

INTRODUCTION
Antibodies produced by immunizing animals with foreign antigens have been invaluable tools for various detection methods. While the importance of their utility is widely accepted, there are limitations regarding the production, and reproducibility of monoclonal and polyclonal antibodies (Bradbury, 2015; Jones, 2016). To overcome these limitations, recombinant antibody development has received increasing attention. Antibody fragment or scaffold protein libraries can be displayed on bacteriophage and binders identified through affinity selection (Kehoe, 2005; McCafferty, 2015; Shim, 2016). Because the DNA sequences of isolated, recombinant antibodies are easily obtained, reagents can be expressed in Escherichia coli and their affinity and specificity can be improved through directed mutagenesis to yield high-quality, specific affinity reagents. By this approach, affinity reagents have successfully been generated to recognize a wide range of targets, including cell signaling proteins, membrane proteins, transcription factors, peptides, and post-translational modifications (Kim 2011; Kummer, 2012; Pershad, 2012; Koide, 2013; Horsby, 2015; Jones, 2016; Gustafsson, 2017).
Successful generation of affinity reagents is highly dependent on the choice of library (Hosse, 2006). Fibronectin type III (FN3) domain scaffold libraries serve as valuable options. Protein engineering experiments have shown that it is possible to randomize residues within three loops (BC, DE, FG) on one side of the FN3 94-amino acid domain (Fig. 1) without loss of stability or folding (Koide, 1998; Batori, 2002). FN3 sequence variants, also known as ‘monobodies,’ have been selected from phage display libraries that bind tightly and selectively to a wide variety of proteins through these randomized regions, such as Abl (Wojcik, 2010), β-catenin (Yeh, 2013), EphA2 (Park, 2015), estrogen receptor (Koide, 2002; Huang, 2006), Fyn (Huang, 2012), integrin (Richards, 2003), Pak1 (Huang, 2012), Ras (Spencer-Smith, 2017), VEGF-R (Fellouse, 2007), and several other human cell-signaling proteins (Huang, 2015). In addition to its target recognition versatility, the FN3 has many practical advantages. It lacks cysteines, it can be overexpressed (≥ 50 mg/L culture) in E. coli, it is thermally stable (Tm = 88°C), and it retains binding when absorbed onto microtiter plate wells (unlike 95% of monoclonal antibodies, which lose functionality when adsorbed onto plastic (Butler, 1993)). These make the FN3 an ideal scaffold for robust affinity reagent generation and production. This article describes the procedure for isolating FN3 monobodies via phage display, expressing and purifying soluble SUMO-tagged variants, and removing the SUMO-tag to produce affinity reagents that are ready for use in future immunoassay applications.
Figure 1. PyMol representations of the FN3 monobody.
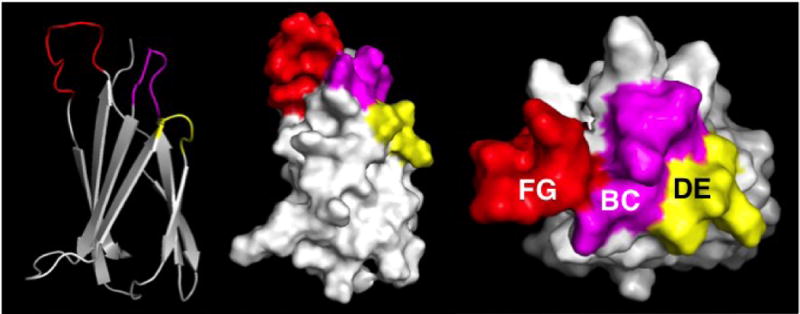
Left: cartoon, with β-sheets and loops (color). Middle: surface representation. Right: surface representation, viewed from the top. Red, purple, and yellow correspond to FG, BC, and DE loops that have been randomized.
BASIC PROTOCOL 1 AFFINITY SELECTION OF A PHAGE LIBRARY DISPLAYING VARIANTS OF THE FN3 MONOBODY
The first step in generating monobodies that bind the target of interest is to screen a phage-library displaying variants of the FN3 monobody in a process known as affinity selection. The protocol described here utilizes a large phage-display library containing 1.0 × 1011 members (Scholle, 2005; Gorman, 2017) to affinity select for FN3 monobodies against a fully-folded, soluble protein target. After two to three rounds of affinity selection, the output pool of clones is screened to identify clones that recognize the target of interest. See Figure 2 for an overview of the selection process.
Figure 2. Generation of affinity reagents through phage display.
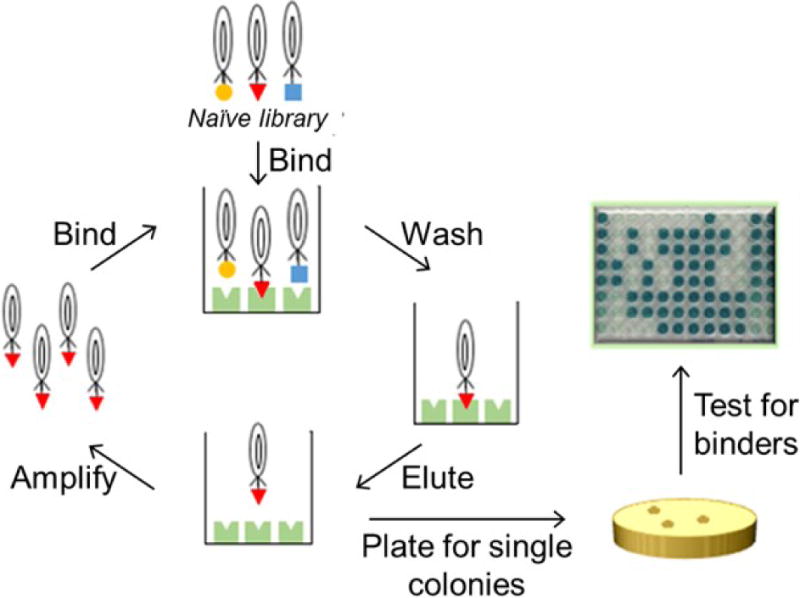
A phage library is incubated with immobilized target protein. Non-binding phage are washed away, the remaining phage are eluted, amplified, and subjected to further rounds of selection. Then individual clones are amplified and analyzed for binding.
