

Abstract
Since 2003, with the FDA approval of bortezomib, proteasome inhibitors have changed the management of hematologic malignancies and dramatically improved outcomes for patients with multiple myeloma and mantle cell lymphoma. Since that time, two additional proteasome inhibitors, carfilzomib and ixazomib have been approved, with other agents and combinations currently under investigation. Proteasomes degrade ubiquitinated proteins or substrates through the ubiquitin-proteasome pathway, a pathway that is utilized in multiple myeloma due to the high protein turnover with immunoglobulin production. Proteasome inhibitors exploit dependence on this pathway, halting protein degradation that ultimately results in apoptosis and cell death. Here, we will discuss the structure of the proteasome and the mechanisms of action for proteasome inhibitors to further understand their role in hematologic malignancies.
Keywords: Proteasome inhibitors, Multiple Myeloma, Bortezomib, Ubiquitin-Proteasome Pathway
Introduction
Proteasomes play a necessary role in cell survival, DNA repair and the proliferation of malignant cells. Choreographed degradation of cyclin dependent kinase (CDK) activators or inhibitors is needed for the cell to progress through all steps of the cell cycle, from DNA replication to mitosis (1). Proteasomes also play an active role in normal cellular functions, and in the degradation of misfolded or mutated proteins.
Despite the essential role of the ubiquitin-proteasome pathway, proteasome inhibitors (PIs) are well tolerated in the clinic, with a limited spectrum of side effects. PIs are effective in hematologic malignancies including multiple myeloma and mantle cell lymphoma, improving progression free survival (PFS) and overall survival (OS). Multiple myeloma is the ideal target for proteasome inhibitors due to the large amount of IgG production in plasma cells. The high protein turnover in myeloma cells, results in a favorable therapeutic window for proteasome inhibitors in this disease with preferential susceptibility of the malignant cells relative to normal cells. In 2003, the FDA approved the first PI, bortezomib (Velcade, PS-341) for the treatment of relapsed and refractory multiple myeloma (Figure 1). Since then, two other agents, carfilzomib (Kyprolis) and ixazomib (Ninlaro) have secured regulatory approval for multiple myeloma, and the indication for bortezomib has expanded to include first line treatment for multiple myeloma and mantle cell lymphoma. Other proteasome inhibitors are still under investigation, including marizomib that may be beneficial in patients with glioblastoma. Here, we will discuss the proteasome structure and function and explore the mechanisms that allow proteasome inhibitors to be so effective in hematologic malignancies.
Figure 1. Chemical Structures of bortezomib, carfilzomib and ixazomib.

The circles highlight the active moieties of the individual proteasome inhibitors
The Ubiquitin-Proteasome Pathway
The ubiquitin-proteasome pathway (UPP) regulates cellular functions, removing proteins that are damaged, misfolded or otherwise not wanted in the cell in a well-orchestrated manner. The protein is first targeted for degradation through ubiquitination. Ubiquitination requires three distinct steps to achieve a polyubiquitinated product that is easily identified by the proteasome. The first step occurs when ubiquitin becomes activated by E1, the ubiquitin activating enzyme. Activated ubiquitin is then transferred to E2, the ubiquitin-conjugating enzyme and E3, the ubiquitin-protein ligase that transfers ubiquitin to proteins. This process is repeated multiple times until a polyubiquitin chain emerges, targeting the protein for degradation in the proteasome (1, 2).
The 26S proteasome is a multiprotein complex made of a 20S catalytic core and one or two 19S regulatory subunits on either end of the 20S core (Figure 2). The 19S subunits bind the polyubiquitin chain, cleaving it from the target protein. The protein then passes through the 20S core where it is degraded to small oligopeptides, less than 25 amino acids. The 19S subunits commonly flanks the 20S core. However, the 20S core can additionally act alone to cause ubiquitin independent protein degradation. This core is a barrel structure composed of 4 heptameric rings. The two alpha rings sandwich the two beta rings. The beta rings each contain 3 active sites for protein degradation: chymotrypsin-like (β5), trypsin-like (β2) and caspase-like (β1). The chymotrypsin-like site on β5 is the primary target of proteasome inhibitors (3) although at higher drug concentrations of drug, the other trypsin- and caspase-like two sites are also inhibited as well.
