

Abstract
The stereoselective oxidation of hydrocarbons represents one of the most significant advances in synthetic chemistry over the last fifty years1–3. Inspired by nature, chemists have developed enantioselective dihydroxylations, epoxidations, and other oxidations of unsaturated hydrocarbons. More recently, the catalytic enantioselective allylic C–H oxidation of alkenes has emerged as a powerful chemical strategy, streamlining the production of pharmaceuticals, natural products, fine chemicals and other functional materials4–7. Allylic functionalization provides a direct path to chiral synthons with a newly formed stereocenter from petrochemical feedstocks while preserving the olefin functionality as a handle for further elaboration. Various metal-based catalysts have been discovered for the enantioselective allylic C–H oxidation of simple alkenes with cyclic or terminal double bonds8–16. However, a general and selective allylic oxidation remains elusive with the more common internal alkenes. Here, we report the enantioselective, regioselective, and E/Z selective allylic oxidation of unactivated internal alkenes via a catalytic asymmetric hetero-ene reaction with a chalcogen-based oxidant. This method represents the first example of selectively converting unsymmetrical internal alkenes into allylic functionalized products with high stereoselectivity and regioselectivity. Stereospecific transformations of the multifunctional allylic oxidation products highlight the potential for rapidly converting internal alkenes into a broad range of enantioenriched structures that can be utilized in the synthesis of complex target molecules.
Unlike terminal alkenes (1), which only contain a single set of enantiotopic allylic protons (Fig. 1a), internal alkenes (2) possess two sets of protons on either side of the olefin moiety, thereby posing the additional challenge of regioselectivity for unsymmetrical substrates (Fig. 1b). Furthermore, when the resulting product is an internal alkene, the issue of E/Z selectivity subsists. The inability to control indiscriminate C–H functionalization of electronically and sterically similar allylic protons, therefore, has the potential to produce a mixture of regio-, diastereo-, and enantiomeric isomers that are difficult to separate via preparative methods.
Figure 1. Catalytic enantioselective oxidation of unactivated terminal and internal alkenes.
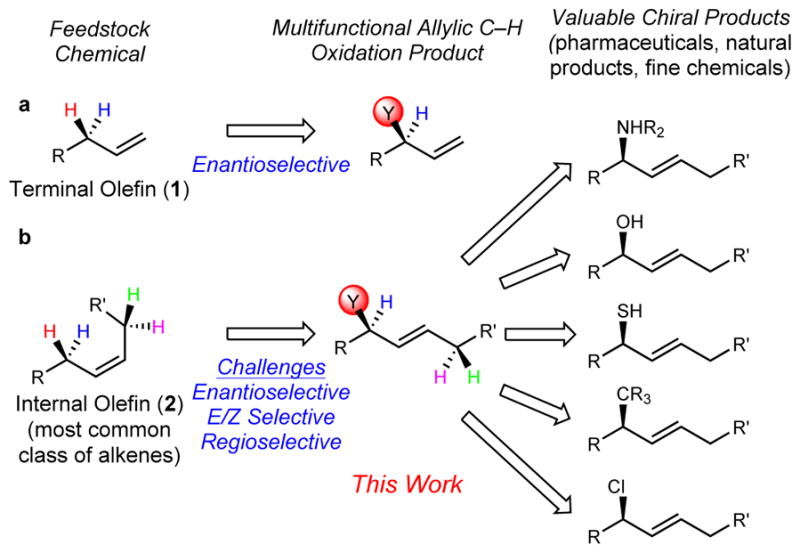
a, Traditional approaches for allylic oxidation of terminal alkenes. b, Challenges of allylic oxidation of internal alkenes.
Our studies initiated with the ultimate goal of developing a general platform for the construction of chiral olefinic building blocks from inexpensive commodity internal alkenes. To this aim, we set out to develop a catalytic enantioselective oxidation that would provide access to a multifunctional intermediate capable of stereospecific differentiation toward a variety of products. This strategy would provide chemists with the ability to selectively introduce allylic C-N, C-O, C-S, C-C, and C–halogen bonds, enabling rapid library synthesis of analogous enantioenriched products (Fig. 1b).
