) (Invitrogen/Gibco)
OptiMEM (Life Technologies)
Gelatin (Sigma)
Penicillin/streptomycin (Sigma)
IFN-γ, murine, recombinant (IFN-γ, Invitrogen/Gibco)
Heat inactivated FBS (Invitrogen/Gibco)
Chick embryo extract (US Biological Life Sciences)
Horse serum (ThermoFisher Scientific)
1× Phosphate Buffered Saline pH 7.4 (PBS, ThermoFisher Scientific)
Trypsin EDTA (Sigma)
Lipofectamine®2000 (ThermoFisher Scientific)
Chloroform, Molecular Biology Grade (Fisher Scientific)
Isopropanol, Molecular Biology Grade (Fisher Scientific)
Ethanol, Molecular Biology Grade (Fisher Scientific)
Nuclease-free water (ThermoFisher Scientific)
Vacuum Aspirator system (VWR)
Cellometer Auto T4 Cell Viability Counter (Nexcelom)
25-mm, 0.2μ-syringe sterile filters (MilliporeSigma)
5-, 10-mL plastic pipettes (Fisher Scientific)
15-mL Falcon Centrifuge tubes (Fisher Scientific)
0.5-mL sterile microcentrifuge tubes (Thomas Scientific)
Flat-bottom, 24-well-plates (Corning/Costar)
Pipettor (Corning)
Centrifuge (Beckman Coulter)
37°C-Water bath (Thermo Scientific)
Vacuum aspirator system (Thermo Scientific)
33°C, 10% CO2 humidified incubator (VWR)
Culture myogenic H-2Kb-tsA58 (H2K) cells
These myogenic cells harbors a temperature sensitive immortalising T-antigen gene (tsA58) under the control of a gamma interferon inducible MHC Class I promoter (H-2Kb). These cells either exhibit continuous mitosis or alternatively terminally differentiate into myotubes depending on culture conditions (growth and proliferation at 33°C and differentiation at 37°C).
-
1
Coat a T-75 flask with 0.4% gelatin so that it covers its entire surface. Place the flask in an incubator kept at 33°C for 30 min.
0.4% gelatin is prepared by mixing 0.4 g of gelatin in 100 mL water. The mixture is autoclaved and then stored at 4°C until needed. -
2
Remove the excess gelatin from the flask using a vacuum aspirator and immediately pipet into the flask 10 mL of a culture medium composed of DMEM supplemented with 20% heat inactivated FBS, 2% chick embryo extract, 1% penicillin/streptomycin (100 U/mL) and 20 μL of a freshly prepared stock solution of 10 μg/mL IFN-γ. Place the flask into a 33°C-incubator for 30 min to warm up the culture medium.
100 μg of IFN-γ is placed in a 25-mL plastic beaker. Add 100 μL of PBS using a pipettor. 10 mL of DMEM high glucose pyruvate supplemented with 2% chick embryo extract and 1% penicillin/streptomycin (100 U/mL) is added to the beaker, using a plastic 10-mL pipette, to provide a 10 μg/mL stock solution of IFN-γ. The solution is sterile-filtered and aliquoted into 0.5-mL sterile microcentrifuge tubes for long-term storage at − 80°C. -
3
Quickly thaw a vial containing ~1×106 of frozen H2K cells in a 37°C water bath. Pipet 4 × 105 cells into the gelatin-coated flask containing the pre-warmed culture medium (step 2). Incubate the flask overnight at 33°C and 10% CO2 to allow cell adherence to the surface of the flask.
H2K cells should be counted prior to be frozen; this will ensure more accurate post-thawing cell number measurements. -
4
Verify cell attachment and whether the cells are visually less than 70% confluent. Remove the medium using a vacuum aspirator and pipet 10-mL of fresh culture medium into the gelatin-coated flask using a 10-mL pipette.
Removal of the culture medium is necessary to eliminate residual dimethyl sulfoxide (DMSO) from the thawed cells. -
5
Allow cells to proliferate at 33°C and 10% CO2 until the cells are visually ~60–70% confluent.
Cells need to be passaged before they reach 70% confluency;this will take ~ 1 week at the rate of three passages per week. -
6
Vacuum-aspirate the culture medium. Wash the cell with 10-mL 1× PBS using a 10-mL pipette.
-
7
Vacuum-aspirate the PBS. Pipet 3 mL of 0.25% Trypsin-EDTA into the flask using a 5-mL pipette. Incubate at 33°C for 5 min.
Check whether the cells are detached from the surface of the flask. If not, place the flask in the 33°C-incubator for an additional 2 min. Repeat until the cells are completely detached from the flask. -
8
Deactivate trypsin by pipetting 9 mL of warm (33°C) culture medium into the flask using a 10-mL pipette. Transfer the cell suspension to a 15-mL centrifuge tube using a 10-mL pipette.
-
9
Pellet the cells by centrifugation at 250 × g for 5 min at ~25°C. Remove carefully the culture medium using a 10-mL pipette without disturbing the cell pellet.
-
10
Re-suspend the cell pellet in 2 mL of culture medium. Count the cells using a cell counter according to the manufacturer’s instructions.
-
11
Using a pipettor, pipet an appropriate volume of culture medium to the cells obtained in step 10 to provide a cell density of 52×103 cells/mL.
Differentiate H2K cells
-
12
Coat a 24-well plate with 0.4% gelatin so as to cover the entire surface area of the wells. Place the plate into an incubator kept at 33°C for 30 min.
-
13
Vacuum-aspirate the gelatin and add ~26 × 103 cells (0.5 mL of the cells obtained in step 11) per well of the 0.4% gelatin-coated 24-well plate using a pipettor. Incubate the plate at 33°C and 10% CO2 over a period up to 24 hr.
-
14
Place a differentiation medium and PBS in a 37°C-water bath.
The differentiation medium is composed of DMEM supplemented with 5% Horse serum and 1% Penicillin/streptomycin. -
15
Vacuum-aspirate the culture medium from the 0.4% gelatin-coated 24-well plate. Using a pipettor, wash cells with 500 μL 1× PBS/per well. Vacuum-aspirate the PBS from each well.
-
16
Add 0.5-mL of pre-warmed differentiation medium per well of the 0.4% gelatin-coated 24-well plate using a pipettor. Place the plate in a 37°C-incubator and 5% CO2 for five days to promote differentiation.
Within the next 24 hours, cells will begin to line up and fuse to form myotubes. The differentiation medium should be replaced with fresh medium every 3 days. However, myotubes become increasingly fragile after 5 days in culture; care should therefore be exercised when refreshing the differentiation medium beyond 5 days of culture.
Prepare PNA or PMO sequence:2′-OMeUtaPS complexes
-
17
To a 1.5-mL microcentrifuge tube containing 20 μL OptiMEM, add 3 μL of a 100 μM PNA sequence 15 or 100 μM PMO sequence 14 stock solution (Basic Protocol 2, annotation of step 1) using a pipettor.
-
18
Prepare a 1 mM 2′-OMeUtaPS stock solution in 1:60:39 (v/v/v) triethylamine/acetonitrile/water. Add 0.6 μL of this solution to PNA sequence 15 or PMO sequence 14 solution of step 17 followed by 27 μL of OptiMEM using a pipettor.
-
19
Repeat step 4 of Support Protocol 2 prior to immediate use in myotube transfection.
When used in cellular uptake experiments, this 2× stock solution of the complexes will provide an extracellular concentration of 1.0 μM PNA or PMO sequence and 2.0 μM in 2′-OMeUtaPS.
Transfect myotubes
-
20
On day 5 of the myoblast differentiation culture (Step 16), remove the medium of each well of the plate by vacuum-assisted suction. Using a pipettor, add successively 150 μL of DMEM containing 10% Horse serum and 150 μL of 2× stock solution of the PNA or PMO sequence:2′-OMeUtaPS complexes (Step 19). Incubate in a 37°C-incubator and 5% CO2 for 24 hr.
