

Significance Statement
The reconstruction of cancer phylogeny trees and quantifying the disease evolution is a challenge task. LICHeE and BAMSE are two computational tools designed and implemented recently for this purpose. They both utilize estimated variant allele fraction of somatic mutations across multiple samples to infer the most likely cancer phylogenies. This unit provides extensive guidelines for installing and running both LICHeE and BAMSE.
Keywords: Cancer Phylogeny, Somatic Variations, Next Generation Sequencing, Deep Sequencing, Tumor Heterogeneity
1 Introduction
Evolution of somatic mutations that accumulate over time and confer a fitness advantage to tumor cells is the main driver of cancer progression as described in several recent studies. Furthermore, the heterogeneity of somatic variants within subpopulations of cells within the tumor has been increasingly characterized in multiple cancer types [1, 2, 9, 10]. These subpopulations are referred to as subclones with distinct sets of mutations. The strides that have been made in next-generation DNA sequencing technologies have allowed for many large-scale efforts aimed at characterizing the genetic profile of tumors and matched normal DNA in order to identify driver events in tumors. Using this genomic data, it is necessary to infer the evolutionary relationship between subclones as this information is increasingly recognized as having value when making clinical decisions.
With the emphasis being placed on characterizing tumor heterogeneity, computational tree-building methods are required for cancer phylogeny reconstruction. Here, we present two protocols that define the use of LICHeE (Lineage Inference for Cancer Heterogeneity and Evolution) and BAMSE (BAyesian Model Selection for tumor Evolution), two states-of-the-arts computational methods for inference of tumor lineage trees in the context of heterogeneous multi-region tumor samples. LICHeE and BAMSE identify the set of lineage trees that are consistent with variant allele frequencies (VAFs) profiles obtained from deep-sequencing from somatic single nucleotide variants (SSNVs). LICHeE is a combinatorial approach. It utilizes an evolutionary constraint network that represents all possible trees that can potentially be obtained from the data and evaluates constraints of evolution in order to reduce the search space. In contrast, BAMSE is a probabilistic approach to building lineage trees and inferring subclonal history. It uses read counts taken from standard sequencing of heterogeneous tumor samples and employs a novel Bayesian model selection to identify the models that most accurately fit the observed data.
2 BASIC PROTOCOL 1: Running LICHeE on tumor data
LICHeE is a computational approach aimed at the reconstruction of tumor phylogenies and the characterization of the subclonal composition of tumor samples using variant allele frequencies (VAFs) of somatic single nucleotide variants (SSNVs) from multiple samples of the same patient (e.g. extracted from different regions of the tumor) sequenced at high depth. LICHeE relies on the perfect phylogeny model to define a set of constraints on the evolutionary ordering of SSNVs using their presence patterns and VAFs across input samples and finds all the lineage trees that satisfy these constraints. LICHeE can efficiently find all such trees by formulating the problem as a search for all valid spanning trees in an evolutionary constraint network, which encodes all valid pairwise ancestral relationships among the SSNVs. Given each such tree, LICHeE also provides estimates of the subclonal mixtures of each input sample. LICHeE is freely available at https://github.com/viq854/lichee.
At a macroscopic level, LICHeE’s execution can be broken down into the following steps: (1) SSNV calling across input samples, (2) SSNV clustering using VAFs (each group of SSNVs present in the same set of samples is clustered separately), (3) construction of the evolutionary constraint network (where the nodes are the clusters obtained in step (2) and the edges represent valid pairwise ancestry relationships), (4) search for lineage trees embedded in the network that satisfy all the phylogenetic constraints, and (5) output visualization. In this protocol we describe how various parameters can control the execution of the program at each step, providing some insights into their tuning, and comment on the interpretation of the results.