Materials
Phage library
Biotinylated, soluble protein target
Phosphate-buffered saline (PBS) (see recipe)
4% skim milk solution (diluted in PBS, w/v) (see recipe for buffer)
Streptavidin-coated paramagnetic beads (Promega)
Phosphate-buffered saline with 0.1% Tween 20 (PBST) (see recipe)
Elution solution (see recipe)
Neutralization solution (see recipe)
E. coli strain TG1 (Lucigen)
15 cm by 1.5 cm 2×YT agar plates supplemented with carbenicillin (see recipe)
2×YT liquid media (see recipe)
Carbenicillin (1000×)
M13-KO7 helper phage (New England BioLabs)
Kanamycin (1000× concentrated stock solution)
75% glycerol
PEG solution (see recipe)
NeutrAvidin (Thermo Fisher Scientific)
Anti-FLAG-Biotin conjugate antibody (mouse, Sigma-Aldrich)
Anti-M13-HRP conjugate antibody (Sigma-Aldrich)
Sodium Citrate buffer
2,2′-Azinobis (3-ethylbenzothiazoline-6-Sulfonic Acid) diammonium salt tablets (Thermo Fisher Scientific)
Hydrogen Peroxide 3% (Walgreens)
1.5 mL centrifuge tubes
Rotator
Magnetic bead stand
50 mL conical tubes
Glass spreader beads
Shaking incubator
Cabinet incubator
Centrifuge with conical tube rotor and 96 well plate rotor
Vortexer
96-Well DeepWell™ Polypropylene Microplate (Fisher Scientific)
Nunc™ MicroWell™ 96-Well Microplates (Thermo Fisher Scientific)
Absorbance plate reader (BMG Labtech)
Affinity selection via phage-display (Round 1)
-
1Block four 1.5 mL centrifuge tubes with 4% skim milk solution for ≥ one hour at room temperature.
- This step can be completed the night before, with the filled tubes stored at 4°C overnight.
-
2Remove the soluble, biotinylated target and phage-library aliquot from the freezer and thaw on ice.
- Target can be biotinylated in vivo or in vitro (Kay, 2011).
-
3
Separately, remove streptavidin-coated paramagnetic beads from 4°C storage. Mix thoroughly by shaking to ensure all beads are suspended in solution.
-
4
Once the tubes have been blocked, remove the liquid content from one of the blocked 1.5 mL centrifuge tubes, and add 500 μL of streptavidin-coated paramagnetic beads.
-
5Add 500 nmol of biotinylated target to the beads, and bring to a volume (BTV) of 1 mL with PBS. Mix for at least 30 minutes on a rotator.
- Thirty minutes is the minimum; otherwise mix for 1 hour to ensure complete capture of the target to the beads
-
6
Next, place the tube containing the target/bead mixture on the magnetic separator stand. Wait until the supernatant appears clear. Decant the supernatant using a micropipette, taking great care not to aspirate any beads from the bottom of the tube.
-
7Resuspend the beads with the phage library. Using the Kay monobody library (estimated size is 1011), add 1.2 × 1012 phage particles, stored in PBS/16% glycerol. BTV 1 mL with PBS.
- Determine the phage concentration according to the M13 Titer Protocol (New England BioLabs).
-
8Transfer the phage/target/bead mixture to the rotator, and incubate for 1.5–2 hours.
- During this step, phage particles displaying variants that bind the target will be captured to the beads.
-
9Place the mixture on the magnetic separator stand. Wait until the supernatant is clear, and again carefully aspirate the supernatant using a micropipette.
- It may take longer for the magnet to attract the beads with phage-libraries that are stored with glycerol due to the viscosity.
-
10Resuspend the beads in PBS to begin the wash steps. Pipette up and down a few times using a micropipettor. Then place back onto the magnetic separator stand and aspirate the supernatant.
- During this step, non-bound virions are washed away.
-
11
Repeat step 10 until the phage/target/bead mixture has been washed 3 times.
-
12After the last washing step, resuspend the beads in 500 μL of the elution solution and rotate for 10 minutes.
- The acid will denature the displayed reagents, thus releasing them from the beads. Phage particles can be subjected to this pH for ~ 10 minutes before they lose infectivity.
-
13
During the elution step, take a new, previously-blocked 1.5 mL centrifuge tube and add 25 μL of the neutralization solution.
-
14Place the eluted virions/beads mixture onto the magnetic separator stand. Remove the supernatant and place it into the tube containing the neutralization solution.
- The neutralized phage can be stored on ice for >1 hour if necessary.
-
15Add the neutralized phage to 2.5 mL of TG1 cells at mid-log growth phase, determined by an optical density at 600 nm wavelength (OD600nm) of 0.4–0.6, for infection. Mix gently at 150 revolutions per minute (RPM) on a shaking incubator for 1 hour at 37°C.
- If a shaking incubator is not available, place in a 37°C environment and gently mix the tube manually every 10 minutes so that the cells do not settle to the bottom.
-
16
Spread the infected cells onto the 15 cm by 1.5 cm 2×YT plates supplemented with carbenicillin (CB). Add a maximum of 800 μL of cells to each plate. Incubate the plates at 30°C overnight for 15–18 hours. Also plate some of the uninfected TG1 cells on a 2×YT/CB plate as a negative control.
Preparing virions for subsequent rounds of selection
-
17Remove the 2×YT plates from the 30°C incubator. The negative control plate should be void of bacteria as uninfected TG1 lack CB resistance.
- The large plate should contain a lawn of cells.
-
18
Mix 4 mL of 2×YT liquid media, 1 mL of 75% glycerol, and 5 μL of carbenicillin and add the mixture to the large plate. Use a glass spreader or metal inoculating loop to scrape the lawn of cells from the surface of the plate into the overlying liquid.
-
19
When the bacteria are completely dispersed into the mixture (i.e., no clumps), use a pipette to transfer the solution into a chilled 15 mL conical tube. Repeat the scraping procedure for each large plate, each time placing the scraped cells into the same tube.
-
20Vortex the scraped cells well. Measure the OD of the mixture.
- Typically, the OD is between 30 and 60, well above the limit for most spectrophotometers. Thus, dilute the cells 1:100 in the scraping solution, measure the OD, and calculate the actual OD.
-
21
Using the scraped cells, inoculate 60 mL of 2×YT media supplemented with CB such that the starting OD is 0.0125. Use a beveled, 250 mL Erlenmeyer flask and place in a 37°C shaking incubator (RPM 250–280). Allow the culture to grow until the OD reaches 0.4–0.6.
-
22
While the culture is growing, aliquot the remainder of the scraped cells into labelled 1.5 mL centrifuge tubes. Freeze in liquid nitrogen or dry ice/ethanol and store at -80°C.
-
23Once the culture reaches mid-log phase, dump 30 mL of the culture and infect with the helper phage, M13-KO7, such that the ratio of virions to cells is 10:1. Infect for 1 hour at 37°C with shaking (150 RPM).
- This ratio is also referred to as the “multiplicity of infection,” or MOI. Calculate the number of cells present in the culture based on the OD and use that value to determine how many M13-KO7 phage particles to add. As before, if a shaker is not available simply place the infection culture in a 37°C environment, and manually mix the solution every 10 minutes.
-
24After infection, spin down the culture for 10 minutes at 10,000 xg and 4°C, discard the supernatant and resuspend the cells in 30 mL of 2×YT media supplemented with carbenicillin and kanamycin.
- The infected cells will now contain two antibiotic resistance genes, one for carbenicillin (provided by the phagemid), and one for kanamycin (provided by the M13-KO7 phage).