Figure 2. Proteasome Structure.

The 20S catalytic core binds to the 19S regulatory complex to form the 26S proteasome structure. Proteins that are tagged with ubiquitin bind to the 19S complex and are degraded at the proteolytic β subunits. Bortezomib, carfilzomib and ixazomib all inhibit the β5 subunit thereby inhibiting the catalytic activity of the proteasome.
Mechanism of Proteasome Inhibitor-Mediated Cellular Cytotoxicity (Figure 3)
Figure 3. Mechanism of Proteasome Inhibitors.
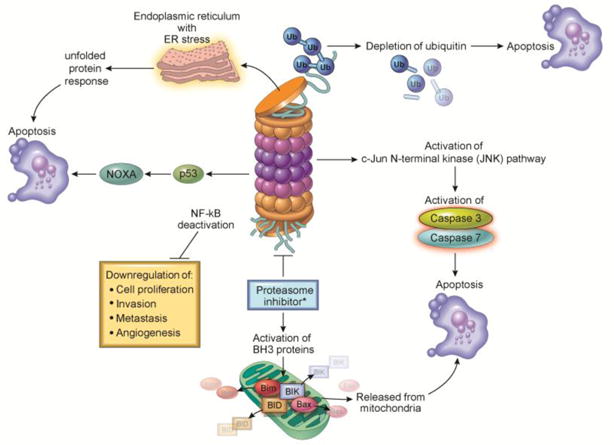
Proteasome inhibition acts through multiple mechanisms to induce cell death. Inhibition of NF-κB results in downregulation of multiple pro-neoplastic pathways. Activation of the JNK pathway leads to caspase activation and apoptosis. Additionally, interference with the degradation of pro-apoptotic proteins such as Bim, BIK, BID, or Bax and an increase in NOXA activation promotes apoptosis. The depletion of ubiquitin and ER stress with activation of the unfolded protein response can promote apoptosis as well.
The mechanism by which proteasome inhibitors lead to cell death is diverse and affects many pathways utilized in cancer cells. One putative mechanism of cytotoxicity is inhibition of the NF-kB pathway, a pro-survival pathway for many cell types, especially those of hematopoetic lineages. IκBα is an endogenous protein inhibitor of NF-κB that is degraded by the proteasome when the cell receives a stimulus to activate the pathway. Its degradation is necessary for the p50/p65 NF-kB transcription factors to become active and translocate to the nucleus (4). When the proteasome is inhibited, IκBα remains intact and bound to the p50/p65 NF-kB heterodimer, preventing activation of the NF-kB pathway. Inhibition of the NF-kB pathway was initially thought to be the principal mechanism of anti-cancer activity of proteasome inhibitors as this pathway plays a role in cell proliferation, invasion, metastasis and angiogenesis. However, a potent IκB kinase inhibitor, PS-1145, that blocks NF-kB activation proximal to the IκBα step, does not emulate the cellular toxicity profile of proteasome inhibitors, suggesting other mechanisms are equally, if not more, important (5).
Multiple putative mechanisms of cellular toxicity have been proposed for proteasome inhibitors (PIs). Included amongst these is direct induction of apoptosis through c-Jun NH2-terminal kinase (JNK) and p53. For example, proteasome inhibition leads to activation of JNK, resulting in programmed apoptotic cell death via caspase 8 and caspase 3. The expression of p53, a known tumor-suppressor important in activating cell death under diverse circumstances, is induced by treatment with PIs, which can induce apoptosis even in the presence of mutant p53 (6). Alternatively, proteasome inhibition can induce interaction of p53 with MDM2, followed by activation of the JNK pathway, shifting the cell towards apoptosis (7). Additionally PIs can act indirectly to cause apoptosis by preventing degradation of pro-apoptotic family proteins. Normally, the pro-apoptotic proteins Bim, Bid and Bik are regulated through rapid ubiquitination and proteasomal degradation. With proteasomal inhibition, these proteins accumulate, triggering caspase activation and cell death (8–10). NOXA, a pro-apoptotic member of the Bcl-2 family that interacts with p53 in the setting of DNA damage (11), can be induced by hypoxia, cytokine signaling or mitogenic stimulation, but is normally rapidly degraded through the proteasome (12). Proteasome inhibition increases levels of NOXA, activates caspase-9 and consequently leads to apoptosis (13). Proteasome inhibition can also induce expression of NOXA independently of p53, inducing further cell death (14).