Early reports of racemic allylic oxidation of unfunctionalized alkenes via hetero-ene reactions with chalcogen-based oxidants indicated that this class of enophile might serve as a suitable starting point for the development of a highly selective allylic oxidation of internal alkenes17–22. These reactions proceed through spontaneous thermal hetero-ene reactions that are not enantioselective but exhibit oxidant-controlled regioselectivity and E/Z selectivity for allylic oxidation of unsymmetrical internal alkenes.
Selection of electrophilic chalcogen-based oxidant 3d was an essential step in the development of this new approach to the stereoselective allylic oxidation of internal alkenes (Extended Data Fig. 1a). We sought to identify a chiral Lewis acid that could catalyze the ene reaction between internal alkenes and oxidant 3d via LUMO-lowering activation of the electrophilic oxidant. In the absence of a background reaction at −70 °C, a combination of SbCl5 and (R)-BINOL proved competent to catalyze the ene reaction between cis-5-decene 4 and oxidant 3d with regio- and stereochemical control (Extended Data Fig. 1b, entry 10). After an extensive exploration of reaction parameters, SbCl5 and co-catalyst 6 were deemed optimal for the formation of allylic oxidation product 8a in 84% isolated yield and 92.5:7.5 enantiomeric ratio with complete E-olefin selectivity (Fig. 2a). The BINOL-based co-catalysts can be recovered by aqueous workup upon completion of the reaction (Fig. S5, Supporting Information).
Figure 2. Substrate scope of the catalytic enantioselective and regioselective allylic oxidation of internal unactivated alkenes.

Reaction conditions. Cis-5-decene (1 equiv), sulfurimide reagent 3d (1.5 equiv), solvent (0.13 M). Isolated yield (>20:1 initial dr). a, Enantioselective oxidation of functionalized symmetrical internal alkenes. [a] When BINOL was used instead of co-catalyst 6 in a solvent mixture of CH2Cl2:PhMe (1:2), product 8a was formed in 81% yield and 89:11 er. [b] Yields were determined by 1H NMR using 1,4-dimethoxybenzene as an internal standard. b, Regio- and chemoselectivity trends in the enantioselective allylic oxidation of unsymmetrical internal alkenes. EWG = electron-withdrawing group. [a] BINOL was used instead of co-catalyst 6, in a solvent mixture of CH2Cl2:PhMe (1:2). [b] Yields were determined by 1H NMR using 1,4-dimethoxybenzene as an internal standard. [c] Isolated yield was determined by methylating the allylic oxidation product in the presence of Me2SO4 and NEt3.
With the ultimate goal of developing an allylic C–H oxidation that can be successfully employed in many complex molecular settings, we explored the efficiency of this enantioselective reaction with a diverse range of symmetrical internal alkenes. Acyclic cis-internal alkenes with varying chain length and functionality all afforded the oxidized product with comparably good yield and stereoinduction (8a–8c, Fig. 2a), representing the most general strategy for this class of substrate to date. We synthesized chiral enantioenriched allylic sulfinamide products with various functional groups such as aromatic rings (8d), chlorides (8e), bromides (8f), iodides (8g), trifluoromethyl groups (8h), trifluoroacetates (8i), and tosylated indoles (8j). In general, the allylic oxidation products were formed in good yields and enantiomeric ratios, with exclusive E-olefin selectivity. X-ray crystallographic analysis of the major diastereomer formed initially for allylic oxidation product 8e defined the absolute and relative stereochemistry of the ene reaction products. A preliminary functional group robustness screen23 revealed that this reaction is compatible with nitro groups, fluorides, boronic acids, carboxylic acids, alkynes aldehydes, acetates, cyanides, furans, thiophenes, and protected amines (see supporting information). Products from acyclic internal alkenes were generated in greater enantioenrichment than the corresponding products from cyclic alkenes (8k–8l), presumably due to unfavorable steric interactions encountered with more rigid cyclic olefins.
Initial studies have identified general trends in regioselectivity for the oxidation of unsymmetrical internal alkenes (Fig. 2b). Under optimized reaction conditions, an internal olefin flanked by a methylene and methine group selectively undergoes enantio- and regioselective ene reactions with the methylene proton (indicated in blue, entry 1). Methylene protons react preferentially to methyl protons (entries 2–4). Competition between methyl and methine protons reveals the more facile reactivity of methyl protons (entry 5), albeit in diminished yield.
