

Abstract
Breast cancer is the most common type of cancer and leading cause of cancer mortality among women worldwide. However, the majority of breast malignancies are of sporadic etiology. Therefore, identifying risk-mitigating factors may significantly decrease the burden of breast cancer. Diet can have both a predisposing and protective role in breast tumorigenesis. However, establishing efficacy of dietary constituents for cancer prevention has been limited by suboptimal dietary assessment. There is a need to acquire new experimental evidence that can be used to discriminate beneficial from harmful dietary constituents. The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that is recognized as the mediator of halogenated and polycyclic aromatic hydrocarbon toxicities. Importantly, evidence points to a breast tumor-promoting role for the AhR. Preclinical and clinical studies suggest that the AhR is overexpressed in advanced and triple negative breast cancers. Several dietary constituents, namely flavonoid compounds, have demonstrated inhibitory effects on AhR activation. Given this background, in this paper we elaborate on the working hypothesis that a diet rich in AhR food agonists favors breast tumor development, whereas a diet rich in AhR food antagonists is protective. As an initial approach to developing an AhR diet hypothesis, we conducted a review of published studies reporting on the association between intake of AhR inhibitory foods and risk of breast cancer. To assist the reader with interpretation of the concepts leading to the AhR diet hypothesis, we have preceded this review with an overview of AhR biology and its role in breast cancer development.
Keywords: aryl hydrocarbon receptor, BRCA1, breast cancer, dietary bioactives, dioxin, epigenetics, flavonoids
Introduction
Breast cancer (BC) is the most common type of cancer and leading cause of cancer mortality among women worldwide [1]. Recent statistics indicate the global prevalence of BC is increasing [2]. However, only ~5 to 10 percent of BC cases result from germline DNA mutations [3,4] and the remainder of cases are considered sporadic. This suggests the continued need to develop and refine preventive approaches against sporadic BC. Evidence suggests both a predisposing and protective role of dietary factors in BC etiology [5]. The American Institute for Cancer Research (AICR) and World Cancer Research Fund (WCRF) have identified some dietary practices with limited evidence to suggest a protective effect in regard to BC development [6]. For example, higher intakes of starchy vegetables may reduce the risk of hormone receptor negative BC, foods containing carotenoids may decrease overall BC risk, and diets high in calcium and dairy may decrease premenopausal BC risk [6]. However, there is a critical need for more extensive investigations and to identify both protective and harmful foods in addition to their bioactive components. Comprehensive investigation into this topic will undoubtedly aid in the development of precise dietary protocols to mitigate BC risk.
Gene expression clustering analysis has revealed distinct molecular subtypes of BC, which are based on the tumor immunoprofile for estrogen receptor-alpha (ERα referred to throughout the paper as ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) [7]. Molecular subtype classifications provide tumor profiles that account for heterogeneity among cases in regard to tumor behavior, response to therapy, and overall prognosis [8-10]. Luminal A tumors express both ER (ER+) and PR (PR+) but lack HER2 expression (HER2-). Although they are the most prevalent form of sporadic BC (~40 percent of cases) [11], they tend to have the most favorable prognosis [9] and respond well to anti-endocrine therapy such as selective estrogen receptor modulators (SERM, i.e., Tamoxifen) and aromatase inhibitors [12,13]. Luminal B tumors (ER and/or PR+, HER2+ or -) are slightly more aggressive than luminal A [9], however they are less prevalent (~20 percent of cases) and can be targeted with anti-endocrine and anti-HER2 therapy (depending on HER2 status) [7]. Tumors that have amplified expression of HER2 but lack the hormone receptors (HR) are referred to as HER2-enriched and represent ~10 to 15 percent of cases [11]. Although these cases are more aggressive than HR+ cases, treatment with Herceptin (trastuzumab), a monoclonal antibody against HER2, has significantly improved clinical outcomes for these patients [14]. Triple negative breast cancers (TNBC; i.e., those that do not express ER, PR, or HER2) represent ~15 to 20 percent of BC cases and are associated with higher growth rates, increased susceptibility to visceral metastasis, and worse overall survival [7]. There is no targeted therapy currently available for TNBC [15]. As a result, cytotoxic chemotherapy is the only therapeutic option for these patients and current treatment methods often fail to slow tumor progression [16].