Transfection of the positive control 2′-O-methyl RNA sequence 16 has been performed using Lipofectamine®2000, as a transfection reagent, in serum free-medium at a concentration recommended by the supplier.
BASIC PROTOCOL 6. A QUALITATIVE NESTED-PCR ANALYSIS OF THE EXCISION OF EXON 23 FROM DYSTROPHIN PRE-mRNA UPON 2′-O-MeUtaPS-MEDIATED DELIVERY OF ANTISENSE PNA AND PMO SEQUENCES IN mdx MOUSE MYOTUBES
This protocol provides the details of a nested-PCR method for qualitative analysis of exon 23 skipping induced by the 2′-OMeUtaPS-mediated delivery of PNA sequence 15 and PMO sequence 14 in mdx mouse myotubes.
Materials
High Capacity cDNA Reverse Transcription Kit (Life Technologies)
Platinum TaqPCRx DNA Polymerase (Life Technologies)
Nuclease-free water (ThermoFisher Scientific)
-
1× Phosphate buffered saline, pH 7.4 (PBS, ThermoFisher)
Outer Forward primer amplifying Exon 20 (5′-TTCTTCAGCTTGTGTCATCC, Integrated DNA Technologies)
Outer Reverse primer amplifying Exon 26 (5′-TTCTTCAGCTTGTGTCATCC, Integrated DNA Technologies)
Inner Forward primer amplifying Exon 20 (5′-CCCAGTCTACCACCCTATCAGAGC, Integrated DNA Technologies)
Inner Reverse primer amplifying Exon 24 (5′-CAGCCATCCATTTCTGTAAGG, Integrated DNA Technologies)
TRIzol reagent (Invitrogen)
Ethanol (Fisher)
Chloroform (Fisher)
Propan-2-ol (isopropanol, Fisher Scientific)
RNase-free water (ThermoFisher)
Agarose (Bio-Rad)
-
1× TAE Running Buffer (Bio-RAD)
40 mM Tris, 20 mM acetic acid, 1 mM EDTA, pH 8.0
-
1× nucleic acid sample buffer (Bio-RAD)
50 mM Tris-HCl, pH 8.0, 25% glycerol, 5 mM EDTA, 0.2% bromophenol blue, 0.2% xylene cyanole FF
Ethidium Bromide Solution (10 mg/mL, Bio-Rad)
500-mL Erlenmeyer (Kimble)
1.5 mL microcentrifuge tube (Thomas Scientific)
0.2-mL PCR 8 strip tube (USA Scientific)
7900HT Fast Real-Time PCR system (Applied Biosystems)
Heat Block (VWR)
Pipettor (Corning)
Vacuum aspirator system (VWR)
Vortex mixer (VWR)
Centrifuge (Eppendorf)
Microwave oven (Panasonic)
Horizontal gel system (Fisher Scientific)
Gel combs (Fisher Scientific)
UV/Vis spectrometer (Agilent Technologies)
Prepare total RNA
-
1
Vacuum-aspirate the medium from each well of the plate (Basic Protocol 5, Step 20). In each well of the plate, sequentially pipet 0.5 mL of PBS using a pipettor, vacuum-aspirate the PBS and pipet 0.5 mL of TRIzol reagent. Pipet-in and expel-out the reagent three times to homogenize the mixture.
Precautions should be taken when vacuum-aspirating medium and PBS to avoid a significant loss of myotubes. -
2
Pool duplicate wells together, so that each sample is in 1 mL of TRIzol. Transfer the content into a 1.5 mL microcentrifuge tube using a pipettor. Vortex to homogenize. Incubate 5 min at ~25°C.
-
3
Pipet 0.2 mL of chloroform in the microcentrifuge tube. Securely cap the tube. Manually shake the tube for 15 sec until the solution turns to a uniform chalky pink color. Store the tube at ~25°C for 3 min.
-
4
Centrifuge at 12,000 × g at 4°C for 15 min. Remove the tube from the centrifuge. Ensure that the mixture has separated into a lower red phenol-chloroform phase, an interphase, and a colorless upper aqueous phase.
RNA remains exclusively in the aqueous phase
Isolate RNA
Always use the appropriate precautions to avoid RNase contamination when preparing and handling RNA. When precipitating RNA from small samples (i.e., <106 cells), it is recommended to add 5–10-μg of RNase-free glycogen, as a carrier, to the aqueous phase. Glycogen co-precipitates with the RNA, but does not interfere at concentrations <4 mg/mL with first-strand cDNA synthesis and does not interfere with PCR. The use of a carrier was not needed in this experiment.
-
5
Pipet 0.5 mL of isopropanol into a new 1.5-mL microcentrifuge tube using a pipettor. Carefully pipet only ~200 μL from the upper aqueous phase of the centrifuged mixture (step 4). Avoid drawing any of the interphase or organic layer. Expel the aqueous solution into the isopropanol-containing microcentrifuge tube.
-
6
Repeat pipetting ~200 μL from the upper aqueous phase of the centrifuged mixture until ~500 μL of the aqueous phase is collected and transferred to the isopropanol-containing microcentrifuge tube. Cap and gently invert the tube five times to ensure mixing. Store the microcentrifuge tube at −20°C overnight to precipitate the RNA.
-
7
Centrifuge the RNA precipitate at 12,000 × g for 10 min at 4°C. Pipet and discard the supernatant using a pipettor without disturbing the RNA pellet.
-
8
Wash the RNA pellet by adding 1 mL of 75% ethanol into the microcentrifuge tube using a pipettor. Slowly pipet-in and expel-out the aqueous ethanol, in order to gently dislodge the RNA pellet.
-
9
Centrifuge at 7,500 × g for 5 min at 4°C. Pipet and discard the supernatant using a pipettor without disturbing the RNA pellet. Draw residual ethanol from the tube by capillarity using a 200-μL pipette tip. Leave the microcentrifuge tube uncapped to air-dry for ~10 minutes at ~25°C.
Do not allow the RNA pellet to dry completely as it can lose solubility -
10
Resuspend the RNA pellet in 20 μL of nuclease-free water by pipetting-in and expelling-out the water until complete solubility is achieved. Heat the solution at 55°C for 10 min using a heat block.
-
11
Measure the RNA concentration at 260 nm using UV/Vis spectrometer. Store the RNA solution frozen at −80°C or proceed to cDNA synthesis immediately.
The total amount of RNA isolated is in the range of 1–2 μg or 50–100 ng/μL.
Prepare primary cDNA products
-
12
Place 350 ng of RNA in 20 μL nuclease-free water into a 0.2-mL PCR 8 strip tube using a pipettor. Add 30 μL of High Capacity cDNA Reverse Transcription Kit, as reported in the manufacturer’s protocol.
Modification of the High Capacity cDNA Reverse Transcription Kit protocol has been made regarding the DNA primers to be used, without changing the metrics; the outer forward and outer reverse primers are forward (5′-CAGAATTCTGCCAATTCGTGAG) and reverse (5′-TTCTTCAGCTTGTGTCATCC) DNA primers for amplification of exons 20 to 26. -
13
Seal and centrifuge the PCR tube (325× g, 15 sec, 25°C) to eliminate air bubbles. Store the tube on ice until ready to initiate reverse transcription.
-
14
Using a 7900HT Fast Real-Time PCR system, reverse-transcribed the RNA mixture with a hot start of 2 min at 94°C followed by 30 cycles, each of which, consisting of 30 sec at 95°C, 1 min at 55°C, 2 min at 72°C. Upon completion of the 30 cycles, store the primary cDNA products at 4°C.
Prepare secondary cDNA products
-
15
Place ~3-μL (~20 ng/μL) of primary cDNA products into a 0.2-mL PCR 8 strip tube using a pipettor. Add 47 μL of High Capacity cDNA Reverse Transcription Kit, as reported in the manufacturer’s protocol, using a pipettor.
Modification of the High Capacity cDNA Reverse Transcription Kit protocol has been made regarding the DNA primers to be used, without changing the metrics; the inner forward and inner reverse primers are forward (5′-CCCAGTCTACCACCCTATCAGAGC) and reverse (5′-CAGCCATCCATTTCTGTAAGG) DNA primers for amplification of exons 20 to 24. -
16
Repeat step 13.