We also observed the effect of electronic perturbation on the regioselectivity of the allylic oxidation (entries 6–11). An electron-withdrawing group such as a chloride disfavors the enantioselective, regioselective ene reaction of the proximal allylic protons (entries 6–7). Instead, the oxidation favors reactivity through the alternative remote allylic protons (indicated in red). The inductive effect of the chloride functionality is modulated by the distance between the electron-withdrawing group and the olefin, which directly impacts the regioselectivity of the reaction. If the chloride or other electron-withdrawing group (e.g., bromide or trifluoroacetate) is positioned too close to the olefin, it diminishes the π-nucleophilicity of the olefin and shuts down the catalytic ene reaction (entry 8). Similar outcomes in regioselectivity were observed for other electron-withdrawing groups, such as iodide (entry 9), trifluoroacetate (entry 10), and phenyl (entry 11). These results suggest emerging trends that may be exploited in the enantioselective and regioselective allylic oxidation of unsymmetrical alkenes in more complex molecular settings. For example, internal competition experiments with certain classes of dienes highlight the chemoselective allylic oxidation of an internal olefin in the presence of a styrene (entry 12) or an allylic halide (entry 13).
With a selective allylic oxidation of internal alkenes in hand, we explored the generality of these products as synthetically versatile chiral building blocks for further chemical transformations (Fig. 3). Although the diversification of allylic oxidation products with a broad range of reagents has been reported for terminal alkenes24, this strategy has not yet been demonstrated with internal alkenes. The multiple atoms in the allylic sulfinamide provide a unique opportunity to convert the allylic oxidation product to a variety of functional groups. Our approach represents a general platform for formal enantioselective allylic C–X bond formation of internal olefins, where X can be carbon, nitrogen, oxygen, sulfur, or halogen based functional groups.
Figure 3. Multiple synthetic derivitizations of the synthetically versatile product of the catalytic enantioselective and regioselective allylic oxidation of internal alkenes.

a: PhSO2N=S=O (3d), (1.5 equiv), SbCl5 (20 mol%), co-catalyst 6 (25 mol%), TFA (0.5 equiv), CH2Cl2, −70 °C, 16 h. b (C–C bond formation): CuBr2·SMe2 (5 mol%) EtMgCl, DME, −70 °C to 0 °C. c (C–S bond retention): LiAlH4, Et2O, 0 °C to 23 °C. d (C–O bond formation): (i) Me2SO4, Et3N, CH2Cl2, 23 °C; (ii) PhMgBr, THF, 0 °C; P(OMe)3, MeOH, 23 °C. e (C–N bond formation): TiCl(Oi-Pr)3 (20 mol%), PhMe, 60 °C; P(OMe)3, MeOH, 23 °C. f (C–Cl bond formation): SO2Cl2, Et2O, −70 ºC to 0 ºC.
Branched hydrocarbon 10 was generated through an allylic substitution of sulfinamide 8a with ethylmagnesium chloride. Although the allylic sulfinamide exists as a mixture of diastereomers (because of the configurationally labile sulfur stereogenic center), the stereochemistry at the oxidized allylic carbon in the starting material dictates the stereochemistry of product 10, which is formed with complete E-olefin selectivity, high enantiospecificity (es), and moderate regioselectivity. The regioselective allylic substitution of unbiased internal allylic electrophiles (e.g., halides, acetates, carbonates) with organometallic reagents remains a challenging synthetic problem25. Therefore, even the moderate regioselectivity of 3:1 (γ:α) represents a significant advance.
Next, we sought to diversify allylic sulfinamide 8a to a variety of chiral heteroatom-containing products with the goal of enabling efficient library synthesis of thiol, alcohol, amine and halogenated building blocks. Enantioenriched allylic thiol 11 was isolated with exclusive E-olefin selectivity after reduction of 8a. Allylic alcohol 12 and allylic amine 13 formed regioselectively with high E-olefin selectivity and enantiospecificity through stereoselective [2,3]-rearrangements of allylic sulfinamide 8a via either the oxygen atom or nitrogen atom, respectively. The presence of two diastereomers of allylic sulfinamide (epimeric at sulfur) did not affect the stereochemical outcome in the formation of products 12 and 13. Most notably, sulfuryl chloride mediated the formation of allylic chloride 14 in excellent yield, with regioselectivity and stereospecificity.
