

Abstract
Retroviral (RV) expression of genes of interest (GOIs) is an invaluable tool and has formed the foundation of cellular engineering for adoptive cell therapy in cancer and other diseases. However, monitoring of transduced T cells long term (weeks to months) in vivo remains challenging because of the low frequency and often poor durability of transduced T cells over time when transferred without enrichment. Traditional methods often require additional overnight in vitro culture after transduction. Moreover, in vitro-generated effector CD8+ T cells enriched by sorting often have reduced viability, making it difficult to monitor the fate of transferred cells in vivo. Here, we describe an optimized mouse CD8+ T-cell RV transduction protocol that uses simple and rapid Percoll density centrifugation to enrich RV-susceptible activated CD8+ T cells. Percoll density centrifugation is simple, can be done on the day of transduction, requires minimal time, has low reagent costs and improves cell recovery (up to 60%), as well as the frequency of RV-transduced cells (~sixfold over several weeks in vivo as compared with traditional methods). We have used this protocol to assess the long-term stability of CD8+ T cells after RV transduction by comparing the durability of T cells transduced with retroviruses expressing each of six commonly used RV reporter genes. Thus, we provide an optimized enrichment and transduction approach that allows long-term in vivo assessment of RV-transduced T cells. The overall procedure from T-cell isolation to RV transduction takes 2 d, and enrichment of activated T cells can be done in 1 h.
INTRODUCTION
T cells have a key role in combating cancer and infection by intracellular pathogens. Therapeutically improving the effectiveness of T cells for vaccines or immunotherapies requires a detailed understanding of the molecular mechanisms of T-cell differentiation. A major challenge of these studies is that many of the key events involved in development of highly functional T cells do not occur in vitro and can be analyzed in detail only by using in vivo models1–3. Thus, to interrogate GOIs in T-cell differentiation in vivo, it is critical to be able to trace T cells with manipulation of the GOIs in vivo over long time frames (weeks to months), during which T-cell memory or exhaustion, in the case on chronic infections or cancer, can form. Genetic manipulation of mouse genomes has been a mainstay of research on T-cell memory and exhaustion, and it has become even more facile with the development of CRISPR technologies. However, developmental concerns, the cost of maintaining large animal colonies, concerns about controlling for systemic effects and the speed with which manipulations on the genetic level can be performed are still limiting factors when designing such experiments.
Retroviral transduction approaches have several advantages, including rapid construction, methods to control gene expression or function, the ability to incorporate reporter genes to monitor only transduced cells and the ability to be applied to multiple genetic backgrounds (e.g., transduction of wild-type versus genetic knockout cells)4–7. A major advantage of such approaches for experimental models of effector, memory and exhausted T-cell biology is the ability to adoptively transfer RV-transduced T cells in vivo and monitor their differentiation (Fig. 1; refs.8–10). There are several methods or protocols describing RV transduction of T cells in the context of adoptive T-cell transfer therapy using human peripheral CD8+ T cells and general protocols for mouse T cells5–7,11–15. However, few publications describe details of RV transduction for CD8+ T cells for long-term use in vivo. Indeed, monitoring of RV-transduced T cells long term in vivo remains challenging for several reasons. First, the frequency of RV-transduced T cells often decreases after adoptive transfer in vivo, making detailed studies at late time points challenging (Fig. 2). Often, high numbers of RV-transduced T cells are transferred to circumvent this limitation. However, in some settings, the precise number of transferred CD8+ T cells is critical to the pathogenesis of infection16. In addition, adoptively transferring unphysiologically high numbers of CD8+ T cells can distort the normal pattern of memory differentiation17. Therefore, approaches that allow RV-transduced T cells to be used at numbers that approximate the endogenous antigen-specific T-cell response and also allow long-term monitoring would be a major advantage. Enriching for RV-transduced T cells before transfer can improve these approaches. However, flow-cytometric-based or magnetic-bead-based enrichment methods usually require additional overnight in vitro culture to allow reporter gene expression, potentially contributing to some of the inefficiencies described above. Moreover, in vitro-generated effector CD8+ T cells that are enriched by flow-cytometric sorting often show significantly reduced cell viability (P < 0.0005; see Fig. 3e for data), potentially due to mechanical stress and/or surface staining with antibodies that could cause rejection. A second challenge for in vivo studies using RV-transduced T cells is the choice of reporter genes/proteins for transduction. Multiple genes, including GFP, violet-excited fluorescent protein (VEX), monomeric Kusabira Orange 2 (mKO2), mCherry, Thy1.1 and human nerve growth factor receptor (hNGFR), have been used as RV reporters. However, there is the potential for these reporter genes and the proteins they encode to serve as rejection antigens, leading to deletion of RV-transduced cells by the host immune system18. Therefore, compatibility of markers used as reporters of RV transduction with long-term in vivo T-cell persistence is essential, but a systematic comparison of reporter genes for use in T-cell-memory studies is lacking. Thus, there is a need for an optimized, flexible and efficient RV transduction approach that allows efficient manipulation of the GOI for the study of long-term T-cell biology, T-cell durability and memory differentiation in vivo.
Figure 1.
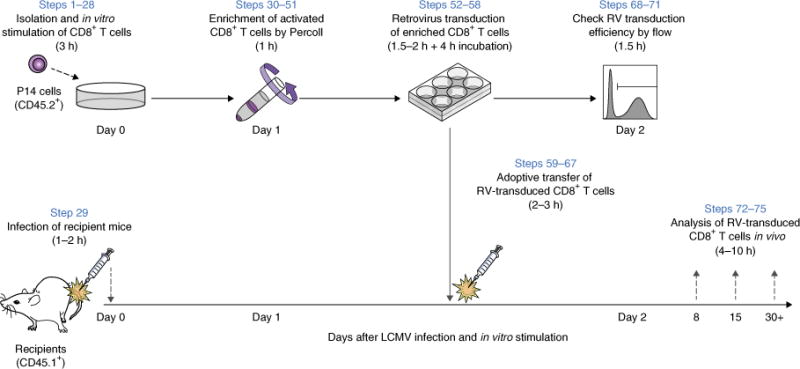
Schematic overview of retroviral transduction experiments in mouse T cells. Overview of in vitro RV transduction of mouse CD8+ T cells (P14 T-cell receptor transgenic (TCR Tg) cells specific for LCMV GP33-41 presented by H-2Db), followed by in vivo adoptive transfer. (Steps 1–28) P14 cells are harvested from the spleen, enriched using a CD8-negative-selection kit, and stimulated with anti-CD3ε and CD28 antibodies in the presence of recombinant human IL-2. (Step 29) On the same day, recipient mice are infected with a model pathogen (here, the LCMV Arm strain was used as acute viral infection model). (Steps 30–51) One day after in vitro stimulation, activated and RV-susceptible P14 cells are enriched by Percoll density centrifugation. (Steps 52–58) Enriched P14 cells are transduced with RV and incubated for 4 h. (Steps 59–67) After incubation, RV-transduced P14 cells are adoptively transferred into the recipient mice. P14 cells have different congenic markers that distinguish donor cells in recipient animals (CD45.2+ to CD45.1+ is shown). (Steps 68–71) An aliquot of RV-transduced P14 cells is maintained in vitro for an additional day and analyzed for RV transduction efficiency. (Steps 72–75) In vivo differentiation of RV-transduced T cells is assessed at multiple time points (e.g., effector expansion, survival and memory or exhaustion differentiation on days 8, 15 and 30, respectively). To enrich RV-transduced cells, the conventional approach is to select RV-positive cells on day 2 using flow sorting or magnetic beads based on an RV reporter such as GFP or Thy1.1 (not shown here).
Figure 2.
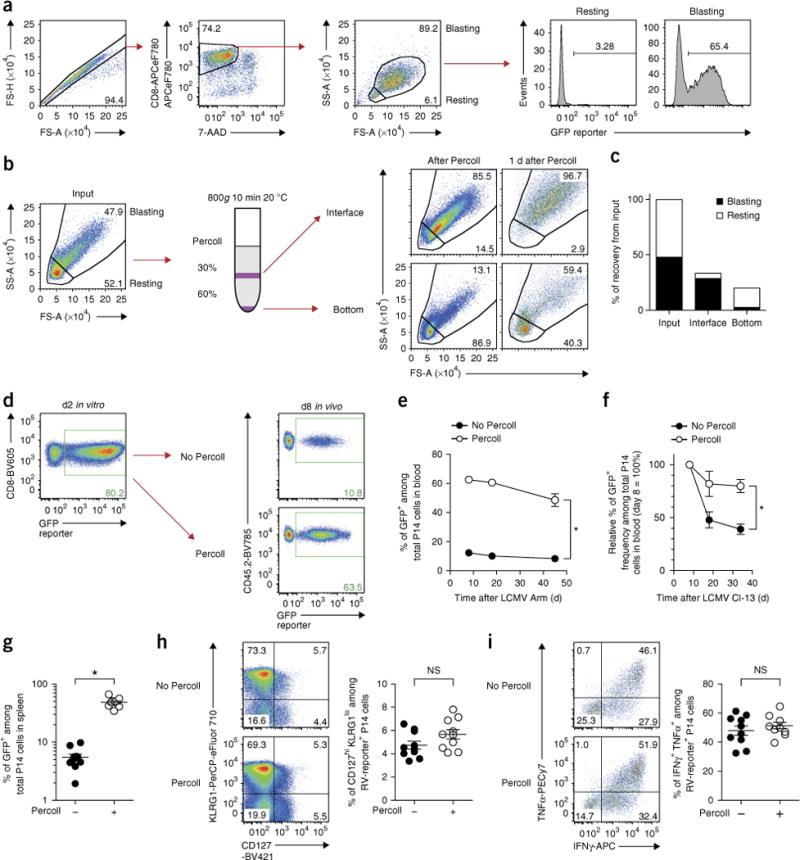
Enrichment of RV-transduced CD8+ T cells before transfer improves the efficiency of retroviral experiments in vivo. (a) Small-sized ‘resting’ CD8+ T cells are mostly RV-negative. As shown in Figure 1, in vitro-stimulated wild-type P14 cells that were transduced with RV (empty GFP) on day 1 were cultured overnight and analyzed on day 2 (1 d after RV transduction). (b) Density centrifugation with 30 and 60% (vol/vol) Percoll layers enriches activated CD8+ T cells on day 1 (22 h after in vitro stimulation). Flow plots that are gated on 7-AAD-negative CD8-positive population show enrichment step from input (left) to shortly after Percoll separation on day 1 (right middle) and 1 d after (right-most). (c) Graph showing recovery of small- (‘resting’) and large (‘blast’)-size cells in each interface and bottom after Percoll centrifugation. Data are representative of three independent experiments (one technical replicate per experiment). See Supplementary Figure 1 for data from two other experiments. (d) Enrichment by Percoll increases the frequency of RV-transduced CD8+ T cells ~sixfold in vivo. 2 × 104 unenriched or Percoll-enriched P14 cells were adoptively transferred into recipient mice that were infected with LCMV Arm a day before. Flow plots gated on CD8+ P14 cells in vitro at 1 d after RV transduction (left) and in blood at 8 d post infection (d.p.i.) with Arm (right) are shown. (e,f). Percoll enrichment maintains a higher and more stable frequency of RV-transduced CD8+ T cells over time in vivo with LCMV Arm acute (e) and clone 13 chronic (f) infection. Circles and whiskers show mean±s.e.m. Data are representative of two independent experiments (n = 5–10 per group). (g) Percoll minimizes the variance of RV-transduced T cells in vivo. Frequencies of GFP+ cells among P14 cells in spleen at 34 d.p.i. with clone 13 are shown. Bar shows mean ± s.e.m. (n = 9–10 per group). (h,i) Effector and memory differentiation was assessed by CD127 and KLRG1 expression on day 8 (h) and by the percentage of IFNγ+ TNF+ cells upon gp33 peptide re-stimulation (i) on day 46 in Arm infection. Representative plots gated on empty-GFP RV-transduced CD8+ T cells are shown. Bar shows mean±s.e.m. *P < 0.0001 (two-tailed t test). Data are representative of two independent experiments (n = 5–10 per group). All animal experiments depicted in this figure were performed in accordance with the Institute Animal Care and Use Guidelines for the University of Pennsylvania. FS-A, forward scatter–area; FS-H, forward scatter–height; SS-A, side scatter–area.