-
17
Using a 7900HT Fast Real-Time PCR system, amplify the single-stranded cDNA products with a hot start of 2 min at 94°C followed by 22 cycles, each of which, consisting of 30 sec at 95°C, 1 min at 55°C, 2 min at 72°C. Upon completion of the 22 cycles store the secondary cDNA products at 4°C.
Analyze the double-stranded cDNA products by electrophoresis on agarose gels
-
18
Mix 8 μL of the amplified cDNA products with 2 μL 5× nucleic acid sample buffer using a pipettor.
-
19
Pipet the cDNA solution obtained from step 18 into a well of a 1.5% agarose gel placed in a horizontal electrophoresis chamber filled with 1× TAE, pH 8.0 buffer. Electrophorese at 100 V until the bromophenol blue marker travels the full length of the gel.
The agarose gel is prepared by mixing 1.5 g of agarose and 100 ml of 1× TAE, pH 8.0 buffer in a 500-mL Erlenmeyer. The Erlenmeyer is heated in a microwave oven until the agarose is completely melted. The agarose solution is allowed to cool for 5 min. 5 μL of a 10 mg/mL solution Ethidium Bromide Solution is added to the melted agarose which is then poured into the horizontal gel electrophoresis apparatus, combed and allowed to gel.The electrophoretogram presented in Fig. 4.xx.8 demonstrates the excision of exon 23 from the mdx mouse dystrophin pre-mRNA
Figure 4.xx.8.
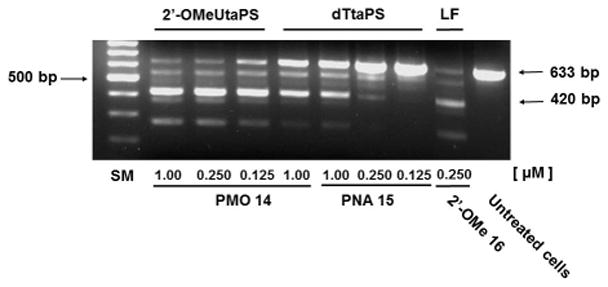
Efficiency of 2′-OMeUtaPS and dTtaPS at inducing the excision of exon 23 from the mdx mouse dystrophin pre-mRNA upon transfection of the PMO or PNA sequence 14 or 15, respectively, in mdx mouse myotubes. The concentration of 2′-OMeUtaPS or dTtaPS is kept at 2 μM in serum containing medium. Lipofectamine®2000 (LF) is used as a transfection reagent in serum free-medium at a concentration recommended by the supplier. Total RNA is extracted from transfected myotubes and amplified by nested RT-PCR using appropriate sets of DNA primers (Basic protocol 6). Two major secondary PCR products have been separated by electrophoresis on a 1.5% agarose gel; the larger 633 bp and shorter 420 bp PCR products correspond to the unspliced and correctly spliced pre-mRNA exon 23, respectively. SM = size marker.
COMMENTARY
Background Information
Duchenne muscular dystrophy (DMD) is a progressive deterioration of muscle function that is predominantly caused by the insertion and/or deletion of a number of nucleotides in the DMD gene encoding the protein dystrophin (Hoffman et al., 1987; Yokota et al., 2007; Aoki et al., 2013; Vila et al., 2015). These mutations cause a shift in the reading frame of the mRNA codons and prevent the correct expression of dystrophin. A viable approach to restoration of the correct mRNA reading frame and production of a functional dystrophin protein would be, as mentioned above, through skipping the exon(s) bearing those genomic mutations (Muntoni and Wood, 2011; Shabanpoor et al., 2015). In this context, the steric interference imparted by RNase H-incompetent oligonucleotide analogues (e.g., uncharged peptide nucleic acids (PNA), phosphorodiamidate morpholino (PMO) sequences or negatively charged 2′-O-methyl RNA phosphorothioates) complementary to specific pre-mRNA splice sites, has been demonstrated to be efficient at redirecting exon splicing during assembly of mature mRNAs (Aartsma-Rus and van Ommen, 2007; Järver et al., 2014; Shabanpoor et al., 2015). Although PMO sequences are more efficient than 2′-O-methyl phosphorothioate RNA sequences at deleting exon 23 from the dystrophin pre-mRNA in the mdx mouse model of muscular dystrophy (Fletcher et al., 2006), internalization of PMO sequences in muscle cells is relatively poor, as low levels of dystrophin production has been reported (Shabanpoor et al., 2015). The conjugation of cationic cell-penetrating peptides (CPP) or a tetraguanidinium-linked nonpeptidic transporter (Hassane et al., 2010; Bhadra et al., 2016) to PMOs has improved cellular uptake of these sequences and induced pre-mRNA splicing correction activities in mammalian cells and animal models (O’Donovan et al., 2015; Bhadra et al., 2016). However, the preparation of PMO-CPP or PMO-tetraguanidinium-linked nonpeptidic conjugate is labor-intensive and, by default, not readily available. Furthermore, CPP-PMO conjugates are sensitive to peptidases (Youngblood et al., 2007) and produce undesirable toxicity (Wu et al., 2012). With the objective of improving the delivery of uncharged nucleic acid sequences in live mammalian cells, amphipathic trans-acting polythymidylic (dTtaPS) and poly-2′-O-methyluridylic (2′-OMeUtaPS) thiophosphate triester elements have recently been implemented (Jain et al., 2015, 2017) for efficient delivery of polyA-tailed PNA and PMO sequences in HeLa pLuc 705 and mdx-mouse myotube cells. The chemical synthesis of dTtaPS or 2′-OMeUtaPS, is easily accomplished using an automated DNA/RNA synthesizer and deoxyribo- or 2′-OMe ribo-nucleoside phosphoramidite monomers functionalized with either a N,N-dimethylaminopropyl or an octyl phosphite triester group (Fig. 4.xx.1). The individual steps of a standard solid-phase synthesis protocol have been optimized to ensure maximal coupling efficiencies (98–99%) of the phosphoramidite monomers and provide an overall synthesis yield of 92 ± 5%, based on the 4,4′-dimethoxytrityl cation assay (Ellington and Pollard, 2000). After release from the CPG support, the identity of diastereomeric dTtaPS or 2′-OMeUtaPS is confirmed by mass spectrometry while the purity of these sequences is determined by C-4 RP-HPLC. As shown in Fig. 4.xx.9, the C-4 RP-HPLC chromatogram of 2′-OMeUtaPS displays a broad product peak accounting for 86% of all peak areas. The broad retention time (12.5 to 15.5 min) and shape of the product peak are consistent with the diastereomeric and amphiphilic properties of the nucleic acid sequence. The purity of 2′-OMeUtaPS is nonetheless acceptable for its intended purpose.
Figure 4.xx.9.
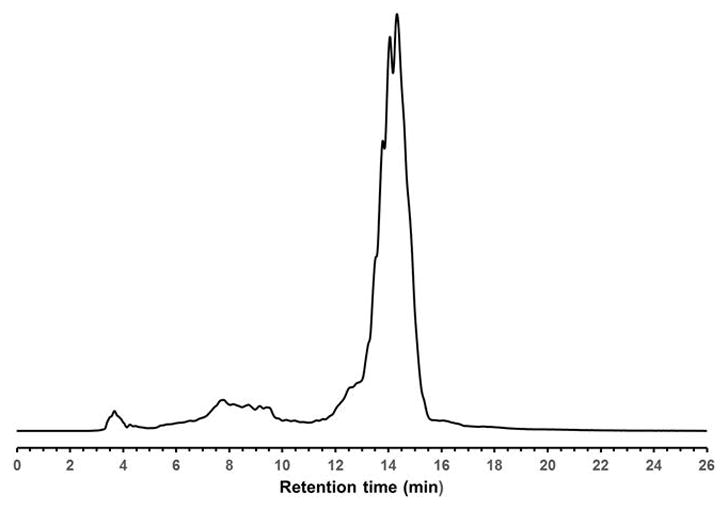
RP-HPLC profile of unpurified of 2′-OMeUtaPS. RP-HPLC analysis is performed employing a 5-μm Phenomenex Jupiter C-4 HPLC Column (25 cm × 4.6 mm) under the following chromatographic conditions: starting from 0.1 M ammonium acetate in 1:1 (v/v) water/acetonitrile, a gradient of acetonitrile is pumped at a flow rate of 1 mL/min to 100% acetonitrile over a period of 20 min.