-
25
Incubate the culture at 25°C with shaking (200 RPM), for 18 to 24 hours.
-
26
Spin down the virion/cell mixture for 15 minutes at 10,000 xg and 4°C, and decant the supernatant into a fresh 50 mL conical tube. Discard the cells. Repeat this process two times to ensure all cells have been removed from the supernatant.
-
27Add the PEG solution to the clarified supernatant at a 1:5 ratio. Vortex the solution well and incubate on ice for 1 hour to precipitate the phage particles.
- If pressed for time, 30 minutes should be sufficient.
-
28
Spin the supernatant again. A small, white pellet should be visible; it is comprised of precipitated virions.
-
29
Decant the supernatant and carefully wash the pellet two to three times using PBS to wash away any excess PEG.
-
30
Resuspend the phage particles in 1 mL of PBS, and store on ice.
Affinity selection, Rounds 2 and 3
31. Rounds 2 and 3 of the selection process are performed in the same manner as Round 1 (Steps 1–30). However, instead of using a complete library, one uses virions recovered through each round of affinity selection. Due to amplification of these recovered clones, stringency of conditions can be increased to eliminate weak binders. The conditions for each round are shown below.
Round 2
50 nmol target
50 μL streptavidin-coated beads
- Wash steps, in order:
-
○3 times with PBS
-
○3 times with PBST
-
○
Elute in 200 μL of Elution buffer
Neutralize in 10 μL of Neutralization buffer
Infect 1 mL of mid-log phase TG1 cells
Plate 800 μL of TG1 infected cells
Round 3
5 nmol target
50 μL streptavidin-coated beads
- Wash steps, in order:
-
○3 times with PBS
-
○3 times with PBST
-
○
Elute in 200 μL of Elution buffer
Neutralize in 10 μL of Neutralization buffer
Infect in 1 mL of mid-log phase TG1 cells
Plate 800 μL of TG1 infected cells
Screening the selection output
-
32Along with plating the 800 μL of undiluted, infected TG1 cells from the third round of selection, plate dilutions of 10−7, 10-8, and 10-9.
- Dilution plates are made to obtain single colonies that are needed to screen individual clones to identify variants that recognize the selection target.
-
33
Add 200 μL of 2×YT/CB solution into a 96 deep-well plate using a multichannel pipettor. Pick individual colonies from the dilution plates using sterile toothpicks or pipette tips, swirl the tip in the solution briefly, and discard. Repeat 93 times. Also inoculate a negative and a positive control well.
-
34Grow the cultures in a shaking incubator at 37°C for three hours.
- Before moving to the next step, hold the plate up to the light and check that each well has sufficient growth (i.e., the solution appears cloudy).
-
35
When ready, infect the cells with M13-KO7 helper virus, with an MOI of 10, assuming the cells are at the mid-logarithmic growth phase. Dilute the M13-KO7 in 2×/CB, and add the appropriate amount to each well. Incubate for 1 hour with minimal shaking.
-
36
After, spin the cells down 10 minutes at 3000 ×g and 4°C and decant the supernatant. Resuspend the pellets in 400 μL of 2×YT/CB/KN. Grow at 30°C for 18–24 hours.
-
37
The same day, coat three MicroWell plates. Prepare 30 mL of NeutrAvidin (5 ng/μL in PBS), and add 100 μL to each well on all three plates. Store overnight at 4°C.
-
38
The next day, remove the MicroWell plates and dispose of the coating solution. Prepare a 4% skim milk (in PBS) blocking solution, and add 150 μL to each well on all three plates. Incubate at room temperature for 1 hour.
-
39
During the block step, remove the selection target from the freezer and thaw on ice. Prepare a 10 mL solution of the target in PBS (5 ng/μL). Store on ice.
-
40
Also prepare a 1:10,000 dilution of the anti-FLAG-biotin conjugated antibody using PBS. Store on ice.
-
41
Once the blocking step is complete, dispose of the solution from the MicroWell plates. If necessary, wash once with PBST to remove any leftover blocking solution.
-
42
To one plate, add the biotinylated target solution (5 ng/μL, 100 μL per well). To the second, add the anti-FLAG-biotin conjugate antibody (1:10,000 dilution, 100 μL per well). To the last plate, add PBS. Incubate with shaking for 1 hour at room temperature (RT).
-
43
When the virions are ready, spin down the DeepWell plate for 10 minutes at 3000 xg and 4°C to pellet the cells. Store at 4°C.
-
44
When ready, remove the microtiter plates from the shaker and dispose of the solution from each plate. Wash each plate once with PBST.
-
45
Add 50 μL of PBST to each well on all three plates. Carefully pipette 50 μL of supernatant from the DeepWell plate, and add to the corresponding row of the ELISA plates.
-
46
When the phage supernatant has been added to all three MicroWell plates, incubate for 1 hour at RT with shaking.
-
47
Remove the phage supernatant from the MicroWell plates, and wash 3 times with PBST. Then, add 100 μL of HRP/Anti-M13 Monoclonal Conjugate antibody (1:5,000 in PBST) to all wells. Incubate for 1 hour at RT with gentle shaking.
-
48With 5 minutes remaining in the incubation step, mix 30 mL of sodium citrate buffer, 300 μL of 3% H2O2, and three 2,2′-Azinobis (3-ethylbenzothiazoline-6-Sulfonic Acid) diammonium salt tablets. Vortex well, so that the tablets are completely dissolved.
- Use the solution within the 10 minutes.
-
49
When ready, remove the HRP/Anti-M13 Monoclonal Conjugate antibody solution from all MicroWell plates, and wash again three times with PBST. Then, add the substrate. The solution in the well will turn a noticeable shade of green over time (i.e., 5 to 60 minutes). For each plate, measure the optical absorbance at 405 nm using a microplate reader.
-
50Graph and interpret the data.
- The NeutrAvidin only plate serves as negative control signals for each plate and can be subtracted from the experimental and α-FLAG plates. The α-FLAG plate denotes the relative level of display for each clone and can be used to normalize the experimental signals.
BASIC PROTOCOL 2 EXPRESSION AND PURIFICATION OF MONOBODIES USING A pET14B-SUMO VECTOR
After the selection and screening process, the next step is to use a cloning method to move the FN3 coding region for selected binders into an expression vector. Our lab uses a SLiCE cloning scheme (Fig. 3, Zhang 2012) to accomplish subcloning, but more traditional, endonuclease-based methods are effective as well. To express the proteins in E. coli, our lab uses a pET14b vector which has been modified to contain a SUMO fusion partner at the N-terminus (Fig. 4a; Huang, 2015). In our experience, fusing the monobody to the SUMO protein maximizes the solubility and expression of the recombinant FN3 protein in E. coli. This protocol details the steps necessary for expressing and purifying the FN3 monobodies after they have been subcloned into the expression vector and subsequently transformed into the BL21 strain of E. coli, which has been modified to over-express recombinant proteins.
Figure 3. Subcloning FN3 monobody coding regions into the pET14b-SUMO vector by SLiCE.