Necessary Resources
Hardware: Computer running Linux, Mac OS or Windows
System Requirements: Java Runtime Environment (JRE) 1.6
Files: Text file with SNVs with specified per-sample VAFs
-
Prepare the input file
LICHeE requires a tab-delimited text file with one SSNV entry per line. The columns include information about the SSNV (e.g. position in the genome) and its VAF in each sample according to the following required header:#chr position description <sample names separated by tabs>
An example of a valid input file can be found in LICHeE’s repository here: https://github.com/viq854/lichee/tree/master/LICHeE/data/ccRCC/RK26.txt
Given this information, LICHeE will first call the SSNVs in each sample using a simple heuristic method and then cluster them according to their VAFs. However, if the SSNV calls are already available to the user from a different specialized program, they can be directly provided to LICHeE, bypassing the first step. This can be achieved by enabling the -sampleProfile flag and extending the input file to include a binary string denoting SSNV presence and absence in each sample as an additional column (added before the sample frequency information). If available, the user can also directly substitute VAFs in this file with the more informative cell prevalence (CP) values (and enable the -cp flag).
Similarly, pre-computed SSNV cluster assignments can also be specified by the user, bypassing the default clustering step, by providing an additional tab-delimited input file containing the clusters (with the corresponding centroid VAFs per sample and the member SSNVs): one cluster per line. This file should be of the form:profile <cluster VAFs per sample separated by tabs> <comma-separated list of SSNVs>
-
Identify and set relevant parameters and flags
Let’s assume that LICHeE is installed in the $LICHEE_ROOT_DIR directory. LICHeE is designed to be run from the command-line console using the following guidelines.
From $LICHeE_ROOT_DIR/LICHeE/release, the command template is:$ ./lichee -build -i <input_file_path> [-minVAFPresent <VAF1> -maxVAFAbsent < VAF2> -n <normal_sample_id>] [other options]
These parameters control the initial SSNV filtering and calling and are required if the -sampleProfile flag is not specified (i.e. when SSNV calling is executed). Additional parameters can be supplied from the following list:Parameter Description Input/output and display options -i <arg> Input file path (required) -o <arg> Output file path where the results should be written -cp Input data represents cell prevalence (CP) values (as opposed to default VAF values) -n,−normal <arg> Normal sample column id in the list of samples, 0-based (e.g. 0 is the first column) (required*) -clustersFile <arg> SSNV clusters file path -s,−save <arg> Maximum number of output trees to save, if any (default: 1) -showNetwork,−net Display the constraint network -showTree,−tree <arg> Display the top ranking lineage tree(s) (default: 0) -color Enable lineage tree visualization in color mode -dot Enable DOT file export of the top-scoring tree for Graphviz visualization (saved by default to: input file with suffix.dot) -v,−verbose Verbose mode, prints more information about each step of the algorithm SSNV filtering and calling -maxVAFAbsent,−absent <arg> Maximum VAF to consider an SSNV as robustly absent from a sample (required*) -minVAFPresent,−present <arg> Minimum VAF to consider an SSNV as robustly present in a sample (required*) -maxVAFValid <arg> Maximum allowed VAF in a sample (default: 0.6) -minProfileSupport <arg> Minimum number of robust SSNVs required for a group presence-absence profile to be labeled robust during SNV calling: SNVs from non-robust groups can be re-assigned to existing robust groups (default: 2) SSNV clustering -maxClusterDist <arg> Maximum mean VAF difference on average per sample up to which two SSNV clusters can be collapsed (default: 0.2) -minClusterSize <arg> Minimum number of SSNVs required per cluster (default: 2) -minPrivateClusterSize <arg> Minimum number of SSNVs required for a private cluster (i.e. with SSNVs occurring only in one sample) (default: 1) Constraint network construction and tree search -e <arg> VAF error margin (default: 0.1) -minRobustNodeSupport <arg> Minimum number of robust SSNVs required for a node to be labeled robust during tree search: non-robust nodes can be automatically removed from the network when no valid lineage trees are found (default: 2) -c,−completeNetwork Add all possible edges to the constraint network, by default private nodes are connected only to closest level parents and only nodes with no other parents are descendants of root -nTreeQPCheck <arg> Number of top-ranking trees on which the QP consistency check is run, we have not seen this check to fail in practice (default: 0, for best performance) *these parameters are required unless the -sampleProfile option is specifiedThe defaults for some of these parameters are set fairly conservatively, assuming noisy real data. For best results, or when testing on simulated data, users are advised to customize these parameters to their datasets. For example, lowering -maxClusterDist, which controls the collapsing of nearby clusters, can order additional SSNVs by keeping them in separate clusters (originally obtained by the GMM clustering algorithm); similarly, lowering -minClusterSize will keep single-SSNV clusters in the network.