The selection of the P(III) ester functional groups is based on the cellular uptake of an uncharged thermosensitive DNA sequence, which had been performed earlier (Jain et al., 2013) in mammalian cells (e.g., Vero, GC-2 and HeLa). This experiment has led to the discovery of replacing four neutral thiophosphate triester functions with four positively charged ones results in a 40-fold increase in uptake of the modified thermosensitive DNA sequence in Vero and GC-2 cells (Jain et al., 2013). While the N,N-dimethylaminopropyl thiophosphate protective groups are positively charged at physiological pH, the hydrophobic octyl thiophosphate protecting groups of the thermolabile DNA sequence are necessary to impart amphiphilicity, which has been reported to improve in vitro cellular delivery of nucleic acid sequences (Zhi et al., 2010; Maslov et al., 2012).
Flow cytometry measurements have confirmed the efficiency of dTtaPS in mediating the cellular uptake of polyA-tailed PNA or polyA-tailed PMO sequences into several mammalian cell lines (Jain et al., 2015, 2016). Indeed, when assayed in HeLa pLuc 705 cells, the dTtaPS-assisted delivery of PNA sequence 9 (Fig. 4.xx.4) is more efficient (8–10-fold) than that of the PMO sequence 10 based on luciferase activity production measurements (Jain et al., 2015). However, when 2′-OMeUtaPS is used for the delivery of each sequence in HeLa pLuc 705 cells, uptake of the PMO sequence 10 (Fig. 4.xx.4) is more efficient (8–10 fold) than that of the PNA sequence 9 (Jain et al., 2017) as substantiated by luciferase activity production measurements (Fig. 4.xx.7). Flow cytometry analysis of the dTtaPS-mediated cellular uptake of the control PMO sequence 6 in HeLa pLuc 705 cells is significantly less than that of the control PNA sequence 8 (Fig. 4.xx.10) as determined by the level of fluorescence per cell; this is consistent with the luciferase assay results obtained from the delivery of the PNA and PMO sequences 9 and 10, respectively (Fig. 4.xx.11).
Figure 4.xx.10.
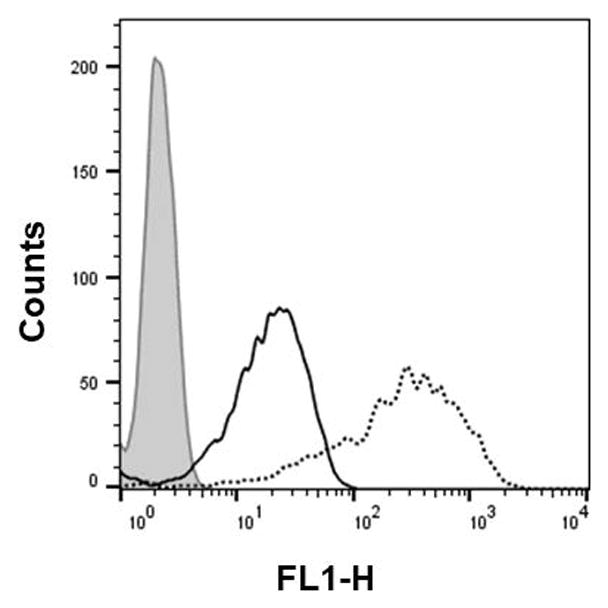
Flow cytometr analysis of the dTtaPS-assisted cellular uptake of the control PMO sequence 6 (peak area with black border) and PNA sequence 8 (peak area with a black dotted border) in HeLa pLuc 705 cells.
Figure 4.xx.11.
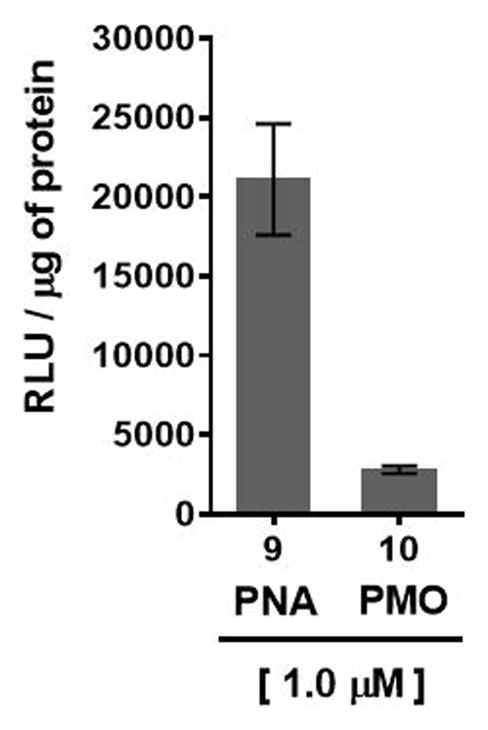
dTtaPS-mediated delivery of polyA-tailed PNA and PMO sequences 9 and 10, respectively, in HeLa pLuc 705 cells cultured in serum-containing medium. The concentration of each sequence is kept at 1.0 μM whereas the concentration of dTtaPS or 2′-OMeUtaPS is kept at 2.0 μM. Error bars represent the mean ± SD of three independent experiments. RLU = relative light units.
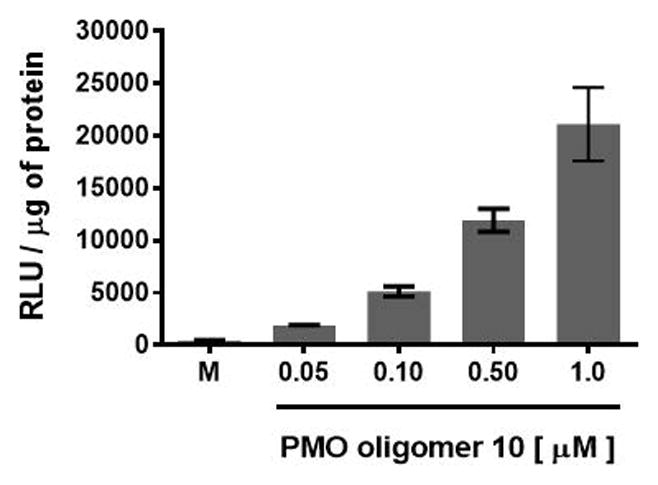
However, flow cytometry measurements of the 2′-OMeUtaPS-assisted delivery of polyA-tailed PMO and PNA sequences 6 and 8, respectively, show only a slightly higher level of fluorescence per cell for the PNA sequence 8 relative to that of the PMO sequence 6 (Fig. 4.xx.5). The outcome of this experiment suggest that the 2′-OMeUtaPS-assisted delivery of polyA-tailed PNA sequence 8 is as efficient as that of the PMO sequence 6. However, it appears that the affinity of 2′-OMeUtaPS for the polyA-tail of PNA sequence 9 interferes with either endosomal release of the sequence or its nuclear membrane penetration; both events can and will lead to poor exon skipping activity in HeLa pLuc 705 cells when compared to the results obtained with the 2′-OMeUtaPS-mediated delivery of polyA-tailed PMO sequence 10 (Fig. 4.xx.7). As presented in Fig. 4.xx.12, the 2′-OMeUtaPS-mediated internalization of PMO sequence 10 in HeLa pLuc 705 cells is concentration-dependent; the PMO sequence induces splice correction of the pre-mRNA encoding luciferase and results in an increased production of luciferase activity that is commensurate with the concentration of PMO sequence 10.
Figure 4.xx.12.