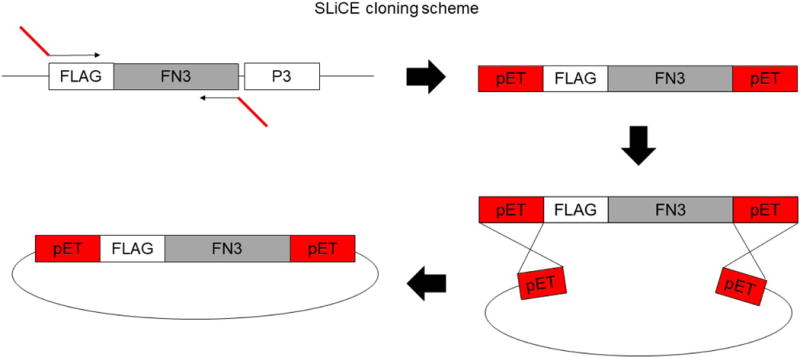
The FN3 coding sequence is amplified with a set of primers that add external sequences homologous to the regions bordering the cloning sites on the pET14b-Sumo vector. The insert is gel-purified and cloned into the destination vector via the SLiCE reaction.
Figure 4. Expression and purification of FN3-SUMO fusion monobodies.

a) Cartoon of FN3-SUMO fusion constructs. b) SDS-PAGE gel of FN3-SUMO expression and purification at lysate, flow through, and elution steps.
Materials
2×YT (See recipe)
Carbenicillin (1000× concentrated stock solution)
D-(+)-Glucose (Sigma-Aldrich)
Overnight Express™ Autoinduction System 1 (Novagen)
Equilibration buffer (see recipe)
complete, EDTA-FREE protease inhibitor cocktail tablet (Sigma-Aldrich)
His60 Ni Superflow Resin (Clontech)
Wash buffer 1 (see recipe)
Wash buffer 2 (see recipe)
Phosphate buffered saline (PBS; see recipe)
Elution buffer (See recipe)
2× Laemmli sample buffer (Bio-Rad)
Protein standard ladder (Bio-Rad)
12% polyacrylamide gel (Bio-Rad)
1× SDS-PAGE running buffer (see recipe)
SimplyBlue™ SafeStain (Thermo Fisher)
Shaking incubator
Centrifuge
Centrifuge tubes
Sonicator
IMAC Column
Rotator
Nanodrop spectrophotometer
Protein Gel apparatus
Power supply
Gel electrophoresis system with white light setting
Recombinant expression of FN3 monobodies
-
1
Using a 200 μL pipette tip, scrape a small amount of frozen, transformed BL21 cell stock. Inoculate 6 mL of 2×YT/CB/1% glucose. Grow overnight @ 37°C with shaking (250 RPM).
-
2The next day, use the 6 mL overnight culture to inoculate 300 mL of autoinduction media supplemented with CB.
- Alternatively, overnight cells can be added to 300 mL of 2×YT media and induced with IPTG.
-
3Grow the culture for ~24 hours @ 30°C with shaking (using a 2000 mL, beveled flask).
- If IPTG is used, conditions will need to be optimized.
-
4The next day, spin down the culture at 6000 ×g for 10 minutes at 4°C.
- At this point, pellets can be frozen until ready to proceed.
Purification of FN3 monobodies
-
5
Dissolve one cOmplete, EDTA-FREE protease inhibitor cocktail tablet in 50 mL of equilibration buffer.
-
6
Resuspend the pellet in 25 mL of equilibration buffer.
-
7
On ice, sonicate the resuspended cells for 5 minutes at 50% amplitude, 10 seconds on - 10 seconds off.
-
8
During sonication, prepare the IMAC column. Add 1 mL of His60 Resin to the column. Wash the column three times with 1 mL of equilibration buffer. Leave the beads suspended in 1 mL of binding buffer.
-
9
When sonication has finished, spin the crude lysate at 6000 × g for 15 minutes at 4°C.
-
10
Set aside 100 μL of the supernatant (lysate) for future analysis. Add the remaining supernatant (lysate) to the column and tumble on the rotator at 4°C for two hours.
-
11After tumbling for two hours, drain the column and set aside 100 μL of the flow through for future analysis.
- Begin the wash steps immediately after the column has drained so that it does not run dry.
-
12
Apply 30 mL of Wash buffer 1 to column and let drain. Collect 100 μL of the drained supernatant for future analysis. Repeat two additional times.
-
13
Repeat step 12 using Wash buffer 2.
-
14
Check the protein concentration of the final drained wash using the NanoDrop spectrophotometer. If no protein is present, proceed to step 15. If there is protein present, continue to wash until no protein remains.
-
15
Wash the column two times with PBS.
-
16Once the PBS has completely run through the column, add 1 mL of Elution buffer. Let sit for 2–3 minutes. Then drain the column into a fresh, labeled microfuge tube and measure the protein concentration. Repeat until no protein remains.
- The first elution will most likely contain little recombinant protein and so it is important to repeat the elution step.
SDS-PAGE
-
17
Using the supernatant samples that had previously been set aside, separately mix 20 μL of the lysate, flow through, each wash, and each elution with 20 μL of 2× Laemmli sample buffer. Heat samples at 95°C for 5 minutes.
-
18Load the protein standard ladder and 20 μL of each sample on a 12% polyacrylamide gel at 35 milliamps in 1× SDS-PAGE running buffer until the dye reaches the bottom of the gel.
- This will take ~90 minutes.
-
19
To stain the gel, cover with SimplyBlue™ SafeStain for 1 hour with gentle shaking.
-
20
Destain the gel for an additional 1–3 hours in water with gentle shaking.
-
21
After destaining, visualize the gel under white light (Fig. 4b).
-
22If appropriate, combine the elution fractions and concentrate if necessary, measure concentration with a spectrophotometer, and make 50–100 μL aliquots. Snap-freeze in liquid nitrogen and store at −80°C.
- If liquid nitrogen is not available, proteins can be snap frozen in a dry ice/ethanol bath.
Support Protocol 1 SUMO PROTEASE CLEAVAGE
The SUMO fusion tag has not been found to alter the specificity or affinity of monobody variants for their targets. However, one may wish to remove the tag when taking biophysical measurements or for other immunoassays in which the tag may interfere. The protocol for cleaving the SUMO tag by SUMO protease is quick and efficient. The Sumo protease is derived from the Ubi1 protein from Saccharomyces cerevisiae (Li, 1999). It recognizes the tertiary structure of SUMO, rather than an amino acid sequence, and cleaves C-terminal to SUMO (Mossessova, 2000; Müller, 2001). It is a highly active enzyme that is active from 2°C to 37°C.
