

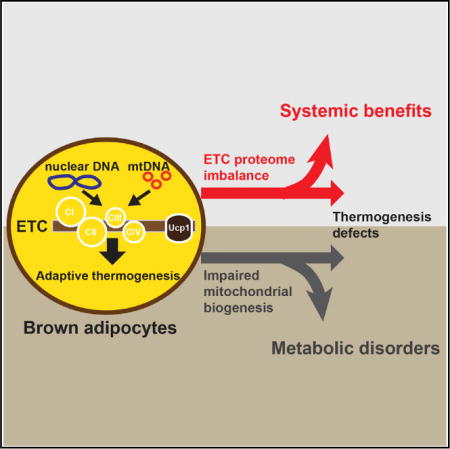
SUMMARY
Brown adipose tissue (BAT) thermogenesis is critical for thermoregulation and contributes to total energy expenditure. However, whether BAT has non-thermogenic functions is largely unknown. Here, we describe that BAT-specific liver kinase b1 knockout (Lkb1BKO) mice exhibited impaired BAT mitochondrial respiration and thermogenesis but reduced adiposity and liver triglyceride accumulation under high-fat-diet feeding at room temperature. Importantly, these metabolic benefits were also present in Lkb1BKO mice at thermoneutrality, where BAT thermogenesis was not required. Mechanistically, decreased mRNA levels of mtDNA-encoded electron transport chain (ETC) subunits and ETC proteome imbalance led to defective BAT mitochondrial respiration in Lkb1BKO mice. Furthermore, reducing mtDNA gene expression directly in BAT by removing mitochondrial transcription factor A (Tfam) in BAT also showed ETC proteome imbalance and the trade-off between BAT thermogenesis and systemic metabolism at room temperature and thermo-neutrality. Collectively, our data demonstrate that ETC proteome imbalance in BAT regulates systemic metabolism independently of thermogenesis.
Graphical abstract
In Brief Masand and Paulo et al. demonstrate that Lkb1 and Tfam regulate mtDNA-encoded ETC gene expression, and their deficiencies in BAT lead to ETC proteome imbalance, a mismatch between proportional ETC complexes. This ETC proteome imbalance locally in BAT can cause systemic adaptive responses, which ultimately result in metabolic fitness.
INTRODUCTION
Energy balance requires equivalent energy intake and energy expenditure. Energy is expended primarily through basal metabolism, physical activity, and adaptive thermogenesis. Adaptive thermogenesis, which is the process of body heat production in response to environmental changes, occurs in the mitochondria of brown adipose tissue (BAT) (Cannon and Nedergaard, 2004, 2011). BAT contains specialized mitochondria-rich brown adipocytes whose thermogenic functionality is conferred by the uncoupling protein 1 (Ucp1) (Feldmann et al., 2009; Golozoubova et al., 2001, 2006; Krauss et al., 2005; Shabalina et al., 2013). It has been demonstrated that cold-induced sympathetic activity can trigger lipolysis and acutely activate Ucp1, along with promoting mitochondrial biogenesis chronically through the induction of peroxisome proliferator-activated receptor gamma coactivator 1 α (Pgc1 α) (Lowell and Spiegelman, 2000). Ucp1-mediated adaptive thermogenesis in BAT plays a crucial role in energy homeostasis in rodents. Dysfunction of adaptive thermogenesis precedes the development of obesity in ob/ob mice (Trayhurn et al., 1977). Furthermore, genetic ablation of BAT or inactivation of β-adrenergic receptors (β-ARs) in mice leads to defective thermoregulation and obesity (Bachman et al., 2002; Klaus et al., 1998; Lowell et al., 1993), while BAT transplant improves whole-body energy metabolism in mice (Gunawardana and Piston, 2012; Liu et al., 2013; Stanford et al., 2013).
The presence of thermogenic UCP1-positive fat depots in adult humans (human brown fat) has been recognized using 18F-fluoro-deoxy-glucose positron emission tomography (18F-FDG PET) with computer-assisted tomography (Cypess et al., 2009; Saito et al., 2009; van Marken Lichtenbelt et al., 2009; Virtanen et al., 2009). Human brown fat activity gradually declines with aging and metabolic diseases (Cannon and Nedergaard, 2004; Cypess et al., 2009; Saito et al., 2009; van Marken Lichtenbelt et al., 2009). However, whether the increase of adaptive thermogenesis in humans is quantitatively sufficient to explain the association between brown fat abundance/activity and metabolic health remains under debate (Dulloo et al., 2012; Major et al., 2007; Rosenbaum and Leibel, 2010).
The contribution of BAT adaptive thermogenesis to total energy expenditure is dependent on ambient temperature (Abreu-Vieira et al., 2015; Cannon and Nedergaard, 2011; Ganeshan and Chawla, 2017; Maloney et al., 2014). At room temperature (RT), BAT-mediated adaptive thermogenesis is already active, and defective adaptive thermogenesis is often associated with reduced energy expenditure and development of obesity in many animal models. At thermoneutrality (~30°C for mouse), BAT-mediated adaptive thermogenesis is not needed to maintain core body temperature and contributes little to total energy expenditure. Hence, metabolic assessment at both RT and thermoneutrality is required to determine whether any genetic or pharmacological means regulates energy metabolism through manipulating BAT adaptive thermogenesis. Indeed, the pan adipocyte-specific carnitine palmitoyltransferase 2 (Cpt2, an obligate step in mitochondrial long-chain fatty acid oxidation) knockout mice exhibited defective fatty acid oxidation and adaptive thermogenesis at both RT and thermoneutrality (Lee et al., 2015, 2016). However, these mice only exhibited increased adiposity at RT, suggesting that decreased adaptive thermogenesis in BAT indeed contributed to the development of obesity in these mice at RT (Lee et al., 2015, 2016). Moreover, since the thermoneutral zone for humans is around 22°C, metabolic studies in mouse models at thermoneutrality would be more relevant to human physiology.
Sympathetic activity and βAR signaling drive adaptive thermogenesis in adipose tissue (Lowell and Spiegelman, 2000). Previously, we have demonstrated how liver kinase b 1 (Lkb1) suppressed βAR-induced transcription response and regulated the formation of cold-induced brown-like adipocytes in subcutaneous adipose tissue (Wang et al., 2017). Here, we show that brown adipocyte-specific Lkb1 knockout mice (Lkb1BKO) had improved metabolic performance under a high-fat diet (HFD) at both RT and thermoneutrality, despite impaired mitochondrial respiration in BAT and defective adaptive thermogenesis. The disassociation between BAT thermogenic function and energy homeostasis was due to a specific blockage of mtDNA gene expression and then electron transport chain (ETC) proteome imbalance. This conclusion was further confirmed in an additional mouse model, the brown adipocyte-specific mitochondrial transcription factor A knockout mice (TfamBKO). Therefore, BAT can promote organismal metabolic fitness at the expense of its own adaptive thermogenic function. Exploiting this non-thermo-genic function of BAT may provide novel means to tackle metabolic disorders.
RESULTS
Lkb1 Regulates Mitochondrial Respiration in BAT and Adaptive Thermogenesis
We have generated brown adipocyte-specific Lkb1 knockout mice, Ucp1-Cre; Lkb1f/f (Lkb1BKO), to investigate the roles of Lkb1 in brown adipocytes. Immunoblots confirmed that Lkb1 was efficiently deleted in BAT of Lkb1BKO mice (Figures 1A and 1B). AMPK is a known substrate of Lkb1 (Alessi et al., 2006). BAT AMPK activity was also diminished in Lkb1BKO mice (Figures 1A and 1B), further confirming the loss of Lkb1 in BAT. In addition, hematoxylin and eosin (H&E) staining of BAT showed that brown adipocytes were bigger in size (hypertrophy), and a higher percentage of brown adipocytes exhibited unilocu-lar lipid droplet morphology in Lkb1BKO mice (Figures S1A–S1C), consistent with histological observations in the pan adipocyte-specific Lkb1 knockout mice (Lkb1AKO) (Shan et al., 2016; Wang et al., 2017). Lkb1 deficiency in BAT did not affect overall fat mobilization/utilization in these mice; the basal and Forskolin-induced lipolytic activities in BAT and epididymal WAT (eWAT) were similar between control and Lkb1BKO mice (Figures S1D and S1E). In addition, there was no significant difference in in vivo lipolytic response to β3-adrenergic stimulation (mimicked by CL316,243/CL administration) (Figure S1F).
Figure 1. Lkb1BKO Mice Have Defective Adaptive Thermogenesis Regardless of Ucp1 Levels.
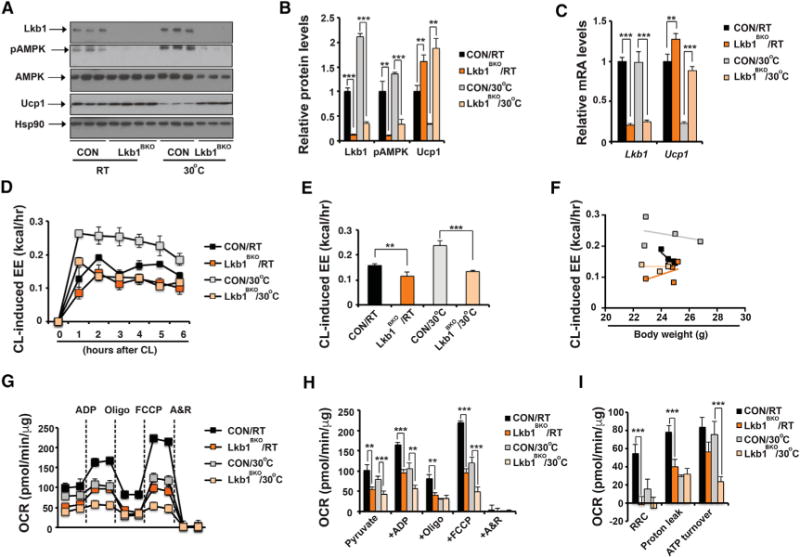
(A) Immunoblots showing amounts of Lkb1, pThr172-Ampk, total Ampk, Ucp1, and Hsp90 in BAT of ~8- to 10-week-old male control (CON) and Lkb1BKO mice housed at room temperature (RT) and thermoneutrality (30°C).
(B) Quantitative analysis of western blot in (A).
(C) qPCR of Lkb1 and Ucp1 mRNA levels in BAT of ~8- to 10-week-old male CON and Lkb1BKO mice housed at RT and 30°C. Sample size: CON/RT (n = 13), Lkb1BKO/RT (n = 9), CON/30°C (n = 6), and Lkb1BKO/30°C (n = 6).
(D and E) Recordings of energy expenditure (EE) increases in response to CL (D) and hourly average of CL-induced EE (E) in ~8- to 10-week-old male CON and Lkb1BKO mice housed at RT and 30°C. CL injected at 0 hr.
(F) Regression plot of CL-induced EE as a function of mouse weight in ~8- to 10-week-old male CON and Lkb1BKO mice housed at RT and 30°C. Sample size: CON/RT (n = 4), Lkb1BKO/RT (n = 3), CON/30°C (n = 4), and Lkb1BKO/30°C (n = 4).
(G) Seahorse experiments measuring oxygen consumption rates (OCR) of isolated mitochondria from BAT of ~8- to 10-week-old male CON and Lkb1BKO mice housed at RT and 30°C, upon addition of ADP, Oligo, FCCP, and A&R.
(H and I) Average OCR (H) and RRC, proton leak, and ATP turnover rates (I) in seahorse measurements. Sample size: CON/RT (n = 4), Lkb1BKO/RT (n = 6), CON/30°C (n = 9), and Lkb1BKO/30°C (n = 9).
Oligo, oligomycin; FCCP, carbonyl cyanide-p-trifluromethoxyphenylhydrazone; A&R, antimycin and rotenone; RRC, reserve respiratory capacity. Data are means and error bars are ±SEM. Student’s t test, **p < 0.05 and ***p < 0.01.