Figure 3.
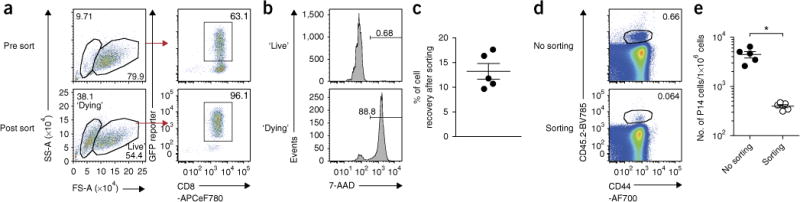
Reduced cell viability after flow-cytometric sorting leads to instability of transferred T cells in vivo. Wild-type P14 cells that were activated in vitro were transduced with empty-GFP RV on day 1, as depicted in Figure 1. The following day (day 2 after in vitro stimulation), GFP+ P14 cells were enriched by flow-cytometric sorting using the BD ARIA II with a 100-μm nozzle and 20-p.s.i. condition. After sorting, viable cell numbers were carefully determined by 7-AAD and trypan blue staining. As a control, P14 cells without sorting were used, and 1.5 × 105 live cells of each cell type were adoptively transferred into LCMV-Arm-infected recipients. On day 8, expansion of P14 cells was determined in blood. (a) Flow plots showing physical cell size and purity of GFP+ CD8+ cells before and after flow sorting on day 2. Purity of GFP+ CD8+ cells was over 95%. (b) 7-AAD histograms gated on ‘live’ or ‘dying’ cells based on forward scatter (FS) versus side scatter (SS) plot in a, showing that the majority of small cells after sorting are dying. (c) Cell recovery (%) based on the number of total live (trypan blue negative) cells in sorting. (d) Flow plots showing frequencies of P14 cells among total CD8+ cell gate in blood at 8 d.p.i. (e) Graph showing the number of P14 cells per 1 × 106 cells in blood at 8 d.p.i. *P< 0.0005 (two-tailed t test). Data are representative of two experiments (n = 5–15 per group). All animal experiments depicted in this figure were performed in accordance with the institutional animal care and use guidelines of the University of Pennsylvania.
Here, we describe a step-by-step protocol for RV transduction of mouse CD8+ T cells that uses Percoll density centrifugation to enrich RV-susceptible cells. This approach improves the viability of RV-transduced cells and enables more efficient long-term T-cell memory and exhaustion experiments with RV-transduced T cells in vivo (Figs. 1 and 2). In previously published research from our group9,10, we have applied this optimized RV transduction protocol to track and analyze RV-transduced CD8+ T cells in both acute and chronic viral infection mouse models for more than 10 weeks after transfer in vivo.
Development and overview of the protocol
Two keys to improving the efficiency of in vivo CD8+ T-cell RV-transduction studies are to enrich RV-transduced (or RV-susceptible) T cells while minimizing cell stress and to adoptively transfer cells to recipient mice as soon as possible (ideally within a few hours) after RV transduction. RV are typically used for gene delivery into mouse T cells, as most lentiviruses transduce mouse T cells poorly7. However, unlike lentiviruses, which can transduce resting human T cells, RV transduction is restricted to proliferating cells4. Thus, an in vitro T-cell activation strategy is used to induce susceptibility of CD8+ T cells to RV transduction (Fig. 1)5–7,15. To begin to develop an optimized RV approach for long-term in vivo studies, we initially examined RV-transduced CD8+ T cells 1 d after transduction (day 2 after in vitro stimulation; Figs. 1 and 2a). At this stage, RV-transduced cells are found almost entirely within the large-sized ‘blasting’ (i.e., more activated and susceptible to RV transduction) gate by flow cytometry (Fig. 2a). By contrast, very few RV-transduced CD8+ T cells in the population of live CD8+ T cells remain as small-sized ‘resting’ cells after 48 h of in vitro stimulation with soluble anti-CD3ε and CD28 antibodies (Fig. 2a). T cells that are primed later in vivo and receive less stimulation during initial activation have been reported to have an advantage in differentiating into memory T-cell populations in vivo1,19. Alternatively, weakly activated or nonactivated (i.e., still-naive T cells) might be efficiently primed upon adoptive transfer to infected mice. Thus, we hypothesized that the residual small-sized cells might engraft and proliferate in vivo more efficiently compared with the blasting population. If true, this could result in a greater decline in the RV-transduced blasting cell population at later time points. To test this idea and circumvent this potential problem, we devised a simple strategy to separate these two T-cell populations. Previously activated human lymphocytes have been separated by density centrifugation because of their lower overall density20,21. Thus, we tested whether Percoll density centrifugation could also enrich the larger-sized ‘blasting’ T cells on day 1 after in vitro stimulation (Fig. 2b)22. As expected, large-sized ‘blasting’ cells that have the capability to differentiate into true effector T cells after additional overnight in vitro culture are efficiently enriched in the thin layer between the 30 and 60% (vol/vol) Percoll in the Percoll density gradient on day 1 (Fig. 2b,c, Supplementary Fig. 1). By contrast, small-sized ‘resting’ cells that are not fully able to differentiate into blasting effectors are removed into the bottom fraction (Fig. 2b,c, Supplementary Fig. 1). We find that the recovery of the large-sized cells by Percoll is up to 60% (Fig. 2c, Supplementary Fig. 1b,d, black bar), higher than typical sorting-based enrichment for this blasting population (normally 10–20%, Fig. 3c). Next, we examined whether this enrichment of blasting cells on day 1 can improve the frequency of RV-transduced T cells in vivo. After in vivo transfer, ~60–65% of the donor CD8+ T-cell population was RV-transduced on day 8 as compared with only 10–12% in the case of the unenriched population (Fig. 2d; see Supplementary Fig. 2 for staining and gating strategy used to detect antigen-specific CD8+ T cells in vivo in this study). In separate experiments, we directly compared the frequency of RV-transduced T cells on day 8 in vivo for the following groups of donor cells: nonenriched, interface only, bottom only or a re-mixture of interface and bottom cells, all isolated from the same activated P14 CD8+ T-cell source (Supplementary Fig. 3a). As expected, enrichment and adoptive transfer of resting cells results in recovery of a low frequency of RV-transduced T cells, based on the RV-reporter+ frequency from day 2 to day 8 (Supplementary Fig. 3b,c). Moreover, add-back of resting cells to blasting cells before adoptive transfer decreases the frequency of RV-transduced T cells among the donor T-cell pool on day 8, supporting the idea that resting cells in the cell transfer mixture outcompete blasting cells and reduce efficiency of detection of RV-transduced cells at later time points in vivo (Supplementary Fig. 3b,c). Percoll density centrifugation also improves the stability and durability of RV-transduced cells over several weeks in vivo (Fig. 2e–g). Moreover, Percoll-based enrichment minimizes the variance of frequency of RV-transduced T cells in vivo, contributing to more statistically reliable results (Fig. 2g). Centrifugation does not appear to alter T-cell differentiation, as effector and memory CD8+ T cells derived from Percoll enrichment are similar to those from non-Percoll-treated controls in terms of surface phenotype and cytokine production after lymphocytic choriomeningitis virus (LCMV) Armstrong (Arm) infection (Fig. 2h,i). These data in Figure 2h also demonstrate identification of memory precursor populations (i.e., CD127hiKLRG1lo) in the RV-transduced T-cell pool. These data indicate that although we are purifying a more activated fraction of in vitro-activated CD8+ T cells, at least some cells in this pool are capable of giving rise to memory precursor cells. Overall, use of a simple Percoll density centrifugation approach enriches for RV-transduced CD8+ T cells after in vitro activation while minimizing cellular stress. This approach substantially improves the in vivo efficiency of RV-transduced memory or exhausted CD8+ T-cell experiments by increasing transduced cell yields and improving the in vivo durability of transduced CD8+ T-cell populations.
Application of the protocol
The protocol described here was developed specifically for CD8+ T-cell studies in a viral infection mouse model. However, it is likely that the basic concept of Percoll separation has multiple applications and a wider interdisciplinary utility, including those for (i) T-cell responses in other disease contexts such as cancer immunology or autoimmunity; (ii) other hematopoietic subsets, such as CD4+ T cells (see below), B cells and bone-marrow-progenitor cells; and (iii) broader biological fields, in which RV transduction target cells are heterogeneous in terms of cell proliferation and size (i.e., density). Of note, this protocol is independent of specific molecules (e.g., GFP) derived from target cells or RV for T-cell enrichment. Thus, we believe that this protocol can be easily applied to general RV transduction experiments.
Advantages
Flow-cytometric sorting or magnetic-bead-enrichment approaches have traditionally been used to enrich RV-transduced CD8+ T cells before adoptive transfer into recipient mice. Compared with these two approaches, enrichment using Percoll density centrifugation has several advantages. First, flow-cytometric sorting requires the expression of RV-derived reporter genes, and additional days of cell culture are required for these genes to be expressed after RV transduction (Fig. 3). By contrast, enrichment using Percoll can be done on the same day as RV transduction, reducing the time CD8+ T cells are cultured in vitro before adoptive transfer to mice. For optimal CD8+ T-cell differentiation, particularly for long-lived memory CD8+ T-cell studies, in vitro-stimulated CD8+ T cells perform better when adoptively transferred into recipient mice early after initial activation, perhaps because of preservation of memory precursor cells in the early activated pool. Thus, the ability to minimize the duration of in vitro culturing by using density centrifugation on day 1 after activation is likely to have advantages for long-term in vivo T-cell studies. Second, enrichment by flow sorting can lead to low overall experimental efficiency (i.e., cell recovery and engraftment after transfer) for RV-transduced T cells (Fig. 3). This inefficiency could be due to mechanical damage to the blasting T cells during sorting due to high pressure and/or shear stress. In addition, some antibodies used to stain reporter markers (e.g., Thy1.1) could potentially cause antibody-dependent cell depletion after transfer. Because enrichment by Percoll does not involve any shear stress or staining, Percoll density centrifugation maintains T-cell integrity and, as a result, yields highly reproducible and stable results with better cell recovery (Figs. 2b–g and 3c–e). Third, Percoll density centrifugation is substantially less expensive and faster than alternative approaches. Density centrifugation does not require any special or expensive equipment, and reagent cost is minimal. In addition, enrichment by Percoll can be done in 1 h, whereas flow sorting typically requires several hours and additional technical expertise to complete.
Limitations
Although overall cell recovery is improved using Percoll density centrifugation as compared with flow-cytometric sorting, we have found that increasing the initial number of T cells is useful in minimizing cell loss (Fig. 4; see optimal number of transferred T cells for detail). This requirement typically results in using more TCR transgenic mice as donors for T cells to be transduced. In a typical experiment for which one might need five to ten recipient mice per RV, ~107 starting T-cell-receptor (TCR) transgenic T cells (i.e., ~1 donor spleen) per RV appear optimal. An additional limitation to this protocol is that although Percoll enriches highly activated T cells, it is possible that the purified T cells could show slightly modified differentiation due to either subtleties in the population purified or due to the effects of Percoll itself. Although this issue has not been observed in our initial studies, it is, of course, valuable to use the same enrichment protocol for a control population (i.e., an empty RV containing only the reporter gene) when analyzing the effects of the RV-expressed GOI in vivo. A third limitation is overall the purity of transduced populations. Although the efficiency of in vivo recovery and persistence is improved with the Percoll approach, if highly purified populations are needed, flow-cytometric sorting (for which purity can reach over 95%, Fig. 3a) will still have an advantage. RV non-transduced, but activated blasting cells, will be contained in the Percoll-purified populations (purity of RV-transduced cells is ~50–60%; Fig. 2d). In some settings, these nontransduced populations can be a useful control (in addition to the empty RV control). However, in other situations in vivo readouts such as pathogen burden or tumor control will be affected by the total number of adoptively transferred cells, especially when TCR transgenic populations are used. Thus, purity of RV-transduced populations will be a major consideration in some settings. One advantage, of course, of such RV approaches is the ability to introduce the GOI into T cells from different genetic backgrounds, such as re-introducing mutant forms of a protein into T cells genetically deficient in that protein. In some settings, genetic deficiency might alter initial T-cell activation, changing the efficiency of this Percoll approach. In such settings, it may be possible to transduce bone marrow T-cell progenitors, followed by adoptive transfer to recipient mice to allow T-cell development to occur for the generation of mature T cells that can be used to address the question of interest23,24.