The BRCA1 gene encodes a 220 kDa nuclear phosphoprotein that functions as a tumor suppressor through maintenance of genomic stability via DNA homologous recombination [17]. Other critical functions of BRCA1 include involvement in cell cycle checkpoint control, transcriptional regulation, apoptosis, and mRNA splicing. Women who inherit germline BRCA1 mutations carry a 60 to 80 percent lifetime risk of developing BC [18]. The majority (> 80 percent) of BRCA1-related familial tumors are triple negative [19-22] and cluster with sporadic basal-like TNBC cases in gene expression analyses [23], suggesting a similar etiology between these two distinct BC types [24]. Although somatic mutations in BRCA1 are rare (1.4 to 5 percent [25]), epigenetic silencing has been proposed as an alternative mechanism for loss of BRCA1 expression in sporadic BC [26]. This appears to be of particular relevance to TNBC, as a high frequency (~20 to 65 percent, depending on study population [26-28]) of cases harbor hypermethylated BRCA1 and some studies [26] indicate BRCA1 hypermethylation is specific to the TNBC subtype.
Our group has identified and extensively characterized a key role of the activated aryl hydrocarbon receptor (AhR) in the epigenetic silencing of BRCA1 [29-36]. We recently reported significantly higher levels of BRCA1 hypermethylation in primary tumor samples from TNBC patients compared to healthy tissue and samples representative of the other BC subtypes [37]. This was observed in parallel with overexpression of AhR mRNA among TNBC samples. The AhR is a ligand-activated transcription factor of the basic helix loop helix (bHLH) superfamily [38,39] that regulates genes involved in metabolism and conjugation of drugs and other xenobiotic compounds [40-42]. Perhaps the most well-studied function of AhR is its involvement in mediating the bioactivation and toxicological effects of several xenobiotic contaminants, namely planar halogenated aromatic hydrocarbons (HAH; i.e., dioxins, dibenozofurans, biphenyls) and non-halogenated polycyclic aromatic hydrocarbons (PAH; i.e., benzo[a]pyrene, benzanthracene) [43]. However, evidence is continuing to emerge suggesting a tumor-promoting role of AhR activation in various stages of carcinogenesis [43] and across several tissue types [44]. There is also evidence to suggest involvement of the AhR in BC initiation [29-34,45]and promotion of a particularly aggressive TNBC phenotype [46-48].
Exogenous activating ligands of the AhR are ubiquitous and have been detected in foods, spices, environmental contaminations, and among commercial and consumer products [43]. Many endogenous ligands of the AhR have also been identified [49-53] and their accumulation within tumors [54,55] and the inflammatory tumor microenvironment [56] may contribute to sustained AhR activation and thus tumor promotion. The tumor-promoting role of activated AhR [57,58] and its particular relevance to BC, suggests inhibiting its activation by agonistic ligands from environmental, physiological, and pathological sources may have a preventive effect on mammary tumorigenesis. Several foods [59] and food bioactives, namely flavonoids, have demonstrated antagonistic effects on AhR activity [32-34,60-71], thus we are developing the working hypothesis that diets high in these constituents (“anti-AhR diets”) have an inverse association with BC risk. As an initial exploration of this working hypothesis we present a literature review of observational studies investigating the association between BC risk and intakes of foods reported to have AhR inhibitory properties. As a supplement, we precede this exploration of our hypothesis with a brief highlight of AhR biology and the role of AhR in BC.
Aryl Hydrocarbon Receptor Biology
According to the classical mechanism of AhR activation, under basal conditions, the AhR exists in the cytosol as part of a complex containing a heat shock protein (HSP)90 homodimer, the co-chaperone X-associated protein (XAP)2, and p23 [72-74]. Upon ligand binding, the AhR/HSP90/XAP2/p23 complex undergoes a conformational change, which exposes the AhR nuclear localization signal and leads to its nuclear translocation. The interaction of AhR with aryl hydrocarbon receptor nuclear translocator (ARNT) in the nucleus displaces the chaperone complex and leaves the AhR/ARNT heterodimer, which binds xenobiotic response elements (XRE) in regulatory regions of target genes [75]. Binding of AhR/ARNT to XRE regulates gene transcription through recruitment of coactivator proteins [76,77], bending of enhancer DNA [78], and mediating increased promoter accessibility [79].