Our findings that a catalytic antimony-BINOL system affects remarkably pronounced enantio-, regio-, diastereo- and E-olefin selectivity in the hetero-ene reaction between oxidant 3d and simple internal alkenes prompted us to better understand the mechanism by which this selectivity is derived (Fig. 4).
Figure 4. Mechanistic studies of the catalytic enantioselective allylic oxidation of internal alkenes.

a, Lewis acid assisted Brønsted acid mode of catalysis. b, Correlation study between the enantiomeric excess of the BINOL co-catalyst and the enantiomeric excess of the product. c, Stereochemical support for a closed transition state. d, Proposed transition state for the catalytic enantioselective allylic oxidation. e, Rationalization of trends in regioselectivity based on the closed transition state.
We observed a markedly diminished yield of sulfinamide 8a in the absence of BINOL 6 (Fig. 4a), which we attribute to the Lewis acid assisted Brønsted acidity (LBA) of BINOL26. This form of activation was utilized by Corey and co-workers for the development of enantioselective polycyclizations mediated by antimony-BINOL27, wherein the acidified proton of BINOL activated an alkene. Our reaction represents a unique application of antimony-BINOL as a catalytic chiral Brønsted acid28–32 for the activation of a heteroatomic electrophile. The lack of product formation in the presence of the sterically hindered Brønsted base 2,6-(t-Bu)2pyridine (which does not exhibit any interaction with SbCl5 via low temperature 1HNMR analysis) is consistent with the proposed LBA mechanism. To examine the structure of the active chiral Brønsted acid catalyst, we conducted a correlation study between the enantiomeric excess of BINOL 6 and the enantiomeric excess of product 8a (Fig. 4b). This experiment revealed a linear relationship, which suggests a 1:1 stoichiometry between the BINOL co-catalyst and the alkene substrate in the transition state. The role of trifluoroacetic acid may be to stabilize the protonated SbCl5-BINOL complex that is necessary for LBA activation.
Stereochemical analysis of the oxidation products obtained from the cis and trans isomers of 5-decene supports a closed transition state for the chiral Brønsted acid catalyzed hetero-ene reaction (Fig. 4c). Stereoisomeric alkenes (Z)-4 and (E)-4 yielded different diastereomeric major products under the SbCl5-BINOL conditions, where the absolute configurations of the carbon stereocenters were opposite (5a and ent-5b). This observation suggests that olefin configuration dictates π-face selectivity in this transformation. Interestingly, the major diastereomers for products obtained from alkenes (Z)-4 and (E)-4 were opposite to the corresponding products obtained from the thermal ene reactions. In addition, unreacted alkene (Z)-4 did not isomerize at lower conversions under the optimized reaction conditions to the more stable (E)-4 isomer.
Based on our mechanistic analysis, we propose that the hetero-ene reaction occurring between sulfurimide reagent 3d and internal alkenes proceeds through a closed transition state in which the chiral Brønsted acid catalyst is activating the sulfurimide reagent through a LUMO lowering effect (Fig. 4d). Transition states 15 and 16 benefit from stabilizing π-interactions between the coordinated oxidant 3d and the naphthyl backbone of co-catalyst 6. π-Face selectivity may be ascribed to steric shielding by the adjacent naphthyl group. Additionally, the switch in diastereoselectivity between the thermal endo-selective hetero-ene reaction and the catalytic reaction suggests that the antimony-BINOL catalyzed transformation is proceeding through an exo transition state.
The observed regiopreference in the allylic oxidation of unsymmetrical internal alkenes is consistent with the proposed closed transition state (Fig. 4e). The trends in regioselectivity can be rationalized on the basis of steric strain in the developing transition state, where the lowest energy chair conformation is favored. These mechanistic studies will enable the improvement of the efficiency of this process, while also generalizing this mode of catalysis to other unprecedented reactions of π-nucleophiles.
Extended Data
Extended Data Figure 1. Development of an enantioselective and regioselective allylic oxidation of internal unactivated alkenes via an ene reaction.