Figure 4.
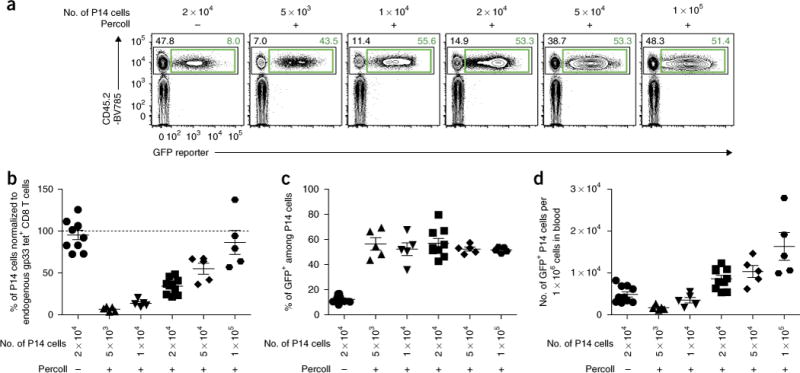
Optimal number of transferred P14 cells in Percoll density enrichment in LCMV Arm infection. Differential numbers of empty-GFP RV-transduced CD45.2+ wild-type P14 cells with or without Percoll enrichment were adoptively transferred into LCMV-Arm-infected CD45.1+ recipient mice as shown in Figure 1. LCMV gp33 antigen-specific CD8+ T cells in the blood were analyzed at day 8 post infection. (a) Representative flow plots gated on gp33-tetramer+ CD8+ T cells. Numbers adjacent to the outlined areas indicate percentage of P14 cells among total gp33-specific CD8+ T cells (top left; black) or percentage of GFP+ cells among total P14 cells (top right; green). (b) Frequency of P14 cells is normalized to endogenous gp33-specific CD8+ T cells in the blood of the same individual mouse. (c) Enhanced in vivo stability of RV-transduced cells using the Percoll approach is independent of the number of transferred P14 cells. Frequencies of GFP+ cells among P14 cells are shown. (d) Numbers of RV-transduced P14 cells per 1 × 106 cells in blood (mean±s.e.m.). Data are representative of two independent experiments (n = 5–10 per group). All animal experiments depicted in Figure 4 were performed in accordance with the institutional animal care and use guidelines of the University of Pennsylvania.
Experimental design
This T-cell enrichment and RV transduction protocol involves seven parts: (i) isolation and in vitro stimulation of CD8+ T cells (Steps 1–28), (ii) infection of recipient mice (Step 29), (iii) enrichment of activated CD8+ T cells by Percoll density centrifugation (Steps 30–51), (iv) RV transduction of enriched T cells (Steps 52–58), (v) adoptive transfer of RV-transduced T cells (Steps 59–67), (vi) checking of RV transduction efficiency (Steps 68–71), and (vii) analysis of RV-transduced T cells in vivo (Steps 72–75).
CD4+ versus CD8+ T cells
In this protocol, we mostly focused on CD8+ T cells. However, we also tested key observations in CD4+ T cells and found that activated ‘blasting’ CD4+ T cells can also be enriched by Percoll centrifugation when the same stimulation method with anti-CD3ε, anti-CD28 antibodies and rhIL-2 was used (Supplementary Fig. 4a–c). Further optimization will be needed, for example, to compare different CD4+ T-cell-polarization culture conditions.
Preparation of RV stock
As there is a tropism between RV pseudotype and type of target mammalian cells, it is critical to use the appropriate RV system in each experimental setting4,15. To balance biosafety and higher transduction efficiency, we have used an eco-tropic murine stem-cell virus-based RV system in our work that can transduce only rodent cells. Detailed protocols for preparation and titration of RV have been described previously4–7,14,23,25. Although use of freshly prepared RV supernatant is recommended in other fields, frozen RV stocks can be used for transduction of CD8+ T cells5. RV stocks can be stored at −80 °C for up to 2 years without noticeable loss of transduction efficiency. In addition, as there can be variability in RV titers among production batches, using frozen stocks of virus for which the titer has been previously verified is recommended.
Isolation and in vitro stimulation of CD8+ T cells
As the frequency of antigen-specific T cells in a polyclonal setting is extremely low, use of T-cell-receptor transgenic (TCR Tg) animals facilitates many aspects of these studies. In our studies, we used LCMV infection and LCMV glycoprotein H-2Db/gp33–41-specific ‘P14’ TCR Tg CD8+ cells9,10,26. In addition, the spleen is an ideal source for T cells; however, the frequency of CD8+ T cells among splenic mononuclear cells is only ~15–20%. Moreover, it was reported that the purity of target cells at RV transduction affects transduction efficiency5. Thus, purification of CD8+ T cells before in vitro stimulation is important (Fig. 1).
For T-cell activation, there are multiple options, including phorbol myristate acetate and ionomycin, concanavalin A, soluble antibodies and cognate peptide, in the case of TCR Tg cells5,6,15,27. In our experience, stimulation with soluble anti-CD3ε and anti-CD28 is a highly efficient and easy method of T-cell activation for RV transduction purposes (Fig. 1). As an alternative method, stimulation with cognate peptide has been described14. In our studies, we have found that cognate peptide stimulation results in lower T-cell survival after transfer (data not shown), perhaps because T cells themselves can acquire peptides and recognize and kill each other or because of the poor availability of professional antigen-presenting cells, such as dendritic cells, in the purified CD8+ T-cell cultures.
T-cell enrichment for RV transduction
Detailed information on Percoll and density centrifugation has been provided previously22,28. To obtain the optimal combination of Percoll concentrations for activated T-cell separation, we have compared different combinations of Percoll solutions for two layers from 10 to 90% in 10% steps and found that a combination of 30% (vol/vol) for the upper and 60% (vol/vol) for the lower Percoll layer results in the best separation (data not shown). However, the type of centrifuge and/or rotor, temperature and production lot of the reagents and plastic tubes could influence separation efficiency28. Thus, slight modification by changing Percoll concentrations in 5% steps may be helpful for further optimization. Furthermore, the activation status of target T-cell populations, which can directly influence cell density, has a strong impact on gradient separation efficiency. Cell density during in vitro stimulation and, more importantly, the timing since initial stimulation in culture are major factors for T-cell activation status. In our experience, even a few hours’ difference from in vitro stimulation (e.g., 18 h versus 22 h versus 25 h) can result in a large change in activation status (see cell size in forward scatter versus side scatter flow plots in Supplementary Fig. 1a (18 h), Fig. 2b (22 h) and Supplementary Fig. 1c (25 h)), and, as a result, in subsequent Percoll separation and RV-transduction efficiency in vivo. Therefore, the conditions of in vitro stimulation and timing should be carefully determined.
In our experience, whether Percoll enrichment is used before or after RV transduction does not affect RV-transduction efficiency if the same number of T cells is used for the RV-transduction step (i.e., if the T-cell/RV ratio is fixed) (data not shown). It may be possible to increase the efficiency of transduction slightly by reducing the number of blasting cells per well (i.e., a higher multiplicity of infection). However, this will also result in greater consumption of RV supernatants. As the overall cell recovery after Percoll density enrichment is ~30%, our approach has been to perform Percoll density centrifugation first to limit the amount of RV used. In addition, when RV transduction precedes Percoll enrichment, activated T cells are often divided into multiple groups (i.e., for each different RV), resulting in smaller cell numbers per condition. In such settings, it may be more challenging to efficiently recover RV-transduced T cells after Percoll treatment because of low starting cell numbers.
Conditions of RV transduction
The efficiency of RV transduction can be improved by modifying three factors. (i) Timing: once stimulated in vitro, CD8+ T cells start to divide within ~24–30 h and continue to proliferate for ~ 4–5 d, provided conditions are optimal. However, the susceptibility of mouse CD8+ T cells to RV transduction peaks at 24 h after in vitro stimulation (Fig. 5a)5–7,15,27. Thus, for the highest efficiency, RV transduction of mouse CD8+ T cells should be performed at 1 d after in vitro stimulation (Figs. 1 and 5a). We also found that mouse CD4+ T cells have the highest susceptibility to RV transduction at 48 h after in vitro stimulation (Supplementary Fig. 4d). (ii) Repetition and incubation time: to maximize RV-transduction efficiency, multiple rounds of transduction or extended incubation time (>4 h) have been described in some protocols5,15. However, provided that RV titers are sufficiently high and target CD8+ T cells are appropriately activated in vitro, the efficiency reaches 60–80% with single-RV-transduction and 4-h incubation in most cases (Fig. 2d)15. Thus, in this protocol we use a single round of spin-transduction and 4 h for incubation, and we transfer T cells into recipients as early as possible (Fig. 1). (iii) Facilitators: electronic charge repulsion between RV and sialic acid on the cell surface can reduce RV-transduction efficiency29. To facilitate adsorption of RV to target cells, multiple reagents, including Polybrene (hexadimethrine bromide) and RetroNectin (recombinant human fibronectin fragment) have been used5,15,29. We have directly compared Polybrene and RetroNectin in the context of CD8+ T-cell RV experiments and found that RetroNectin results in higher transduction efficiency (Fig. 5b, top four samples); however, RetroNectin requires additional time for preparation of coated plates and is more expensive than Polybrene. Alternatively, we also found that, in the case of Polybrene, use of non-tissue-culture-treated plates yields slightly better transduction efficiency with increased reporter gene (GFP) signal intensity (Fig. 5b, bottom two samples).
Figure 5.
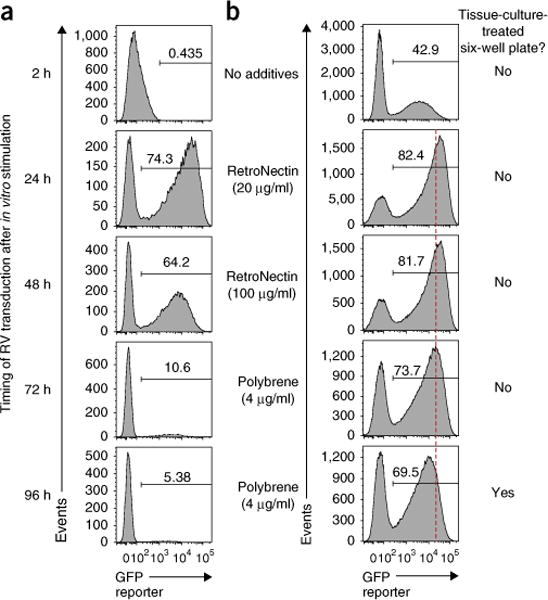
Optimal timing and facilitator for RV transduction to CD8+ T cells. (a,b) Wild-type P14 cells were transduced with empty-GFP RV, at the indicated time after in vitro stimulation in the presence of Polybrene (a), or on day 1 in the absence or presence of Polybrene (4 μg/ml) or RetroNectin (coated at 20 or 100 μg/ml) using six-well plates with or without tissue culture treatment (b), and analyzed for GFP expression 1 d after RV transduction. Plots were gated on live P14 cells. (a) RV transduction efficiency to P14 cells peaks at 24 h after in vitro stimulation. (b) RetroNectin showed better RV transduction efficiency as compared with Polybrene. Data are representative of two independent experiments (duplicate to triplicate per condition).