Concentration-dependence of the 2′-OMeUtaPS-mediated delivery of PMO sequence 10 on luciferase production in serum-containing media. The concentration of 2′-OMeUtaPS is kept at 2.0 μM in all experiments. Error bars represent the mean ± SD of three independent experiments. RLU = relative light units.
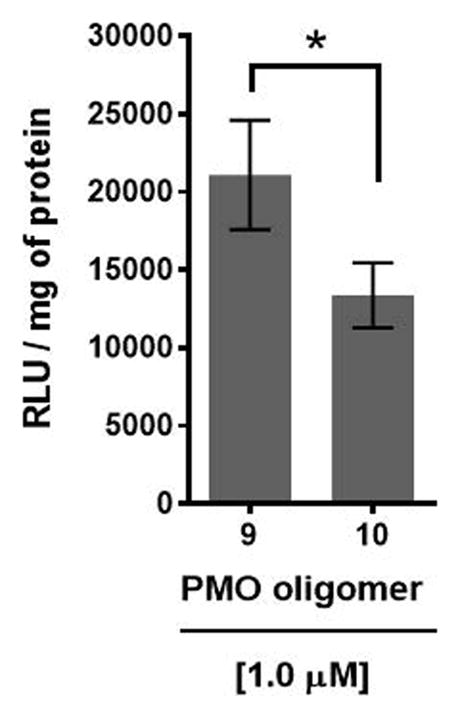
Recognition of the PMO-polyA tail of sequence 10 by either dTtaPS or 2′-OMeUtaPS is required for cellular uptake in mammalian cells (Jain et al., 2015, 2016, 2017). This is demonstrated by incorporating a PMO-T into the polyA stretch of sequence 10, which results in a statistically significant decrease in luciferase activity that is consistent with a weaker binding of 2′-OMeUtaPS to the modified polyA stretch of PMO sequence 11 (Fig. 4.xx.13).
Figure 4.xx.13.
Reduction of luciferase production upon 2′-OMeUtaPS-mediated internalization of PMO sequence 11 in HeLa pLuc 705 cells, as a consequence of an A→T substitution in the polyA tail of PMO sequence 10. The concentrations of 2′-OMeUtaPS is kept at 2.0 μM. Error bars represent the mean ± SD of three independent experiments. RLU = relative light units.
Additional evidence supporting the recognition of polyA-tailed PMO sequences by 2′-OMeUtaPS is provided by replacing the polyA stretch of PMO sequence 5 with a PMO polyT or a polyC tail to yield PMO sequence 12 or 13 (Fig. 4.xx.4), respectively. As measured by flow cytometry (Fig. 4.xx.6), the results of these experiments show that the 2′-OMeUtaPS-mediated internalization of PMO sequence 12 or 13 in pLuc 705 cells is comparable to that of PMO sequence 7 with no polyA tail, or that of the polyA-tailed PMO sequence 5 in the absence of 2′-OMeUtaPS. In spite of those convincing experiments, the 2′-OMeUtaPS-mediated internalization of polyA-tailed PMO sequence 5 in HeLa pLuc 705 cells has been carried out in the presence of urea, which is expected to interference with the ability of 2′-OMeUtaPS to recognize the polyA tail of the PMO sequence given the potent denaturing properties of urea. As anticipated, a significant decrease (~80–90%) in cellular uptake has been detected by flow cytometry (Fig. 4.xx.14) when the 2′-OMeUtaPS-assisted internalization of the PMO sequence 5 is performed in the presence of 2.0 M urea, when compared to that measured from the same experiment performed in the absence of urea. These results strongly suggest that the recognition of polyA-tailed PMO sequences by 2′-OMeUtaPS is sequence-specific, necessary and sufficient for efficient cellular uptake in live HeLa pLuc 705 cells.
Figure 4.xx.14.
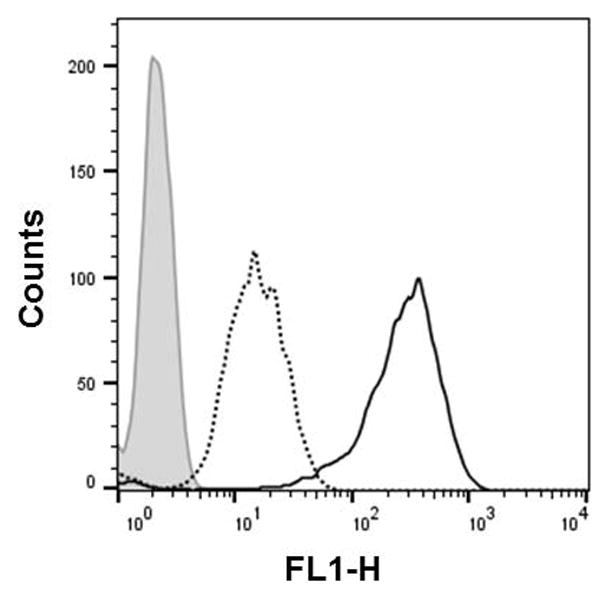
Flow cytometry analysis of the 2′-OMeUtaPS-mediated internalization of PMO sequence 5 (peak area with a solid black border) in the presence of urea (peak area with a dotted black border) in HeLa pLuc 705 cells. Pale gray peak area accounts for untreated HeLa pLuc 705 cells. The concentration of 5 and 2′-OMeUtaPS is 1.0 μM and 2.0 μM, respectively, while the concentration of urea is 2.0 M during the course of PMO sequence 5:2′-OMeUtaPS complex formation.
Although it is likely that the dTtaPS- and 2′-OMeUtaPS-mediated internalization of PNA or PMO sequences in HeLa pLuc 705 cells proceeds through an energy dependent endocytic mechanism, it is prudent to verify whether this is indeed the case. Cellular uptake experiments have been carried out at 37°C and 4°C, respectively. Fig. 4.xx.15 shows that when the cellular uptake experiment is carried out at 4°C, restoration of luciferase activity is decreased by more than 40% relative to that measured when the experiment is carried out at 37°C. These findings are in agreement with an endocytic uptake mechanism (Khalil et al., 2006). With the intent of identifying the most probable endocytic pathway leading to the 2′-OMeUtaPS-mediated uptake of PMO sequence 10, an investigation has been initiated employing endocytic pathway inhibitors at concentrations known to not significantly cause cell cytotoxicity (Ivanova et al. 2008). For this purpose, chlorpromazine for inhibition of clathrin-coated pits-mediated endocytosis (Khalil et al., 2006; Ivanova et al. 2008), nystatin and cytochalasin D for inhibition of caveolae-mediated endocytosis (Khalil et al., 2006; Ivanov, 2008), latrunculin for inhibition of both clathrin-coated pits- and caveolae-mediated endocytosis (Ivanov, 2008) and 5-(N-ethyl-N-isopropyl)amiloride (EIPA) for inhibition of macropinocytosis (Khalil et al., 2006; Ivanova et al. 2008) have been utilized. Although, the clathrin-coated pits-mediated endocytosis pathway appears to modestly inhibit the 2′-OMeUtaPS-assisted internalization of PMO sequence 10 in HeLa pLuc 705 cells, macropinocytosis is clearly the predominant endocytic pathway used for this internalization process (Fig. 4.xx.15).
Figure 4.xx.15.
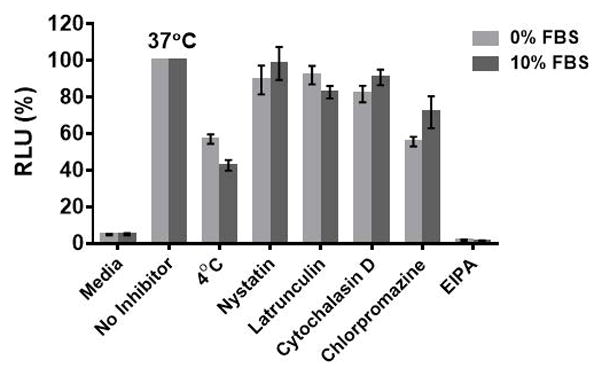
Inhibition of luciferase activity production at 4°C or 37°C and by endocytic pathway inhibitors upon 2′-OMeUtaPS-mediated internalization of PMO sequence 10 in both serum-containing and serum-free media. For each experiment, the extracellular concentrations of 2′-OMeUtaPS and sequence 10 are 2.0 μM and 1.0 μM, respectively, whereas the extracellular concentrations of nystatin, latrunculin, cytochalasin D, chlorpromazine and EIPA are 30 μM, 2 μM, 2 μM, 50 μM and 0.5 mM, respectively. Error bars represent the mean ± SD of three independent experiments. EIPA = 5-(N-ethyl-N-isopropyl)amiloride; RLU = relative light units; FBS = Fetal Bovine Serum.