Housing temperature affects BAT thermogenic capacity in mice (Cannon and Nedergaard, 2011; Ganeshan and Chawla, 2017). We therefore performed experiments at both RT and thermoneutrality. For the thermoneutrality experiments, ~5-week-old mice were acclimatized at 30°C for an additional 3–4 weeks before experiments. We first measured Ucp1 expression and thermogenic activity in control and Lkb1BKO mice housed at RT and thermoneutrality. Ucp1 mRNA and protein levels were upregulated at both RT and thermoneutrality (Figures 1A–1C). Indirect calorimetry analysis indicated that Lkb1BKO mice had similar energy expenditure (EE), basal oxygen consumption (VO2), respiratory exchange ratio (RER), food intake and physical activity at both RT and thermoneutrality (Figures S1K–S1P). Scatterplots showed that there were no differences in night and day EE between control and Lkb1BKO mice at either RT or thermoneutrality (Figures S1H and S1I). Post hoc pairwise comparisons of mean difference in EE, using body weight as a covariate in the ANCOVA, further showed that ambient temperatures, not genotypes, caused the difference in EE in CON and Lkb1BKO mice (Figure S1J). However, the EE increase in response to CL stimulation in Lkb1BKO mice was reduced at RT and thermoneutrality (Figures 1D and 1E). Linear regression analysis indicated that the slopes of change of CL-induced EE per unit change of body weight were not overlapping between CON and Lkb1BKO mice at both RT and 30°C (Figure 1F). We also performed an acute 4°C cold tolerance test to determine the contribution of BAT adaptive thermogenesis to total thermogenic capacity. Lkb1BKO mice singly housed at RT maintained their core temperature initially, but succumbed to the 4°C cold exposure despite elevated Ucp1 levels (Figures S1G and 1A–1C). Both control and Lkb1BKO mice housed at thermoneutrality failed to maintain their core temperature. Therefore, BAT Ucp1 abundance alone does not determine thermogenic capacity in Lkb1BKO mice.
Adaptive thermogenesis in BAT requires mitochondrial respiration followed by the Ucp1-mediated uncoupling of mitochondrial respiration from ATP production. We therefore sought to determine whether the defective adaptive thermogenesis observed in Lkb1BKO mice was due to mitochondrial respiration defects in the BAT. Respiration was measured in isolated mitochondria from BAT of Lkb1BKO mice housed at RT or thermoneutrality, which eliminates potential differences in mitochondrial numbers. BAT mitochondria from Lkb1BKO mice housed at both temperatures exhibited reduced respiration rates (Figures 1G and 1H). Notably, these mitochondria also showed significant defects in uncoupled respiration, induced by the uncoupler FCCP (carbonyl cyanide-p-trifluoro-methoxyphenylhydrazone) (Figures 1G and 1H). The reserve respiratory capacity (RRC), also known as spare respiratory capacity, reflects the mitochondrial ability to respond to increased energy demand under stress. RRC is calculated as the difference between maximum uncoupled respiration and state3 respiration in response to ADP. RRC in the BAT mitochondria of Lkb1BKO mice was reduced (Figure 1I), indicating that these mitochondria were constantly operating close to their bioenergetic limit. The proton leak was also reduced by half in the BAT mitochondria of Lkb1BKO mice at RT without a difference in Ucp1 expression and remained unchanged at thermoneutral conditions despite more abundant Ucp1 (Figure 1I). ATP turnover rate was largely unaffected in Lkb1BKO mice at RT (Figure 1I), but it was reduced under thermoneutral conditions. From these results, we conclude that BAT mitochondria from Lkb1BKO mice exhibit respiratory abnormalities, which accounts for the defective adaptive thermogenesis observed in the Lkb1BKO mice. Notably, the mitochondrial respiration defects were not determined in the BAT of the pan adipocyte-specific Lkb1-deficient mice (Shan et al., 2016). The increased VO2 per body weight in these pan adipocyte-specific Lkb1-deficient mice could be due to their smaller body weight under HFD.
Lkb1BKO Mice Show ETC Proteome Imbalance in BAT
Mitochondrial respiration is dependent on the activity of the ETC, consisting of five protein complexes (C-I to C-V). A proton gradient across the mitochondrial membrane generated by C-I to C-IV drives heat production through Ucp1-mediated uncoupling in brown adipocytes, rather than producing ATP through C-V (ATP synthase). Since BAT mitochondria from Lkb1BKO mice had respiratory defects, we questioned whether the Lkb1 deficiency was causing ETC defects in these mice. We first investigated if the composition of the ETC proteome was affected in the BAT of Lkb1BKO mice. Blue-native PAGE revealed no structural anomalies; all the complexes and super-complexes were readily detected in BAT mitochondria of these mice (Figure S2A). However, immunoblots showed reductions of mt-Co1 (a C-IV subunit) and Uqcrc2 (a C-III subunit) at both RT and thermoneutrality (Figures S2B and S2C).
To obtain a global view of the BAT mitochondrial proteome, we performed mass spectrometry analysis of isolated mitochondria from BAT of control and Lkb1BKO mice at RT and thermoneutrality (Table S2) (Cox et al., 2014; Cox and Mann, 2008). In each condition, we identified approximately 760 mitochondrial proteins (roughly 70% of the mitochondrial proteins listed in MitoCarta2.0) (Figure S2D). Only the mitochondrial proteins were selected for further analyses. Principal component analysis revealed variation between the mitochondrial proteomes of controls at both RT and thermoneutrality but no variation between the mitochondrial proteomes in the Lkb1BKO mice at the two temperature conditions (Figure S2E). Using 1.5-fold difference and adjusted p value (false positive rate) <0.1 as the cutoff, more proteins were found deregulated in Lkb1BKO mice at thermoneutrality (Figures S2F and S2G). The differences between control and LBK1BKO mice found at RT (18 of 35 upregulated proteins and 28 of 41 downregulated proteins) were maintained at thermoneutrality (Figures S2H and S2I). Protein-complex enrichment analysis revealed that C-III and C-IV subunits were among the most downregulated proteins in Lkb1BKO mice (Figure 2A). We found lower protein abundance for most C-IV protein subunits in Lkb1BKO mice, while most C-IV assembly factors were upregulated (Figure 2B). Indeed, immunoblots confirmed that C-IV subunits mt-Co1, mt-Co2, Cox4, Cox5b, and Cox6b1 were reduced, while C-II subunit Sdhb remained unchanged in the BAT of Lkb1BKO mice (Figure 2C). On the other hand, Surf1 protein, an assembly factor for C-IV (Zhu et al., 1998), was increased in the BAT of Lkb1BKO mice (Figure 2C), which might reflect a failed attempt to rescue the C-IV deficiency. Consistently, in vitro C-III and C-IV enzyme activities were also attenuated in the BAT of Lkb1BKO mice (Figure 2D).
Figure 2. BAT Mitochondria of Lkb1BKO Mice Show ETC Proteome Imbalance due to Reduced mtDNA Gene Expression.
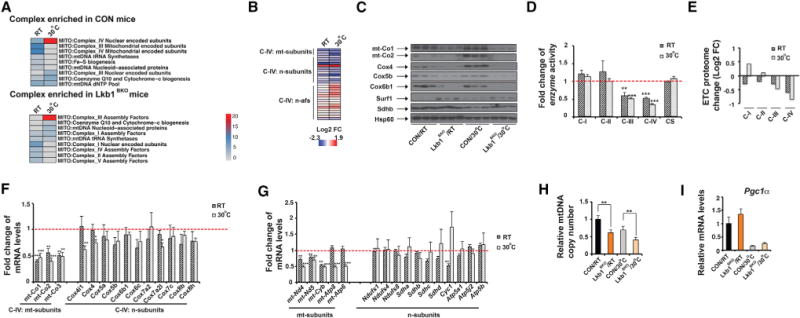
(A) Heatmap showing adjusted p values of protein complex Gene Ontology terms enriched in BAT mitochondria from ~8- to 10-week-old male CON and Lkb1BKO mice housed at RT and 30°C.
(B and C) Heatmap (B) of protein abundance measured by mass spectrometry and immunoblots (C) of C-IV subunits (mt-Co1, mt-Co2, Cox4, Cox5b, and Cox6b1) and assembly factor Surf1 in BAT mitochondria from male CON and Lkb1BKO mice housed at RT and 30°C. mt-subunits, mtDNA-encoded subunits; n-subunits, nuclear-encoded subunits; n-afs, nuclear-encoded assembly factors. Immunoblots of C-II subunit Sdhb and Hsp60 are also shown.
(D) Relative in vitro enzyme activities of C-I to C-IV and citrate synthase (CS) in BAT of ~8- to 10-week-old male and female CON and Lkb1BKO mice housed at RT and 30°C. Sample size: CON/RT (n = 6), Lkb1BKO/RT (n = 6), CON/30°C (n = 6), and Lkb1BKO/30°C (n = 6).
(E) Log2 fold change values of each ETC proteome from BAT of Lkb1BKO mice housed at RT and 30°C.
(F) Fold change of steady-state mRNA levels of mtDNA-encoded and nuclear-encoded C-IV subunits in BAT of ~8- to 10-week-old male and female CON and Lkb1BKO mice at RT and 30°C.
(G) Fold change of mRNA levels of mtDNA-encoded and nuclear-encoded C-I, C-II, C-III, and C-V subunits in the BAT of ~8- to 10-week-old male and female CON and Lkb1BKO mice housed at RT and 30°C. Sample size: CON/RT (n = 5), Lkb1BKO/RT (n = 7), CON/30°C (n = 6), and Lkb1BKO/30 C (n = 6).
(H) Relative mtDNA copy number in BAT of ~8- to 10-week-old male Lkb1BKO mice at RT and 30°C. Sample size: CON/RT (n = 5), Lkb1BKO/RT (n = 5), CON/30°C (n = 8), and Lkb1BKO/30°C (n = 8).
(I) Relative mRNA levels of Pgc1a in BAT of ~8- to 10-week-old male and female CON and Lkb1BKO mice housed at RT and 30°C. Sample size: CON/RT (n = 5), Lkb1BKO/RT (n = 7), CON/30°C (n = 6), and Lkb1BKO/30°C (n = 6).
Data are means and error bars are ±SEM. Student’s t test, *p < 0.1, **p < 0.05 and ***p < 0.01.
Optimal ETC activity requires precise control of the composition between C-I to C-V, and its proteome imbalance leads to respiratory defects. To quantitate this BAT ETC proteome in the Lkb1BKO mice, we evaluated proteome changes of an individual complex by calculating the average log2 fold change (log2FC) values for all identified proteins within each complex from our mass spectrometry dataset. These values reflect proteome change of each ETC complex as a whole, rather than the change of an individual protein, in the BAT of Lkb1BKO mice. Notably, C-IV was most affected at both RT and thermoneutrality, while C-II, which is entirely nuclear encoded, was least affected (Figure 2E). For instance, BAT mitochondria of Lkb1BKO mice had about a 2-fold relative reduction of the C-IV proteome (compared with the C-II proteome) particularly at thermoneutrality (Figure 2E). This quantitative analysis reveals that significant alteration in the ETC composition, especially reduction of the C-IV proteome, occurs in the BAT of Lkb1BKO mice.
Lkb1 Deficiency in BAT Specifically Regulates mRNA Levels of mtDNA-Encoded ETC Subunits
To determine where these ETC defects stem from, we looked at steady-state mRNA levels of C-IV subunits in the BAT of Lkb1BKO mice. C-IV is a large protein complex consisting of 13 different subunits, which are encoded by both the mitochondrial (mtDNA) and nuclear genomes. Strikingly, steady-state mRNA levels of mtDNA-encoded C-IV subunits (mt-Co1, mt-Co2, and mt-Co3) were downregulated in BAT of Lkb1BKO mice at RT and thermoneutrality, while the steady-state mRNA levels of nuclear-encoded subunits remained largely unchanged (Figure 2F). Interestingly, this mitochondrion-specific regulation of ETC subunit gene expression was not restricted to C-IV. For example, steady-state mRNA levels of mtDNA-encoded ETC subunits of C-I (mt-Nd4 and mt-Nd5), C-III (mt-Cyb) and C-V (mt-Atp8 and mt-Atp6) were also downregulated in the BAT of Lkb1BKO mice, without significant changes in steady-state mRNA levels of nuclear-encoded subunits (Figure 2G). This was consistent with the reduction of mtDNA copy number in the BAT of Lkb1BKO mice at both temperatures (Figure 2H). Therefore, Lkb1 deficiency in BAT specifically decreases steady-state mRNA levels of the mtDNA-encoded ETC subunits, thus leading to an ETC proteome imbalance.