Optimal number of adoptively transferred CD8+ T cells
As mentioned above, one key factor to consider for studies using RV-transduced TCR Tg T-cell populations is the number of cells to be adoptively transferred. In some settings, it may be critical to match the number of adoptively transferred TCR Tg cells as closely as possible to that of endogenous antigen-specific T cells because of the effects on memory CD8+ T-cell differentiation17 or impacts on pathogenesis16. Many variables may impact how T cells behave in vivo and thus influence the choice of the number of RV-transduced T cells to be adoptively transferred. These variables include the following: the RV-expressed GOI itself; the nature of donor TCR Tg cells (e.g., affinity of TCR, wild type versus knockout); viability of donor TCR Tg cells (e.g., cell density during in vitro stimulation, method of in vitro stimulation and toxicity of Polybrene); and type and dose of antigen challenge in vivo (i.e., infection versus cancer, virus versus bacteria and acute versus chronic infection). Moreover, Percoll enriches more activated T cells, suggesting that T-cell response kinetics could differ between non-Percoll and Percoll purification approaches. Thus, the number of transferred TCR Tg cells should be optimized in an individual experimental setting. For example, we have titrated the number of P14 cells in the LCMV Arm acute virus infection model and found that when enriched by Percoll, transferring 1 × 105 wild-type P14 cells results in a response that is similar in magnitude to the endogenous host gp33-specific CD8+ T-cell response (Fig. 4a,b). Increasing the number of transferred P14 cells does not affect the frequency of RV-transduced cells, but it does minimize variance (Fig. 4c). As a result, transferring 1 × 105 Percoll-enriched P14 cells increases the number of RV-transduced CD8+ T cells by several fold as compared with 2 × 104 nonenriched P14 cells, while keeping the total P14 cell response similar to the endogenous level (Fig. 4b,d).
RV reporter genes (proteins) for long-term in vivo analysis
Flexibility of reporter markers is a key advantage of RV experiments. For example, whereas cytosolic fluorescent protein markers (e.g., GFP, VEX, mKO2 and mCherry) are suitable for live-cell enrichment by sorting, membrane protein markers (e.g., Thy1.1 and hNGFR) have flexibility in the choice of staining reagents and have stronger resistance to fixation and permeabilization. GFP cannot be used as an RV reporter gene for cells already expressing GFP or for some analytic applications that require specific fluorescent reagents with emission spectra that overlap or interfere with GFP (e.g., a mitochondrial analysis dye such as Mito Tracker Green). In addition, combining multiple RVs allows double or triple transduction within individual cells. Therefore, having multiple options for RV reporter genes expands the experimental applications and opportunities. However, some reporter genes used to monitor RV-transduced cells may become rejection antigens and/or cytotoxic (e.g., aggregation of reporter proteins in cytoplasm or altered cell signaling can damage cell viability), leading to loss of RV-transduced cells over time18. However, there is little direct comparative analysis of this topic for long-term T-cell biology in vivo. Thus, we compared the use of six RV reporter genes (GFP, VEX, mKO2, mCherry, Thy1.1 and hNGFR) by monitoring cell number over time and memory differentiation for RV-transduced CD8+ T cells30–33 (Fig. 6). We found that CD8+ T cells transduced with empty RV vectors expressing GFP, VEX, mKO2 or Thy1.1 RV showed stable frequencies of transduced cells over time once transferred in vivo (Fig. 6a–c). By contrast, the RV reporters mCherry and hNGFR resulted in loss of RV-transduced cells over time, suggesting rejection or other mechanisms of poor durability for these reporter genes (Fig. 6a–c). In addition, we found that GFP, VEX, mKO2, mCherry or Thy1.1 RV-transduced CD8+ T cells showed similar surface phenotype and cytokine production on day 36 after infection (Fig. 6d,e). Taken together, GFP, VEX, mKO2 and Thy1.1 allow long-term stability of RV-transduced T cells in C57Bl/6 mice, facilitating detailed experiments of T-cell memory or exhaustion over time in vivo.
Figure 6.
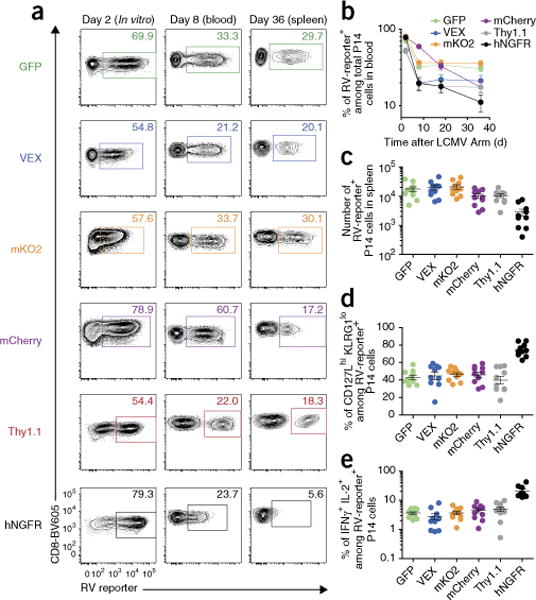
Comparing the utility of six different RV reporter genes for the study of CD8+ T-cell stability and memory differentiation in vivo. CD45.2+ wild-type P14 cells that were transduced with empty-GFP, VEX, mKO2, mCherry, Thy1.1 or hNGFR RV after Percoll enrichment were adoptively transferred into LCMV-Arm-infected CD45.1+ recipients as shown in Figure 1. (a) Representative flow plots gated on P14 cells showing transduction efficiency on day 2 in vitro (1 d after RV transduction, left), and frequencies of RV-transduced cells on day 8 in blood (center) and on day 36 in the spleen (right). Note that mean fluorescence intensity of hNGFR was already decreased on day 8. (b) Graph showing longitudinal frequencies of RV-reporter+ cells among total P14 cells in blood. (c) Number of RV-transduced P14 cells in the spleen on day 36. (d,e) Memory differentiation was assessed by CD127 and KLRG1 expression (d) and percentage of IFNγ+ IL-2+ cells upon gp33 peptide re-stimulation (e) on day 36. All data in Figure 6 are representative of two independent experiments (n = 9–10 per group). Mean ± s.e.m. All animal experiments depicted in Figure 6 were performed in accordance with the institutional animal care and use guidelines of the University of Pennsylvania.
Improved detection of RV-transduced cells by additional fixation
One challenge when analyzing RV-transduced T cells using intracellular staining (ICS) for flow cytometry is that loss of signal for some fluorescent proteins derived from RV (e.g., GFP, VEX or mKO2) after permeabilization of the cell membrane makes it difficult to detect all of the RV-transduced cells. A previous study by Heinen et al. showed that commonly used permeabilization kits for ICS assays have weak fixation reagents, thus resulting in the loss of some cytoplasmic molecules, including fluorescent proteins34. This same study also showed that an additional fixation step increased GFP retention in the cytoplasm of permeabilized cells. Thus, we tested whether an additional fixation before permeabilization with commercial kits could improve the detection of fluorescent proteins derived from RV transduction in T cells. As shown in Figure 7, pre-fixation with 2% (wt/vol) PFA improves the detection of RV-transduced cells ~tenfold, and a similar frequency as compared with no permeabilization is observed.
Figure 7.
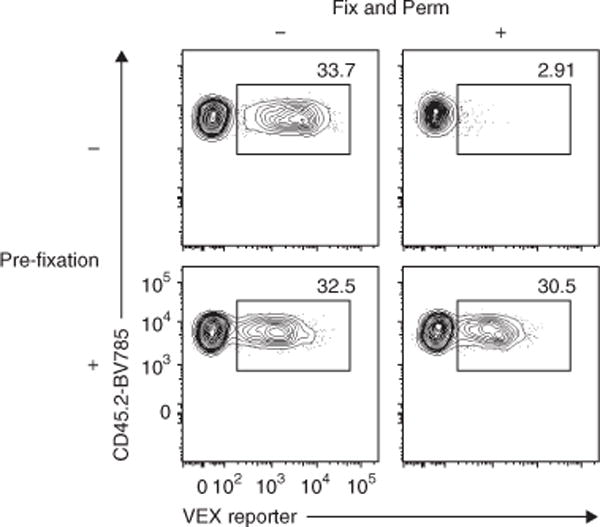
Pre-fixation of RV-transduced T cells prevents the loss of RV-derived marker detection. Spleens containing empty-VEX+ RV-transduced wild-type P14 cells were harvested on day 36 after Arm infection. After staining with surface CD8, TCR Vα2, CD44, CD45.1 and CD45.2, spleen cells were treated with mock PBS or 2% (vol/vol) PFA in PBS at 4 °C for 20 min. Then half of the cells were fixed and permeabilized (Fix and Perm) using an eBioscience Foxp3 staining kit according to the manufacturer’s instructions. The remaining half of the samples were kept in PBS at 4 °C during the Fix and Perm steps. All samples were analyzed immediately after Fix and Perm procedures. Representative flow plots gated on P14 cells are shown. Numbers in the plots indicate percentage of VEX+ cells among total P14 cells. Data are representative of three independent experiments (1–2 technical replicate(s) per condition in each experiment). All animal experiments depicted in Figure 7 were performed in accordance with the institutional animal care and use guidelines of the University of Pennsylvania.
Level of expertise, reagents and equipment needed to implement the protocol
In general, the procedure described in this protocol can be done by a single person with standard cellular immunology and mouse experiment skills. However, extra attention to detail may be required to implement the Percoll density centrifugation, because forming and maintaining the two layers during the various procedures and collecting the cells of interest from interface layer after centrifugation are all key points for best recovery and purity. All antibodies (Supplementary Table 1) and reagents used in this protocol are commercially available. As for the facility and equipment, no particular special lab equipment is required, but access to animal husbandry is essential, and biological safety level 2 (BSL-2) laboratory environments will be needed for studies using infectious agents.
MATERIALS
REAGENTS
Mouse and infectious agents
! CAUTION Appropriate institutional regulatory board permission for animal infection experiments must be obtained. All animal work in this study was performed in accordance with the institutional animal care and use guidelines for the University of Pennsylvania.
TCR Tg mice (e.g., CD45.2+ P14 mice (from H. Pircher (University of Freiburg)) for CD8+ T-cell response) ▲ CRITICAL To avoid potential rejection, it is recommended to test the compatibility of donor TCR Tg cells and recipient mice without RV transduction. In our case, we adoptively transferred 1–5 × 106 naive P14 cells to congenically disparate recipient mice after CD8+ T-cell enrichment. The frequency and number of donor CD8+ T cells was monitored for at least 30 d after transfer.
Recipient mice (e.g., CD45.1+ (B6.SJL-Ptprca Pepcb/BoyJ) from the Jackson Laboratory)
Pathogen for in vivo infection experiments (e.g., LCMV Arm or clone 13 for P14 cells. LCMV stocks were grown and used as described35, and viral seed stocks were originally obtained from R. Ahmed (Emory University))
! CAUTION Growing and use of live pathogens must be performed according to all relevant governmental and institutional guidelines and regulations, in compliance with protocols approved by local animal ethics committees. All animal and infection work in this study was performed in accordance with the institutional animal care and use guidelines for the University of Pennsylvania.
Cell culture
EasySep Mouse CD8+ T Cell Isolation Kit (StemCell, cat. no. 19853)
RPMI-1640 with L-glutamine (300 mg/l; Cellgro; Corning, cat. no. 10-040-CM)
PBS, 1× (Cellgro; Corning, cat. no. 21-031-CM)
EDTA, 0.5 M, pH 8.0 (Gibco, cat. no. 15575-020)
FBS, heat-inactivated (Gemini Bio-Products, cat. no. 100-106)
▲ CRITICAL As the culture of primary T cells is sensitive to serum quality, it is recommended that several lots of serum be tested for culture optimization.
Penicillin–streptomycin (Gibco, cat. no. 15140-122)
Sodium pyruvate (Gibco, cat. no.11360-070)
Nonessential amino acids (Gibco, cat. no. 11140-050)
HEPES (N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid; Gibco, cat. no. 15630-080)
2-Mercaptoethanol (Sigma-Aldrich, cat. no. M3148)
! CAUTION 2-Mercaptoethanol is toxic upon inhalation or contact with skin and is harmful if swallowed. Handle it with protective gloves under a fume hood.