The efficiency of 2′-OMeUtaPS-assisted delivery of PMO sequence 14 (Fig. 4.xx.4) at skipping exon 23 from the mdx mouse dystrophin pre-mRNA has been qualitatively demonstrated (Jain et al., 2017) through a nested RT-PCR assay (Goyenvalle et al., 2015) consisting of reverse-transcribing total RNA isolated from mdx mouse myotubes and amplifying the cDNA encoding exons 20–26 with appropriate DNA primers. After re-amplification of the primary PCR product encoding exons 20–24 through the use of another set of DNA primers, the secondary PCR products are separated by electrophoresis on a 1.5% agarose gel (Fig. 4.xx.8). The 633 bp secondary PCR product corresponds to the unspliced pre-mRNA exon 23, whereas the shorter 420 bp PCR product corresponds to the correctly spliced exon 23, which is consistent with the 213 bp difference between the secondary PCR products reported by others (Mann et al., 2001; Le et al, 2017). Thus, the electrophoretic data presented Fig. 4.xx.8 provide evidence that the 2′-OMeUtaPS-mediated internalization of PMO sequence 14 in mdx mouse myotubes is dose-dependent and results in the excision of exon 23 from approximately 60% of the pre-mRNA transcripts at a PMO sequence concentration of 250 nM in serum-containing medium. The outcome of this exon skipping experiment is comparable to that obtained with the Lipofectamine®2000-mediated transfection of the positive control 2′-OMe RNA sequence 16 at a concentration of 250 nM in serum-free medium (Fig. 4.xx.8) and vastly more significant than that obtained with the 2′-OMeUtaPS-assisted transfection of PNA sequence 15 at a sequence concentration of 1.00 μM in serum-containing medium. As expected, the dTtaPS-mediated transfection of PMO sequence 14 in mdx mouse myotubes is considerably less efficient at correctly splicing exon 23, even at a PMO sequence concentration of 1.00 μM. Contrary to expectations, the dTtaPS-mediated transfection of PNA sequence 15 in mdx mouse myotubes is relatively inefficient at correctly splicing exon 23 even at a PNA sequence concentration of 1.00 μM (Fig. 4.xx.8). A quantitative RT-qPCR assay protocol has been developed and implemented for measuring the extent of exon 23 skipping in mdx mouse myotubes; this assay uses genomic DNA cloned from a plasmid to establish a standard curve. The details of this protocol are provided in Novak et al., 2017.
The cytotoxicity of PMO sequence 14 and 2′-OMeUtaPS in HeLa pLuc 705 cells has then been evaluated in serum containing (10% FBS) DMEM medium over a period of 18 h using a commercial cell-counting kit. Increasing the concentration of PMO 14 from 100 nM to 2.0 μM (Fig. 4.xx.16A) or 2′-OMeUtaPS from 0.5 μM to 5.0 μM (Fig. 4.xx.16B) did not induce significant cytotoxicity at concentrations optimal for transfection experiments.
Figure. 4.xx.16.
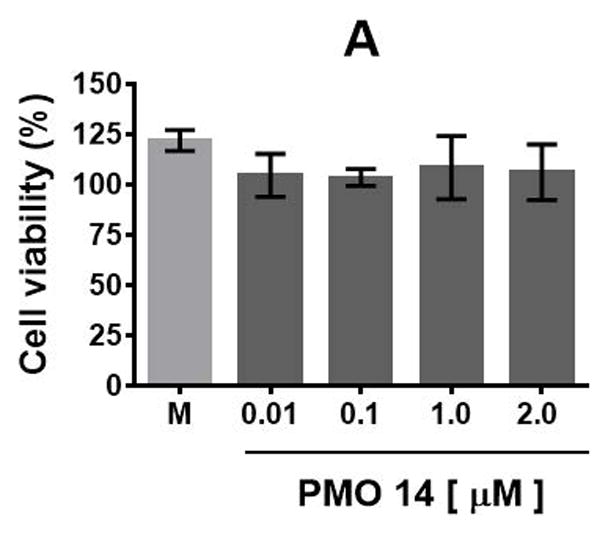
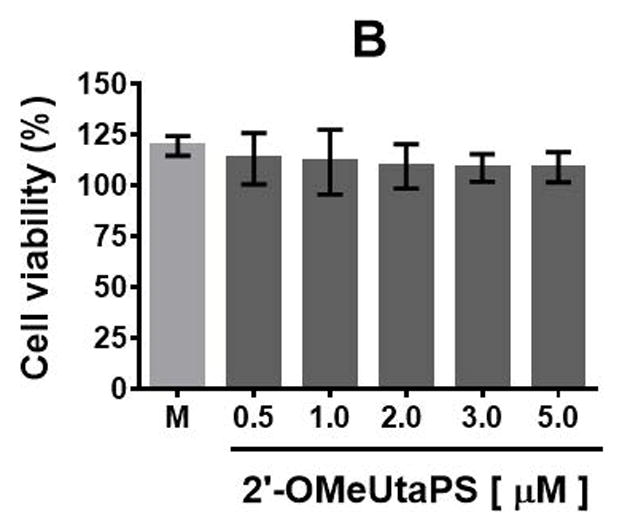
(A) Cytotoxicity of PMO sequence 14 and (B) 2′-OMeUtaPS in HeLa pLuc 705 cells when increasing their respective concentrations. Each cytotoxicity study has been evaluated over a period of 18 h in serum-containing (10% FBS) DMEM medium, using a commercial cell-counting kit, and expressed as a percentage of the viable cells. Error bars represent the mean ± SD of three independent experiments. M = medium.
The trans-acting phosphorothioate RNA element 2′-OMeUtaPS is a unique nucleic acid-based transfection reagent for the delivery of uncharged nucleic acid sequences to either HeLa pLuc 705 cells or mdx mouse myotube cells. The findings reported herein strongly underscore the versatility of synthetic nucleic acid-based transfection reagents for the delivery of antisense nucleic acid-based drugs for potential therapeutic applications while encouraging further investigations on the parameters associated with the endosomal trafficking pathways taken by those drugs.
Critical Parameters and Troubleshooting
All glassware is flame-dried and allowed to cool in a desiccator over phosphorus pentoxide prior to use.
The deoxyribonucleoside phosphoramidite 3 and 4 (Basic Protocol 1, Fig. 4.xx.1) must be prepared under strictly anhydrous conditions and must not be exposed to any acid, which would render these compounds prone to hydrolysis. With the objective to preventing this undesirable outcome, readers are referred to the Critical Parameters and Troubleshooting sections of UNITS 2.7 & 3.17, which address critical issues associated with the preparation and use of nucleoside phosphoramidites. The precipitation of 3 and 4 from cold hexane after silica-gel purification facilitates the removal of H-phosphonate impurities, which often co-elute with the desired phosphoramidite monomers. Also removed during the course of the precipitation step is residual triethylamine, which if not removed, can neutralize 1H-tetrazole and reduce the coupling efficiency of phosphoramidites during automated solid-phase DNA synthesis. The presence of moisture can also compromise the coupling efficiency of phosphoramidites and should be taken into consideration; this concern has also been addressed in UNIT 2.7.