Following synthesis on ribosomes, proteins are usually folded and assembled in the endoplasmic reticulum (ER) before being released. In multiple myeloma, for example, plasma cells produce large quantities of immunoglobulins resulting in a high protein burden for the ER. The ER has a quality control mechanism to monitor for misfolded proteins that cannot be properly refolded, and targets those proteins for degradation by the proteasome (15). When the cell produces large amounts of proteins, the ER becomes stressed. This ER stress initiates the unfolded protein response (UPR), activating intracellular signal transduction pathways to maintain homeostasis in the ER by reducing protein synthesis; alternatively, the UPR can cause cell cycle arrest and induce apoptosis, depending on the severity of the ER stress (16). PIs prevent the degradation of ubiquitinated proteins, effectively blocking the translocation of misfolded proteins out of the ER to the proteasome. The accumulation of misfolded proteins in the ER effectively increases ER stress and activates the UPR, cell cycle arrest, and subsequent apoptosis (17).
Clinical Development of Proteasome Inhibitors
Initially, proteasome inhibitors were developed to prevent cancer-related cachexia. Their role in promoting apoptosis in cancer cell lines in preclinical studies, however, quickly led to the trials that resulted in the 2003 approval of bortezomib for multiple myeloma followed by carfilzomib in 2012 and ixazomib in 2015. These proteasome inhibitors all work primarily at the chymotrypsin-like, β5 subunit. At higher concentrations, these proteasome inhibitors also inhibit the trypsin-like (β2) and caspase-like (β1) subunits as well. These three compounds have all shown activity in multiple myeloma with bortezomib additionally indicated for treatment in mantle cell lymphoma (Table 1). While in multiple myeloma the PIs can be given as single agents, with dexamethasone, they are more effective given as a triplet therapy in combination with dexamethasone and an immunomodulatory drug (lenalidomide, thalidomide, or pomalidomide).
Table 1.
Proteasome inhibitors and their mode of action
| Name | Kinetics | Active moiety | Indication | Route of administration | Common toxicities |
|---|---|---|---|---|---|
| Bortezomib [Velcade®] | Slowly reversible inhibitor β5>β1>β2 | Boronate | First line, relapsed or refractory MM and MCL | IV/SC | Peripheral neuropathy, nausea, vomiting, diarrhea, cytopenias, infection |
| Carfilzomib [Kyprolis®] | Irreversible inhibitor β5>β2/β1 | Epoxyketone | Relapsed or refractory MM | IV | Dyspnea, cytopenias, nausea, vomiting, diarrhea, fatigue, headache, peripheral edema |
| Ixazomib [Ninlaro®] | Reversible inhibitor β5>β1 | Boronate | MM after having received one line of treatment | Oral | Diarrhea, constipation, cytopenias, peripheral neuropathy, nausea, vomiting, peripheral edema, back pain |
In 2003, Richardson et al. reported a single arm phase 2 clinical trial in 202 patients with relapsed, refractory multiple myeloma and found that treatment with bortezomib resulted in a 35% response rate (18). This was followed by a randomized controlled trial comparing bortezomib to high dose dexamethasone for relapsed multiple myeloma. Here, the response rate was 38% for bortezomib compared to 18% for dexamethasone (p<0.001) with a median time to progression of 6.22 months and 3.49 months, respectively (hazard ratio 0.55; p<0.001) (19). Since then, bortezomib has been used in multiple combinations for first line therapy and in relapsed, refractory multiple myeloma with continued improvement in progression free survival and overall survival.