DMSO (Sigma-Aldrich, cat. no. D8418)
Recombinant human IL-2 (rhIL-2; PeproTech, cat. no. 200-0)
▲ CRITICAL As human and murine recombinant IL-2 have differential biological effects on mouse CD8+ T cells, an optimally titrated concentration should be determined if murine IL-2 is used.
Acetic acid, 99% (vol/vol) (Sigma-Aldrich, cat. no. A6283)
Trypan blue solution in PBS (Corning, cat. no. 25-900-Cl)
Purified anti-mouse CD3ε antibody (145-2C11 clone; eBioscience, cat. no. 14-0031) ▲ CRITICAL Anti-CD3ε clone 145-2C11 has resulted in more robust results as compared with clone 17A2 in our experience (data not shown). As the culture medium containing CD3ε and CD28 antibodies is removed in the later steps, it is not necessarily to use an in vivo functional-grade antibody for in vitro stimulation.
Purified anti-mouse CD28 antibody (37.51 clone, eBioscience, cat. no. 14-0281)
Ethanol, 200 proof (100% (vol/vol)) (Decon Labs, cat. no. 2716)
Density centrifuge and RV transduction
Percoll (GE Healthcare, cat. no. 17-0891-01)
PBS, 10×, no calcium, no magnesium (Gibco, cat. no. 14200-075)
NaHCO3, 7.5% (wt/vol) (Gibco, cat. no. 25080-094)
Polybrene (hexadimethrine bromide; Sigma-Aldrich, cat. no. H9268)
RetroNectin (Takara-Clontech, cat. no. T100A or T100B)
Refrigerator
Retrovirus supernatant ! CAUTION Universal precautions must be taken while handling RV supernatants and RV-transduced samples, and all experiments must be carried out in at least class II biological safety cabinets and using appropriate protection equipment (e.g., lab coat, eye protection and gloves). ▲ CRITICAL It is important to determine the titer of RV stock in advance. The RV titer can be determined as described in previous protocols13,15. RV titer can vary among production batches and will affect downstream experiments.
Flow cytometry
7-AAD (BioLegend, cat. no. 420403)
4% (wt/vol) Paraformaldehyde (PFA) in PBS (Affymetrix, cat. no. 19943) ! CAUTION PFA is toxic and irritating to the skin, eyes and respiratory system, and it may be carcinogenic; wear protective gloves and work under a chemical hood while preparing the solution.
Fluorescently labeled anti-mouse CD8, CD45.1 and CD45.2 (if applicable) antibodies (all antibodies used in this study are listed in Supplementary Table 1) ▲ CRITICAL Each antibody should be titrated before use. ▲ CRITICAL All antibodies must be kept at 4 °C and protected from exposure to direct light. ▲ CRITICAL Choose an appropriate fluorescent color and avoid FITC (for GFP RV), AmCyan/BV510 (for VEX RV), PE (for mKO2 RV) or PE Texas Red (for mCherry) for specific RV reporter genes.
Fluorescently labeled anti-mouse/rat CD90.1 (Thy1.1) or anti-human CD271 (NGFR) antibodies if Thy1.1 or hNGFR RV is used ▲ CRITICAL Each antibody should be titrated before use.
Foxp3 Transcription Factor Staining Buffer Set (eBioscience, cat. no. 00-5523-00) ! CAUTION Fixation buffer contains PFA and is toxic; wear protective gloves and work under a chemical hood while preparing the solution.
EQUIPMENT
Cell culture and animal experiments
Biological safety level 2 (BSL-2) tissue culture and animal facilities
Class II biological safety cabinet (The Baker Company, cat. no. SG403A-HE)
Water vacuum pump
Cell incubator (37 °C, 5% CO2) (Sanyo-Panasonic, cat. no. MCO-17AC)
Inverted light microscope (Jenco, cat. no. CP-2A1)
Hemocytometer (Fisher Scientific, cat. no. 02-671-5) or another cell counter
Water bath (Sheldon Manufacturing, cat. no. SWB7)
Tweezers and scissors
Restrainer (Plas Labs, cat. no. 551-BSRR)
Heat lamp
Automatic pipette (Pipet Aid XP; Drummond Scientific)
Manual pipette (e.g., Gilson)
500-ml and 1,000-ml autoclavable glass bottles (Pyrex, cat. nos. 00413612 and 10053295)
Easystep Magnet (15 ml; StemCell, cat. no. 18001)
Vortex mixer (Scientific Industries, cat. no. SI-0236)
Microcentrifuge (Eppendorf, cat. no. 5424)
Centrifuge (Eppendorf, cat. no. 5810R, 15-Amp version)
Rotor for swing buckets (Eppendorf, cat. no. A-4-81)
MTP/Flex bucket for Rotor A-4-81 (Eppendorf, cat. no. 022638866)
500-ml rectangular bucket, for Rotor A-4-81 (Eppendorf, cat. no. 022638629)
Adaptor for five conical 50-ml tubes (Eppendorf, cat. no. 022638769)
Adaptor for 12 conical 15-ml tubes (Eppendorf, cat. no. 022638742)
Flow cytometric analyzer (BD Biosciences, model no. LSR II. running DIVA 8.0)
Flow cytometric sorter (BD Biosciences, model no. ARIA II, running DIVA 8.0)
FlowJo v9 software for flow cytometry data analysis (Tree Star, https://www.flowjo.com/solutions/flowjo/downloads)
Prism v5 (GraphPad, https://www.graphpad.com/how-to-buy/)
Consumables
50- and 15-ml polypropylene conical centrifuge tubes (Bioexpress, GeneMate, cat. nos. C-3394-3 and C-3394-1, respectively)
75-cm2 cell culture flask (Greiner Bio-One, cat. no. 658175)
Six-well plate, tissue-culture-treated (Greiner Bio-One, cat. no. 657165)
24-Well plate, tissue-culture-treated (BD Falcon, cat. no. 353047)
48-Well plate, tissue-culture-treated (BD Falcon, cat. no. 353078)
Six-well plate, non-tissue-culture-treated (Corning, cat. no. 351146)
60-mm tissue culture dish (Thermo Fisher Scientific, cat. no. 130181)
1,000-μl, 200-μl and 20-μl barrier tips (GeneMate; Bioexpress, cat. nos. P1247-1250, P1237-200 and P1237-20)
25-ml, 10-ml, 5-ml and 1-ml serological sterile pipettes (Fisher Scientific, cat. nos. 13-678-11, 13-678-11E, 13-678-11D and 13-678-11B)
10-ml and 5-ml syringes (BD, cat. nos. 309604 and 309646)
28-gauge × 1/2 inch and 29-gauge × 1/2 inch insulin syringes (Exelint International, cat. nos. 26027 and 26028)
18-gauge needle (BD, cat. no. 305196)
0.2-μm bottle top filter (Corning, cat. no. CLS431174)
0.2-μm syringe filter (Corning, cat. no. 431224)
70-μm cell strainer (BD Falcon, cat. no. 352350)
Glass Pasteur pipette, 230-mm length (Wheaton, cat. no. 357335)
5-ml polystyrene tube for flow cytometry (BD Falcon, cat. no. 352008)
Microcentrifuge tubes (1.5 ml; Eppendorf, cat. no. 022363204)
Titertube micro test tubes (Bio-Rad, cat. no. 223-9391)
REAGENT SETUP
2-Mercaptoethanol (50 mM)
Add 0.3 ml of 100% 2-mercaptoethanol (14.3 M) to 85.8 ml of water. Prepare 1-ml aliquots of 2-mercaptoethanol in 1.5-ml microcentrifuge tubes, and store the solution at −20 °C for up to 2 years.
IL-2 (104 U/μl master stock and 102 U/μl working stock)
Given that the specific activity of rhIL-2 produced by PeproTech is at least 1 × 107 units per mg, we use 1 × 107 units per mg. Briefly (300g at room temperature (24 °C) for 30 s), centrifuge the vial containing lyophilized recombinant human IL-2 (rhIL-2) before opening. Reconstitute 100 μg of rhIL-2 in 100 μl of 100 mM acetic acid to make a 1 μg/μl master stock (104 U/μl). Prepare 10-μl aliquots of the master stock in 1.5-ml microcentrifuge tubes and store them at −80 °C for up to 1 year. To make the 102 U/μl working stock, add 990 μl of sterile PBS to 10 μl of 104 U/μl master stock. Prepare 100-μl aliquots of working stock in 1.5-ml microcentrifuge tubes, and store them at −20 °C for up to 1 year.
Complete RPMI-1640 (1,000 ml)
Add 100 ml of FBS (final = 10% (vol/vol)), 1 ml of 50 mM 2-mercaptoethanol (final = 0.05 mM), 10 ml of 10,000 U/ml penicillin and 10,000 μg/ml streptomycin (final = 100 U/ml and 100 μg/ml, respectively), 20 ml of 1 M HEPES (final = 20 mM), 10 ml of 100 mM sodium pyruvate (final = 1 mM), 10 ml of 10 mM nonessential amino acids (final = 100 μM) to 1,000 ml of RPMI-1640 medium. Filter-sterilize the medium through a 0.22-μm bottle top filter unit, and store it at 4 °C for up to 2 weeks.
T-cell stimulation medium (50 ml)
Add 50 μl of 1 mg/ml anti-mouse CD3ε antibody (final = 1 μg/ml), 25 μl of 1 mg/ml anti-mouse CD28 antibody (final = 0.5 μg/ml), and 50 μl of 100 U/μl rhIL-2 (final = 100 U/ml) to 50 ml of complete RPMI-1640 medium. Store the medium at 4 °C for up to 2 weeks. ▲ CRITICAL It is important to add IL-2 and 2-mercaptoethanol to the T-cell stimulation medium. Transduction efficiency is <10% in the absence of IL-2.
Isotonic Percoll (270 ml)
Transfer 231.25 ml of Percoll to an autoclaved glass bottle. Add 36 ml of 10× PBS and 3.26 ml of 7.5% (wt/vol) NaHCO3. Store the solution at 4 °C for up to 1 year. ▲ CRITICAL As Percoll can form precipitates during long-term storage, shake the bottle vigorously before using.
30 and 60% (vol/vol) Percoll
Add 2.8 ml of complete RPMI-1640 medium to 1.2 ml of isotonic Percoll to make 4 ml of 30% (vol/vol) Percoll solution. Add 1.2 ml of complete RPMI-1640 medium to 1.8 ml of isotonic Percoll to make 3 ml of 60% (vol/vol) Percoll solution. ▲ CRITICAL To avoid any risk of Percoll sedimentation and bacterial contamination, it is advised to use freshly prepared Percoll solutions.
Polybrene (800 and 8 mg/ml)
Dissolve 5 g of Polybrene in 6.25 ml of water to make an 800 mg/ml master stock. Take 1 ml of master stock and add 99 ml of water to make an 8 mg/ml working stock. Stocks can be stored in a 1.5-ml or 2-ml tube at −20 °C for at least 1 year.
RetroNectin coating of a six-well plate
For coating one well of a six-well plate, add 40 μl of RetroNectin (1 μg/μl) to 1,960 μl of PBS to make 2 ml of 20 μg/ml solution. Apply 2 ml of 20 μg/ml RetroNectin solution per well of the non-tissue-culture-treated six-well plate. Incubate the plate at 4 °C overnight. Remove the RetroNectin solution. Wash the plate twice with ice-cold PBS and use it immediately.
2% (vol/vol) PFA (100 ml)
Add 50 ml of PBS to 50 ml of 4% (vol/vol) PFA in PBS. Store the solution at 4 °C for up to 1 month. ! CAUTION PFA is toxic; wear protective gloves and work under a chemical hood while preparing and using this solution.
MACS buffer (1,000 ml)
Add 10 ml of serum (final = 1% (vol/vol)) and 4 ml of 0.5 M EDTA (final = 2 mM) to 1,000 ml of PBS. Store the buffer at 4 °C for up to 1 month.