When preparing the 2× stock solution of PNA or PMO sequence:2′-OMeUtaPS complexes (Support protocol 2) it is critically important to use freshly made 2′-OMeUtaPS solution from solid 2′-OMeUtaPS stored at −20°C. It is also imperative to add the 2′-OMeUtaPS solution to the PNA or PMO sequence solution, as stated in step 2 of Support Protocol 2. The use of PNA or PMO sequence:2′-OMeUtaPS complexes immediately after the 15-min storage at 4°C is strongly recommended for optimal cellular uptake experiments.
With the intent of ensuring consistent measurements of luciferase activity production, it is critical to employ a Bright Glow reagent (Basic Protocol 4, step 6) that has been stored frozen at −20°C or −80°C and measure luminescence immediately after addition of the reagent to cell lysates.
The H2K cells may cease to differentiate at high passage; it is therefore prudent to freeze multiple vials of cells at a low passage number so that differentiation can be resumed.
Precautions must be taken when isolating and working with RNA; reference is made to relevant URLs in the Internet Resources section.
Anticipated Results
The synthesis of phosphordiamidites (Support Protocol 1) is straightforward and consistently provides post-purification yields exceeding 80%. Similarly, the protocol for the preparation of ribonucleoside phosphoramidites that are required for solid-phase synthesis of 2′-OMeUtaPS is easy and proceeds in high yields (>90%) based on 31P NMR spectroscopy data. However, the isolated yield of each ribonucleoside phosphoramidite is variable and depends on the solubility of the phosphoramidite precipitate in cold hexane.
When every step of the solid-phase synthesis of 2′-OMeUtaPS is performed as delineated in Basic Protocol 2, an optimal yield 92 ± 5%, based on spectrophotometric measurement of the DMTr cation released upon completion of the last coupling step of the synthesis cycle.
In order to obtain optimal cellular uptake of PNA or PMO sequence:2′-OMeUtaPS complexes in live mammalian cells, Support Protocol 2 must be carefully executed. Indeed, when preparing 2× stock solutions of PNA or PMO sequence:2′-OMeUtaPS complexes, fresh 2′-OMeUtaPS solutions made from solid 2′-OMeUtaPS stored at −20°C should be used. The addition of the 2′-OMeUtaPS solution to the PNA or PMO sequence solution, as stated in step 3 of Support protocol 2 should be performed. Also recommended for optimal cellular uptake experiments is the use of PNA or PMO sequence:2′-OMeUtaPS complexes immediately after the mandatory15-min storage at 4°C.
As mentioned above, careful execution of the steps delineated in Support Protocol 2 will ensure consistent cellular uptake results by FACS analysis (Basic Protocol 3) or luciferase activity measurements (Basic Protocol 4), where appropriate. Likewise, meticulous execution of the steps outlined in Basic protocol 5 will lead to the production and efficient transfection of mdx mouse myotube muscle cells.
Should the extraction and isolation of total RNA from transfected mdx mouse myotube muscle cells be carried out in a RNase-free environment, the agarose gel analysis of the deletion of exon 23 from mdx mouse dystrophin pre-mRNA, will reproducibly provide the data presented in Fig. 4.xx.8.
Time Considerations
The preparation and purification of either phosphordiamidite 1 or 2 needed for the synthesis of deoxyribonucleoside phosphoramidites takes approximately 20 hr; lyophilization of silica gel-purified 1 or 2 is complete within 14 hr. The synthesis, purification and hexane precipitation of deoxyribonucleoside phosphoramidite 3 or 4 is achieved within 11 hr. Subsequent, lyophilization of each phosphoramidite precipitate is accomplished within 14 hr.
The automated solid-phase synthesis of the amphipathic trans-acting phosphorothioate RNA element (2′-OMeUtaPS) including its release and elution from the solid support, followed by complete evaporation of the eluate, require 8 hr to complete.
Prior to performing cellular uptake experiments, formation of complexes between 2′-OMeUtaPS and PNA or PMO sequences is achieved within 1 hr. Culture of HeLa pluc 705 cells to 70% confluency typically takes 3 days. Cell plating and performing the 2′-OMeUtaPS-assisted internalization of fluorescently labelled PNA or PMO sequence in HeLa pluc 705 cells is done within 43 hr. Measurements of the number of cells that have internalized PNA or PMO sequences by FACS take 1 hr. Luciferase activity production resulting from the 2′-OMeUtaPS-mediated internalization of PNA or PMO sequences in HeLa pLuc 705 cells can be measured within 30 h. As graphically presented in Fig. 16A and 16B, studies made to assess the cytotoxicity of the PMO sequence 14 and 2′-OMeUtaPS in a serum-containing medium require 20 hr for completion.
Culture and passage H2K mdx mouse myoblast cells to ~70% confluency is achieved within one week. Differentiation of H2K myoblasts into myotube muscle cells is typically complete within 5 days. The 2′-OMeUtaPS-mediated internalization of PNA or PMO sequences in mdx mouse myotube cells takes 24 hr. Extraction of total RNA from the myotube cells and isolation of total RNA by precipitation is complete within 24 hr. The preparation and amplification of primary and secondary cDNA products is completed within 5 hr. Qualitative analysis of the amplified cDNA products by agarose gel electrophoresis is obtained within 3 hr.
Significance Statement.
A method for the chemical synthesis of a short RNA sequence, acting as a transporter element for the delivery of a specific neutral nucleic acid sequence into mouse muscle cells, is described. Duchenne muscular dystrophy (DMD) is a lethal disease characterized by a progressive decline of muscle function caused by mutations in the gene encoding dystrophin; a neutral nucleic acid sequence has been designed to eliminate those mutations. When delivered by the transporter RNA element, this specific nucleic acid sequence induces the deletion of the genetic mutations responsible for the DMD condition, thereby restoring reading frame and dystrophin production in mouse muscle cells. The transporter RNA element may potentially find applications in the delivery of nucleic acid-based drugs indicated for the treatment of DMD in humans.
Acknowledgments
The authors thank Prof. Rudolph L. Juliano (University of North Carolina, Chapel Hill) and Dr Terence A. Partridge (Children’s National Medical Center, Washington DC) for kindly providing the HeLa pLuc 705 and H-2Kb-tsA58 (H2K) mdx myoblast cell lines, respectively. FDA intramural funds to S.L.B supporting this research are gratefully acknowledged. K. N. is supported by the National Institutes of Health (R56NS097229-01, 5U54HD053177, P50AR060836-01, 2R24HD050846-06), the Muscular Dystrophy Association and the US Department of Defense (W81XWH-05-1-0616, W81XWH-11-1-0782, W81XWH-11-1-0330).
Footnotes
Internet Resources
https://lifescience.roche.com/en_us/articles/precautions-for-handling-of-rna.html
http://genomics.no/oslo/uploads/PDFs/workingwithrna.pdf
Both URLs provide useful information on how to work with RNA and maintain an RNase-free environment.