The canonical AhR pathway described above was largely elucidated through studies investigating the capacity of high affinity agonists to induce AhR-dependent transactivation of target genes [73,77]. However, studies have demonstrated involvement of AhR in transcriptional repression and corepressor recruitment [73,80]. It is now recognized that the downstream effects of AhR activation are pleiotropic and ligand-specific [73]. This is likely owing to the promiscuity of the AhR with regard to its ligandome [81]. While a myriad of studies investigated biological outcomes associated with AhR activation by the prototypical high affinity HAH 2,3,7,8‑tetrachlorodibenzo-p‑dioxin (TCDD), many classes of AhR ligands, and even structurally similar ligands within the same class of compound, have been shown to exert differential effects on AhR activity and downstream outcomes [43,81]. These ligand-specific effects are thought to be resultant of ligand-dependent modifications of the AhR, which lead to interactions with different binding partners or nuclear factors, binding to non-canonical XRE or DNA sequences, or differential recruitment of transcriptional coactivator/corepressor proteins [81].
The AhR favors interactions with planar aromatic compounds that have approximate van der Waals dimensions of 14 X 12 X 5 Å and lack bulky substituent groups [82]. Compounds within the HAH (i.e., TCDD) and PAH (i.e. benzo[a]pyrene] classes of environmental contaminants are among the most well-studied high affinity AhR ligands. Other classes of AhR ligands (Figure 1) include naturally derived compounds such as indoles, substituted flavonoids, and other dietary compounds [82]. Several endogenous AhR ligands have also been identified [83] and include tryptophan metabolites [49-51], arachidonic acid metabolites [84], and intermediates of heme degradation [52]. There is evidence to suggest endogenous AhR ligands may accumulate within cancer cells and the tumor microenvironment [54-56]. A landmark study by Opitz and colleagues [54] demonstrated malignant progression and poor survival in glioma correlates with levels of AhR and tryptophan-2,3-dioxygenase (TDO)2, a rate-limiting enzyme in the generation of the endogenous AhR ligand kynurenine. Subsequent studies have revealed TDO2 is upregulated in TNBC [85,86] and representative cell-lines produce intracellular concentrations of kynurenine that are sufficient for AhR activation [86].
Figure 1.
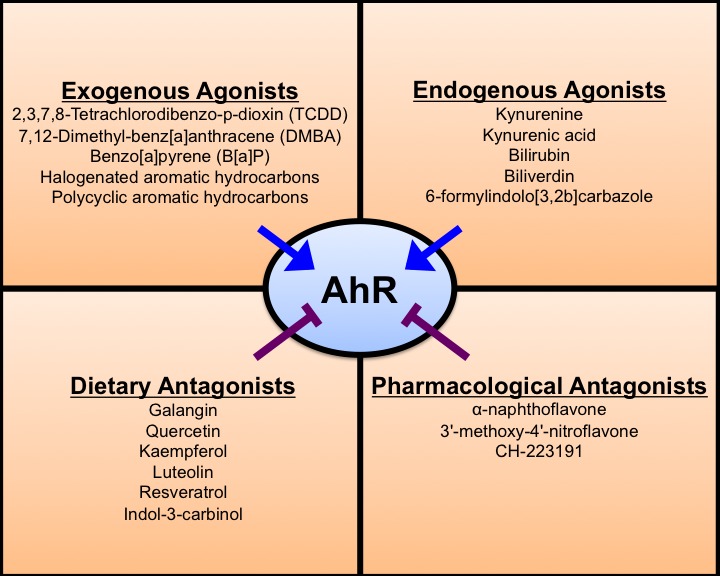
Known ligands of the AhR. The AhR ligandome consists of endogenous and exogenous compounds. Compounds with the highest affinity for the AhR include the exogenous compounds of the halogenated aromatic hydrocarbon (HAH) and polycyclic aromatic hydrocarbon (PAH) classes. Intermediates of tryptophan metabolism, such as kynurenine and kynurenic acid, and heme metabolism are well-studied endogenous agonists. Dietary compounds, namely flavonoids (i.e., galangin, quercetin, kaempferol, etc.) have demonstrated inhibitory effects on AhR activation. Several pharmacological antagonists of AhR have also been identified.