-
Run LICHeE to infer lineage trees and tumor heterogeneity
We provide several examples of running LICHeE on the following two publicly-available data sets included in the LICHeE repository (https://github.com/viq854/lichee/tree/master/LICHeE/data) for testing while varying several parameters:To display the top ranking lineage tree for the ccRCC RK26 patient dataset:$ ./lichee -build -i ../data/ccRCC/RK26.txt -maxVAFAbsent 0.005 -minVAFPresent 0.005 -n 0 -showTree 1
LICHeE will use the -maxVAFAbsent and -minVAFPresent arguments as thresholds when calling SSNVs in each sample. The -showTree argument will display the top ranking tree.
To infer trees that don’t contain private clusters (clusters with SSNVs present in only one sample) with fewer than 2 SSNVs, while also increasing the error margin used in phylogenetic constraint enforcement, and save the top ranking tree (for the ccRCC RMH008 patient dataset):$ ./lichee -build -i ../data/ccRCC/RMH008.txt -maxVAFAbsent 0.005 - minVAFPresent 0.005 -n 0 -minPrivateClusterSize 2 -e 0.2 -s 1
To prevent initial clusters from being collapsed (for the hgsc case6 patient dataset):$ ./lichee -build -i ../data/hgsc/case6.txt -maxVAFAbsent 0.005 -minVAFPresent 0.01 -n 0 -maxClusterDist 0 -showTree 1
-
Save and visualize results
The resulting trees and sample decomposition information produced by LICHeE can be written to a text file (using the -s option that specifies up to how many top trees should be saved) and visualized via the interactive LICHeE Lineage Tree Viewer GUI (using the -showTree option that specifies how many trees should be displayed). It is also possible to export the best-scoring tree as a DOT file for Graphviz visualization (using the -dot or -dotFile options).
If Graphviz is installed, you can visualize the dot file as in Fig.1B using:
$ dot -Tpdf ../data/ccRCC/RK26.txt.dot -O The output file (default ending in trees.txt) lists each node in the tree(s), the binary presence profile of SSNVs in that node, a vector representing the VAF centroid of that node and members of that node. It also reports the adjacencies of the nodes in the tree(s) and how much each node contributes to the decomposition of each sample within the tumor.
-
Tune additional parameters for lineage tree generation
In some cases, LICHeE may not be able to infer a valid tumor lineage tree for the input dataset under the default parameter setting. In addition, alternative lineage trees might be valid under a different set of parameters. Therefore, it might be useful to tune parameters and explore the possible set of solutions. For instance, reliable SSNV calling results are imperative for lineage tree reconstruction. Since LICHeE uses a heuristic method to call SSNVs that heavily relies on appropriate values of the -maxVAFAbsent and -minVAFPresent parameters, adjusting these parameters to reflect the expected noise levels in the data, or supplying pre-computed calls can be very useful.
As an example, the trees obtained for the ccRCC RK26 patient dataset differ as follows based on the values of these parameters (e.g. the higher -maxVAFAbsent threshold of 0.01 will filter out many more SSNVs as noise).
$ ./lichee -build -i ../data/ccRCC/RK26.txt -maxVAFAbsent 0.01 -minVAFPresent 0.2 -n 0 -showTree 1 -color $ ./lichee -build -i ../data/ccRCC/RK26.txt -maxVAFAbsent 0.005 -minVAFPresent 0.005 -n 0 -showTree 1 -color -
Adjust criteria for incorporating clusters into the constraint network for futher refinement (optional)
As a further refinement, it might be useful to adjust the criteria for incorporating clusters into the constraint network. For example, clusters that contain only a few SSNVs are more likely to represent mis-called presence patterns and can be filtered out by increasing the -minClusterSize and -minPrivateClusterSize parameters. The parameter -minRobustNodeSupport (which determines how many robustly-called SSNVs are required for a node to be non-removable) can be increased to iteratively remove nodes from the network while no valid trees are found automatically. For very noisy data (e.g. due to low coverage), nearby clusters can also be collapsed using the -maxClusterDist parameter as described above (or, additionally, the -e parameter can be increased). On the other hand, adjusting these parameters in the opposite direction can result in more granular trees and is advisable on less noisy datasets in order to get the most informative results.