In order to determine whether ETC proteome imbalance in BAT is uniquely affected by Lkb1 deficiency, rather than a universal response associated with defective adaptive thermogenesis, we analyzed ETC proteome in the BAT of thermogenesis defective betaless mice (Bachman et al., 2002). In fact, the C-IV ETC proteome was not affected by βAR deficiency, as protein levels of C-IV subunits were not significantly altered in isolated BAT mitochondria from betaless mice (Figures S2J and S2K). The mRNA levels of Pgc1α, the master regulator of mitochondrial biogenesis, were only reduced in the BAT of betaless mice (Figures 2I and S2O). Consequently, mRNA levels of mtDNA- and nuclear-encoded ETC subunits and Ucp1 were downregulated in the BAT of betaless mice (Figures S2L–S2O), suggesting there was impaired mitochondrial biogenesis in the BAT of betaless mice. This was in sharp contrast with the mitochondrion-specific regulation of ETC subunit gene expression observed in Lkb1BKO mice. Thus, mitochondrial ETC proteome imbalance, rather than impaired mitochondrial biogenesis, causes the adaptive thermogenesis defect in the Lkb1BKO mice.
Since the Lkb1 deficiency in BAT led to reduced mtDNA copy number and mtDNA gene expression, we surveyed mRNA and protein levels of key factors involved in mtDNA replication (Twinkle, Tfam, and Ssbp1), mtDNA transcription (Gfm1, Gfm2, Tfb2m, Mterf2, Mtpap, Polg, and Polrmt), and mitochondrial RNA processing and stability (Grsf1, Lrpprc, and Slirp). None of these factors were significantly downregulated in the BAT of Lkb1BKO mice at both RT and thermoneutrality, although the mRNA levels of some factors were reduced (Figure S2P). In fact, Polrmt, Tfb2m, and Grsf1 proteins were even elevated, which might reflect a compensatory response to impaired mtDNA-encoded gene expression.
Lkb1BKO Mice Exhibit Improved Metabolic Performance Despite Defective Adaptive Thermogenesis
Defective adaptive thermogenesis is often associated with obesity. For example, the betaless mice were prone to the development of obesity and hepatic steatosis (Bachman et al., 2002). Lkb1BKO mice did not show any body weight difference up to 5 months of age under normal chow (Figures S3A–S3C); neither did the Lkb1AKO mice (Wang et al., 2017). We therefore investigated the metabolic phenotypes of Lkb1BKO mice in response to HFD feeding. At RT, BAT of Lkb1BKO mice was hypertrophic and contained an increased percentage of unilocular brown adipocytes (Figures S3D–S3F). However, Lkb1BKO mice gained less body weight after 4-week HFD (Figures 3A, 3B, and S3G). In addition, they had smaller inguinal (iWAT) and epididymal (eWAT) white adipose tissue depots (Figures 3C and 3D), along with smaller adipocytes in eWAT (Figures 3E and 3F). Lkb1BKO mice also showed decreased triglyceride (TG) accumulation in the liver (Figures 3G and 3H), lower fasting glucose levels and a minor reduction of glucose disposal rate in insulin tolerance test (ITT) (Figures S3L and S3N). There were no differences in serum insulin levels, glucose disposal rates in glucose tolerance test (GTT), and insulin-induced Akt phosphorylation in the muscle (Figures S3M and S3O–S3Q). The mRNA levels of macrophage markers (Cd68, F4/80, and Cd11c) and adipokines (Fgf21, adiponectin, and leptin) in eWAT were not changed (Figure S3R). Taken together, Lkb1BKO mice exhibit metabolic benefits under short-term HFD, mainly reduced adiposity and liver TG contents, despite thermogenic defects in BAT.
Figure 3. Lkb1BKO Mice Exhibit Improved Systemic Metabolism after 4-Week HFD.
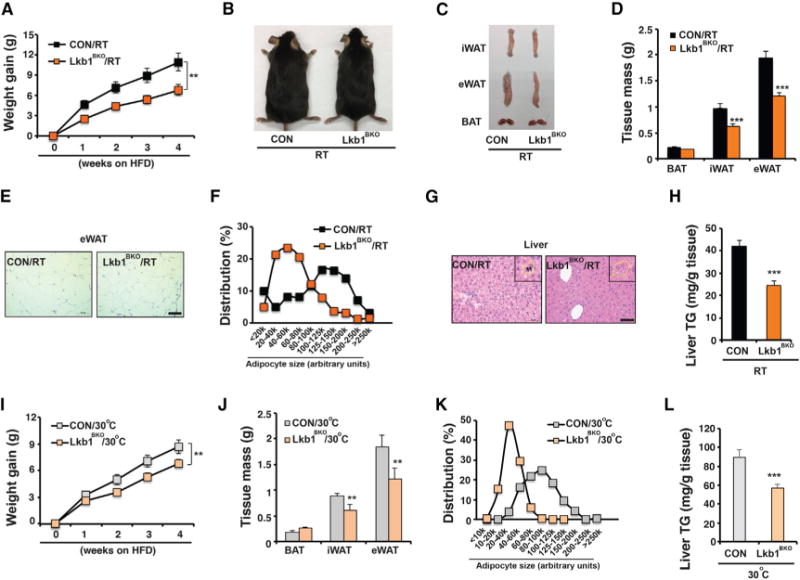
(A) Body weight gain of male CON and Lkb1BKO mice housed at RT after 4-week HFD feeding (started at ~6–8 weeks of age). Sample size: n = 14 for each genotype.
(B and C) Representative images of mice (B) and dissected iWAT, eWAT, and BAT (C) from male CON and Lkb1BKO mice housed at RT after HFD feeding.
(D) Tissue mass of iWAT, eWAT, and BAT from male CON and Lkb1BKO mice housed at RT after HFD. Sample size: CON/RT (n = 4) and Lkb1BKO/RT (n = 6).
(E) Representative H&E staining of eWAT from male CON and Lkb1BKO mice housed at RT after HFD. Scale bar, 100 μm.
(F) Adipocyte size distribution in eWAT from male CON and Lkb1BKO mice housed at RT after HFD. Total adipocytes counted: CON/RT (n = 294) and Lkb1BKO/RT (n = 539).
(G) Representative H&E staining of liver from male CON and Lkb1BKO mice housed at RT after HFD. Scale bar, 50 μm. Insert: hepatocyte circled with a dashed yellow line. Black arrowheads, lipid droplets.
(H) Liver triglyceride content of male CON and Lkb1BKO mice housed at RT after HFD. Sample size: CON/RT (n = 7) and Lkb1BKO/RT (n = 8).
(I) Body weight gain in male CON and Lkb1BKO mice housed at 30°C. Sample size: CON/30 C (n = 17) and Lkb1BKO/30 C (n = 23).
(J–L) Fat tissue mass (J), adipocyte size distribution in eWAT (K), and liver triglyceride content (L) in male CON and Lkb1BKO mice housed at 30°C after HFD.
Sample size: CON/30°C (n = 12) and Lkb1BKO/30°C (n = 15). Total adipocytes counted in eWAT: CON/30°C (n = 476) and Lkb1BKO/30°C (n = 632). Data are means and error bars are ±SEM. Student’s t test, **p < 0.05 and ***p < 0.01.
These metabolic benefits observed in Lkb1BKO mice at RT could be due to certain compensatory thermogenic mechanisms that were more metabolic insufficient (Enerback et al., 1997; Feldmann et al., 2009; Liu et al., 2003). To circumvent this possibility, we investigated whether these metabolic benefits were also present in Lkb1BKO mice at thermoneutrality, since BAT mitochondria remained dysfunctional at thermoneutrality. We found that, at thermoneutrality, Lkb1BKO mice still gained less body weight, showed reduced adipose mass and smaller adipocytes under HFD (Figures 3I–3K and S3H–S3J), despite a complete loss of multilocular morphology in brown adipocytes (Figures S3D–S3F). At thermoneutrality, Lkb1BKO mice also had reduced triglyceride accumulation in the liver (Figures 3L and S3K), although their glucose metabolism (in both GTT and ITT) and marker expression in eWAT were not affected (Figures S3L–S3O and S3R). Lkb1BKO mice showed a mild decrease in insulin-induced Akt phosphorylation in skeletal muscle (Figures S3P and S3Q). Indirect calorimetry experiments showed that Lkb1BKO mice after 4-week HFD exhibited similar EE, VO2, and RER as control mice at either RT or thermoneutrality (Figures S3S–S3Y). There were slight reductions of food intake at RT and physical activity at thermoneutrality in Lkb1BKO mice (Figures S3Z–S3AA). Therefore, contributions of BAT thermogenic function to organismal thermoregulation and metabolic performance are uncoupled in the Lkb1BKO mice.
TfamBKO Mice Exhibit Mitochondrial Respiration Defects Due to Decreased mtDNA Gene Expression
Lkb1 regulates many cellular functions besides mtDNA gene expression (Alessi et al., 2006); thus, ETC proteome imbalance in BAT may not contribute to systemic metabolism in the Lkb1BKO mice. In order to establish the causal relationship between BAT ETC proteome imbalance and systemic metabolic benefits, we further investigated whether reducing mtDNA-encoded gene expression directly in BAT could lead to ETC proteome imbalance and subsequently to systemic metabolic benefits.
Mitochondrial transcription factor A (Tfam) is essential for mtDNA replication and stability as well as transcription of mtDNA-encoded genes, and Tfam deletion in mice led to reduced mtDNA copy number and mtDNA gene expression (Larsson et al., 1998). We therefore generated BAT-specific Tfam knockout mice (Ucp1-Cre; Tfamf/f, TfamBKO) and examined the effect of Tfam deficiency on mtDNA gene expression in BAT. Tfam mRNA and proteins levels were significantly reduced in BAT of TfamBKO mice (Figures S4A and S4B). At RT, BAT from TfamBKO mice was also hypertrophic and contained a higher percentage of unilocular brown adipocytes (Figures S4C–S4E), similar to Lkb1BKO mice. Lipolytic activity was not affected in TfamBKO mice in vitro and in vivo (Figures S4F–S4H). As expected, BAT of TfamBKO mice showed reduced steady-state mRNA levels of mtDNA-encoded C-IV subunits at both RT and thermoneutrality, and mRNA levels of nuclear-encoded C-IV subunits and Ucp1 were not altered in these mice (Figures 4A and S4B). The BAT of TfamBKO mice had reduced expression of most mtDNA-encoded C-I, C-III, and C-V genes, while nuclear-encoded ETC genes were largely unchanged (Figure 4B). Immunoblots further confirmed the reduction of mtDNA- and nuclear-encoded C-IV proteins (mt-Co1, mt-Co2, Cox4, Cox5b, and Cox6b1) (Figures 4C, S4J, and S4K), and the BAT of TfamBKO mice exhibited reduced C-IV activity at RT and thermo-neutrality (Figure 4F). The mtDNA copy number was also reduced in the TfamBKO mice (Figure S4I). Due to the defects of mtDNA gene expression, TfamBKO mice housed at RT and thermoneutrality also exhibited reduced respiration rates, as well as reduced RRC and ATP turnover rates (Figures 4G–4I). Indirect calorimetry analysis further showed that TfamBKO mice had reduced CL-induced EE (Figures 4J and 4K). Similar linear regression analysis indicated that the slopes of change of CL-induced EE per unit change of body weight were not overlapping between CON and TfamBKO mice at both RT and 30°C (Figure 4L). Basal EE, VO2, RER, food intake, and physical activity remained largely unaffected (Figures S4L–S4T). Collectively, Tfam deficiency leads to reduced expression of mtDNA-encoded ETC genes and respiratory defects in the BAT.
Figure 4. TfamBKO Mice Exhibit ETC Proteome Imbalance and Defective Mitochondrial Respiration in BAT.