a, Our approach to generating one allylic oxidation product from unactivated internal alkenes and chalcogen-based oxidants. Sulfurimide reagent 3d was chosen for several reasons. First, compared to diimide oxidants 3b–c, sulfurimide 3d is considerably less electrophilic and therefore less reactive in thermal hetero-ene reactions, affording greater opportunity for a catalyst-controlled process. Second, the ene adducts generated between internal olefins and oxidants 3a-c undergo spontaneous [2,3]-rearrangements, which preclude the capability to diversify the resulting oxidation products. Lastly, the presence of distinct nitrogen and oxygen moieties on the central sulfur atom in the allylic oxidation product provides an opportunity for further chemistry to access synthetically diverse products via C–N and C–O bond formation (see Fig. 1B). b, Optimization of the enantioselective allylic oxidation of cis-5-decene. Reaction conditions: Cis-5-decene (1 equiv), sulfurimide reagent 3d (1.5 equiv), solvent (0.13M). Yields were determined by 1H NMR using 1,4-dimethoxybenzene as an internal standard. [a] 0.5 equiv. trifluoroacetic acid added to reaction. [b] 10 mmol scale. [c] Isolated yield. [d] >20:1 initial dr (5a:5b). At −70 °C, reagent 3d did not undergo a background thermal ene reaction with cis-5-decene 4 in the absence of a catalyst (entry 1). Achiral Lewis acids such as TiCl4, SnCl4, and SbCl5 catalyzed the ene reaction at −70 °C in CH2Cl2 to furnish the allylic oxidation product 5 in low yields (entries 2–4). While coordination of BINOL to titanium and tin provided ene-adduct 5 in low enantiomeric excess (entries 5–6), the antimony-BINOL complex gave the oxidized product in considerably higher enantiomeric excess with enhanced yield (entry 7). Addition of 50 mol% trifluoroacetic acid (TFA) improved the efficiency of the reaction (entry 8). Examination of several solvents revealed the beneficial effects of CH2Cl2 on the yield of the reaction (entry 8) and PhMe on the enantioselectivity of the reaction (entry 9). In concert, these two solvents improved the stereoselectivity of the transformation, which was performed on 10 mmol scale with commercially available (R)-BINOL (entry 10). Based on the observed effect of the aromatic solvent on the stereoselectivity of the reaction, we evaluated a series of aryl-substituted BINOL-based diols. After an extensive exploration (see supporting information), co-catalyst 6 was deemed optimal for this process, with slightly improved enantioselectivity (entry 11). While the ene adduct was formed as a >20:1 mixture of epimers at sulfur (5a and 5b), indicating that this process is also highly diastereoselective at −78 °C, this mixture equilibrated over several hours at ambient temperature to a 4:1 mixture of epimers.
Supplementary Material
Fig. S1. Commercially available alkene starting materials.
Fig. S2. Commercially available BINOL co-catalysts.
Fig. S3. Known BINOL co-catalysts synthesized in our laboratory.
Fig. S4. Screen of acid additives.
Fig. S5. Scale up of enantioselective allylic oxidation on preparative scale (10 mmol).
Fig. S6. Functional group robustness screen.
Fig. S7. X-ray structural analysis of 8e. View of 8e showing the atom labeling scheme. Displacement ellipsoids are scaled to the 50% probability level.
Table S1. Extended screen of BINOL co-catalysts (Part 1)
Table S2. Extended screen of BINOL co-catalysts (Part 2)
Table S3. Catalyst loading screen.
Table S4. Correlation study between the enantiomeric excess of co-catalyst 6 and the enantiomeric excess of product 5
Acknowledgments
Financial support was provided by the W. W. Caruth, Jr. Endowed Scholarship, the Robert A. Welch Foundation (Grant I-1748), the National Institutes of Health (R01GM102604), the National Science Foundation CAREER Award (1150875), and the Sloan Research Fellowship. We thank Dr. Vincent Lynch for X-ray structural analysis.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions L.B., P.Q.L., and U.K.T. conceived the work and designed the experiments. L.B. and P.Q.L. performed the laboratory experiments. U.K.T. oversaw the project. All authors analysed the data and wrote the manuscript.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of this article at www.nature.com/nature.
Data Availability Statement The data supporting the findings of this study are available within the paper and its supplementary information files, or are available from the corresponding author upon reasonable request. Crystallographic data is available at the Cambridge Crystallographic Data Centre (CCDC 1543728).