Indoleamine 2,3-dioxygenase (IDO) is another rate-limiting enzyme in kynurenine generation that has been shown to correlate with poor prognosis in BC when upregulated in tumor stroma [87]. The metabolism of tryptophan by IDO also derives the neurologically active metabolites kynurenic acid, 3-hydroxykynurenine, quinolinic acid, and serotonin, some of which have been implicated in neurological disorders [88]. For example, elevated levels of 3-hydroxykynurenine in the brain has been associated with neurodegenerative conditions such as Huntington’s [89] and Parkinson’s [90]. In humans, IDO1 is expressed in various tissues in response to cytokine signaling [91] and has been shown to be involved in the regulation of several immunological cells including T cells, dendritic cells, monocytes, macrophages, and microglia [88]. In the context of cancer, it has been suggested that IDO1 facilitates creation of an immunosuppressive microenvironment by depleting tryptophan and forming kynurenine [91], which has been shown to inhibit T cell and Natural Killer cell function and activate regulatory T cells [92]. Several factors are involved in IDO expression and activation including IFN-γ, TNF-α, IL-1β, and IL-6 [88], however IDO induction requires the AhR [93]. Given that kynurenine is an AhR agonist, this indicates a potential positive feedback system of sustained AhR activation. Interestingly, overexpression of cyclooxygenase (COX)-II, which is an AhR target gene, in BC cells promotes upregulation of IDO in co-cultured fibroblasts and accumulation of kynurenine in conditioned media [87]. This suggests another potential positive feedback loop for sustained AhR hyperactivation where intratumoral AhR induces COX-II expression, which in turn upregulates IDO in the tumor stroma, leading to kynurenine accumulation and further AhR activation.
Flavans are derivatives of benzopyran and have a 2-phenyl-3,4-dihydro-2H-chromene skeleton consisting of two phenyl rings (A and B) and a heterocyclic ring (C). Flavans are divided into flavonoids and isoflavonoids, which use 2-phenylchromen-4-one and 3-phenylchromen-4-one skeletons, respectively. These two groups are further classified into subgroups based on chemical substitutions on these skeletons and include flavones (which are based on a 2-phenylchromen-4-one skeleton), isoflavones (3-phenylchromen-4-one), flavonols (3-hydroxy-2-phenylchromen-4-one skeleton), flavanones (2,3-dihydro-2-phenylchromen-4-one), and flavan-3-ols or catechins (2-phenyl-3,4-dihydro-2H-chromen-3-ol). Several flavonoids and isoflavones fulfill the chemical characteristics of preferred AhR ligands described above and have demonstrated the capacity to inhibit AhR activity [63,67,70,94].
One study used gel mobility shift assays to assess the capacity for a myriad of flavan compounds to inhibit activation of rat hepatic AhR by 1 nM TCDD [67]. This study reported a wide range (0.14 - > 200 µM) of antagonistic IC50 concentrations across the different compounds tested and revealed flavones and flavonols had the highest affinity, followed by flavanones, isoflavones, then catechins. Flavone (0.14 µM), flavonol (0.42 µM), galangin (a flavonol 0.22 µM), and flavanone (0.65 µM) had IC50 values similar to the known AhR antagonist drug α-naphthoflavone (aka 7,8-benzoflavone, 0.39 µM). Structural considerations for these compounds include small molecular size, hydrophobicity, and planar structures. With the exception of galangin, which is found in certain species of ginger and propolis, these unsubstituted flavan derivatives are rarely found in foods. However, several ubiquitous flavones (i.e. apigenin, 3.2 µM; luteolin 6.5 µM), and flavonols (i.e., quercetin, 1.5 µM; kaempferol, 2.1 µM; myricetin, 7.6 µM) demonstrated modest antagonistic effects in this assay.