-
Account for CNVs (Copy Number Variants)
Since LICHeE does not detect and incorporate CNVs explicitly into its model, CNVs can be a major cause for the violation of phylogenetic constraints applied to VAF values, and the resulting absence of valid lineage trees. While some of the above parameters can remedy the effect of CNVs (e.g. clustering-related parameters and the -e parameter), providing pre-computed CP values instead of VAFs would be the most effective approach to overcome this limitation. These can be provided to LICHeE directly instead of VAFs and enabled using the -cp flag.
Finally, orthogonal data and information about the dataset can be helpful in analyzing the resulting lineage trees when multiple trees are produced.
Figure 1.
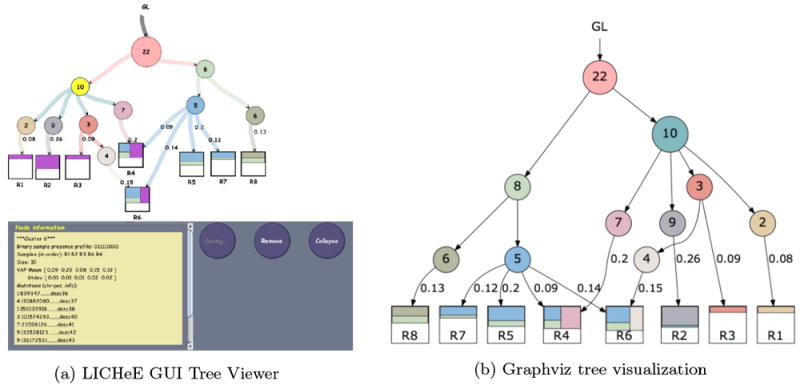
Visualization of lineage tree
3 BASIC PROTOCOL 2: BAMSE
In this section, we present a new probabilistic technique, BAMSE, for inferring cancer phylogenetic trees. Considering possible evolutionary scenarios for tumor evolution as models, BAMSE performs a search over the model space and reports the models that best fit the read count profile for the somatic mutations. BAMSE requires the number of reference and variant read counts for each SSNV across the tumor samples. The output is the description of top scoring models and one DOT file per model for visualization. BAMSE is freely available at https://github.com/HoseinT/BAMSE.
Necessary Resources
System Requirements: A computer with Python 2.7 Interpreter
Files: text file with SSNVs and their variant and reference read counts across the samples
-
Install BAMSE dependencies:
Add scikit-learn [14] and CVXPY [15] to your python installation
-
Prepare the Data
Make a tab delimited file including somatic mutations as rows and including columns named <sample_name>.ref for reference reads and <sample_name>.var for each sample.
-
Run BAMSE
use this syntax to run BAMSE:
$ python2 bamse.py [-e sequencing_error] [-s sparsity] [-nc num_candidates] [-n maximum_subclones] [-t top_trees] <inputfile> Input Description input_file A tab delimited input file sparsity A number between zero and one that represents the prior probability that any subclone is absent at any sample, negative values are interpreted as zero num_candidates Number of initial clusterings to pick for tree analysis maximum_subclones Maximum number of subclones top_trees Number of top solutions to show The results are saved in the bamse.pickle file and also in a text file for each top model. The entries of the list inside the pickle file is a sorted list of top trees. Text file names are numbered and the number 0 represents the model with highest score. Each text file (equivalently a pickle file entry) provides additional information on a model. i.e. its tree structure, its score and maximum likelihood subclone fractions.