Carfilzomib with dexamethasone is approved for the therapy of relapsed or refractory multiple myeloma and acts similarly to bortezomib except that it binds irreversibly and selectively to the proteasome. The toxicity profile is slightly different with less peripheral neuropathy and more cytopenias observed with rare pulmonary and cardiac toxicity. As a single agent in patients with relapsed, refractory multiple myeloma, the overall response rate was 23.7% with a median overall survival of 15.6 months in a phase II study with 266 patients (20). Carfilzomib was also shown effective in combination therapy, both as first line and second line, with either lenalidomide or pomalidomide and dexamethasone.
Ixazomib is the only oral proteasome inhibitor and it is used in combination with lenalidomide and dexamethasone for patients with relapsed, refractory multiple myeloma. In 722 patients randomly assigned to receive either ixazomib with lenalidomide and dexamethasone or placebo with lenalidomide and dexamethasone, the median progression free survival (PFS) was 20.6 months compared to 14.7 months (HR 0.74 p<0.01) (21). Similar to bortezomib, it is a reversible inhibitor with observed cytopenias and peripheral neuropathy.
Future studies
Since proteasome inhibitors have been impressively effective in the treatment of hematologic malignancies, they continue to be an area of interest for new indications and to improve existing PI regimens. Clinical trials of PIs in combination with other agents are being conducted in leukemias, lymphomas, solid tumors and GVHD. PIs continue to be developed that might target the proteasome at different sites to overcome resistance or have activity in different malignancies. Areas of resistance include hyperactivation of the trypsin-like (β2) and caspase-like (β1) proteolytic sites to compensate for inactivation at the chymotrypsin-like (β5) site. Newer PIs such as marizomib irreversibly bind to all three proteolytic sites on the 20S preventing this compensatory activation of the β2 and β5 subunit. Marizomib is currently being studied in glioblastoma and multiple myeloma (22). Inhibition of the ubiquitination process may additionally overcome the resistance seen in PIs and offer a novel form of treatment in the future. NEDD8 activating enzyme inhibitors such as pevonedistat inhibit the E3 ubiquitin ligase pathway and may show benefit in hematologic malignancies (23). Alternatively, Protacs or proteolysis chimeric molecules are currently being developed to selectively target cancer-promoting proteins for degradation. This process utilizes the mechanism of the E3 ubiquitin ligase to ubiquitinate and rapidly degrade the target of interest with promising preclinical results in different solid tumor models (24).
Conclusion
In conclusion, proteasome inhibitors target cellular mechanisms of protein degradation by blocking the β proteolytic subunits of the 20s proteasome. Their main mechanisms of cytotoxicity are due to turning off cell survival pathways, turning on apoptotic pathways, and increasing ER stress. To date, PIs have shown the most clinical activity in treating hematologic malignancies especially multiple myeloma and mantle cell lymphoma. They have impacted our treatment of multiple myeloma most impressively, with improvements in progression free survival and overall survival. Ongoing studies in the laboratory and in clinical trials will show us how to optimize their use in other settings as well.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.King RW, Deshaies RJ, Peters J-M, Kirschner MW. How Proteolysis Drives the Cell Cycle. Science. 1996;274(5293):1652–9. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- 2.Adams J. The proteasome: structure, function, and role in the cell. Cancer Treatment Reviews. 2003;29(Supplement 1):3–9. doi: 10.1016/s0305-7372(03)00081-1. [DOI] [PubMed] [Google Scholar]
- 3.Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nature Reviews Clinical Oncology. 2017;14:417. doi: 10.1038/nrclinonc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell. 1994;78(5):773–85. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 5.Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, et al. NF-κB as a Therapeutic Target in Multiple Myeloma. Journal of Biological Chemistry. 2002;277(19):16639–47. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 6.Hideshima T, Mitsiades C, Akiyama M, Hayashi T, Chauhan D, Richardson P, et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood. 2003;101(4):1530–4. doi: 10.1182/blood-2002-08-2543. [DOI] [PubMed] [Google Scholar]