Although this study used rat AhR, a luciferase-based approach using ER+ MCF-7 human BC cells demonstrated the capacity of quercetin, kaempferol, and luteolin to inhibit AhR activation by 5 nM TCDD at both 1 and 10 µM concentrations [63]. The flavonol myricetin had antagonistic effects at 10 µM only. Although quercetin has demonstrated AhR agonistic properties in MCF-7 cells in other studies [95], it has been suggested these types of compounds act as partial agonists/antagonists depending on concentration and context [63]. Moreover, it is possible that certain compounds may act as agonists alone but competitively inhibit the capacity for high affinity compounds (i.e., TCDD) to activate AhR. In line with this speculation is a study in MCF-7 cells that demonstrated galangin treatment alone induces CYP1A1 expression but inhibits the capacity for 7,12-Dimethylbenz(a)anthracene (DMBA)- and TCDD-induced CYP1A1 expression [64]. An important consideration in extending these studies to our hypothesis of AhR antagonism in cancer is: how some AhR antagonists are ligand-specific and inhibit the activating capacity of some AhR ligands (such as TCDD) but have no impact on others [96]. This potential needs to be tested in the case of flavonoid antagonists, particularly against endogenous AhR ligands (i.e., kynurenine) that may be pathologically upregulated in cancer. However, there may not be a strict requirement for AhR activating ligands in carcinogenesis, as constitutively active AhR has been detected in breast [46] and other types of cancer [97,98] and is reported to elicit different molecular outcomes than AhR induced by exogenous compounds [99].
The Role of AhR and Bioactive Antagonists in Breast Cancer
In this section we discuss the role of AhR in BC and highlight key findings from studies investigating dietary AhR antagonists. This is not meant to represent a comprehensive evaluation of the preclinical evidence for dietary AhR antagonists and BC prevention. Rather, we selected key studies/data, including our own, that we considered relevant for the development of our working hypothesis. The AhR has an essential role in normal mammary gland development and has been suggested to drive mammary gland tumorigenesis in a similar fashion (reviewed in detail in [48]). Multiple laboratories, including our own, have detected AhR overexpression in human mammary tumors and in DMBA-induced mammary tumors in rodents [37,46,99,100]. Knock-in studies reveal high levels of AhR protein are sufficient to induce malignant transformation in normal human mammary epithelial cells (HMEC), characterized by epithelial-to-mesenchymal transition (EMT), increased growth rates, abrogated cell cycle control, and increased migration and invasive potential [45]. These effects are likely due in part to AhR-dependent activation of genes with pro-oncogenic activity, such as SLUG, a key mediator of EMT [101]. On the other hand, knockdown of constitutively active AhR induces apoptosis and inhibits growth and migration of TNBC cells [47]. These AhR-depleted TNBC cells also have reduced capacity for orthotopic xenograft growth and lung metastasis [47].
Indole metabolites formed in plants, such as indol-3-carbinol (I3C), have demonstrated potent anti-carcinogenic effects [102-105]. Interactions of these compounds with AhR [106] and the kynurenine pathway [107] are well established. In the low pH of the stomach, I3C undergoes condensation to derive 3,3’-diindolylethane (DIM) and indolo[3,2-b]carbazole (ICZ) [108]. Although DIM is the major metabolic product, the relative binding affinity of ICZ for the AhR (Kd= 1.9 x 10-10 M) [109] is much stronger than both DIM (Kd= 9 x 10-8 M) and I3C (Kd= 2.7 x 10-5 M) [110]. Treatment of MCF-7 and MDA-MB-468 BC cell lines with I3C leads to the upregulation of BRCA1 and thus potentiation of DNA damage response [111]. Dietary intake of I3C and DIM prior to DMBA treatment had an inhibitory effect on mammary tumor formation in rats [112]. However, the overall concentration of these compounds is an important consideration. In MCF-7 cells, DIM at low levels (~10-50 µM) induced nuclear localization of AhR, however higher concentrations (> 50 µM) were needed to activate transcription of the AhR target gene CYP1A1 [113]. Interestingly, DIM fed to rats (5 mg/kg every other day) inhibited DMBA-induced mammary tumor formation, and these effects were observed in the absence of changes to hepatic CYP1A1 activity. Owing to the differential outcomes exerted by different AhR ligands, it was found that DIM exerts its bioactivity in MCF-7 cells independent of cytochrome p450 (CYP) signaling but strongly inhibits ERα expression and signaling [114]. This was in contrast to TCDD, which had strong effects on CYP gene expression with weak effects on ERα signaling. Many groups have investigated the anti-BC effects of DIM. As a reference, we turn readers to an excellent comprehensive review on this subject by Thompson and colleagues [115].