PROCEDURE
Isolation and in vitro stimulation of CD8+ T cells ● TIMING 3 h
! CAUTION Appropriate institutional regulatory board permission for animal infection experiments must be obtained before you start mouse experiments. All animal work in this study was performed in accordance with the institutional animal care and use guidelines for the University of Pennsylvania.
1| Ensure that you have sufficient numbers of donor mice.
▲ CRITICAL STEP Because adoptively transferred T cells will be analyzed over several weeks to months in vivo in some experiments, it is important to match the genetic backgrounds of donor CD8+ T cells and recipient mice to avoid potential rejection issue. See Reagents for details.
▲ CRITICAL STEP Generally, consider preparing one donor mouse per RV. See ‘Limitations’ and ‘ANTICIPATED RESULTS’ for more detail.
? TROUBLESHOOTING
2| Euthanize mice according to institutional guidelines (e.g., we use a lethal dose of CO2 narcosis, followed by cervical dislocation).
! CAUTION Experiments involving mice should be performed according to all relevant governmental and institutional guidelines and regulations, in compliance with protocols approved by local animal ethics committees. All animal work in this study was performed in accordance with the institutional animal care and use guidelines for the University of Pennsylvania.
▲ CRITICAL STEP Time from in vitro stimulation on day 0 to RV transduction on day 1 is an important parameter for optimal RV transduction efficiency. A stimulation time of at least 18 h, but no more than 48 h, is required for optimal CD8+ T-cell activation and subsequent RV transduction (Fig. 5). In addition, the entire experimental procedure on day 1, from enrichment of activated T cells to adoptive transfer by injection, can typically take at least 9 h. Thus, to start and finish day 1 procedures efficiently (e.g., between 9 a.m. and 6 p.m.), we recommend starting in vitro stimulation early on day 0.
? TROUBLESHOOTING
3| Spray 70% (vol/vol) ethanol over the mouse body, and collect the spleens in a 70-μm sterile cell strainer immersed in 2 ml of MACS buffer (Reagent Setup) and placed in a sterile 60-mm cell culture dish on ice.
4| Dice the spleens into 2- to 3-mm pieces before smashing them on the 70-μm cell strainer.
▲ CRITICAL STEP This step is important to maximizing cell recovery and viability.
5| Prepare a single-cell suspension by crushing the spleen through the strainer in MACS buffer.
▲ CRITICAL STEP For the best T-cell viability, we recommend not using a red blood cell (RBC) lysis buffer such as ammonium–chloride–potassium (ACK) buffer.
6| Transfer the spleen cell suspension to a 15-ml conical tube.
7| Wash the 70-μm cell strainer by passing 8 ml of MACS buffer over the 60-mm cell culture dish.
8| Combine the MACS buffer containing residual spleen cells from the 60-mm cell culture dish with the spleen cell suspension in the 15-ml tube (from Step 6).
9| Centrifuge the 15-ml tube at 300g for 5 min at 4 °C.
10| Aspirate the supernatant.
11| Resuspend the cell pellet in 1 ml of MACS buffer per spleen.
12| Enrich CD8+ T cells by CD8 negative selection. As we normally use a kit from StemCell Technologies, we describe the protocol for the EasySep Mouse CD8+ T Cell Isolation Kit (Reagents) here.
▲ CRITICAL STEP Most commercial kits are likely to work. However, inclusion of anti-RBC antibody (e.g., Ter-119) in the kit is essential if RBC lysis buffer is not used during spleen processing.
13| Add 50 μl of rat serum (provided in the kit) per 1 ml of spleen cell suspension.
14| Add 50 μl of Isolation Cocktail per 1 ml of spleen cell suspension.
15| Mix the contents and incubate them at room temperature for 10 min.
16| Vortex the RapidSpheres (provided in the EasySep Mouse CD8+ T Cell Isolation Kit) for 30 s.
17| Add 125 μl of RapidSpheres per 1 ml of spleen cell suspension.
18| Mix the contents and incubate them at room temperature for 5 min.
19| If the cell suspension volume is <4 ml, bring the volume to 5 ml by adding MACS buffer; if the cell suspension volume is >4 ml, bring the volume to 10 ml by adding MACS buffer.
20| Mix the contents by gently pipetting up and down two to three times. Remove the cap from the 15-ml tube.
21| Place the tube without the cap into the magnet, and leave it for 2.5 min at room temperature. In the meantime, prepare a new 15-ml tube.
22| Pick up the magnet, and, in one continuous motion, invert the magnet and tube, and pour the enriched CD8+ T-cell suspension into the new 15-ml tube.
23| Count the number of cells using a hemocytometer or any other cell counter. Normally, at least 5 × 106 cells per spleen are recovered.
24| On the basis of the number of cells, prepare an appropriate volume (1 ml per 1 × 106 cells) of T-cell stimulation medium in a separate tube or flask by adding anti-CD3ε (final = 1 μg/ml), anti-CD28 (final = 0.5 μg/ml) antibodies and rhIL-2 (final = 100 U/ml) to complete RPMI-1640 medium (Reagent Setup).
▲ CRITICAL STEP Testing the serum lot for T-cell culture in advance is recommended.
▲ CRITICAL STEP Make sure to add 2-mercaptoethanol to the complete RPMI-1640 medium. 2-mercaptoethanol is essential to keep IL-2 activity in the medium and facilitate CD8+ T-cell activation.
? TROUBLESHOOTING
25| Centrifuge the enriched CD8+ T-cell suspension at 300g for 5 min at 4 °C.
26| Aspirate the supernatant and resuspend the cell pellet with stimulation medium in order to obtain a cell density of 1 × 106 cells per ml.
27| Plate the cells in six-well culture plates at a concentration of 1 × 106 cells per ml in a volume of 3–4 ml per well (or use a T-75 flask for larger cell numbers).
▲ CRITICAL STEP Cell density is critical to optimal activation of CD8+ T cells in our tests. Using a cell concentration <1 × 106 cells per ml results in lower RV transduction efficiency.
? TROUBLESHOOTING
28| Incubate the cells at 37 °C and 5% CO2 for 18–24 h. In the meantime, continue with the infection of the recipient mice (Step 29).
Infection of recipient mice ● TIMING 1–2 h
29| On the same day as when the CD8+ T cells are stimulated in vitro (Step 24), infect recipient mice with the desired pathogen (e.g., we use LCMV for P14 TCR Tg cells). We inject 2 × 105 plaque-forming units (p.f.u.) of Armstrong strain i.p. and 4 × 106 p.f.u. of clone 13 strain intravenously (i.v.) for acute and chronic infection, respectively. We use 28-gauge and 29-gauge insulin syringes for i.p. and i.v. injections, respectively.
Enrichment of activated CD8+ T cells by Percoll ● TIMING 1 h
30| Leave 100% (vol/vol) isotonic Percoll and complete RPMI-1640 medium at room temperature for at least 1 h or use a water bath to bring the temperature up to 24 °C. Set the centrifuge at 25 °C.
▲ CRITICAL STEP The temperatures of the Percoll, medium, centrifuge and cell suspension are critical to optimal separation and recovery in density centrifugation. Make sure that all reagents and the centrifuge are prewarmed to room temperature before starting cell harvest. Always keep the cell suspension at room temperature during enrichment steps.
? TROUBLESHOOTING
31| Examine the T-cell morphology using an inverted light microscope.
▲ CRITICAL STEP At least 50% of the cells should be blasting, with an enlarged round shape and glossy appearance. In addition, there should be some cell aggregates, as activated T cells form ball-shaped aggregates in vitro.
32| Harvest stimulated T cells from the six-well plate or a 75-cm2 flask into a 50-ml conical tube. Before transferring to a tube, pass the cell suspension through a 70-μm sterile cell strainer to remove the cell aggregates.
▲ CRITICAL STEP Pipette the suspension up and down intensively until all non-adherent cells have been removed, as in vitro-stimulated day 1 effector cells are often firmly attached and form some cell aggregates. We usually do this 10–20 times with a 5-ml pipette with no negative effect on cell viability.
▲ CRITICAL STEP To maximize cell recovery, grind the cell aggregates on a 70-μm strainer using the plunger of a 5-ml syringe.
33| Count the number of cells.
▲ CRITICAL STEP Recovery from Step 23 to Step 33 is normally ~40–60%.
34| Prepare 4 ml of 30% (vol/vol) and 3 ml of 60% (vol/vol) Percoll solution with 100% (vol/vol) isotonic Percoll and complete RPMI medium in 15-ml conical tubes (Reagent Setup).
▲ CRITICAL STEP Up to 2 × 107 cells can be separated with 4 ml of 30% (vol/vol) and 3 ml of 60% (vol/vol) Percoll solution in a 15-ml tube. For a larger cell number, consider increasing the number of tubes, rather than increasing the Percoll volume.
▲ CRITICAL STEP Use freshly made 30 and 60% (vol/vol) Percoll solutions for density centrifugation.
▲ CRITICAL STEP Although we have carefully determined the best combination of Percoll solutions, adjusting the concentration by 5% may be needed, depending on the experimental setting. See Reagent Setup.
? TROUBLESHOOTING
35| Take an aliquot of cells (~5 × 105) into a separate tube, and keep this at 4 °C as a non-Percoll separation control (unfractionated sample).
36| Centrifuge the conical tube containing the activated cells at 300g for 5 min at 25 °C.
37| After centrifugation, transfer the culture supernatant to a new 50-ml tube. The culture supernatant containing anti-CD3ε, anti-CD28 and IL-2 can be used for RV transduction (Steps 49 and 53) and to check the transduction efficiency (Steps 68–71).
38| Resuspend the cell pellet with 4 ml of 30% (vol/vol) Percoll solution, and transfer this to a new 15-ml conical tube.
▲ CRITICAL STEP Because the interface cell layer after Percoll density centrifugation is thin, we recommend using smaller tubes (15 ml) rather than wide tubes (50 ml) for the best visualization.
39| Place an autoclaved Pasteur glass pipette into the 15-ml tube.
40| Underlay 3 ml of 60% (vol/vol) Percoll solution beneath the 30% (vol/vol) Percoll cell suspension by carefully pouring 60% (vol/vol) Percoll solution into the Pasteur glass pipette. Let the 60% (vol/vol) Percoll solution flow by gravity.
▲ CRITICAL STEP For the best separation result, it is essential to add the 60% (vol/vol) Percoll solution gently to form two layers.
? TROUBLESHOOTING
41| Slowly and carefully remove the Pasteur pipette from the 15-ml conical tube.
42| Centrifuge the 15-ml conical tube with the Percoll gradient and activated T cells at 800g for 20 min at 25 °C, with no acceleration or brake, using a swing bucket rotor.
▲ CRITICAL STEP Centrifugation force, time and temperature are all critical. Acceleration and the brake must be off to ensure that the Percoll gradient is not disturbed. After centrifugation, dead cells and fragments accumulate on the top of the 30% (vol/vol) Percoll layer, activated large-sized T cells form a thin, whitish layer at the interface between the 30 and 60% (vol/vol) Percoll layers and nonactivated small-sized T cells form a cell pellet in the bottom of the 60% layer (Fig. 2b).
? TROUBLESHOOTING
43| Aspirate the upper 3 ml of the 30% (vol/vol) Percoll layer and discard. This fraction contains dead cells and debris.
44| Transfer the interface layer to a new 15-ml conical tube along with the remaining 1 ml of the 30% (vol/vol) Percoll layer and the top 0.5 ml of the 60% (vol/vol) Percoll layer. Bring the final volume to 15 ml with complete RPMI-1640 medium. This fraction contains activated T cells that will be used for RV transduction (interface sample).
45| Carefully aspirate the upper half of the remaining 60% (vol/vol) Percoll. Resuspend the cell pellet with complete RPMI-1640 medium, and bring the final volume to 15 ml (bottom sample).
46| Centrifuge the two 15-ml conical tubes at 300g for 5 min at 25 °C.
47| Aspirate the supernatant and resuspend the cell pellet in 10–15 ml of complete RPMI-1640 medium.
▲ CRITICAL STEP As residual Percoll in cell suspension can reduce transduction efficiency in later steps, it is important to wash the activated cells at least twice with fresh medium.