References
- 1.Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis I–III. Springer; 1999. [Google Scholar]
- 2.Ojima I, editor. Catalytic Asymmetric Synthesis, Third Edition. John Wiley & Sons, Inc; 2010. [Google Scholar]
- 3.Bonini C, Righi G. A critical outlook and comparison of enantioselective oxidation methodologies of olefins. Tetrahedron. 2002;58:4981–5021. [Google Scholar]
- 4.Page PCB, McCarthy TJ. 2.1 - Oxidation Adjacent to C=C Bonds. In: Fleming I, editor. Comprehensive Organic Synthesis. Pergamon; Oxford: 1991. pp. 83–117. [Google Scholar]
- 5.Grennberg H, Bäckvall J-E. Transition Metals for Organic Synthesis. Wiley-VCH Verlag GmbH; 2008. Allylic Oxidations; pp. 243–265. [Google Scholar]
- 6.Liu G, Wu Y. Palladium-Catalyzed Allylic C–H Bond Functionalization of Olefins. In: Yu J-Q, Shi Z, editors. C-H Activation. Springer Berlin Heidelberg; Berlin, Heidelberg: 2010. pp. 195–209. [DOI] [PubMed] [Google Scholar]
- 7.Andrus MB. Allylic and benzylic oxidation. In: Evans PA, editor. Stereoselective Synthesis 3. Vol. 3 Georg Thieme Verlag; Stuttgart: 2011. [Google Scholar]
- 8.Johannsen M, Jørgensen KA. Allylic Amination. Chemical Reviews. 1998;98:1689–1708. doi: 10.1021/cr970343o. [DOI] [PubMed] [Google Scholar]
- 9.Zalatan DN, Bois JD. Metal-Catalyzed Oxidations of C–H to C–N Bonds. In: Yu J-Q, Shi Z, editors. C-H Activation. Springer Berlin Heidelberg; Berlin, Heidelberg: 2010. pp. 347–378. [DOI] [PubMed] [Google Scholar]
- 10.Collet F, Lescot C, Dauban P. Catalytic C-H amination: the stereoselectivity issue. Chemical Society Reviews. 2011;40:1926–1936. doi: 10.1039/c0cs00095g. [DOI] [PubMed] [Google Scholar]
- 11.Covell DJ, White MC. A Chiral Lewis Acid Strategy for Enantioselective Allylic C–H Oxidation. Angewandte Chemie International Edition. 2008;47:6448–6451. doi: 10.1002/anie.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramirez TA, Zhao B, Shi Y. Recent advances in transition metal-catalyzed sp3 C-H amination adjacent to double bonds and carbonyl groups. Chemical Society Reviews. 2012;41:931–942. doi: 10.1039/c1cs15104e. [DOI] [PubMed] [Google Scholar]
- 13.Andrus MB, Zhou Z. Highly Enantioselective Copper–Bisoxazoline-Catalyzed Allylic Oxidation of Cyclic Olefins with tert-Butyl p-nitroperbenzoate. Journal of the American Chemical Society. 2002;124:8806–8807. doi: 10.1021/ja026266i. [DOI] [PubMed] [Google Scholar]
- 14.Eames J, Watkinson M. Catalytic Allylic Oxidation of Alkenes Using an Asymmetric Kharasch–Sosnovsky Reaction. Angewandte Chemie International Edition. 2001;40:3567–3571. doi: 10.1002/1521-3773(20011001)40:19<3567::AID-ANIE3567>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 15.Davies HML, Manning JR. Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature. 2008;451:417–424. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trost BM, Donckele EJ, Thaisrivongs DA, Osipov M, Masters JT. A New Class of Non-C2-Symmetric Ligands for Oxidative and Redox-Neutral Palladium-Catalyzed Asymmetric Allylic Alkylations of 1,3-Diketones. Journal of the American Chemical Society. 2015;137:2776–2784. doi: 10.1021/jacs.5b00786. and references therein. [DOI] [PubMed] [Google Scholar]
- 17.Sharpless KB, Lauer RF. Selenium dioxide oxidation of olefins. Evidence for the intermediacy of allylseleninic acids. Journal of the American Chemical Society. 1972;94:7154–7155. [Google Scholar]
- 18.Schonberger N, Kresze G. Chemistry of Sulfur Diimides. 6. Ene Reactions and [2+2] Cycloadditions of N,N′-Ditosyl Sulfur Diimide and N-Sulfinyl-Para-Toluenesulfonamide. Annalen Der Chemie-Justus Liebig. 1975:1725–1731. [Google Scholar]