Materials
Purified, FN3-SUMO fusion protein
10× SUMO protease buffer + salt (Thermo Fisher Scientific)
SUMO protease (Thermo Fisher Scientific)
His60 Ni Superflow Resin (Clontech)
Phosphate buffered saline (PBS) (See recipe)
2× Laemmli sample buffer (Bio-Rad)
Protein standard ladder (Bio-Rad)
12% polyacrylamide gel (Bio-Rad)
1× SDS-PAGE running buffer (See recipe)
SimplyBlue™ SafeStain (Thermo Fisher)
1.5 mL centrifuge tubes
Rotator
Microcentrifuge
Protein Gel apparatus
Power supply
Gel electrophoresis system with white light setting
NanoDrop spectrophotometer
Cleaving the SUMO tag
-
1
Assemble the digestion mixture on ice: 20 μg of the FN3 fusion protein, 20 μL of 10× SUMO protease buffer + salt, bring mixture to 190 μL with water, and then add 10 μL of SUMO protease (1 U/μL).
-
2
Once reaction is assembled, incubate at 4°C for two hours.
Purifying the tag-cleaved monobody
-
3Add the digested mixture to 10 μL of His60 Ni2+ Superflow Resin and tumble for 1 hour at 4°C.
- The FN3 monobodies purified from the pET14b-SUMO vector contain an N-terminal His tag followed by the N-terminal SUMO tag. This allows the cleaved SUMO tag to be removed from the mixture by nickel beads. The SUMO protease also contains a His tag and can be removed by this method as well.
-
4
Once the incubation is complete, spin down the mixture for 10 minutes at 1000 RPM.
-
5
Collect the supernatant and discard the beads
Assess purity
-
6
Separately mix 2 μg of controls: SUMO protease and non-cleaved SUMO-monobody fusion; and experimental: cleaved monobody; with 20 μL of 1× Laemmli sample buffer (dilute to 1× with PBS). Heat samples at 95°C for 5 minutes.
-
7
Load the protein standard ladder and 10 μL of each sample on a 12% polyacrylamide gel at 35 milliamps in 1× SDS-PAGE running buffer until the dye reaches the bottom of the gel.
-
8
To stain the gel, cover with SimplyBlue™ SafeStain for 1 hour with gentle shaking.
-
9
Destain the gel for an additional 1–3 hours in water with gentle shaking.
-
10After destaining, visualize the gel under white light.
- Pure, digested monobody will run at ~13 kD. If other bands are present such as the SUMO protease band (~25 kD), the SUMO tag (~17 kD), or non-cleaved SUMO-monobody fusion (~30 kD), the sample is not pure. Repeat the purification process.
-
11
Repeat the purification process until the cleaved monobody sample is pure. Once pure, determine the protein concentration using the NanoDrop spectrophotometer, snap freeze in liquid nitrogen, and store at −80°C.
Reagents and Solutions
Use Milli-Q purified water or equivalent for all solutions.
2×YT liquid media
1.6% (w/v) Bacto Tryptone
1% (w/v) Bacto Yeast Extract
0.5% (w/v) NaCl
Sterilize by autoclaving
Store indefinitely at RT or 4°C
2×YT agar plates supplemented with carbenicillin
Add 1.5% (w/v) agar to pre-autoclaved 2×YT broth
Sterilize by autoclaving
Let cool to 55°C-60°C and add carbenicillin at 1× concentration, swirl the flask carefully
Pour petri plates using sterile technique. Store plates upside down at 4°C for up to one month.
Elution Buffer
50 mM glycine
Adjust to pH 2 with HCl
Store indefinitely at RT
Equilibration Buffer
300 mM NaCl
50 mM NaP04
20 mM imidazole
Filter sterilize and store at 4°C indefinitely.
Neutralization Buffer
2 M Tris
Adjust to pH 10 with HCl
Store indefinitely at room temperature.
Phosphate Buffered Saline (PBS)
137 mM NaCl
2.7 mM KCl
10 mM Na2HPO4
1.8 mM KH2PO4
Adjust to pH 7.4 with HCl
Sterilize by filtrating
Store indefinitely at room temperature or 4°C.
Phosphate Buffered Saline with 0.1% Tween 20 (PBST)
To 999 mL of PBS, add 1 mL of Tween 20
Sterilize by filtrating
Store indefinitely at room temperature or 4°C.
PEG Solution
24% (w/v) PEG 8000
Stir mixture and heat at 80°C until the PEG is completely dissolved.
Once cooled, add 3 M NaCl
Filter sterilize and store at 4°C indefinitely
SDS-PAGE running buffer (10×)
30.0 g Tris
144.0 g glycine
10.0 g of SDS
Bring to 1000 ml with H2O.
Store the running buffer at room temperature and dilute to 1× before use.
Sodium Citrate buffer
50mM sodium citrate
Adjust to pH 4 with citric acid
Filter sterilize and store indefinitely at 4°C
Wash buffer 1
50 mM imidazole
0.5% Tween 20
Filter sterilize and store at 4°C indefinitely.
Wash buffer 2
50 mM imidazole
Filter sterilize and store at 4°C indefinitely.
COMMENTARY
Background Information
Since its description in 1985 (Smith, 1985), researchers have spent a great amount of effort to improve and expand upon the phage display technique. It is now a tool that can be used to generate binding partners (i.e., peptides, antibody fragments, scaffold proteins, protein fragments) for a wide variety of targets. The M13 virion is the most popular vehicle for display because its structure is well understood (Marvin, 1998). The virion houses its genetic code as circular, single-stranded DNA and is covered with five coat proteins: 2700 copies of pVIII, and five copies of pVII, pIX, pIII, and pVI (van Wezenbeek, 1980; Barbas, 2001). Upon their introduction, phage particles bind the F pilus of E. coli strains containing and coded by Fertility Factor genes (Barbas, 2001). When this occurs, the virion’s DNA enters the bacteria, which is converted into double-stranded DNA, and synthesis of the M13 proteins begins (Nakamura, 2003). Coat proteins, which have inserted into the outer bacterial membrane, transfer to the inner bacterial membrane while new, single-stranded phage DNA is synthesized and coated by pV, a single-stranded DNA-binding protein (Barbas, 2001; Nakamura, 2003). The protein-coated, single-stranded DNA interacts with export machinery, including other M13 proteins, to assemble and secrete phage particles by utilizing the coat proteins that had previously inserted into the membrane (Feng, 1997; Rakonjac, 1998; Marciano, 1999).
Each of the coat proteins have been documented to successfully display fusion scaffold proteins, peptides, or cDNA libraries, with pIII and pVIII fusions being the most commonly used (Smith, 1985; Gao, 1999; Hayhurst, 1999; Hufton, 1999; Fuh, 2000; Weiss, 2000; Gao, 2002). It is important to select the proper coat protein fusion as each has its advantages and disadvantages. Because pVIII contains 2700 copies, fusions can cause an avidity effect in which multiple copies of a variant give the appearance that they have a stronger affinity for the target than is the case. This is useful for displaying peptides intended to bind difficult targets with low affinity. However, only small peptides can be displayed without steric hindrance. pIII is useful for displaying larger peptides or proteins, as there are only five copies on each virion. This also helps in the discrimination of tight binders because the avidity effect is greatly reduced (Kehoe, 2005).