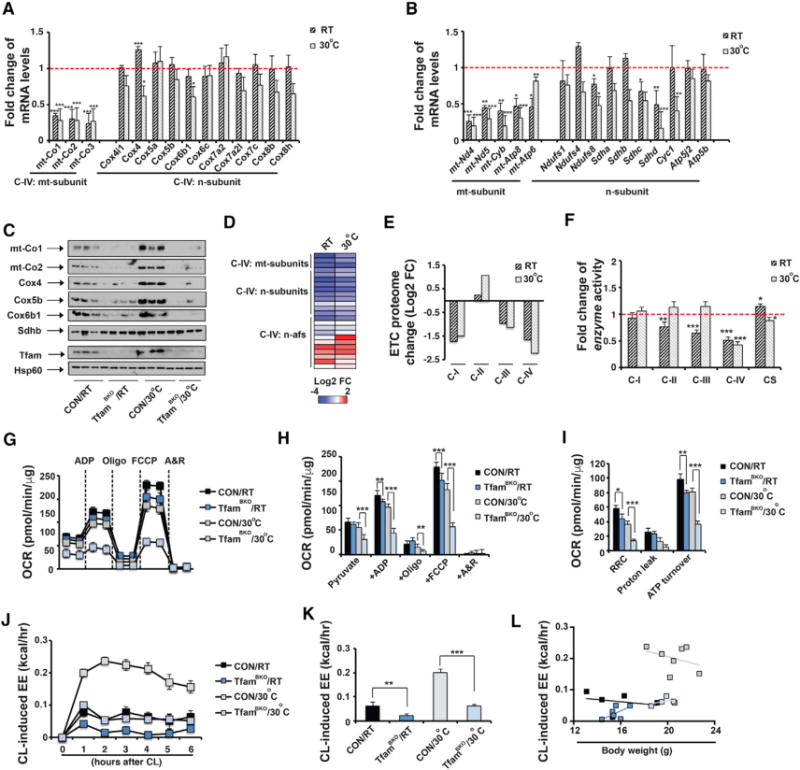
(A) Fold change in steady-state mRNA levels of C-IV subunits in BAT from ~8- to 10-week old male and female TfamBKO mice housed at RT and 30°C.
(B) Fold change in mRNA levels of mtDNA-encoded C-I, C-III, and C-V subunits and selected nuclear-encoded C-I, C-II, C-III, and C-V subunits in ~8- to 10-week-old male and female TfamBKO mice housed at RT and 30°C. Sample size: TfamBKO and control at RT (n = 6 each) and TfamBKO and control at 30°C (n = 7 and 10).
(C) Immunoblots of C-IV subunits (mt-Co1, mt-Co2, Cox4, Cox5b, and Cox6b1), C-II subunit Sdhb, Tfam, and Hsp60 in BAT mitochondria from male and female CON and TfamBKO mice housed at RT and 30°C.
(D) Heatmap of protein abundance of C-IV subunits and assembly factors in BAT mitochondria from male CON and TfamBKO mice housed at RT and 30°C.
(E) Log2 fold change values of each ETC proteome from BAT of male TfamBKO mice housed at RT and 30°C.
(F) Fold change of in vitro enzyme activities of C-I to C-IV and CS in BAT from ~8-week-old male and female CON and TfamBKO mice housed at RT and 30°C. Sample size: TfamBKO and control at RT (n = 11 each), TfamBKO and control at 30°C (n = 16 and 13).
(G) Seahorse experiments measuring OCRs in isolated mitochondria from BAT of ~8-week-old male and female CON and TfamBKO mice housed at RT and 30°C, upon addition of ADP, Oligo, FCCP, and A&R.
(H and I) Average OCR (H) and RRC, proton leak, and ATP turnover rates (I) in seahorse measurements. Sample size: CON/RT (n = 8) and TfamBKO/RT (n = 12), CON/30°C (n = 9), and TfamBKO/30°C (n = 6). Oxygen consumption in response to CL in ~8-week-old male CON and TfamBKO mice housed at RT (H) and 30 C (I). CL injected at 0 hr. Sample size: CON/RT (n = 5), TfamBKO/RT (n = 8), CON/30°C (n = 5), and TfamBKO/30°C (n = 7).
(J and K) Recordings of energy expenditure (EE) increases in response to CL (J) and hourly average of CL-induced EE (K) in ~8- to 10-week-old male CON and TfamBKO mice housed at RT and 30°C. CL injected at 0 hr.
(L) Regression plot of CL-induced EE as a function of mouse weight in ~8- to 10-week-old old male CON and TfamBKO mice housed at RT and 30°C. Sample size: CON/RT (n = 5), TfamBKO/RT (n = 6), CON/30°C (n = 7), and TfamBKO/30°C (n = 5).
Data are means and error bars are ±SEM. Student’s t test, *p < 0.1, **p < 0.05, and ***p < 0.01.
The C-IV deficiency in the BAT of TfamBKO mice was further confirmed by mass spectrometry analysis of BAT mitochondrial proteome (Figures S5A–S5F; Table S3). Similar to Lkb1BKO mice, TfamBKO mice BAT mitochondrial proteomic profiling showed that C-IV was one of the most profoundly affected ETC complexes; all mtDNA- and nuclear-encoded C-IV protein were reduced while many C-IV assembly factors were upregulated at both RT and thermoneutrality (Figures 4D and 4E). C-IV subunits (Cox5a, Cox6b1, Cox6c, Cox7c, and Ndufa4) were downregulated in both Lkb1BKO and TfamBKO mice at both RT and thermo-neutrality with p values <0.1 (Figure S5G). Other C-IV proteins, such as mt-Co2 and Cox4i1, did not reach the 0.1 cutoff, possibly due to the small number used in the mass spectrometry analysis (n = 3). C-I subunits (mtDNA-encoded mt-Nd1, mt-Nd3, and most nuclear-encoded subunits) and mitochondrial ribosome subunits were specifically downregulated in TfamBKO mice (Figure S5G). In addition to changes in ETC, several proteases and chaperons (Dnajc15, Afg3l1, Afg3l2, Clpx, and Lonp2) were selectively upregulated in Lkb1BKO and/or TfamBKO mice (Figure S5H), suggesting that mitochondrial proteostasis might be activated in response to the ETC proteome imbalance. We also compared expression levels of key components of mitochondrial fusion/fission, glycolysis, tricarboxylic acid (TCA), and beta-oxidation enzymes (Figures S5I–S5K). Interestingly, Mfn1 was reduced and Hk1 was elevated in Lkb1BKO and TfamBKO mice at both temperatures, which might suggest mitochondrial dynamics and metabolic reprogramming might occur in order to cope with the respiratory defects. Although no consistent changes in protein levels of TCA and beta-oxidation enzymes were observed, we could not rule out the possibility that TCA and beta-oxidation activities are altered in Lkb1- or Tfam-deficient BAT.
TfamBKO Mice Are Protected against HFD-Induced Obesity, Insulin Resistance, Adipose Inflammation, and Hepatic Steatosis
Because Tfam only regulates mtDNA stability and gene expression while Lkb1 regulates additional processes outside mitochondria, we reasoned that TfamBKO mice would be the better model system to investigate the metabolic consequences of BAT ETC proteome imbalance. Under normal chow, TfamBKO mice weighed less than controls at about 6 months of age, due to the decreases of both lean and fat mass (Figures S6A–S6C), which was similar to the pan adipocyte-specific Tfam knockout mice (Adiponectin-Cre; Tfamf/f, TfamAKO) (Vernochet et al., 2014). However, TfamBKO mice did not show insulin resistance as observed in the TfamAKO mice at a similar age (Vernochet et al., 2014). Serum TG levels were reduced in TfamBKO mice only at thermoneutrality (Figure S6H). The glucose disposal rates in both GTT and ITT assays and serum insulin levels in TfamBKO mice were not altered (Figures S6D–S6F), and neither were the liver TG levels and macrophage infiltration in eWAT (Figures S6G and S6I), suggesting these metabolic activities are differentially regulated by brown and white adipocytes.
We then examined metabolic performance of TfamBKO mice under HFD at both RT and thermoneutrality. In particular, we characterized their metabolic activities after 4 and 12 weeks on HFD. In BAT of TfamBKO mice, brown adipocytes lost their multilocular morphology completely (Figure 5A). TfamBKO mice were resistant to HFD-induced obesity (Figure 5B), due to reductions in fat mass (Figures 5C–5E). They also showed reduced adipose tissue mass (Figures 5F and 5G) and smaller adipocytes (Figures 5H–5J) at both temperatures. Notably, these differences were obvious as early as 4 weeks on HFD and became more significant along the time course of an HFD regimen.
Figure 5. TfamBKO Mice Show Reduced Adiposity under HFD at RT and Thermoneutrality.
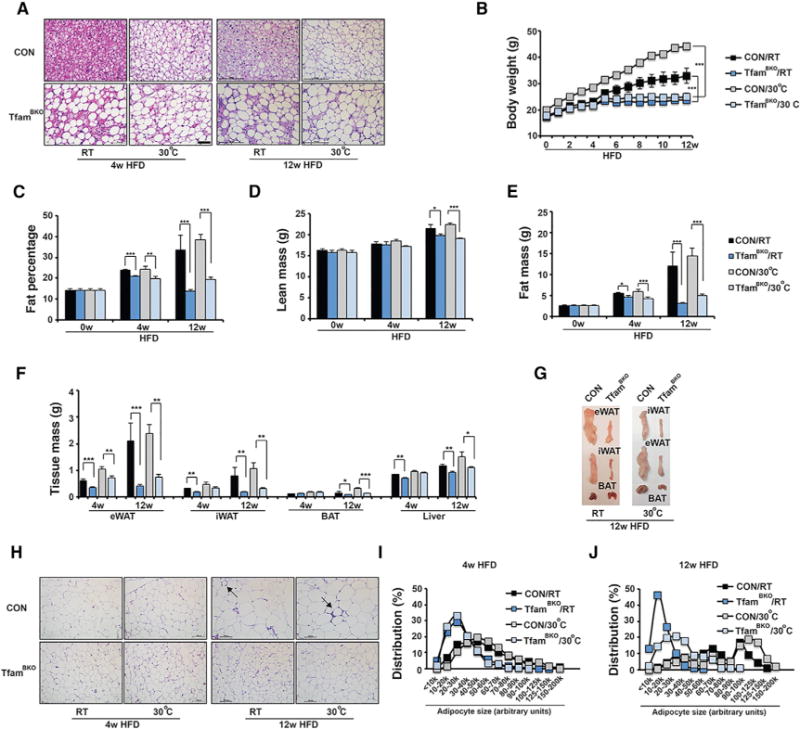
(A) Representative H&E staining of BAT from male CON and TfamBKO mice after 4- and 12-week HFD at both RT and 30°C. Scale bar, 50 μm.
(B) Body weight of male CON and TfamBKO mice under HFD at RT and 30°C. Sample size: CON/RT (n = 7), TfamBKO/RT (n = 13), CON/30°C (n = 3), and TfamBKO/30°C (n = 5).
(C–E) Fat percentage (C), lean mass (D), and fat mass (E) of male CON and TfamBKO mice under HFD at RT and 30°C. Sample size: CON/0w/RT (n = 7), TfamBKO/0w/RT (n = 6), CON/4w/RT (n = 4), TfamBKO/4w/RT (n = 4), CON/4w/30°C (n = 7), TfamBKO/4w/30°C (n = 9), CON/12w/RT (n = 4), TfamBKO/12w/RT (n = 7), CON/12w/30°C (n = 7). and TfamBKO/12w/30°C (n = 13).
(F) Tissue mass of eWAT, iWAT, BAT, and liver of male CON and TfamBKO mice after 4 and 12-week HFD at both RT and 30°C. Sample size: CON/4w/RT (n = 4), TfamBKO/4w/RT (n = 4), CON/4w/30°C (n = 5), TfamBKO/4w/30 C (n = 3), CON/12w/RT (n = 4), TfamBKO/12w/RT (n = 8), CON/12w/30°C (n = 5), and TfamBKO/12w/30°C (n = 9).