? TROUBLESHOOTING
48| Centrifuge the 15-ml conical tubes at 300g for 5 min at 25 °C.
49| Aspirate the supernatant and resuspend the cell pellet in 1 ml (or an appropriate volume) of culture supernatant that was saved at Step 37.
50| Count the number of cells.
▲ CRITICAL STEP Typically, the recovery rate from input (Step 33) to interface (Steps 44 and 50) is ~30% (Fig. 2c).
51| Check the purity of CD8+ T cells and the efficiency of enrichment by comparing the unfractionated (Step 35), interface (Step 44) and bottom (Step 45) samples by flow cytometry. (Fig. 2b) Stain for surface markers (e.g., CD8, congenic markers), and add 7-AAD (or another live/dead discrimination) dye before analysis.
▲ CRITICAL STEP To save time, this purity check can be done during the 1-h spin RV transduction (Step 57) or the 4-h incubation after RV transduction (Step 58).
Retrovirus transduction of enriched CD8+ T cells ● TIMING 1.5–2 h + 4 h incubation
! CAUTION All RV work should be performed in a class II biosafety cabinet with appropriate personal protective equipment (lab coat, eye guard and gloves). RV must be handled and disposed of in accordance with your institutional biohazard control regulations. Waste should be disposed of in biohazard bags and autoclaved.
▲ CRITICAL If you use RetroNectin as an RV transduction facilitator, start RetroNectin coating of the six-well plate on the same day as the in vitro T-cell stimulation.
52| Set the temperature of the centrifuge to 30 °C.
▲ CRITICAL STEP The temperature of the centrifuge is important for optimal RV transduction efficiency, as a lower temperature may reduce T-cell activity and susceptibility to RV transduction.
? TROUBLESHOOTING
53| On the basis of cell count (Step 50), adjust the cell concentration of the interface sample (enriched CD8+ T cells) to 1–3 × 106 cells per ml by adding an appropriate volume of saved culture supernatant (Step 37) (e.g., a final 3-ml volume for total of 9 × 106 cells).
▲ CRITICAL STEP To maximize transduction efficiency and cell recovery after transduction, it is recommended to use 3 × 106 cells per ml if the number of activated T cells is sufficient.
54| Keep an aliquot of the enriched CD8+ T-cell population (~5 × 105 cells) as a non-RV-transduced control at room temperature until other samples are ready (Step 68).
55| Thaw aliquots of RV stock(s) at 37 °C in a water bath. Prepare 1 ml of RV supernatant per well of a six-well plate.
▲ CRITICAL STEP Choose the appropriate RV system for your experimental purpose. See Experimental design for more information.
▲ CRITICAL STEP For transduction to CD8+ T cells, the RV supernatant does not have to be freshly produced at the time of transduction. Check the titer of frozen RV stocks in advance. See Experimental design for more information.
▲ CRITICAL STEP Although GFP, VEX, mKO2 and Thy1.1 were verified as robust RV reporters in our test (Fig. 6), it is advisable to check the stability in your experimental setting.
? TROUBLESHOOTING
-
56| RV transduction can be done with either Polybrene (option A) or RetroNectin (option B)
-
RV transduction using polybrene
Thaw a frozen working stock (8 mg/ml) of Polybrene in a 37 °C water bath.
-
Add Polybrene to the RV supernatant at a concentration of 8 μg/ml (1/1,000 dilution of the 8 mg/ml working stock).
▲ CRITICAL STEP Polybrene can be toxic to T cells. Titrate the concentration of Polybrene if T-cell viability after RV transduction is low.
? TROUBLESHOOTING
-
Dispense 1 ml of the T-cell suspension (1–3 × 106 cells) per well of a non-tissue-culture-treated six-well plate.
▲ CRITICAL STEP Use of non-tissue-culture-treated plates yields better transduction efficiency (Fig. 5b)
? TROUBLESHOOTING
Add 1 ml of RV supernatant containing Polybrene per well of a six-well plate. The total volume per well is now 2 ml, and the final concentration of Polybrene is 4 μg/ml.
-
RV transduction using RetroNectin
Start RetroNectin coating of the six-well plate a day before (Reagent Setup) at 4 °C.
Remove the RetroNectin-coated six-well plate from the refrigerator.
Aspirate the PBS-containing RetroNectin and wash the plate twice with cold PBS.
Remove the PBS and immediately dispense 1 ml of the T-cell suspension (1–3 × 106 cells) per well of a RetroNectin-coated six-well plate. Do not allow the wells to become dry.
Add 1 ml of RV supernatant per well of a six-well plate. Now the total volume is 2 ml per well.
-
57| Spin-transduce CD8+ T cells at 2,000g for 60 min at 30 °C, with no acceleration or brake, using a swinging plate rotor.
▲ CRITICAL STEP Centrifugation time can be extended to 120 min.
58| After spin-transduction, place the plate into the CO2 incubator at 37 °C for at least 4 h.
▲ CRITICAL STEP Incubation may be extended up to overnight if RV transduction efficiency after 4 h is low. However, prolonged incubation in the presence of Polybrene could reduce cell viability, and, in our experience, the increase in efficiency with extended culture is generally moderate. See Experimental design.
? TROUBLESHOOTING
Adoptive transfer of RV-transduced CD8+ T cells ● TIMING 2–3 h
59| After incubation, collect the cells from the six-well plate into 15-ml conical tubes.
60| Centrifuge the cells at 300g for 5 min at 30 °C.
61| Aspirate the supernatant and resuspend the cells in 10 ml of complete RPMI-1640.
62| Centrifuge the cells at 300g for 5 min at 30 °C.
63| Aspirate the supernatant and resuspend the cells in 10 ml of PBS.
64| Centrifuge the cells at 300g for 5 min at 30 °C.
65| Aspirate the supernatant, and resuspend the cells in 0.5–1 ml of PBS.
66| Remove 10 μl of cell suspension and count the number of cells with a hemocytometer. Keep an aliquot (~1 × 106 cells) in a separate tube at room temperature for a transduction efficiency check (Steps 68–71).
67| Adjust the cell concentration to 5 × 105 cells per ml with PBS, and pass them through a 70-μm strainer. Inject 200 μl (1 × 105 cells) per recipient mouse i.v. We use a 29-gauge insulin syringe for tail-vein injection.
▲ CRITICAL STEP The number of transferred CD8+ T cells should be optimized for individual experimental settings (Fig. 4). See optimal number of adoptively transferred CD8+ T cells in Experimental design.
? TROUBLESHOOTING
Checking of RV-transduction efficiency by flow cytometry ● TIMING 1.5 h
68| To determine RV transduction efficiency, use non-RV-transduced (from Step 54, as a negative control) and RV-transduced (from Step 66) CD8+ T cells. Centrifuge the tubes containing non-RV-transduced or RV-transduced cells at 300g for 5 min at 30 °C
▲ CRITICAL STEP A transduction efficiency check in vitro is essential, because this information may help to identify any problems if low RV-transduction efficiency in vivo is observed at later steps (Fig. 1).
? TROUBLESHOOTING
69| Discard the supernatant and resuspend the cell pellets in saved culture supernatant containing rhIL-2 (Step 37) at 1–5 × 105 cells per ml.
70| Transfer the cells to a 24- or 48-well cell culture plate, and incubate the plate overnight at 37 °C, 5% CO2 (Fig. 1).
71| On day 2, harvest the CD8+ T cells from the tissue culture plate. Check the transduction efficiency by flow cytometry. Stain for surface markers (e.g., CD8, congenic markers and Thy1.1 or hNGFR for RV detection, if necessary).
Analysis of RV-transduced CD8+ T cells in vivo ● TIMING 4–10 h
72| Bleed or harvest target organs from recipient mice after the desired time.
▲ CRITICAL STEP For a typical infection model, such as LCMV, the following three time points are preferred for an initial screening experiment: day 8 (up to day 10) to determine the magnitude of effector proliferation, day 15 (up to day 18) to examine contraction, and day 30 and onward to examine development, differentiation and stability of memory or exhausted T cells.
73| Prepare a single-cell suspension using standard cellular immunology protocols, and stain cell-surface markers for flow cytometry.
▲ CRITICAL STEP We have noticed that at some time points (particularly days 6–8), there was substantial loss of RV-transduced CD8+ T cells during single-cell preparation when prepared with Histopaque density centrifugation. It is possible that RV-transduced CD8+ T cells have slightly altered cell density during the effector phase. Use of RBC lysis buffer at this point is recommended.
? TROUBLESHOOTING
74| If intracellular molecules are stained in the cells that were transduced with RV with fluorescent protein marker (e.g., GFP, VEX, mKO2), pre-fix the samples with 2% (wt/vol) PFA in PBS for 20 min at 4 °C before applying fixation buffer from commercial ICS kits.
! CAUTION PFA is toxic. Wear appropriate personal protective equipment.
▲ CRITICAL STEP Additional fixation prevents loss of cytoplasmic fluorescent protein signal and improves the detection of RV-transduced T cells (Fig. 7).
? TROUBLESHOOTING
75| Analyze the samples by flow cytometry.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 2, 24, 27 | Low RV transduction efficiency | Insufficient activation of CD8+ T cells |
|
| 30, 52 | Low RV transduction efficiency | The temperature was not optimal during the procedure | Cells should be kept above room temperature to maintain activity. Do not place cells on ice at any time. Set the centrifuge to 30 °C for spin-transduction |
| 56, 58 | Low RV transduction efficiency | Low viability of T cells during transduction | Excessive exposure to Polybrene: lower the concentration or shorten the time of exposure to viral supernatant. Consider switching to RetroNectin if use of Polybrene continues to result in low viability |
| 47 | Low RV transduction efficiency | Residual Percoll particles in cell suspension | Wash the cells at least twice with complete RPMI-1640 before RV transduction |
| 55 | Low RV transduction efficiency | The wrong RV system was used | Use an ecotropic RV system for standard mouse T-cell experiments |
| 55 | Low RV transduction efficiency | The RV stock titer was too low | Check the RV titer before starting in vitro T-cell stimulation. Use of high-titer stock (>107 reporter-positive focus-forming units/ml) is recommended |
| 56 | Low RV transduction efficiency | The wrong tissue-culture plates were used for spin-transduction and incubation | Noncoated plates should be used for spin-transduction |
| 1 | No visible interface layer or low cell recovery after Percoll separation | The input cell number used was too low | An input cell number of at least 5 × 106 is recommended. Increase the number of spleens if necessary |
| 30, 42 | No visible interface layer or low cell recovery after Percoll separation | The temperature of the Percoll reagents and centrifugation were not optimal | Leave the Percoll reagents at room temperature for at least 1 h or use a water bath. Make sure that the temperature during centrifugation is set to 25 °C |
| 40, 42 | No visible interface layer or low cell recovery after Percoll separation | The 30 and 60% (vol/vol) Percoll layers were disturbed | Care must be taken when underlaying 60% (vol/vol) Percoll and moving the tubes to/from the centrifuge. Accelerator and brake should be off during centrifuge |
| 30, 34 | No visible interface layer or low cell recovery after Percoll separation | 30 and/or 60% (vol/vol) Percoll solutions were not appropriately prepared | Check the reagents. Consider using a density marker if refreshing all reagents fails to solve the problem |
| 34 | No visible interface layer or low cell recovery after Percoll separation | The combination of 30 and 60% (vol/vol) Percoll is not optimal in the experimental setting | Test whether modifying the Percoll concentration by 5% increments works better |
| 1 | Low levels of RV-transduced T cells in vivo | Rejection of transferred T cells due to unmatched genetic background | Check the genetic background of the donor mouse and run a test transfer experiment without RV transduction |
| 55 | Low levels of RV-transduced T cells in vivo | Rejection due to the RV marker | Compare with different RV marker(s) in individual experimental settings. Consider switching RV marker if rejection is suspected |
| 67 | Low levels of RV-transduced T cells in vivo | The number of transferred T cells was too low | Titrate the optimal cell number for transfer in individual experimental settings |
| 68 | Low levels of RV-transduced T cells in vivo | Low RV transduction efficiency | Check the RV transduction efficiency on day 2 in vitro |
| 73 | Low levels of RV-transduced T cells in vivo | Loss of RV-transduced T cells by Histopaque centrifugation during sample preparation | Switch to RBC lysis buffer when preparing single-cell suspension during acute phase |
| 74 | Low levels of RV-transduced T cells in vivo | Leakage of RV-derived fluorescent marker(s) in intracellular staining application | Add a pre-fixation step with 2% (vol/vol) PFA in PBS between staining surface molecules and permeabilizing steps |
● TIMING
Day 0
Steps 1–28, isolation and in vitro stimulation of CD8+ T cells: 3 h
Step 29, infection of recipient mice: 1–2 h
Day 1
Steps 30–51, enrichment of activated CD8+ T cells by Percoll: 1 h
Steps 52–58, retrovirus transduction of enriched CD8+ T cells: 1.5–2 h + 4 h incubation
Steps 59–67, adoptive transfer of RV-transduced CD8+ T cells: 2–3 h
Day 2
Steps 68–71, checking of RV transduction efficiency by flow cytometry: 1.5 h
Days 8–30+
Steps 72–75, analysis of RV-transduced CD8+ T cells in vivo: 4–10 h
ANTICIPATED RESULTS
Provided that target CD8+ T cells have no clear defects in development, typical cell recovery and purity after CD8+ enrichment from a single donor spleen are ~1 × 107 cells and 80%, respectively. Similarly, if target CD8+ T cells have no obvious defects in activation, total cell recovery and frequency of large-sized cells after overnight in vitro stimulation are ~5 × 106 cells and ~50%, respectively (Figs. 1 and 2b, left). After Percoll density centrifugation, cell recovery and frequency of large-sized cells from the interface layer are typically ~2 × 106 cells and >80%, respectively (Fig. 2b, right middle, Fig. 2c, and Supplementary Fig. 1). Given that the cell recovery from RV spin-transduction is 50%, the number of RV-transduced T cells recovered from a single spleen of starting material (~1 ×107 cells) through this protocol is ~1 × 106 cells (Fig. 2c). For our studies, ~1 × 105 enriched CD8+ P14 T cells per recipient mouse were needed to achieve donor T-cell responses of similar magnitude to that of the endogenous response in the LCMV Arm infection model (Fig. 4). Thus, up to ten recipient mice can be prepared from 1 × 106 RV-transduced T cells derived from one donor spleen.