-
Visualize the Results
For each reported model two DOT files are created. One of them visualizes subclone usage per sample, showing the clonal make up of eash sample. The other illustrates details about the subclones and the evolutionary tree connecting them. The details include the number of mutations in that subclone and the percentage of cells harboring those mutations in each sample. These files are easily visualized using Graphviz. For example:
dot -Tpdf tree_number_0_samples.dot -O and dot -Tpdf tree\_number_0_clusters.dot -O For example to generate likely evolutionary scenarios for sample EV006 of the ccRCC study, we assume the number of subclones is not larger than 15, sequencing error rate is 0.001. To inform BAMSE to test 20 initial clusterings for each possible number of subclones and generate output for the top scoring 5 trees with sparsity parameter set to 0.1, we use the command:
$ python2 bamse.py -e 0.001 -s 0.1 -nc 20 -m 15 -t 5 EV006.dat The visualized result for the top tree is in Fig[3,4]:
In Fig[3], each subclone is denoted by a letter followed by the number of mutations in that subclone. For each subclone, the maximum likelihood fraction of cells that carry the mutations originating in that subclone are plotted for each sample. Fig[4] shows the subclonal composition for each of the samples i.e which subclones are present in the sample and what fraction of cells they contain.
4 Guidelines for Understanding Results
4.1 LICHeE
The ‘-s,-save’ parameter allows inferred trees to be saved to a text file. This output file is divided into multiple sections. The first of which describes the nodes present in the tree. Below in an example of the first three lines of this section for the tree shown in Fig 2A.
Figure 2.
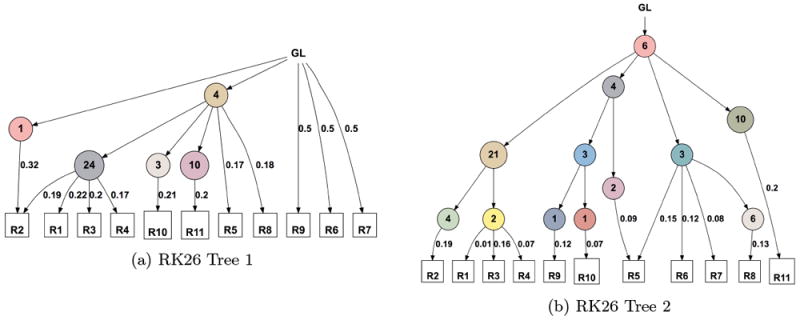
Trees build with different parameters
Nodes:
| 1 011111101001 [ 0.21 0.25 0.23 0.2 0.23 0.19 0.17 0.18] snv1 snv2 snv3 snv4 |
| 2 011110000000 [ 0.19 0.22 0.2 0.17] snv9 snv28 snv5 snv6 snv7 snv8 snv10 snv11 snv12 |
| snv13 snv14 snv15 snv16 snv17 snv18 snv19 snv20 snv21 snv22 snv23 snv24 snv25 |
| snv26 snv27 |
The output gives the node id in the first column followed by the binary presence of mutations found in that node. In the third column is a vector which represents the centroid VAFs for that node followed by all the mutations which were assigned to that node.
The second section of the text output describes connectivity between nodes:
| ****Tree 0**** |
| 0 -> 4 |
| 0 -> 1 |
| 1 -> 5 |
| 1 -> 3 |
| 1 -> 2 |
| Error score: 0.041299345649182535 |
Numbers shown here correspond to node ids from the aforementioned section of the file. Next, the sample decomposition for each sample in the tree is displayed and how much each node contributes to the genomic makeup of the sample. For example see the breakdown of sample R2 in Fig 2A below:
| Sample lineage decomposition: R2 |
| GL |
| …..010000000000: 0.323 [0] |
| …..011111101001: 0.211 [0.032] |
| ……….011110000000: 0.185 [0.049] |
The final section of the output file provides additional information about the mutations which have been placed on the tree.
4.2 BAMSE
In its text output, BAMSE specifies tree structure using asciitree format. For the EV006 solution shoown in Fig[3], the section of text output describing the tree is:
Figure 3.