(G) Representative images of dissected iWAT, eWAT, and BAT from CON and TfamBKO mice after 12-week HFD at RT and 30°C.
(H–J) Representative H&E staining (H) of eWAT from male CON and TfamBKO mice after 4- and 12-week HFD at both RT and 30°C. Scale bar, 100 μm. Arrows indicate crown-like structures. Adipocyte size distribution in eWAT from CON and TfamBKO mice after 4- and 12-week HFD at RT (I) and 30°C (J). Total adipocytes counted: CON/4w/RT (n = 593), TfamBKO/4w/RT (n = 1,031), CON/4w/30 C (n = 495), TfamBKO/4w/30°C (n = 552), CON/12w/RT (n = 224), TfamBKO/12w/RT (n = 692), CON/12w/30°C (n = 280), and TfamBKO/12w/30°C (n = 267).
Data are means and error bars are ±SEM. Student t test, *p < 0.1, **p < 0.05, and ***p < 0.01.
Metabolic syndrome includes abdominal obesity, dyslipidemia, insulin resistance, and inflammatory milieu (Lusis et al., 2008). We then examined whether insulin sensitivity and liver TG metabolism were affected in TfamBKO mice under HFD. Glucose metabolism was normal in TfamBKO mice after 4-week HFD; they showed comparable serum fasting glucose and insulin levels and glucose disposal rate in a GTT assay as control mice (Figures S7A–S7D). TfamBKO mice only showed a slightly accelerated glucose clearance in an ITT assay at thermoneutrality (Figure S7A). After 12-week HFD, however, TfamBKO mice exhibited improved glucose disposal rates in ITT and GTT assays and reduced serum insulin levels (Figures 6A–6C). Immunoblots showed there were increased pAkt activities in response to insulin in muscle, suggesting there was increased muscle insulin sensitivity after 12-week HFD at both RT and thermoneutrality (Figures 6D and 6E). Increased muscle insulin sensitivity was not observed in TfamBKO mice after 4-week HFD (Figures S7I and S7J).
Figure 6. TfamBKO Mice Exhibit Improved Insulin Sensitivity, Reduced Adipose Inflammation, and Attenuated Hepatic Steatosis under HFD at RT and Thermoneutrality.
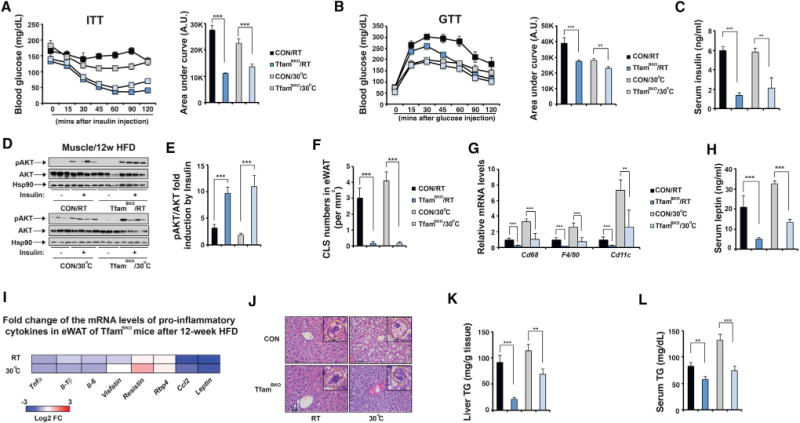
(A) Serum glucose levels during ITT in male CON and TfamBKO mice after 12-week HFD at RT and 30°C. Area under the curve (AUC) values of glucose levels shown. Sample size: CON/RT (n = 4), TfamBKO/RT (n = 8), CON/30°C (n = 8), and TfamBKO/30°C (n = 12).
(B) Serum glucose levels during GTT in male CON and TfamBKO mice after 12-week HFD at RT and 30°C. AUC values of glucose levels shown. Sample size: CON/RT (n = 11), TfamBKO/RT (n = 16), CON/30°C (n = 4), and TfamBKO/30°C (n = 8).
(C) Serum insulin levels in male CON and TfamBKO mice after 12-week HFD at RT and 30°C. Sample size: CON/RT (n = 4), TfamBKO/RT (n = 7), CON/30°C (n = 3), and TfamBKO/30°C (n = 5).
(D) Immunoblots showing amounts of pS473-Akt, total Akt, and Hsp90 in muscle in male CON and TfamBKO mice after 12-week HFD at RT and 30°C. Insulin stimulation indicated in the bottom panel.
(E) Quantitative analysis of pAkt immunoblots in (D).
(F) Quantitative analysis of CLS numbers in eWAT of male CON and TfamBKO mice after 12-week HFD at RT and 30°C.
(G) qPCR analysis of mRNA levels of macrophage markers Cd68, F4/80, and Cd11c in eWAT of male CON and TfamBKO mice after 12-week HFD at RT and 30°C. Sample size: CON/RT (n = 4), TfamBKO/RT (n = 6), CON/30°C (n = 4), and TfamBKO/30°C (n = 4).
(H) Serum leptin levels in male CON and TfamBKO mice after 12-week HFD at RT and 30°C. Sample size: CON/RT (n = 4), TfamBKO/RT (n = 7), CON/30°C (n = 3), and TfamBKO/30°C (n = 5).
(I) Heatmap showing log2 fold change of mRNA levels of pro-inflammatory cytokines in eWAT between male CON and TfamBKO mice after 12-week HFD.
(J) Representative H&E staining of liver from male CON and TfamBKO mice after 12-week HFD housed at RT and 30°C. Scale bar, 50 μm. Insert: hepatocyte circled with a dashed yellow line. Red arrowheads, lipid droplets.
(K) Liver triglyceride contents of male CON and TfamBKO mice after 12-week HFD housed at RT and 30°C. Sample size: CON/RT (n = 4), TfamBKO/RT (n = 8), CON/30°C (n = 3), and TfamBKO/30°C (n = 5).
(L) Serum triglyceride contents of male CON and TfamBKO mice after 12-week HFD housed at RT and 30°C. Sample size: CON/RT (n = 4), TfamBKO/RT (n = 8), CON/30°C (n = 7), and TfamBKO/30°C (n = 13).
Data are means and error bars are ±SEM. Student’s t test, **p < 0.05 and ***p < 0.01.
Adipose tissue inflammation was not evident in control and TfamBKO mice after 4-week HFD, although there were slight reductions in macrophage gene expression in the eWAT of TfamBKO mice (Figure S7E). However, the crown-like structures (CLSs) were abundant in the eWAT of control mice after 12-week HFD at both RT and thermoneutrality, and absent in TfamBKO mice (Figures 5H and 6F). This was correlated with significant reductions of mRNA levels of macrophage markers Cd68, F4/80, and Cd11c in the eWAT of TfamBKO mice (Figure 6G). We also surveyed the mRNA levels of known pro-inflammatory cytokines (Tnfα, Il-1β, Il-6, Visfatin, Resistin, Rbp4, Ccl2, and Leptin) in eWAT (Makki et al., 2013). Among them, Ccl2 and Leptin showed most significant reductions in the eWAT of TfamBKO mice after 12-week HFD (Figure 6I). Indeed, serum lep-tin levels were also reduced in TfamBKO mice (Figure 6H). We next examined lipid metabolism in TfamBKO mice. Liver TG contents were reduced in TfamBKO mice after 12-week HFD (Figures 6J and 6K). In fact, the reduced liver TG content in TfamBKO mice was also observed after 4-week HFD at both temperatures (Figures S7F and S7G). However, the serum TG levels were only reduced after 12-week HFD (Figures 6L and S7H).
These metabolic benefits under HFD in TfamBKO mice were present at both RT and thermoneutrality, suggesting that BAT adaptive thermogenesis was not involved. Indeed, indirect calorimetry analysis of TfamBKO mice after 4-week HFD showed that total EE, VO2, RER, and food intake were not changed significantly at both RT and thermoneutrality (Figures S7K–S7R). TfamBKO mice even exhibited trends of reduced physical activities at both temperatures (Figure S7S). Notably, beige adipocytes in subcutaneous iWAT were expanded at RT but were absent at thermoneutrality in TfamBKO mice (Figures S7T–S7V), indicating that beige adipocyte expansion did not contribute to the metabolic benefits under HFD. There was no compensatory expansion of beige adipocytes in iWAT of Lkb1BKO mice at RT (Wang et al., 2017). In conclusion, our data indicate that specific reduction of mtDNA-encoded ETC genes can lead to ETC proteome imbalance locally in BAT, and systemic metabolic benefits in a thermogenesis-independent manner.
DISCUSSION
Although BAT is a non-essential organ for organismal survival under normal physiological conditions, promoting BAT development and function has been proposed as an alternative approach to increase EE and ultimately to improve metabolic health in humans. Several key genetic programs of adaptive thermogenesis have been identified. For example, PR domain-containing 16 (Prdm16) controls the bidirectional fate decision between skeletal myoblasts and brown adipocytes (Seale et al., 2008). Also, Pgc1α and β govern mitochondrial biogenesis in brown adipocytes (Uldry et al., 2006). Mitochondrial functions, such as fatty acid oxidation, TCA, and ETC, are all needed for Ucp1-mediated adaptive thermogenesis in brown adipocytes (Ahmadian et al., 2011; Ellis et al., 2010; Lee et al., 2015, 2016; Yang et al., 2016; Zimmermann et al., 2004). All these mitochondrial functions are controlled by the nuclear genome, but mitochondria also have their own mtDNA that encodes 13 catalytic subunits of the ETC. Rare mutations of these mtDNA-encoded ETC genes cause respiration defects, leading to mitochondrial disorders with a broad spectrum of neurological and systemic manifestations in humans. Disruption of either nuclear or mitochondrial ETC components should impair adaptive thermogenesis in BAT. Although contributions of individual mtDNA-encoded ETC subunits to adaptive thermogenesis have not been thoroughly examined, our study shows that reducing expression levels of all mtDNA-encoded ETC genes indirectly (through Lkb1 deficiency) or directly (through Tfam deficiency) affects adaptive thermogenesis in BAT.
Lkb1 is a master kinase that regulates diverse cellular activities through the phosphorylation of 13 downstream AMPK and AMPK-related kinases (Alessi et al., 2006; Shan et al., 2016; Zhang et al., 2013). AMPK has been identified as an essential non-mitochondrial component of oxidative phosphorylation (Arroyo et al., 2016). But pan adipocyte-specific AMPK knockout mice were obese and insulin resistant (Kim et al., 2016; Mottillo et al., 2016), suggesting that other AMPK-related kinases may mediate Lkb1’s effect in BAT. Similarly, Lkb1 can modulate mitochondrial functions in hematopoietic stem cells through AMPK-dependent and -independent mechanisms (Gan et al., 2010; Gurumurthy et al., 2010; Nakada et al., 2010). Although BAT of Lkb1BKO mice had similar mitochondrial respiration defects and ETC gene expression profiles as those of TfamBKO mice, Tfam expression was not altered in the BAT of Lkb1BKO mice (data not shown). Therefore, Lkb1 may regulate mtDNA gene expression and mitochondrial respiration in BAT through Tfam-independent mechanisms. Moreover, given multi-layered regulation of mtDNA-encoded protein expression (Hallberg and Larsson, 2014), it is tempting to speculate that manipulating any step, including mtDNA replication, transcription, RNA processing/maturation, ribosome assembly, or protein translation, can similarly cause reduction of mtDNA-encoded gene expression and mitochondrial ETC proteome imbalance.