Typically, the RV-transduction efficiency of in vitro-activated mouse CD8+ T cells using Polybrene peaks on day 1 (Fig. 5a) and is 40–80%, depending on RV titer, length of GOI (RV insert size has an effect) and activation status of target T cells when analyzed in vitro 1 d after transduction (Figs. 2d and 5b). Percoll density centrifugation enriches activated CD8+ T cells that are susceptible to RV transduction and increases the frequency of RV-transduced cells ~sixfold over several weeks in vivo (Fig. 2d–g). RV reporter genes GFP, VEX, mKO2 and Thy1.1 allow the generation of stable long-lived RV-transduced CD8+ T cells in vivo (Fig. 6). For ICS applications, an additional fixation step before permeabilization with commercial kits prevents the loss of cytoplasmic fluorescent proteins derived from RV transduction, resulting in improved detection of RV-transduced T cells (Fig. 7), although this additional fixation step could affect other downstream stains.
Overall, by a combination of optimized steps for in vitro T-cell activation, enrichment of RV-susceptible cells, RV transduction, use of in vivo-compatible RV reporter genes and detection of RV-transduced cells, this protocol enables efficient, long-term, in vivo tracking of the differentiation of RV-transduced CD8+ T cells.
Supplementary Material
Acknowledgments
We thank M.A. Ali for animal care; S. Adamski for technical assistance; K. Lewy for plasmid work; J.E. Wu for protocol verification; T. Yamada and K. Rome for comments on RV transduction of CD4+ T cells; and W.S. Pear (University of Pennsylvania), A. Bhandoola (Center for Cancer Research, National Cancer Institute), S.L. Reiner (Columbia University) and D.P. Beiting (University of Pennsylvania) for plasmids, cell line, template protocols and comments. This work was supported by German Research Foundation fellowship BE5496/1-1 (to B.B.), National Institutes of Health (NIH) grants F30AI129263 (to O.K.) and AI105343, AI112521, AI115712, AI108545, AI117950, AI117718, AI082630, AI095608 (to E.J.W.), U.S. Broad Agency Announcements Grant HHSN272201100018C (to E.J.W.) and funding from the Parker Institute for Cancer Immunotherapy (to E.J.W.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS M.K. and E.J.W. designed the protocol. M.K. and J.K. made the RV stock. J.K. prepared mouse T cells and analyzed flow cytometry data. M.K. optimized density centrifugation. Z.C., J.J. and O.K. optimized in vitro T-cell stimulation and RV transduction methods. M.K., J.K., Z.C., J.J., O.K., B.B., E.S., J.A. and L.M.M. performed the in vivo experiments. J.A. and L.M.M. developed and verified a new fixation method for RV-transduced cells. M.T. and S.U. contributed to the development of a new Kusabira Orange 2 retrovirus. J.K. prepared all figures. M.K. and E.J.W. wrote the manuscript.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol. 2013;13:309–320. doi: 10.1038/nri3442. [DOI] [PubMed] [Google Scholar]
- 3.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cepko C, Pear W. Overview of the retrovirus transduction system. Curr Protoc Mol Biol. 2001 doi: 10.1002/0471142727.mb0909s36. Chapter 9, Unit 9 9. [DOI] [PubMed] [Google Scholar]
- 5.Lee J, Sadelain M, Brentjens R. In: Genetic Modification of Hematopoietic Stem Cells: Methods and Protocols. Baum C, editor. Humana Press; 2009. pp. 83–96. [Google Scholar]
- 6.Zhong S, Malecek K, Perez-Garcia A, Krogsgaard M. Retroviral transduction of T-cell receptors in mouse T-cells. J Vis Exp. 2010;2010:2307. doi: 10.3791/2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kerkar SP, et al. Genetic engineering of murine CD8+ and CD4+ T cells for preclinical adoptive immunotherapy studies. J Immunother. 2011;34:343–352. doi: 10.1097/CJI.0b013e3182187600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kao C, et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8(+) T cell responses during chronic infection. Nat Immunol. 2011;12:663–671. doi: 10.1038/ni.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurachi M, et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8(+) T cells. Nat Immunol. 2014;15:373–383. doi: 10.1038/ni.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bengsch B, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity. 2016;45:358–373. doi: 10.1016/j.immuni.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamers CH, Willemsen RA, Luider BA, Debets R, Bolhuis RL. Protocol for gene transduction and expansion of human T lymphocytes for clinical immunogene therapy of cancer. Cancer Gene Ther. 2002;9:613–623. doi: 10.1038/sj.cgt.7700477. [DOI] [PubMed] [Google Scholar]
- 12.Parente-Pereira AC, Wilkie S, van der Stegen SJC, Davies DM, Maher J. Use of retroviral-mediated gene transfer to deliver and test function of chimeric antigen receptors in human T-cells. J Biol Methods. 2014;1:e7. [Google Scholar]
- 13.Cepko C, Pear W. Retrovirus infection of cells in vitro and in vivo. Curr Protoc Mol Biol. 2001 doi: 10.1002/0471142727.mb0914s36. Chapter 9, Unit 9.14. [DOI] [PubMed] [Google Scholar]
- 14.Engels B, et al. Retroviral vectors for high-level transgene expression in T lymphocytes. Hum Gene Ther. 2003;14:1155–1168. doi: 10.1089/104303403322167993. [DOI] [PubMed] [Google Scholar]
- 15.Zhang T, Tsang TC, Harris DT. Efficient transduction of murine primary T cells requires a combination of high viral titer, preferred tropism, and proper timing of transduction. J Hematother Stem Cell Res. 2003;12:123–130. doi: 10.1089/152581603321210208. [DOI] [PubMed] [Google Scholar]
- 16.Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med. 2015;212:1125–1137. doi: 10.1084/jem.20142237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marzo AL, et al. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat Immunol. 2005;6:793–799. doi: 10.1038/ni1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ansari AM, et al. Cellular GFP toxicity and immunogenicity: potential confounders in in vivo cell tracking experiments. Stem Cell Rev. 2016;12:553–559. doi: 10.1007/s12015-016-9670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Souza WN, Hedrick SM. Cutting edge: latecomer CD8 T cells are imprinted with a unique differentiation program. J Immun. 2006;177:777–781. doi: 10.4049/jimmunol.177.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mozo L, Rivas D, Zamorano J, Gutierrez C. Differential expression of IL-4 receptors in human T and B lymphocytes. J Immunol. 1993;150:4261–4269. [PubMed] [Google Scholar]
- 21.Pistoia V, et al. Production of hematopoietic growth factors by human B lymphocytes: mechanisms and possible implications. Stem Cells. 1993;11:150–155. doi: 10.1002/stem.5530110824. [DOI] [PubMed] [Google Scholar]
- 22.Pertoft H. Fractionation of cells and subcellular particles with Percoll. J Biochem Biophys Methods. 2000;44:1–30. doi: 10.1016/s0165-022x(00)00066-x. [DOI] [PubMed] [Google Scholar]
- 23.Holst J, et al. Generation of T-cell receptor retrogenic mice. Nat Protoc. 2006;1:406–417. doi: 10.1038/nprot.2006.61. [DOI] [PubMed] [Google Scholar]
- 24.Haviernik P, Zhang Y, Bunting KD. Retroviral transduction of murine hematopoietic stem cells. Methods Mol Biol. 2008;430:229–241. doi: 10.1007/978-1-59745-182-6_16. [DOI] [PubMed] [Google Scholar]
- 25.Pear W. Transient transfection methods for preparation of high-titer retroviral supernatants. Curr Protoc Mol Biol. 2001 doi: 10.1002/0471142727.mb0911s36. Chapter 9, Unit 9 11. [DOI] [PubMed] [Google Scholar]
- 26.Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 27.Hagani AB, Riviere I, Tan C, Krause A, Sadelain M. Activation conditions determine susceptibility of murine primary T-lymphocytes to retroviral infection. J Gene Med. 1999;1:341–351. doi: 10.1002/(SICI)1521-2254(199909/10)1:5<341::AID-JGM58>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 28.Dupl’akova N, Dobrev PI, Renak D, Honys D. Rapid separation of Arabidopsis male gametophyte developmental stages using a Percoll gradient. Nat Protoc. 2016;11:1817–1832. doi: 10.1038/nprot.2016.107. [DOI] [PubMed] [Google Scholar]
- 29.Davis HE, Rosinski M, Morgan JR, Yarmush ML. Charged polymers modulate retrovirus transduction via membrane charge neutralization and virus aggregation. Biophys J. 2004;86:1234–1242. doi: 10.1016/S0006-3495(04)74197-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weber BN, et al. A critical role for TCF-1 in T-lineage specification and differentiation. Nature. 2011;476:63–68. doi: 10.1038/nature10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karasawa S, Araki T, Nagai T, Mizuno H, Miyawaki A. Cyanemitting and orange-emitting fluorescent proteins as a donor/acceptor pair for fluorescence resonance energy transfer. Biochem J. 2004;381:307–312. doi: 10.1042/BJ20040321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Y, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 33.Izon DJ, et al. Notch1 regulates maturation of CD4+ and CD8+ thymocytes by modulating TCR signal strength. Immunity. 2001;14:253–264. doi: 10.1016/s1074-7613(01)00107-8. [DOI] [PubMed] [Google Scholar]
- 34.Heinen AP, et al. Improved method to retain cytosolic reporter protein fluorescence while staining for nuclear proteins. Cytometry A. 2014;85:621–627. doi: 10.1002/cyto.a.22451. [DOI] [PubMed] [Google Scholar]
- 35.Ahmed R, et al. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence J Exp Med. 1984;160:521–540. doi: 10.1084/jem.160.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.