To further increase success with this technique, multiple phage display systems have been created. The traditional phage system displays the fusion protein or peptide with every copy of the coat protein to which it is fused (Smith, 1985). To circumvent the avidity effect caused by this system, the hybrid and phagemid systems have been developed. The hybrid system alters the virion genome to contain both the coat protein DNA sequence with the fusion sequence attached and the coat protein sequence alone (Zhong, 1994). This allows for only some copies of that coat protein to display the fusion. The phagemid system is unique in that it is a plasmid containing the capsid fusion sequence, bacteria and phage origins of replication, and the phage packaging signal. However, it lacks coding for all other phage proteins needed for assembly and release. Upon infection of helper phage, the replication and assembly can begin now that all other phage proteins and the capsid protein without the fusion are supplied. Additionally, the capsid fusion protein is synthesized so that the phage will display both wild type and fusion capsid (Lund, 2010).
The phage display technique has been applied to solve a variety of problems. Not only have they been utilized for affinity reagent generation, but also to improve protein stability (Pershad, 2012), generate inhibitors (Huang, 2012), provide insight into potential protein binding interactions (Halperin, 2003), and rapidly map transcription factor-DNA interactions (Freckleton, 2009). As the technology continues to develop, the opportunities involving phage display will as well.
Critical Parameters
Infecting TG1 cells
The state of the TG1 cells should be considered when infecting with phage for amplification purposes. In order to successfully infect, the bacteria cells must contain F pili, coded by the fertility factor genes. These cells must be treated with caution to ensure the pili are not damaged (Holland, 2006). Freeze/thaw cycles should be avoided. It is also extremely important to infect the cells during their mid-logarithmic growth phase while the F pili are still present.
Working with phage
Sterile, aseptic technique should be used while working with bacteriophage as they can easily contribute to contamination. Filter pipette tips should be used and gloves and benches should be wiped down with ethanol after use. Additionally, when working with phage, use extreme caution as they are fragile; avoid vortexing or rapidly pipetting phage particles.
Confirming proper folding of monobodies
Testing for proper folding of monobodies that have been expressed and purified from E. coli cells is very important before applying them to immunoassays, as some variants may be unstable. Denatured or misfolded proteins can be “sticky” and provide inaccurate binding results. To test for proper folding, fluorescence-based thermal shift assays are often used because of their accuracy and ease of use (Lo, 2004; Huynh, 2015). Stable FN3 monobody variants have melting temperatures near 80°C or above. Optimization during protein expression and purification may be required for certain variants to reach this stability threshold.
Troubleshooting
Table 1 describes common problems encountered during selection, possible causes, and suggestions for fixing such problems.
Table 1.
Troubleshooting common problems
| Problem | Possible causes | Solution |
|---|---|---|
| No binders obtained |
|
|
| Only weak binders obtained |
|
|
| Binders are cross-reactive with another antigen |
|
|
| Low display of recombinant phage when testing binding in ELISA (anti-FLAG binding signal is low) |
|
|
Statistical Analyses
Refer to step 50 of Basic protocol 1 for analysis of potential monobody binders. It is desirable to retest potential binders in triplicate to determine the average signal of binding and standard deviation.
Understanding Results
For Basic Protocol 1, the expected results are as follows: Most of the wells on the plate coated with the Anti-FLAG-Biotin conjugate antibody should turn dark green with a high signal (absorbance > 1), signifying that virions display a recombinant scaffold protein. If this is not the case, the amplification step should be repeated with consideration of critical parameters. Some of the corresponding wells on the plate coated with the target protein of interest will darken to various degrees (i.e., different ‘signal’), indicating potential FN3 monobodies that bind the target with different affinities. Note that not all of the wells will turn green, because not all clones will bind the target. Wells on the negative control plate should have a low signal (i.e., OD405nm <0.25); if the OD values are high, then there may have been inefficient washing or a particular clone is displaying a ‘sticky’, denatured variant.
Basic Protocol 2, the expected results are shown in Figure 4b. The lysate sample should contain proteins of all sizes with a high level of expression of the FN3 monobody. The flow-through sample should look like the lysate; however, the amount of FN3 protein should be greatly reduced, as most of it will be captured by the beads. High levels of FN3 protein in the flow-through sample indicate inefficient capture. The elution sample should contain only FN3 protein. Wash steps may vary. Support Protocol 1 expected results are described in step 10 of this protocol.
Time Considerations
Basic protocol 1: This protocol can be completed in 7 days. Each round of selection takes one day to select phage variants that recognize the target followed by one day to amplify those phage clones. After amplifying individual clones isolated from the third round of selection, a final day is devoted to testing for binders by ELISA.
Basic protocol 2: Once the FN3 coding region has been cloned into the protein expression vector and the sequence is confirmed, which will take about a week depending on the cloning method chosen, the protocol will take three days to complete. The first day, an overnight culture is started, the next day the FN3 monobodies are expressed, and the third day the FN3 monobodies and purified from E. coli cells. When removing the SUMO fusion partner, an additional day is required.
Significance Statement.
Antibodies are useful tools for detecting individual proteins in complex samples and learning about their location, amount, binding partners, and function in cells. Unfortunately, generating antibodies is time consuming, laborious, and their affinity and/or specificity is often limited. This protocol offers a fast and inexpensive alternative to generate antibody surrogates through phage display of a library of fibronectin type III (FN3) monobody variants and affinity selection for binders.
Acknowledgments
This work was supported by the National Institutes of Health [Grant number: U54 DK093444]; and by the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust.