Adaptive thermogenesis is the primary function of BAT mitochondria. Here, we describe that BAT mitochondria have the capacity to regulate systemic metabolism independently of their thermogenic function. Two different mouse models (Lkb1BKO and TfamBKO mice) exhibit reduced adiposity and liver TG accumulation at both RT and thermoneutrality despite their defective adaptive thermogenesis. Mechanistically, this trade-off between thermogenic capacity and overall organismal fitness is due to the specific reduction of mtDNA-encoded gene expression in the BAT (Figure 7). In contrast, reducing Pgc1α-mediated mitochondrial biogenesis in brown-adipocyte-specific Irf4 and Hdac3 knockout mice affected the expression of both mtDNA- and nuclear-encoded genes in BAT, leading to thermogenic defects but without metabolic benefits (Emmett et al., 2017; Kong et al., 2014). TfamBKO mice are resistant to long-term HFD-induced insulin resistance, dyslipidemia, and non-alcoholic fatty liver disease, which might be secondary to reduced adiposity. Thus, this unknown non-thermogenic function in BAT originates from stressed mitochondria and is more dominant than Ucp1-mediated uncoupling. It is plausible that stressed mitochondria with ETC proteome imbalance can activate mitochondrial unfolded protein response locally in BAT to affect fat mass. Other processes, such as abnormal hormone secretion or metabolic reprogramming in BAT adapted from the ETC proteome imbalance, are also possible. Investigating the ‘‘altruistic behaviors” of BAT ETC proteome in systemic metabolism will not only improve the understanding of BAT physiology but also provide novel approaches to prevent and treat obesity and associated metabolic disorders.
Figure 7. Proposed Model.
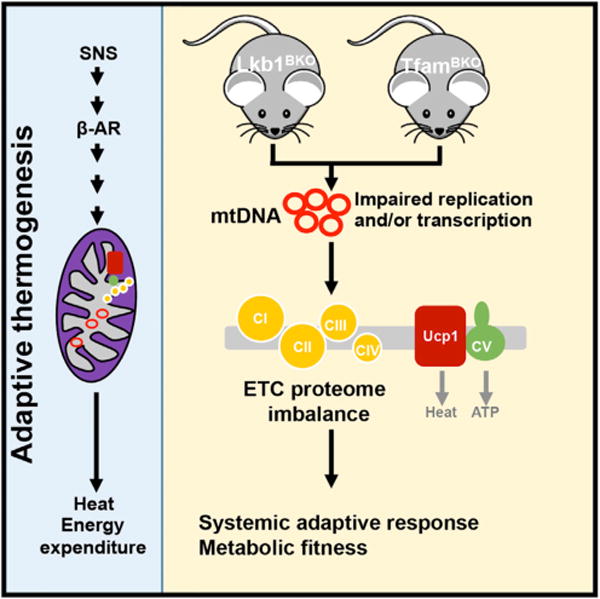
As a major thermoregulatory organ, BAT can generate heat (through Ucp1-mediated adaptive thermogenesis) by sympathetic nervous system-mediated β-AR signaling. Lkb1 and Tfam regulate mtDNA-encoded ETC subunit gene expression, and their deficiencies in BAT lead to ETC proteome imbalance, a mismatch between proportional ETC complexes. This ETC proteome imbalance locally in BAT can cause systemic adaptive responses, which ultimately result in metabolic fitness.
ETC proteome imbalance has been observed in other settings (referred to as mitonuclear protein imbalance). ETC proteome imbalance can activate mitochondrial unfolded protein response (UPRmt) to regulate longevity in Caenorhabditis elegans (Houtkooper et al., 2013). The activation of UPRmt in neurons can induce metabolic fitness in C. elegans in a non-cell autonomous manner (Berendzen et al., 2016; Mardones et al., 2014). Some anti-bacteria antibiotics can cause ETC proteome imbalance in multiple species due to their activities to specifically suppress mtDNA translation (De Silva et al., 2015; Greber and Ban, 2016). For example, doxycycline can inhibit mitochondrial protein synthesis by blocking the recruitment of aminoacyl-tRNA to a small subunit (Wang et al., 2015). Wild-type mice fed with doxycycline in the water for 1 week exhibited mitonuclear prote-ome imbalance in the liver, due to its activity to inhibit mitoribo-some (Moullan et al., 2015). In fact, the Lkb1BKO and TfamBKO mice represent the first mouse models that affect ETC proteome imbalance specifically in BAT.
Mitochondria are essential for all metabolic activities, and they have developed adaptive strategies to communicate with other cellular compartments to endure harsh environmental changes throughout evolution (Ryan and Hoogenraad, 2007; Wallace, 2005; Yun and Finkel, 2014). Therefore, the general principles of the trade-off between BAT thermogenic capacity and systemic metabolism, through non-cell autonomous actions of mitochondria with the ETC proteome imbalance, for instance, may also be applicable in other physiological and pathological conditions.
Limitations
The specific mechanism connecting this BAT dysfunction to systemic benefits is not fully delineated. However, the ETC proteome imbalance mechanism will serve as a cornerstone for us, and other investigators, to identify the unknown “batokines” and/or activating peripheral stress signals and to explore this new BAT biology in the future.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Lkb1 | Santa Cruz Biotechnology | Cat#SC-32245; RRID: AB_627890 |
| Rabbit anti-pT172 AMPK | Cell Signaling | Cat#2535 |
| Rabbit anti-AMPK | Cell Signaling | Cat#2603; RRID: AB_490795 |
| Rabbit anti-Hsp90 | Santa Cruz Biotechnology | Cat#SC-7949 |
| Mouse anti-total OXPHOS rodent | Abcam | Cat#ab110413; RRID: AB 2629281 |
| Rabbit anti-Hsp60 | Bethyl | Cat#A302-846A; RRID: AB_10634219 |
| Rabbit anti-mt-Co2 | Proteintech | Cat#55070-1-AP; RRID: AB_10859832 |
| Rabbit anti-Cox4 | Cell Signaling | Cat#4850 |
| Rabbit anti-Cox5b | Bethyl | Cat#A-305-523A-T |
| Rabbit anti-Cox6b | Abgent | Cat#AP20624a |
| Rabbit anti-Surf1 | Proteintech | Cat#15379-1-AP; RRID: AB_2239968 |
| Goat anti-Tfam | Santa Cruz Biotechnology | Cat#SC-23588; RRID: AB_2303230 |
| Rabbit anti-Ucp1 | Sigma | Cat#U6382 |
| Rabbit anti-pS473 AKT | Cell Signaling | Cat#9271 |
| Rabbit anti-AKT | Cell Signaling | Cat#4691; RRID: AB_915783 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Oligomycin A | Sigma | Cat#75351 |
| Sodium pyruvate | Sigma | Cat#SA636 |
| FCCP | Sigma | Cat#C2920 |
| Antimycin A from Streptomyces sp. | Sigma | Cat#A8674 |
| Rotenone | Sigma | Cat#R8875 |
| B-NADH | Sigma | Cat#N7410 |
| DCIP | Sigma | Cat#D1878 |
| DTNB | Sigma | Cat#D8130 |
| Acetyl Coenzyme A | Sigma | Cat#A2056 |
| Cytochrome C | Sigma | Cat#C3131 |
| Decylubiquinone | Sigma | Cat#D7911 |
| Potassium Borohydride | Sigma | Cat#P4129 |
| Potassium Cyanide | Sigma | Cat#20781 |
| Potassium Ferricyanide | Sigma | Cat#244023 |
| Sodium Hydrosulfite | Sigma | Cat#157953 |
| Insulin solution human | Sigma | Cat#I9278-5ML |
| Forskolin | Sigma | Cat#F6886-10MG |
| CL316243 disodium salt | Tocris bioscience | Cat#1499 |
| Bio-Rad Protein Assay Solution | BioRad | Cat#500-0006 |
| TRIsure | Bioline | Cat#BIO-38033 |
| Critical Commercial Assays | ||
| XFe24 Flux Assay Kit | Agilent Technologies | Cat#102340-100 |
| Infinity Triglycerides Reagent | Thermo Scientific | Cat#TR22421 |
| QIAamp DNA Mini Kit | Qiagen | Cat#51304 |
| ISOLATE II RNA Mini Kit | Bioline | Cat#BIO-52073 |
| iScript cDNA Synthesis Kit | BioRad | Cat#170-8891 |
| Mouse insulin ultrasensitive ELISA kit | Alpco | Cat#80-INSMSV-E01 |
| Mouse leptin ELISA kit | Crystal Chem | Cat#90030 |
| Deposited Data | ||
| BAT mitochondrial mass spectrometry data | ProteomeXchange Consortium | ProteomeXchange: PXD008599 |
| Experimental Models: Organisms/Strains | ||
| Lkb1f/f | JAX | Cat#014143 |
| Tfamf/f | JAX | Cat#026123 |
| Ucp1-Cre | JAX | Cat#024670 |
| Betaless | Bachman et al., 2002 | N/A |
| Oligonucleotides | ||
| Full sequences in Table S1 | Elim Biopharm | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| MaxQuant data analysis | Cox and Mann, 2008 | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Biao Wang (biao.wang@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Models
Lkb1f/f (JAX#014143) and Tfamf/f (JAX#026123) mice were obtained from The Jackson Laboratory. Betaless mice were provided by Dr. Shingo Kajimura. About 8-week-old male and female Lkb1BKO, TfamBKO, betaless mice and their relevant controls were used for biochemical analysis. Male Lkb1BKO, TfamBKO mice and their relevant controls (started at ~8-week of age) were used for thermogenic and metabolic studies. Mice were housed in a temperature-controlled environment at 22 °C under a 12h light:dark cycle with free access to water and food (PicoLab Rodent Diet 20, #5053). There were no inclusion/exclusion criteria for mice studies. Mice were in C57BL/6J background. All animal experiments were approved by the UCSF Institutional Animal Care and Use Committee in adherence to US National Institutes of Health guidelines and policies.
METHOD DETAILS
Indirect Calorimetry Measurements
For thermoneutral experiments, ~5-week-old mice were placed in a 30°C rodent chamber (Power Scientific RIS52SD Rodent Incubator) for an additional 3-4 weeks to reach their thermoneutral zone. Basal energy expenditure (EE) and CL-induced EE were calculated and presented as scatter plots, followed by progression analysis. Oxygen consumption in vivo was quantified using CLAMS (Columbus Instruments). Rates of O2 (VO2) consumption and CO2 (VCO2) release were monitored and expressed per mouse and per body weight (Butler and Kozak, 2010; Tschop et al., 2012). Investigators were blinded to the mouse genotypes for CLAMS, which was performed by the UCSF Diabetes and Endocrinology Research Center Metabolic Research Unit.
Metabolic Studies
About 8-week-old mice were transferred to a 60% fat diet (Research Diets, D12492) housed at RT or 30°C. For HFD at thermoneutrality, 5-week-old mice were housed in a 30°C rodent chamber for 3-4 weeks prior to starting HFD. Body weight was monitored once a week. For insulin tolerance test (ITT), mice were fasted 4-6 hours before intraperitoneal administration of insulin (Humulin; 0.75U kg−1). For glucose tolerance test (GTT), mice were fasted 18 hours before intraperitoneal administration of glucose (0.75U kg−1). Glucose was measured from tail vein at indicated time points with a glucometer (Contour, Bayer). For in vitro lipolysis, mice were fasted for 6 hours and 20 mg of fat tissue was collected and incubated at 37°C in modified Krebs-Ringer buffer (121 mM NaCl, 5 mM KCl, 0.5 mM MgCl, 0.4 mM NaH2PO4, and 1 mM CaCl2) supplemented with 1% fatty acid free BSA, 0.1% glucose, and 20 mM HEPES. Glycerol release before and after Forskolin (FSK) is determined using Infinity Triglycerides Reagent (Thermo, TR22421). For in vivo lipolysis, mice were fasted for 6 hours and serum glycerol levels were measured before and after 1mg kg−1 CL injection.
Cold Tolerance Test (CTT)
About 8-week-old male and female mice were single-housed with free-access to food and water during CTT. Rectal temperature was monitored hourly during 4°C cold challenge with a BAT-12 Microprobe Thermometer (Physitem Instruments). Cold challenge was started at 11am (4 hours after light on).