Literature Cited
- Aartsma-Rus A, Van Ommen GJB. Antisense-mediated exon skipping: A versatile tool with therapeutic and research applications. RNA. 2007;13:1609–1624. doi: 10.1261/rna.653607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Nagata T, Yokota T, Nakamura A, Wood MJA, Partridge T, Takeda S. Highly efficient in vivo delivery of PMO into regenerating myotubes and rescue in laminin-α2 chain-null congenital muscular dystrophy mice. Hum Mol Genet. 2013;22:4914–4928. doi: 10.1093/hmg/ddt341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhadra J, Pattanayak S, Khan PP, Kundu J, Sinha S. Internal oligoguanidinium-based cellular transporter enhances antisense efficacy of morpholinos in in vitro and zebrafish model. Bioconjugate Chem. 2016;27:2254–2259. doi: 10.1021/acs.bioconjchem.6b00252. [DOI] [PubMed] [Google Scholar]
- Ellington A, Pollard JD., Jr . Introduction to the synthesis and purification of oligonucleotides. In: Beaucage SL, Bergstrom DE, Glick GD, Jones RA, editors. Current Protocols in Nucleic Acid Chemistry. I. Hoboken: John Wiley & Sons; 2000. pp. A.3C.1–A.3C.22. [DOI] [PubMed] [Google Scholar]
- Fletcher S, Honeyman K, Fall AM, Harding PL, Johnsen RD, Wilton SD. Dystrophin expression in the mdx mouse after localised and systemic administration of a morpholino antisense oligonucleotide. J Gene Med. 2006;8:207–216. doi: 10.1002/jgm.838. [DOI] [PubMed] [Google Scholar]
- Goyenvalle A, Griffith G, Arran Babbs A, El Andaloussi S, Ezzat K, Avril A, Dugovic B, Chaussenot R, Ferry A, Voit T, Amthor H, Bühr C, Schürch S, Wood MJA, Davies KE, Vaillend C, Leumann C, Luis Garcia L. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat Med. 2015;21:270–275. doi: 10.1038/nm.3765. [DOI] [PubMed] [Google Scholar]
- Hassane FS, Saleh AF, Abes R, Gait MJ, Lebleu B. Cell penetrating peptides overview and applications to the delivery of oligonucleotides. Cell Mol Life Sci. 2010;67:715–726. doi: 10.1007/s00018-009-0186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1517/14712598.7.6.8. [DOI] [PubMed] [Google Scholar]
- Ivanov AI. Pharmacological inhibition of endocytic pathways: Is it specific enough to be useful? In: Ivanov A, editor. Methods in Molecular Biology, Vol. 440: Exocytosis and Endocytosis. Totowa: Humana Press; 2008. pp. 15–33. [DOI] [PubMed] [Google Scholar]
- Iyer RP, Phillips LR, Egan W, Regan JB, Beaucage SL. The automated synthesis of sulfur-containing oligodeoxyribonucleotides using 3H-1,2-benzodithiol-3-one 1,1-dioxide as a sulfur-transfer reagent. J Org Chem. 1990;55:4693–4699. doi: 10.1021/jo00302a039. [DOI] [Google Scholar]
- Ivanova GD, Arzumanov A, Abes R, Yin H, Wood MJA, Lebleu B, Gait MJ. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 2008;36:6418–6428. doi: 10.1093/nar/gkn671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain HV, Takeda K, Tami C, Verthelyi D, Beaucage SL. Assessment of the cellular internalization of thermolytic phosphorothioate DNA oligonucleotide prodrugs. Bioorg Med Chem. 2013;21:6224–6232. doi: 10.1016/j.bmc.2013.04.071. [DOI] [PubMed] [Google Scholar]
- Jain HV, Verthelyi D, Beaucage SL. Amphipathic trans-acting phosphorothioate DNA elements mediate the delivery of uncharged nucleic acid sequences in mammalian cells. RSC Adv. 2015;5:65245–65254. doi: 10.1039/c5ra12038a. [DOI] [Google Scholar]
- Jain HV, Beaucage SL. An amphipathic trans-acting phosphorothioate DNA element delivers uncharged PNA and PMO nucleic acid sequences in mammalian cells. Curr Protoc Nucleic Acid Chem. 2016;64:4.69.1–4.69.22. doi: 10.1002/0471142700.nc0469s64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain HV, Boehler JF, Verthelyi D, Nagaraju K, Beaucage SL. An amphipathic trans-acting phosphorothioate RNA element delivers an uncharged phosphorodiamidate morpholino sequence in mdx mouse myotubes. RSC Adv. 2017;7:42519–42528. doi: 10.1039/c7ra04247g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järver P, O’Donovan L, Gait MJ. A chemical view of oligonucleotides for exon skipping and related drug applications. Nucleic Acid Ther. 2014;24:37–47. doi: 10.1089/nat.2013.0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S-H, Cho M-J, Kole R. Up-regulation of luciferase gene expression with antisense oligonucleotides: Implications and applications in functional assay development. Biochemistry. 1998;37:6235–6239. doi: 10.1021/bi980300h. [DOI] [PubMed] [Google Scholar]
- Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev. 2006;58:32–45. doi: 10.1124/pr.58.1.8. [DOI] [PubMed] [Google Scholar]
- Le BT, Murayama K, Shabanpoor F, Asanuma H, Veedu RN. Antisense oligonucleotide modified with serinol nucleic acid (SNA) induces exon skipping in mdx myotubes. RSC Adv. 2017;7:34049–34052. doi: 10.1039/c7ra06091b. [DOI] [Google Scholar]
- Mann CJ, Honeyman K, Cheng AJ, Ly T, Lloyd F, Fletcher S, Morgan JE, Partridge TA, Wilton SD. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc Natl Acad Sci USA. 2001;98:42–47. doi: 10.1073/pnas.011408598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslov MA, Kabilova TO, Petukhov IA, Morozova NG, Serebrennikova GA, Vlassov VV, Zenkova MA. Novel cholesterol spermine conjugates provide efficient cellular delivery of plasmid DNA and small interfering RNA. J Control Release. 2012;160:182–193. doi: 10.1016/j.jconrel.2011.11.023. [DOI] [PubMed] [Google Scholar]
- Muntoni F, Wood MJA. Targeting RNA to treat neuromuscular disease. Nat Rev Drug Discovery. 2011;10:621–637. doi: 10.1038/nrd3459. [DOI] [PubMed] [Google Scholar]
- Novak JS, Hogarth MW, Boehler JS, Nearing M, Vila MC, Heredia R, Fiorillo AA, Zhang A, Hathout Y, Hoffman EP, Jaiswal JK, Nagaraju K, Cirak S, Partridge TA. Myoblasts and macrophages are required for therapeutic morpholino antisense oligonucleotide delivery to dystrophic muscle. Nat Com. 2017;8:941. doi: 10.1038/s41467-017-00924-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donovan L, Okamoto I, Arzumanov AA, Williams DL, Deuss P, Gait MJ. Parallel synthesis of cell-penetrating peptide conjugates of PMO toward exon skipping enhancement in Duchenne Muscular Dystrophy. Nucleic Acid Ther. 2015;25:1–10. doi: 10.1089/nat.2014.0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabanpoor F, McClorey G, Saleh AF, Järver P, Wood MJA, Gait MJ. Bi-specific splice-switching PMO oligonucleotides conjugated via a single peptide active in a mouse model of Duchenne muscular dystrophy. Nucleic Acids Res. 2015;43:29–39. doi: 10.1093/nar/gku1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila MC, Klimek MB, Novak JS, Rayavarapu S, Uaesoontrachoon K, Boehler JS, Fiorillo AA, Hogarth MW, Zhang A, Shaughnessy C, Gordish-Dressman H, Burki H, Straub V, Lu QL, Partridge TA, Brown KJ, Hathout Y, van den Anker J, Hoffman EP, Nagaraju K. Elusive sources of variability of dystrophin rescue by exon skipping. Skeletal Muscle. 2015;5:44. doi: 10.1186/s13395-015-0070-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Lu P, Cloer C, Shaban M, Grewal S, Milazi S, Shah SN, Moulton HM, Lu QL. Long-term rescue of dystrophin expression and improvement in muscle pathology and function in dystrophic mdx mice by peptide-conjugated morpholino. Am J Pathol. 2012;181:392–400. doi: 10.1016/j.ajpath.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota T, Pistilli E, Duddy W, Nagaraju K. Potential of oligonucleotide-mediated exon-skipping therapy for Duchenne muscular dystrophy. Expert Opin Biol Ther. 2007;7:831–842. doi: 10.1517/14712598.7.6.831. [DOI] [PubMed] [Google Scholar]
- Youngblood DS, Hatlevig SA, Hassinger JN, Iversen PL, Moulton HM. Stability of cell-penetrating peptide-morpholino oligomer conjugates in human serum and in cells. Bioconjugate Chem. 2007;18:50–60. doi: 10.1021/bc060138s. [DOI] [PubMed] [Google Scholar]
- Zhi D, Zhang S, Wang B, Zhao Y, Yang B, Yu S. Transfection efficiency of cationic lipids with different hydrophobic domains in gene delivery. Bioconjugate Chem. 2010;21:563–577. doi: 10.1021/bc900393r. [DOI] [PubMed] [Google Scholar]