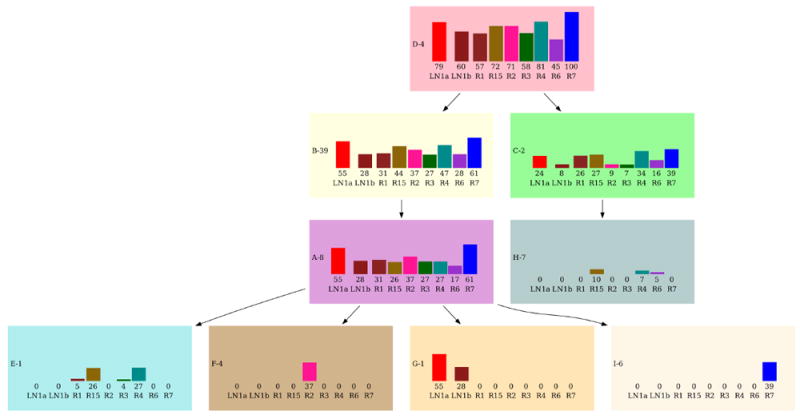
Clonal evolution plot of the top BAMSE solution for EV006
| logscore = 1049.694176 |
| tree = [ 1, 3, 3, -1, 0, 0, 0, 2, 0] |
| D [ 0.79 0.6 0.57 0.72 0.71 0.58 0.81 0.45 1. ] |
| +-- C [ 0.24 0.08 0.26 0.27 0.09 0.07 0.34 0.16 0.39] |
| ∣ +-- H [-0. -0. 0. 0.1 -0. -0. 0.07 0.05 -0. ] |
| +-- B [ 0.55 0.28 0.31 0.44 0.37 0.27 0.47 0.28 0.61] |
| +-- A [ 0.55 0.28 0.31 0.26 0.37 0.27 0.27 0.17 0.61] |
| +-- F [ 0. 0. 0. 0. 0.37 0. 0. 0. 0. ] |
| +-- E [ 0. 0. 0.05 0.26 0. 0.04 0.27 0. 0. ] |
| +-- G [ 0.55 0.28 0. 0. 0. 0. 0. 0. 0. ] |
| +-- I [ 0. -0. 0. -0. -0. -0. -0. -0. 0.39] |
this shows the natural logarthm of tree score, the vector representation of the tree, a text based drawing of the tree along with the maximum likelihood VAF for each subclone at each sample. The vector representation is included for more convinient programmatic access. In this format the nodes are labeled with non-negative integers and T[i] shows the label for the parent of node labeled i. The root node has T[i] = −1. For each subclone, the row number of its mutations in the input file are also listed:
| A [0, 23, 24, 25, 30, 33, 45, 51] |
| B [1, 2, 3, 4, 5, 6, 7, 8, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 26, 27, |
| 28, 29, 31, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 46, 47] |
| C [9, 52] |
| D [32, 48, 49, 50] |
| E [53] |
| F [54, 55, 56, 57] |
| G [58] |
| H [59, 66, 67, 68, 69, 70, 71] |
| I [60, 61, 62, 63, 64, 65] |
this shows the natural logarthm of tree score, the vector representation of the tree, a text based drawing of the tree along with the maximum likelihood VAF for each subclone at each sample. The vector representation is included for more convinient programmatic access. In this format the nodes are labeled with non-negative integers and T[i] shows the label for the parent of node labeled i. The root node has T[i] = −1. For each subclone, the row number of its mutations in the input file are also listed:
| A [0, 23, 24, 25, 30, 33, 45, 51] |
| B [1, 2, 3, 4, 5, 6, 7, 8, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 26, 27, |
| 28, 29, 31, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 46, 47] |
| C [9, 52] |
| D [32, 48, 49, 50] |
| E [53] |
| F [54, 55, 56, 57] |
| G [58] |
| H [59, 66, 67, 68, 69, 70, 71] |
| I [60, 61, 62, 63, 64, 65] |
The maximum likelihood fraction of cells inside each subclone are also provided.