- 19.Sharpless KB, Hori T, Truesdale LK, Dietrich CO. Allylic amination of olefins and acetylenes by imido selenium compounds. Journal of the American Chemical Society. 1976;98:269–271. [Google Scholar]
- 20.Sharpless KB, Hori T. Allylic amination of olefins and acetylenes by imido sulfur compounds. Journal of Organic Chemistry. 1976;41:176–177. [Google Scholar]
- 21.Hori T, Singer SP, Sharpless KB. Allylic deuteration and tritiation of olefins with N-sulfinylsulfonamides. Journal of Organic Chemistry. 1978;43:1456–1459. [Google Scholar]
- 22.Whitesell JK, Carpenter JF. Absolute stereochemical control in allylic oxidation via ene reactions of N-sulfinylcarbamates. Journal of the American Chemical Society. 1987;109:2839–2840. [Google Scholar]
- 23.Collins KD, Glorius F. A robustness screen for the rapid assessment of chemical reactions. Nature Chemistry. 2013;5:597–601. doi: 10.1038/nchem.1669. [DOI] [PubMed] [Google Scholar]
- 24.Sharma A, Hartwig JF. Enantioselective Functionalization of Allylic C–H Bonds Following a Strategy of Functionalization and Diversification. Journal of the American Chemical Society. 2013;135:17983–17989. doi: 10.1021/ja409995w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lauer AM, Mahmud F, Wu J. Cu(I)-Catalyzed, α-Selective, Allylic Alkylation Reactions between Phosphorothioate Esters and Organomagnesium Reagents. Journal of the American Chemical Society. 2011;133:9119–9123. doi: 10.1021/ja202954b. and references therein. [DOI] [PubMed] [Google Scholar]
- 26.Ishibashi H, Ishihara K, Yamamoto H. Chiral proton donor reagents: tin tetrachloride-coordinated optically active binaphthol derivatives. Chemical Record. 2002;2:177–188. doi: 10.1002/tcr.10020. [DOI] [PubMed] [Google Scholar]
- 27.Surendra K, Corey EJ. Highly Enantioselective Proton-Initiated Polycyclization of Polyenes. Journal of the American Chemical Society. 2012;134:11992–11994. doi: 10.1021/ja305851h. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto H, Futatsugi K. Designer Acids”: Combined Acid Catalysis for Asymmetric Synthesis. Angewandte Chemie International Edition. 2005;44:1924–1942. doi: 10.1002/anie.200460394. [DOI] [PubMed] [Google Scholar]
- 29.Rawal VH, Thadani AN. Wiley-VCH Verlag GmbH & Co. KGaA; 2008. pp. 144–148. [Google Scholar]
- 30.Johnston JN, Muchalski H, Troyer TL. To Protonate or Alkylate? Stereoselective Brønsted Acid Catalysis of C-C Bond Formation Using Diazoalkanes. Angewandte Chemie International Edition. 2010;49:2290–2298. doi: 10.1002/anie.200904828. [DOI] [PubMed] [Google Scholar]
- 31.Akiyama T. Stronger Brønsted Acids. Chemical Reviews. 2007;107:5744–5758. doi: 10.1021/cr068374j. [DOI] [PubMed] [Google Scholar]
- 32.Taylor MS, Jacobsen EN. Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angewandte Chemie International Edition. 2006;45:1520–1543. doi: 10.1002/anie.200503132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Commercially available alkene starting materials.
Fig. S2. Commercially available BINOL co-catalysts.
Fig. S3. Known BINOL co-catalysts synthesized in our laboratory.
Fig. S4. Screen of acid additives.
Fig. S5. Scale up of enantioselective allylic oxidation on preparative scale (10 mmol).
Fig. S6. Functional group robustness screen.
Fig. S7. X-ray structural analysis of 8e. View of 8e showing the atom labeling scheme. Displacement ellipsoids are scaled to the 50% probability level.
Table S1. Extended screen of BINOL co-catalysts (Part 1)
Table S2. Extended screen of BINOL co-catalysts (Part 2)
Table S3. Catalyst loading screen.
Table S4. Correlation study between the enantiomeric excess of co-catalyst 6 and the enantiomeric excess of product 5