ETC Complex Activities
Frozen BAT tissue from about 8-week-old male and female mice was homogenized in 250 μL homogenization buffer (120 mM KCl, 20mM HEPES, 1mM EGTA, pH 7.4) by sonication (5 second pulse ×5, 60% power) using a Microson XL2000 Ultrasonic Cell Disruptor (Misonix). Protein was quantitated using the Bradford assay and all samples were diluted to a final concentration of 1μg/μl of protein. The spectrophotometric kinetic assays were performed using a monochromator microplate reader (Tecan M200 Pro). Complex I activity (NADH:ubiquinone oxidoreductase) was determined by measuring oxidation of NADH at 340 nm (using ferricyanide as the electron acceptor) in a reaction mixture of 50 mM potassium phosphate (pH 7.5), 0.2 mM NADH, and 1.7 mM potassium ferricyanide. Complex II activity (Succinate Dehydrogenase) was determined by measuring the reduction of the artificial electron acceptor 2,6-di-chlorophenol-indophenol (DCIP) at 600 nm in a reaction mixture of 50 mM potassium phosphate (pH 7.5), 20 mM succinate, 2 μM DCIP, 10 μM rotenone, and 1 mM potassium cyanide. Complex III activity (Ubiquinol:cytochrome c oxidoreductase) was determined by measuring the reduction of cytochrome c at 550 nm in a reaction mixture of 50 mM potassium phosphate (pH 7.5), 35 μM reduced decylubiquinone, 15 μM cytochrome c, 10 μM rotenone, and 1 mM potassium cyanide. Complex IV activity (Cytochrome c oxidase) was determined by measuring the oxidation of cytochrome c at 550 nm in a reaction mixture of 50 mM potassium phosphate (pH 7.0) and 100 μM reduced cytochrome c. Citrate synthase activity was determined by measuring the reduction of 5,5′-dithiobis(2-nitro-benzoic acid) (DTNB) at 412 nm which was coupled to the reduction of acetyl-CoA by citrate synthase in the presence of oxaloacetate. The reaction mixture consisted of 100 mM Tris-HCl (pH 8.0), 100 μM DTNB, 50 μM acetyl-CoA, and 425 μM oxaloacetate. All activities were calculated as nmoles/min/mg protein, normalized to citrate synthase (CS) activity and finally expressed as the percentage of wild-type activity.
Mitochondria Isolation
Freshly dissected BAT tissue from about 8-week-old male and female mice was homogenized in a Dounce homogenizer with 5ml ice-cold mitochondria isolation buffer (210mM Mannitol, 70mM Sucrose, 1mM EGTA, 5mM HEPES pH7.5, 0.5% BSA). The homogenates were filtered through cheesecloth to remove residual particulates and intact mitochondria were isolated by differential centrifugation using a previously described protocol (Methods Enzymol 1979;55:65-78). The mitochondrial pellet was resuspended in 25μL of isolation buffer and protein was quantitated using the Bradford assay.
Mitochondrial Respiration Assay
The XFe24 extracellular flux analyzer from Seahorse Biosciences was used to measure the rate of mitochondrial oxygen consumption, as previously described (Sadat et al., 2016). Mitochondria were isolated from 6-8-week-old male and female mice and immediately plated on the XFe24 cell culture microplate at a density of 2μg per well. The plate was centrifuged at 2000g for 20 mins at 4°C to enable the mitochondria to adhere to the plate. The XFe24 cartridge was equilibrated with the calibration solution overnight at 37°C. Mitochondrial Assay Solution (1X MAS) (70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1.0 mM EGTA and 0.2% (w/v) fatty acid-free BSA) was freshly prepared and pH adjusted to 7.2. Unless otherwise stated, 0.5 M Pyruvate and 0.5 M Malate were used as substrates in the 1X MAS. 1X MAS was used to prepare cellular stress reagents, 4 mM ADP, 2.5 μg/ml Oligomycin, 4 μM FCCP, 4 μM Antimycin and 4 μM Rotenone (final concentration). All the reagents were loaded in the ports as suggested by Seahorse biosciences. Oxygen consumption rates were measured for 3min with 30 secs of mixing. Oxygen Consumption Rate (OCR) was expressed as pMoles of oxygen/min.
Mass Spectrometry
Purified BAT mitochondria from 10-12-week old male mice housed at RT or thermoneutrality (n=3 for each genotype/condition) were resuspended in 8 M urea, 50 mM Tris, 5 mM CaCl2, 100 mM NaCl, and protease inhibitors. Mitochondria were lysed by probe sonication on ice, and proteins reduced by the addition of 5 mM DTT for 30 min at 37°C, followed cysteine alkylation by the addition of 15 mM iodoacetamide at RT for 45 min in the dark. The reaction was then quenched by the addition of 15 mM DTT for 15 minutes at RT. Proteins were first digested by the addition of endoproteinase LysC (Wako LC) at a 1:50 substrate:enzyme and incubated for 2h at RT. Next, samples were further digested by the addition of trypsin (Promega) at 1:100 substrate:enzyme, and incubated overnight at 37°C. Protein digests were then acidified by the addition of 0.5% triflororacetic acid, and samples desalted on C18 stage tips (Rainin). Peptides were resuspended in 4% formic acid and 3% acetonitrile, and approximately 1μg of digested mitochondria proteins was loaded onto a 75μm ID column packed with 25cm of Reprosil C18 1.9μm, 120Å particles (Dr. Maisch). Peptides were eluted into a Q-Exactive Plus (Thermo Fisher) mass spectrometer by gradient elution delivered by an Easy1200 nLC system (Thermo Fisher). The gradient was from 4.5% to 31% acetonitrile over 165 minutes. All MS spectra were collected with oribitrap detection, while the 15 most abundant ions were fragmented by HCD and detected in the orbitrap. All data were searched against the Mus musculus uniprot database (downloaded July 22, 2016). Peptide and protein identification searches, as well as label-free quantitation, were performed using the MaxQuant data analysis algorithm, and all peptide and protein identifications were filtered to a 1% false-discovery rate (Cox et al., 2014; Cox and Mann, 2008). Proteome changes of each ETC complex were calculated by averaging log2 values of fold change of all identified proteins within individual ETC complex.
Histology
Tissues were fixed in 10% formalin, and processed and stained at AML Laboratories. Cell size was measured using ImageJ. If a single lipid droplet occupied >50% of the cytoplasm, it was counted as a unilocular adipocyte. Adipocyte size distribution was calculated using total adipocyte numbers counted in multiple images.
Blue Native PAGE
To assess ETC complex abundances, blue native polyacrylamide gel electrophoresis (BN-PAGE) was performed as previously described (Wittig et al., 2006) with modifications according to the manufacturer’s protocol (Invitrogen) using equal amounts of protein. Briefly, isolated mitochondria were solubilized with 10% digitonin on ice. After addition of Coomassie G-250, samples were run on 4–16% NativePAGE gels. Following BN-PAGE, gels were placed in a fixed solution containing 40% methanol and 10% acetic acid followed by microwaving for 45 s at 1,100 W. Gels were then washed for 15 min at room temperature, after which the solution was decanted. Destaining was accomplished by the addition of 50 ml of an 8% acetic acid solution and microwaved a second time for 45 s at 1,100 W. The gel was then shaken at room temperature until the desired background was obtained.
Immunoblots
For lysates, tissues from about 8-week-old male and female mice were lysed in ice-cold lysis-buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 6 mM EGTA, 20 mM NaF, 1% Triton X-100, and protease inhibitors) using a TissueLyser II (Qiagen). After centrifugation at 13000 rpm for 15 min, supernatants were reserved for protein determinations and SDS-PAGE analysis. For muscle insulin sensitivity test, mice were fasted for 6 hours, and then leg muscle tissues were collected 5 minutes after intraperitoneal insulin (Humulin; 0.75U kg−1) injection. Mitochondria were lyzed in the above lysis buffer before immunoblotting. Antibodies used are listed in Key Resources Table. The immunoblots were quantified in ImageJ.
Q-PCR
Total RNA was extracted from tissues homogenized in TRIsure (Bioline) reagent. Isolated RNA was reverse transcribed using iScript cDNA Synthesis Kit (Biorad), and the resulting cDNA was used for quantitative PCR on a CFX384 real-time PCR detection system (Bio-Rad). Relative mRNA expression level was determined using the 2(-Delta Ct) method with 36B4 as the internal reference control. Primer sequences are listed in Table S1. Both males and females were used.
mtDNA Quantification
The relative mtDNA content was measured using real-time qPCR. The β2 microglobulin gene (B2M) was used as the nuclear gene (nDNA) normalizer for calculation of the mtDNA/nDNA ratio. A 322bp region of the mouse mtDNA was amplified using forward primer mtDNAF (CGACCTCGATGTTGGATCA) and the reverse primer mtDNAR (AGAGGATTTGAACCTCTGG). A fragment of the B2M gene was amplified using forward primer, B2MF (TCTCTGCTCCCCACCTCTAAGT), and reverse primer, B2MR (TGCTGTCTCGATGTTT GATGTATCT), giving an amplicon of 106 bp. The relative mtDNA content was calculated using the formula: mtDNA content = 1/2ΔCt, where ΔCt = CtmtDNA − Ctβ-Tubulin.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data was presented as average ± SEM. Statistical significance was determined by Student t-test. *: p<0.1, **: p<0.05 and ***: p<0.01. Statistical parameters can be found in the figure legends. Sample sizes for animal experiments were selected based on numbers typically used in similar published studies. No methods were used to determine whether the data met assumptions of the statistical approach. No randomization of animals or predetermination of sample sizes by statistical methods was performed. In vivo metabolic experiments, such as body weight and ITT, were repeated 2-3 times, and in vitro measurements of glycerol were performed with 2 technical replicates.
DATA AND SOFTWARE AVAILABILITY
The mass spectrometry data files (raw and search results) have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository under accession number ProteomeXchange: PXD008599.
Supplementary Material
Highlights.
Lkb1 regulates mitochondrial respiration and adaptive thermogenesis in BAT
BAT-specific Lkb1 KO mice exhibit improved metabolic performance under HFD
Lkb1-deficient BAT shows reduced mtDNA gene expression and ETC proteome imbalance
BAT-specific Tfam KO mice show a similar trade-off between thermogenesis and metabolism
Acknowledgments
This work is supported by NIH grants DK105175 (B.W.), U54NS100717 (N.J.K.), and P50GM082250 (N.J.K.); UCSF Diabetes Research Center P30DK063720 (B.W.); UCSF Nutrition Obesity Research Center P30DK098722 (B.W.); and research grants from Hillblom Foundation (B.W.) and Juvenile Diabetes Research Foundation (B.W.). E.P. is supported by a fellowship grant from the Hillblom Foundation. We thank C. Paillart, K. Ganeshan, and A. Chawla for assistance with CLAMS experiments. We thank Y. Zhang for assistance with mouse colony management and genotyping.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and three tables and can be found with this article online at https://doi.org/10.1016/j.cmet.2018.01.018.