5 Commentary
5.1 Background information
Recently, several studies have been directed at comparing multiple tumor samples from the patient with samples representing different time points in tumor progression or distinct regions of the same tumor [7, 8]. To study these multi-sample datasets, tumor phylogenies are normally inferred manually from a small number of SSNVs and their VAFs along with presence/absence profiles [9, 10], and the ability to scale such studies to handle large, highly heterogeneous samples per patients was lacking in the available approaches. LICHeE was developed to address this need to build phylogenies from large numbers of SSNVs and to resolve the evolutionary timing of these mutations.
LICHeE also represents an improvement over developed methods which exist in a similar functional space. LICHeE is able to handle a large number of samples unlike SubcloneSeeker [11] which takes clusters of variant cell prevalence (CP) values and outputs all possible subclone structures and finds a compatible tree across samples but cannot do this for more than two samples. PhyloSub [12] is another approach to inferring phylogenies but is ideal for datasets with few mutations that resolve to relatively simple topologies as performance dwindles on larger multi-sample datasets. CITUP [13] is a combinatorial solution to this problem that uses an integer programming formulation to find the optimal lineage tree given the VAF data observed in samples, however its optimization problem may become exceedingly difficult with an arbitrarily large lineage tree. LICHeE was developed to provide a useful alternative to these approaches in order to infer tumor phylogenies.
BAMSE is also a unique approach to building tumor phylogenies when multiple samples are available. It is able to assess the fit of multiple models to the data in order to arrive at the most probable tumor phylogeny. While probabilistic, BAMSE is able to avoid the computational burden that is associated with using Markov chain Monte Carlo (MCMC) sampling by integrating out continuous parameters. The discrete parameters remaining are the assignment of mutations to subclones and arrangement of subclones on the evolutionary tree. BAMSE uses heuristic clustering to choose candidates for the clustering, and uses a branch and bound algorithm to get top scoring trees for each configuration. While many tools also rely on external clustering tools to obtain the number of subclones, BAMSE has its own built-in clustering method which allows it to be independent. CNVs can also be considered by BAMSE as it allows for the incorporation of copy number information into the frequency probability distribution for mutations that fall within a CNV region.
5.2 Critical Parameters
In cases where binary presence/absence profiles of somatic mutations are not provided to LICHeE and LICHeE is required to calculate these profiles, special attention must be paid to the selected values for ‘-maxVAFAbsent’ and ‘-minVAFPresent’ as these affect the clustering of mutations and the ability to find a valid tree within the dataset. If ‘-e’ is used to adjust the error margin, too wide of a margin will affect the ability of LICHeE to produce an informative tree based on constraints of perfect phylogeny so caution must be exercised in relaxing this parameter.
For BAMSE, the maximum number of subclones, the number of candidates for each are very important. Higher these numbers, the algorithm will take more time to run but will test more candidates and the outputs are more accurate.
5.3 Troubleshooting
LICHeE outputs a log detailing the execution of each step of the algorithm (with more information provided using the verbose, -v, flag). This log can be very useful when trying to diagnose the performance of the program and view any of its intermediate results. In particular, it provides information about SSNV calling and filtering, clustering, the structure of the resulting constraint network, tree scoring, and any other operations controlled by various parameter settings. For example, any cluster removal or collapse operations triggered by the specified parameter settings are recorded in the log as shown in the following snippet (extracted from the log of the ccRCC RK26 patient):
| … |
| Clustering results for group: 010000010000 |
| Size: 1 |
| VAF Mean: [ 0.08 0.01 ] Stdev: [ 0 0 ] |
| **Filtered due to cluster size constraint (010000010000 size 1): |
| chr6 7246926 desc43 0.00 0.083786725 0 0 0 0 0 0.009090909 0 0 0 0 |
| Collapse clusters: group = 000001000000 cluster 0 and 1 distance = 0.0659543605. |
| New cluster: Size: 10 |
| VAF Mean: [ 0.2 ] Stdev: [ 0.03 ] |
| … |
Using the log, the user can also trace why a particular SSNV was not included in the final output tree(s) (e.g. due to filtering based on the maximum VAF allowed or due to cluster size constraints) by searching the log for the “Filtered” keyword or for the unique descriptor of the SSNV (the log will output the SSNV entry line for each filtered SSNV as it appears in the input file). Furthermore, when no valid trees are found, examining the cluster centroid VAFs and the topology of the constraint network can be helpful to determine why at least one phylogenetic constraint is violated in each candidate embedded spanning trees.