- 7.Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, et al. The Proteasome Inhibitor PS-341 Inhibits Growth, Induces Apoptosis, and Overcomes Drug Resistance in Human Multiple Myeloma Cells. Cancer Research. 2001;61(7):3071–6. [PubMed] [Google Scholar]
- 8.Breitschopf K, Zeiher AM, Dimmeler S. Ubiquitin-mediated Degradation of the Proapoptotic Active Form of Bid: A FUNCTIONAL CONSEQUENCE ON APOPTOSIS INDUCTION. Journal of Biological Chemistry. 2000;275(28):21648–52. doi: 10.1074/jbc.M001083200. [DOI] [PubMed] [Google Scholar]
- 9.Marshansky V, Wang X, Bertrand R, Luo H, Duguid W, Chinnadurai G, et al. Proteasomes Modulate Balance Among Proapoptotic and Antiapoptotic Bcl-2 Family Members and Compromise Functioning of the Electron Transport Chain in Leukemic Cells. The Journal of Immunology. 2001;166(5):3130–42. doi: 10.4049/jimmunol.166.5.3130. [DOI] [PubMed] [Google Scholar]
- 10.Duffy MJ, Blaser J, Duggan C, McDermott E, O’Higgins N, Fennelly JJ, et al. Assay of matrix metalloproteinase type 8 and 9 by ELISA in human breast cancer. Br J Cancer. 1995;71 doi: 10.1038/bjc.1995.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. Noxa, a BH3-Only Member of the Bcl-2 Family and Candidate Mediator of p53-Induced Apoptosis. Science. 2000;288(5468):1053–8. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 12.Ploner C, Kofler R, Villunger A. Noxa: at the tip of the balance between life and death. Oncogene. 2009;27:S84. doi: 10.1038/onc.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qin J-Z, Ziffra J, Stennett L, Bodner B, Bonish BK, Chaturvedi V, et al. Proteasome Inhibitors Trigger NOXA-Mediated Apoptosis in Melanoma and Myeloma Cells. Cancer Research. 2005;65(14):6282–93. doi: 10.1158/0008-5472.CAN-05-0676. [DOI] [PubMed] [Google Scholar]
- 14.Pandit B, Gartel AL. Proteasome Inhibitors Induce p53-Independent Apoptosis in Human Cancer Cells. The American Journal of Pathology. 2011;178(1):355–60. doi: 10.1016/j.ajpath.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kostova Z, Wolf DH. For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin–proteasome connection. The EMBO Journal. 2003;22(10):2309–17. doi: 10.1093/emboj/cdg227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walter P, Ron D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science. 2011;334(6059):1081–6. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 17.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–16. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, et al. A Phase 2 Study of Bortezomib in Relapsed, Refractory Myeloma. New England Journal of Medicine. 2003;348(26):2609–17. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 19.Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, et al. Bortezomib or High-Dose Dexamethasone for Relapsed Multiple Myeloma. New England Journal of Medicine. 2005;352(24):2487–98. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 20.Siegel DS, Martin T, Wang M, Vij R, Jakubowiak AJ, Lonial S, et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood. 2012;120(14):2817–25. doi: 10.1182/blood-2012-05-425934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moreau P, Masszi T, Grzasko N, Bahlis NJ, Hansson M, Pour L, et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. New England Journal of Medicine. 2016;374(17):1621–34. doi: 10.1056/NEJMoa1516282. [DOI] [PubMed] [Google Scholar]
- 22.Levin N, Spencer A, Harrison SJ, Chauhan D, Burrows FJ, Anderson KC, et al. Marizomib irreversibly inhibits proteasome to overcome compensatory hyperactivation in multiple myeloma and solid tumour patients. British journal of haematology. 2016;174(5):711–20. doi: 10.1111/bjh.14113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swords RT, Erba HP, DeAngelo DJ, Bixby DL, Altman JK, Maris M, et al. Pevonedistat (MLN4924), a First-in-Class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. British journal of haematology. 2015;169(4):534–43. doi: 10.1111/bjh.13323. [DOI] [PubMed] [Google Scholar]
- 24.Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, et al. Development of Protacs to Target Cancer-promoting Proteins for Ubiquitination and Degradation. Molecular & Cellular Proteomics. 2003;2(12):1350–8. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]