LITERATURE CITED
- Barbas CF, Burton DR, Scott JK, Silverman GJ. Phage Display: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- Batori V, Koide A, Koide S. Exploring the potential of the monobody scaffold: effects of loop elongation on the stability of a fibronectin type III domain. Protein Eng. 2002;15:1015–1020. doi: 10.1093/protein/15.12.1015. [DOI] [PubMed] [Google Scholar]
- Bradbury A, Pluckthun A. Reproducibility: Standardize antibodies used in research. Nature. 2015;518:27–29. doi: 10.1038/518027a. [DOI] [PubMed] [Google Scholar]
- Butler JE, Ni L, Brown WR, Joshi KS, Chang J, Rosenberg B, Voss EWJ. The immunochemistry of sandwich ELISAs–VI. Greater than 90% of monoclonal and 75% of polyclonal anti-fluorescyl capture antibodies (CAbs) are denatured by passive adsorption. Mol Immunol. 1993;30:1165–1175. doi: 10.1016/0161-5890(93)90135-x. [DOI] [PubMed] [Google Scholar]
- Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, Koide S, Sidhu SS. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J Mol Biol. 2007;373:924–940. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Feng JN, Russel M, Model P. A permeabilized cell system that assembles filamentous bacteriophage. Proc Natl Acad Sci USA. 1997;94:4068–4073. doi: 10.1073/pnas.94.8.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freckleton G, Lippman SI, Broach JR, Tavazoie S. Microarray profiling of phage-display selections for rapid mapping of transcription factor-DNA interactions. PLoS Genet. 2009;5:e1000449. doi: 10.1371/journal.pgen.1000449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuh G, Sidhu SS. Efficient phage display of polypeptides fused to the carboxy-terminus of the M13 gene-3 minor coat protein. FEBS Lett. 2000;480:231–234. doi: 10.1016/s0014-5793(00)01946-3. [DOI] [PubMed] [Google Scholar]
- Gao C, Mao S, Lo CH, Wirsching P, Lerner RA, Janda KD. Making artificial antibodies: a format for phage display of combinatorial heterodimeric arrays. Proc Natl Acad Sci USA. 1999;96:6025–6030. doi: 10.1073/pnas.96.11.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Mao S, Kaufmann G, Wirsching P, Lerner RA, Janda KD. A method for the generation of combinatorial antibody libraries using pIX phage display. Proc Natl Acad Sci USA. 2002;99:12612–12616. doi: 10.1073/pnas.192467999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman KT, Roby LC, Giuffre A, Huang R, Kay BK. Tandem phage-display for the identification of non-overlapping binding pairs of recombinant affinity reagents. Nucleic Acids Res. 2017;45:e158. doi: 10.1093/nar/gkx688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MO, Mohammad DK, Ylösmäki E, Choi H, Shrestha S, Wang Q, Nore BF, Saksela K, Smith CI. ANKRD54 preferentially selects Bruton’s Tyrosine Kinase (BTK) from a Human Src-Homology 3 (SH3) domain library. PLoS ONE. 2017;12:e0174909. doi: 10.1371/journal.pone.0174909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halperin I, Wolfson H, Nussinov R. SiteLight: Binding-site prediction using phage display libraries. Protein Sci. 2003;12:1344–1359. doi: 10.1110/ps.0237103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayhurst A, Harris WJ. Escherichia coli skp chaperone coexpression improves solubility and phage display of single-chain antibody fragments. Protein Expr Purif. 1999;15:336–343. doi: 10.1006/prep.1999.1035. [DOI] [PubMed] [Google Scholar]
- Holland SJ, Sanz C, Perham RN. Identification and specificity of pilus adsorption proteins of filamentous bacteriophages infecting Pseudomonas aeruginosa. Virology. 2006;345:540–548. doi: 10.1016/j.virol.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Hornsby M, Paduch M, Miersch S, Sääf A, Matsuguchi T, Lee B, Wypisniak K, Doak A, King D, Usatyuk S, Perry K, Lu V, Thomas W, Luke J, Goodman J, Hoey RJ, Lai D, Griffin C, Li Z, Vizeacoumar FJ, Dong D, Campbell E, Anderson S, Zhong N, Gräslund S, Koide S, Moffat J, Sidhu S, Kossiakoff A, Wells J. A High Through-put Platform for Recombinant Antibodies to Folded Proteins. Mol Cell Proteomics. 2015;14:2833–2847. doi: 10.1074/mcp.O115.052209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosse RJ, Rothe A, Power BE. A new generation of protein display scaffolds for molecular recognition. Protein Sci. 2006;15:14–27. doi: 10.1110/ps.051817606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JX, Bishop-Hurley SL, Cooper MA. Development of Anti-Infectives Using Phage Display: Biological Agents against Bacteria, Viruses, and Parasites. Antimicrob Agents Chemother. 2012;56:4569–4582. doi: 10.1128/AAC.00567-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Fang P, Kay BK. Improvements to the Kunkel mutagenesis protocol for constructing primary and secondary phage-display libraries. Methods. 2012;58:10–17. doi: 10.1016/j.ymeth.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Fang P, Kay BK. Isolation of monobodies that bind specifically to the SH3 domain of the Fyn tyrosine protein kinase. N Biotechnol. 2012;29:526–533. doi: 10.1016/j.nbt.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Gorman KT, Vinci CR, Dobrovetsky E, Gräslund S, Kay BK. Streamlining the Pipeline for Generation of Recombinant Affinity Reagents by Integrating the Affinity Maturation Step. Int J Mol Sci. 2015;16:23587–23603. doi: 10.3390/ijms161023587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Koide A, Nettle KW, Greene GL, Koide S. Conformation-specific affinity purification of proteins using engineered binding proteins: Application to the estrogen receptor. Protein Expr Purif. 2006;47:348–354. doi: 10.1016/j.pep.2005.10.021. [DOI] [PubMed] [Google Scholar]
- Hufton SE, Moerkerk PT, Meulemans EV, de Bruine A, Arends JW, Hoogenboom HR. Phage display of cDNA repertoires: the pVI display system and its applications for the selection of immunogenic ligands. J Immunol Methods. 1999;231:39–51. doi: 10.1016/s0022-1759(99)00139-8. [DOI] [PubMed] [Google Scholar]
- Huynh K, Partch CL. Analysis of Protein Stability and Ligand Interactions by Thermal Shift Assay. Curr Protoc Protein Sci. 2015;79:28.9.1–28.9.14. doi: 10.1002/0471140864.ps2809s79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ML, Alfaleh MA, Kumble S, Zhang S, Osborne GW, Yeh M, Arora N, Hou JJ, Howard CB, Chin DY, Mahler SM. Targeting membrane proteins for antibody discovery using phage display. Sci Rep. 2016;6:26240. doi: 10.1038/srep26240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TD, Carter PJ, Plückthun A, Vásquez M, Holgate RG, Hötzel I, Popplewell AG, Parren PW, Enzelberger M, Rademaker HJ, Clark MR, Lowe DC, Dahiyat BI, Smith V, Lambert JM, Wu H, Reilly M, Haurum JS, Dübel S, Huston JS, Schirrmann T, Janssen RA, Steegmaier M, Gross JA, Bradbury AR, Burton DR, Dimitrov DS, Chester KA, Glennie MJ, Davies J, Walker A, Martin S, McCafferty J, Baker MP. The INNs and outs of antibody nonproprietary names. MAbs. 2016;8:1–9. doi: 10.1080/19420862.2015.1114320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay BK, Thai S, Volgina VV. High-throughput Biotinylation of Proteins. Methods Mol Bio. 2011;498:185–196. doi: 10.1007/978-1-59745-196-3_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehoe JW, Kay BK. Filamentous Phage Display in the New Millennium. Chem Rev. 2005;105:4056–4072. doi: 10.1021/cr000261r. [DOI] [PubMed] [Google Scholar]