AUTHOR CONTRIBUTIONS
B.W., R.M., and E.P. planned the experiments and wrote the manuscript. R.M. performed experiments measuring mitochondrial activities, and E.P. analyzed metabolic phenotypes in animal studies. Y.W. and D.W. participated in the initial studies. D.L.S., D.J.-M., and N.J.K. performed the mass spectrometry experiments and analyzed the data.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Abreu-Vieira G, Xiao C, Gavrilova O, Reitman ML. Integration of body temperature into the analysis of energy expenditure in the mouse. Mol Metab. 2015;4:461–470. doi: 10.1016/j.molmet.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadian M, Abbott MJ, Tang T, Hudak CS, Kim Y, Bruss M, Hellerstein MK, Lee HY, Samuel VT, Shulman GI, et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011;13:739–748. doi: 10.1016/j.cmet.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- Arroyo JD, Jourdain AA, Calvo SE, Ballarano CA, Doench JG, Root DE, Mootha VK. A genome-wide CRISPR death screen identifies genes essential for oxidative phosphorylation. Cell Metab. 2016;24:875–885. doi: 10.1016/j.cmet.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman ES, Dhillon H, Zhang CY, Cinti S, Bianco AC, Kobilka BK, Lowell BB. betaAR signaling required for diet-induced thermo-genesis and obesity resistance. Science. 2002;297:843–845. doi: 10.1126/science.1073160. [DOI] [PubMed] [Google Scholar]
- Berendzen KM, Durieux J, Shao LW, Tian Y, Kim HE, Wolff S, Liu Y, Dillin A. Neuroendocrine coordination of mitochondrial stress signaling and proteostasis. Cell. 2016;166:1553–1563.e10. doi: 10.1016/j.cell.2016.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AA, Kozak LP. A recurring problem with the analysis of energy expenditure in genetic models expressing lean and obese phenotypes. Diabetes. 2010;59:323–329. doi: 10.2337/db09-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. Nonshivering thermogenesis and its adequate measurement in metabolic studies. J Exp Biol. 2011;214:242–253. doi: 10.1242/jeb.050989. [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva D, Tu YT, Amunts A, Fontanesi F, Barrientos A. Mitochondrial ribosome assembly in health and disease. Cell Cycle. 2015;14:2226–2250. doi: 10.1080/15384101.2015.1053672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulloo AG, Jacquet J, Montani JP, Schutz Y. Adaptive thermogenesis in human body weight regulation: more of a concept than a measurable entity? Obes Rev. 2012;13:105–121. doi: 10.1111/j.1467-789X.2012.01041.x. [DOI] [PubMed] [Google Scholar]
- Ellis JM, Li LO, Wu PC, Koves TR, Ilkayeva O, Stevens RD, Watkins SM, Muoio DM, Coleman RA. Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell Metab. 2010;12:53–64. doi: 10.1016/j.cmet.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmett MJ, Lim HW, Jager J, Richter HJ, Adlanmerini M, Peed LC, Briggs ER, Steger DJ, Ma T, Sims CA, et al. Histone deacetylase 3 prepares brown adipose tissue for acute thermogenic challenge. Nature. 2017;546:544–548. doi: 10.1038/nature22819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enerback S, Jacobsson A, Simpson EM, Guerra C, Yamashita H, Harper ME, Kozak LP. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature. 1997;387:90–94. doi: 10.1038/387090a0. [DOI] [PubMed] [Google Scholar]
- Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 2009;9:203–209. doi: 10.1016/j.cmet.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, Fletcher-Sananikone E, Colla S, Wang YA, Chin L, et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010;468:701–704. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganeshan K, Chawla A. Warming the mouse to model human diseases. Nat Rev Endocrinol. 2017;13:458–465. doi: 10.1038/nrendo.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golozoubova V, Cannon B, Nedergaard J. UCP1 is essential for adaptive adrenergic nonshivering thermogenesis. Am J Physiol Endocrinol Metab. 2006;291:E350–E357. doi: 10.1152/ajpendo.00387.2005. [DOI] [PubMed] [Google Scholar]
- Golozoubova V, Hohtola E, Matthias A, Jacobsson A, Cannon B, Nedergaard J. Only UCP1 can mediate adaptive nonshivering thermogenesis in the cold. FASEB J. 2001;15:2048–2050. doi: 10.1096/fj.00-0536fje. [DOI] [PubMed] [Google Scholar]
- Greber BJ, Ban N. Structure and function of the mitochondrial ribosome. Annu Rev Biochem. 2016;85:103–132. doi: 10.1146/annurev-biochem-060815-014343. [DOI] [PubMed] [Google Scholar]
- Gunawardana SC, Piston DW. Reversal of type 1 diabetes in mice by brown adipose tissue transplant. Diabetes. 2012;61:674–682. doi: 10.2337/db11-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurumurthy S, Xie SZ, Alagesan B, Kim J, Yusuf RZ, Saez B, Tzatsos A, Ozsolak F, Milos P, Ferrari F, et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 2010;468:659–663. doi: 10.1038/nature09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg BM, Larsson NG. Making proteins in the powerhouse. Cell Metab. 2014;20:226–240. doi: 10.1016/j.cmet.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Tang T, Abbott M, Viscarra JA, Wang Y, Sul HS. AMPK phosphorylates desnutrin/ATGL and hormone-sensitive lipase to regulate lipolysis and fatty acid oxidation within adipose tissue. Mol Cell Biol. 2016;36:1961–1976. doi: 10.1128/MCB.00244-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus S, Munzberg H, Truloff C, Heldmaier G. Physiology of transgenic mice with brown fat ablation: obesity is due to lowered body temperature. Am J Physiol. 1998;274:R287–R293. doi: 10.1152/ajpregu.1998.274.2.R287. [DOI] [PubMed] [Google Scholar]
- Kong X, Banks A, Liu T, Kazak L, Rao RR, Cohen P, Wang X, Yu S, Lo JC, Tseng YH, et al. IRF4 is a key thermogenic transcriptional partner of PGC-1alpha. Cell. 2014;158:69–83. doi: 10.1016/j.cell.2014.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss S, Zhang CY, Lowell BB. The mitochondrial uncoupling-protein homologues. Nat Rev Mol Cell Biol. 2005;6:248–261. doi: 10.1038/nrm1592. [DOI] [PubMed] [Google Scholar]
- Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- Lee J, Choi J, Aja S, Scafidi S, Wolfgang MJ. Loss of adipose fatty acid oxidation does not potentiate obesity at thermoneutrality. Cell Rep. 2016;14:1308–1316. doi: 10.1016/j.celrep.2016.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Ellis JM, Wolfgang MJ. Adipose fatty acid oxidation is required for thermogenesis and potentiates oxidative stress-induced inflammation. Cell Rep. 2015;10:266–279. doi: 10.1016/j.celrep.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Rossmeisl M, McClaine J, Riachi M, Harper ME, Kozak LP. Paradoxical resistance to diet-induced obesity in UCP1-deficient mice. J Clin Invest. 2003;111:399–407. doi: 10.1172/JCI15737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zheng Z, Zhu X, Meng M, Li L, Shen Y, Chi Q, Wang D, Zhang Z, Li C, et al. Brown adipose tissue transplantation improves whole-body energy metabolism. Cell Res. 2013;23:851–854. doi: 10.1038/cr.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell BB, Spiegelman BM. Towards a molecular understanding of adaptive thermogenesis. Nature. 2000;404:652–660. doi: 10.1038/35007527. [DOI] [PubMed] [Google Scholar]
- Lowell BB, S-Susulic V, Hamann A, Lawitts JA, Himms-Hagen J, Boyer BB, Kozak LP, Flier JS. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature. 1993;366:740–742. doi: 10.1038/366740a0. [DOI] [PubMed] [Google Scholar]
- Lusis AJ, Attie AD, Reue K. Metabolic syndrome: from epidemiology to systems biology. Nat Rev Genet. 2008;9:819–830. doi: 10.1038/nrg2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major GC, Doucet E, Trayhurn P, Astrup A, Tremblay A. Clinical significance of adaptive thermogenesis. Int J Obes. 2007;31:204–212. doi: 10.1038/sj.ijo.0803523. [DOI] [PubMed] [Google Scholar]
- Makki K, Froguel P, Wolowczuk I. Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflamm. 2013;2013:139239. doi: 10.1155/2013/139239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney SK, Fuller A, Mitchell D, Gordon C, Overton JM. Translating animal model research: does it matter that our rodents are cold? Physiology (Bethesda) 2014;29:413–420. doi: 10.1152/physiol.00029.2014. [DOI] [PubMed] [Google Scholar]
- Mardones P, Dillin A, Hetz C. Cell-nonautonomous control of the UPR: mastering energy homeostasis. Cell Metab. 2014;20:385–387. doi: 10.1016/j.cmet.2014.07.009. [DOI] [PubMed] [Google Scholar]
- Mottillo EP, Desjardins EM, Crane JD, Smith BK, Green AE, Ducommun S, Henriksen TI, Rebalka IA, Razi A, Sakamoto K, et al. Lack of adipocyte AMPK exacerbates insulin resistance and hepatic steatosis through brown and beige adipose tissue function. Cell Metab. 2016;24:118–129. doi: 10.1016/j.cmet.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moullan N, Mouchiroud L, Wang X, Ryu D, Williams EG, Mottis A, Jovaisaite V, Frochaux MV, Quiros PM, Deplancke B, et al. Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Rep. 2015;10:1681–1691. doi: 10.1016/j.celrep.2015.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum M, Leibel RL. Adaptive thermogenesis in humans. Int J Obes. 2010;34:S47–S55. doi: 10.1038/ijo.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MT, Hoogenraad NJ. Mitochondrial-nuclear communications. Annu Rev Biochem. 2007;76:701–722. doi: 10.1146/annurev.biochem.76.052305.091720. [DOI] [PubMed] [Google Scholar]
- Sadat R, Barca E, Masand R, Donti TR, Naini A, De Vivo DC, DiMauro S, Hanchard NA, Graham BH. Functional cellular analyses reveal energy metabolism defect and mitochondrial DNA depletion in a case of mitochondrial aconitase deficiency. Mol Genet Metab. 2016;118:28–34. doi: 10.1016/j.ymgme.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, Iwanaga T, Miyagawa M, Kameya T, Nakada K, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58:1526–1531. doi: 10.2337/db09-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument-Bromage H, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina IG, Petrovic N, de Jong JM, Kalinovich AV, Cannon B, Nedergaard J. UCP1 in brite/beige adipose tissue mitochondria is functionally thermogenic. Cell Rep. 2013;5:1196–1203. doi: 10.1016/j.celrep.2013.10.044. [DOI] [PubMed] [Google Scholar]
- Shan T, Xiong Y, Zhang P, Li Z, Jiang Q, Bi P, Yue F, Yang G, Wang Y, Liu X, et al. Lkb1 controls brown adipose tissue growth and thermogenesis by regulating the intracellular localization of CRTC3. Nat Commun. 2016;7:12205. doi: 10.1038/ncomms12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford KI, Middelbeek RJ, Townsend KL, An D, Nygaard EB, Hitchcox KM, Markan KR, Nakano K, Hirshman MF, Tseng YH, et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest. 2013;123:215–223. doi: 10.1172/JCI62308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trayhurn P, Thurlby PL, James WP. Thermogenic defect in pre-obese ob/ob mice. Nature. 1977;266:60–62. doi: 10.1038/266060a0. [DOI] [PubMed] [Google Scholar]
- Tschop MH, Speakman JR, Arch JR, Auwerx J, Bruning JC, Chan L, Eckel RH, Farese RV, Jr, Galgani JE, Hambly C, et al. A guide to analysis of mouse energy metabolism. Nat Methods. 2012;9:57–63. doi: 10.1038/nmeth.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uldry M, Yang W, St-Pierre J, Lin J, Seale P, Spiegelman BM. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006;3:333–341. doi: 10.1016/j.cmet.2006.04.002. [DOI] [PubMed] [Google Scholar]
- van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- Vernochet C, Damilano F, Mourier A, Bezy O, Mori MA, Smyth G, Rosenzweig A, Larsson NG, Kahn CR. Adipose tissue mitochondrial dysfunction triggers a lipodystrophic syndrome with insulin resistance, hepatosteatosis, and cardiovascular complications. FASEB J. 2014;28:4408–4419. doi: 10.1096/fj.14-253971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto NJ, Enerback S, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ryu D, Houtkooper RH, Auwerx J. Antibiotic use and abuse: a threat to mitochondria and chloroplasts with impact on research, health, and environment. Bioessays. 2015;37:1045–1053. doi: 10.1002/bies.201500071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Paulo E, Wu D, Wu Y, Huang W, Chawla A, Wang B. Adipocyte liver kinase b1 suppresses beige adipocyte renaissance through class IIa histone deacetylase 4. Diabetes. 2017;66:2952–2963. doi: 10.2337/db17-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig I, Braun HP, Schagger H. Blue native PAGE. Nat Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- Yang H, Wu JW, Wang SP, Severi I, Sartini L, Frizzell N, Cinti S, Yang G, Mitchell GA. Adipose-specific deficiency of fumarate hydratase in mice protects against obesity, hepatic steatosis, and insulin resistance. Diabetes. 2016;65:3396–3409. doi: 10.2337/db16-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Finkel T. Mitohormesis. Cell Metab. 2014;19:757–766. doi: 10.1016/j.cmet.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang Q, Song P, Zou MH. Liver kinase b1 is required for white adipose tissue growth and differentiation. Diabetes. 2013;62:2347–2358. doi: 10.2337/db12-1229. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhu Z, Yao J, Johns T, Fu K, De Bie I, Macmillan C, Cuthbert AP, Newbold RF, Wang J, Chevrette M, et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Genet. 1998;20:337–343. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.