There is no specific troubleshooting log for BAMSE to report here.
Figure 4.
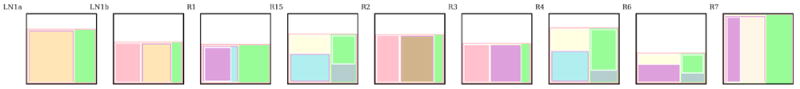
Sublonal composition of the samples for the top BMASE solution for EV006
Acknowledgments
This work was supported by start up funds (Weill Cornell Medicine) to IH. We would also like to acknowledge the support provided by the Tri-Institutional Training Program in Computational Biology and Medicine (CBM) funded by an NIH T32 grant to CR.
References
- 1.Liu Y, Zhang J, Li L, et al. Genomic heterogeneity of multiple synchronous lung cancer. Nature Communications. 2016;7:13200. doi: 10.1038/ncomms13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu M, Liu Y, Di J, et al. Multi-region and single-cell sequencing reveal variable genomic heterogeneity in rectal cancer. BMC Cancer. 2017;17:787. doi: 10.1186/s12885-017-3777-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Popic V, Salari R, Hajirasouliha I, Kashef-Haghighi D, West RB, Batzoglou S. Fast and scalable inference of multi-sample cancer lineages. Genome Biology. 2015;16(1):91. doi: 10.1186/s13059-015-0647-8. http://doi.org/10.1186/s13059-015-0647-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nature genetics. 2014;46:225–233. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bashashati A, Ha G, Tone A, Ding J, Prentice LM, Roth A, Rosner J, Shumansky K, Kalloger S, Senz J. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. The Journal of pathology. 2013;231:21–34. doi: 10.1002/path.4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toosi H, Moeini A, Hajirasouliha I. BAMSE: Bayesian model selection for tumor phylogeny inference among multiple tumor samples. Proceedings of IEEE Advances in Bio and Medical Sciences ICCABS. 2017 doi: 10.1186/s12859-019-2824-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–10. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–26. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newburger DE, Kashef-Haghighi D, Weng Z, Salari R, Sweeney RT, Brunner AL, et al. Genome evolution during progression to breast cancer. Genome Res. 2013;23:1097–108. doi: 10.1101/gr.151670.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green MR, Gentles AJ, Nair RV, Irish JM, Kihira S, Liu CL, et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood. 2013;121:1604–11. doi: 10.1182/blood-2012-09-457283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiao Y, Quinlan AR, Jazaeri AA, Verhaak RG, Wheeler DA, Marth GT, et al. Subcloneseeker: a computational framework for reconstructing tumor clone structure for cancer variant interpretation and prioritization. Genome Biol. 2014;15:443. doi: 10.1186/s13059-014-0443-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiao W, Vembu S, Deshwar AG, Stein L, Morris Q. Inferring clonal evolution of tumors from single nucleotide somatic mutations. BMC Bioinformatics. 2014;15:35. doi: 10.1186/1471-2105-15-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malikic S, McPherson AW, Donmez N, Sahinalp CS. Clonality inference in multiple tumor samples using phylogeny. Bioinformatics. 2015;31:1349–56. doi: 10.1093/bioinformatics/btv003. [DOI] [PubMed] [Google Scholar]
- 14.Pedregosa Fabian, Varoquaux Gaël, Gramfort Alexandre, Michel Vincent, Thirion Bertrand, Grisel Olivier, Blondel Mathieu, Prettenhofer Peter, Weiss Ron, Dubourg Vincent, Vanderplas Jake, Passos Alexandre, Cournapeau David, Brucher Matthieu, Perrot Matthieu, Duchesnay Édouard. Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research. 2011;12:2825–2830. [Google Scholar]
- 15.Diamond Steven, Boyd Stephen. CVXPY: A Python-Embedded Modeling Language for Convex Optimization. Journal of Machine Learning Research. 2016;17(83):1–5. [PMC free article] [PubMed] [Google Scholar]