- Kim J, Stroud RM, Craik CS. Rapid identification of recombinant Fabs that bind to membrane proteins. Methods. 2011;55:303–309. doi: 10.1016/j.ymeth.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimka A, Yu N, Shami EY. Construction of proteolysis resistant human interleukin-2 by fusion to its protective single chain antibody. Cytokine. 2003;22:134–141. doi: 10.1016/s1043-4666(03)00136-4. [DOI] [PubMed] [Google Scholar]
- Koide A, Abbatiello S, Rothgery L, Koide S. Probing protein conformational changes in living cells by using designer binding proteins: application to the estrogen receptor. Proc Natl Acad Sci USA. 2002;99:1253–1258. doi: 10.1073/pnas.032665299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide A, Bailey CW, Huang X, Koide S. The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol. 1998;284:1141–1151. doi: 10.1006/jmbi.1998.2238. [DOI] [PubMed] [Google Scholar]
- Koide S, Huang J. Generation of high-performance binding proteins for peptide motifs by affinity clamping. Methods Enzymol. 2013;523:285–302. doi: 10.1016/B978-0-12-394292-0.00013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer L, Parizek P, Rube P, Millgramm B, Prinz A, Mittl PR, Kaufholz M, Zimmermann B, Herberg FW, Plückthun A. Structural and functional analysis of phosphorylation specific binders of the kinase ERK from designed ankyrin repeat protein libraries. Proc Natl Acad Sci USA. 2012;190:2248–2257. doi: 10.1073/pnas.1205399109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SJ, Hochstrasser M. A new protease required for cell-cycle progression in yeast. Nature. 1999;398:246–251. doi: 10.1038/18457. [DOI] [PubMed] [Google Scholar]
- Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, Ellestad G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem. 2004;332:153–159. doi: 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- Lund PE, Hunt RC, Gottesman MM, Kimchi-Sarfaty C. Pseudovirions as vehicles for the delivery of siRNA. Pharm Res. 2010;27:400–420. doi: 10.1007/s11095-009-0012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciano DK, Russel M, Simon SM. An Aqueous Channel for Filamentous Phage Export. Science. 1999;284:1516–1519. doi: 10.1126/science.284.5419.1516. [DOI] [PubMed] [Google Scholar]
- Marvin DA. Filamentous phage structure, infection and assembly. Curr Opin Struct Biol. 1998;8:150–158. doi: 10.1016/s0959-440x(98)80032-8. [DOI] [PubMed] [Google Scholar]
- McCafferty J, Schofield D. Identification of optimal protein binders through the use of large genetically encoded display libraries. Curr Opin Chem Biol. 2015;26:16–24. doi: 10.1016/j.cbpa.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Mossessova E, Lima CD. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell. 2000;5:865–876. doi: 10.1016/s1097-2765(00)80326-3. [DOI] [PubMed] [Google Scholar]
- Müller S, Hoege C, Pyrowolakis G, Jentsch S. SUMO, ubiquitin’s mysterious cousin. Nature Rev Mol Cell Biol. 2001;2:202–210. doi: 10.1038/35056591. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Tsumoto K, Kumagai I, Ishimura K. A morphologic study of filamentous phage infection of Escherichia coli using biotinylated phages. FEBS Lett. 2003;536:167–172. doi: 10.1016/s0014-5793(03)00050-4. [DOI] [PubMed] [Google Scholar]
- Park SH, Park S, Kim DY, Pyo A, Kimura RH, Sathirachinda A, Choy HE, Min JJ, Gambhir SS, Hong Y. Isolation and Characterization of a Monobody with a Fibronectin Domain III Scaffold That Specifically Binds EphA2. PLoS ONE. 2015;10:e0132976. doi: 10.1371/journal.pone.0132976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pershad K, Pavlovic JD, Graslund S, Nilsson P, Colwill K, Karatt-Vellatt A, Schofield DJ, Dyson MR, Pawson T, Kay BK, McCafferty J. Generating a panel of highly specific antibodies to 20 human SH2 domains by phage display. Protein Eng Des Sel. 2010;23:279–288. doi: 10.1093/protein/gzq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pershad K, Wypisniak K, Kay BK. Directed evolution of the Forkhead-associated domain to generate anti-phosphospecific reagents by phage display. J Mol Biol. 2012;424:88–103. doi: 10.1016/j.jmb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards J, Miller M, Abend J, Koide A, Koide S, Dewhurst S. Engineered fibronectin type III domain with a RGDWXE sequence binds with enhanced affinity and specificity to human alphavbeta3 integrin. J Mol Biol. 2003;326:1475–1488. doi: 10.1016/s0022-2836(03)00082-2. [DOI] [PubMed] [Google Scholar]
- Rakonjac J, Model P. The roles of pIII in filamentous phage assembly. J Mol Biol. 1998;282:25–41. doi: 10.1006/jmbi.1998.2006. [DOI] [PubMed] [Google Scholar]
- Scholle MD, Kehoe JW, Kay BK. Efficient Construction of a Large Collection of Phage-Displayed Combinatorial Peptide Libraries. Comb Chem High Throughput Screen. 2005;8:545–551. doi: 10.2174/1386207054867337. [DOI] [PubMed] [Google Scholar]
- Shim H. Therapeutic Antibodies by Phage Display. Curr Pharm Des. 2016;22:6538–6559. doi: 10.2174/1381612822666160923113714. [DOI] [PubMed] [Google Scholar]
- Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- Spencer-Smith R, Koide A, Zhou Y, Eguchi RR, Sha F, Gajwani P, Santana D, Gupta A, Jacobs M, Herrero-Garcia E, Cobbert J, Lavoie H, Smith M, Rajakulendran T, Dowdell E, Okur MN, Dementieva I, Sicheri F, Therrien M, Hancock JF, Ikura M, Koide S, O’Bryan JP. Inhibition of RAS function through targeting an allosteric regulatory site. Nat Chem Biol. 2017;13:62–68. doi: 10.1038/nchembio.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wezenbeek P, Hulsebos T, Schoenmakers J. Nucleotide sequence of the filamentous bacteriophage M13 DNA genome: comparison with phage fd. Gene. 1980;11:129–148. doi: 10.1016/0378-1119(80)90093-1. [DOI] [PubMed] [Google Scholar]
- Weiss GA, Sidhu SS. Design and evolution of artificial M13 coat proteins. J Mol Biol. 2000;300:213–219. doi: 10.1006/jmbi.2000.3845. [DOI] [PubMed] [Google Scholar]
- Wojcik J, Hantschel O, Grebien F, Kaupe I, Bennett KL, Barkinge J, Jones RB, Koide A, Superti-Furga G, Koide S. A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat Struct Mol Biol. 2010;17:519–527. doi: 10.1038/nsmb.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh JT, Binari R, Gocha T, Dasgupta R, Perrimon N. PAPTi: a peptide aptamer interference toolkit for perturbation of protein-protein interaction networks. Sci Rep. 2013;3:1156. doi: 10.1038/srep01156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Werling U, Edelmann W. SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res. 2012;40:e55. doi: 10.1093/nar/gkr1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G, Smith GP, Berry J, Brunham RC. Conformational mimicry of a chlamydial neutralization epitope on filamentous phage. J Biol Chem. 1994;269:24183–24188. [PubMed] [Google Scholar]