In our study utilizing the DMBA-rat model we observed AhR overexpression in parallel with decreased ER and BRCA1 protein, hypermethylation of BRCA1, and upregulated cyclin D1 and CDK4 mRNA [37]. In the same publication, we reported detecting higher levels of AhR mRNA and BRCA1 methylation in clinical samples from TNBC patients compared to healthy tissue and samples representative of the other molecular subtypes. Tumors from DMBA-induced rats also displayed preferential induction of CYP1B1 over CYP1A1 [37], a hallmark of constitutive AhR activation [48]. The relative expression of CYP1A1 and CYP1B1 and ratio of CYP1A1/CYP1B1 are considered important indicators in BC [116]. This is due to the fact that CYP1B1 catalyzes metabolism of E2 to the genotoxic and carcinogenic 4OH-estradiol metabolite, whereas CYP1A1 generates 2OH-estradiol, which has putative protective roles.
A recent study [117] in 439 clinical BC samples indicated AhR mRNA overexpression is associated with ER and hormone receptor (HR) negativity and HR-/HER2- cases. This is unsurprising, given the AhR antagonizes ER signaling through several mechanisms including by 1) inducing genes involved in estradiol (E2) metabolism [118]; 2) squelching common transcriptional coactivators [119]; 3) directly binding ER and inducing polyubiquitination and proteasomal degradation [120,121] and; 4) hijacking ER away from sites of transcriptional activation [122].
Our group has described a role of the activated AhR in antagonizing E2-dependent expression of BRCA1 through epigenetic silencing. Through ER, E2 stimulates recruitment of unliganded AhR to the BRCA1 promoter, which potentiates ER-dependent activation of BRCA1 transcription [29]. In contrast, upon treatment with TCDD or benzo[a]pyrene, E2-dependent induction of BRCA1 is repressed, which is accompanied by increased recruitment at the BRCA1 promoter of the ligand-bound AhR, histone deacetylase (HDAC)1, de novo and maintenance DNA methyltransferases (DNMT3a, DNMT3b, DNMT1,), and methyl-binding domain protein (MBD)2 [29,32,33]. These responses are observed in parallel with diminution of transcriptionally permissive acetylated histone (AcH)-4 and AcH3 at lysine 9 (AcH3K9) [33], enrichment of repressive H3K9 trimethylation (H3K9me3), and DNA hypermethylation at the BRCA1 promoter [32]. In rats we showed that gestational exposure to TCDD decreases BRCA1 protein levels in adult mammary glands, and this is associated with increased occupancy of DNMT1 and DNA hypermethylation at the BRCA1 promoter, presumably in an AhR-dependent manner [34].
We have demonstrated the capacity of resveratrol, a phytoalexin found in red wine and grapes, to antagonize AhR-dependent epigenetic silencing of BRCA1 in cell lines treated with TCDD [32,33]. In our rat model we have also found that resveratrol during TCDD gestational exposure prevents loss of BRCA1 protein in adult mammary glands and decreases promoter occupancy of DNMT1 and DNA methylation [34]. In our recent studies we’ve shown epigallocatechin gallate (EGCG) and genistein prevent TCDD-induced epigenetic silencing of BRCA1 in ER+ MCF-7 cells [36]. In the same study we showed genistein and α-naphthoflavone reverse constitutive BRCA1 hypermethylation in ER- UACC-3199 cells, which overexpress AhR. These effects were observed in parallel with preferential expression of CYP1A1 compared to CYP1B1. Other groups have reported similar effects on CYP expression elicited by quercetin in non-transformed MCF10F mammary epithelial cells [123]. COX-II is a target of AhR transactivation that has been shown to enhance invasive capacity and metastatic potential of mammary tumors [124]. In MCF-7 cells, we showed conjugated linoleic acid [61] and 3,3’-diindolylmethane (DIM) [60] prevent TCDD-induced activation of COX-II expression by AhR. Other groups have shown α-naphthoflavone and resveratrol antagonize benzo[a]pyrene-dependent induction of COX-II and an invasive phenotype in MDA-MB-231 cells [62]. This evidence suggests these compounds may antagonize AhR-dependent processes involved in mammary tumorigenesis (Figure 2).
Figure 2.
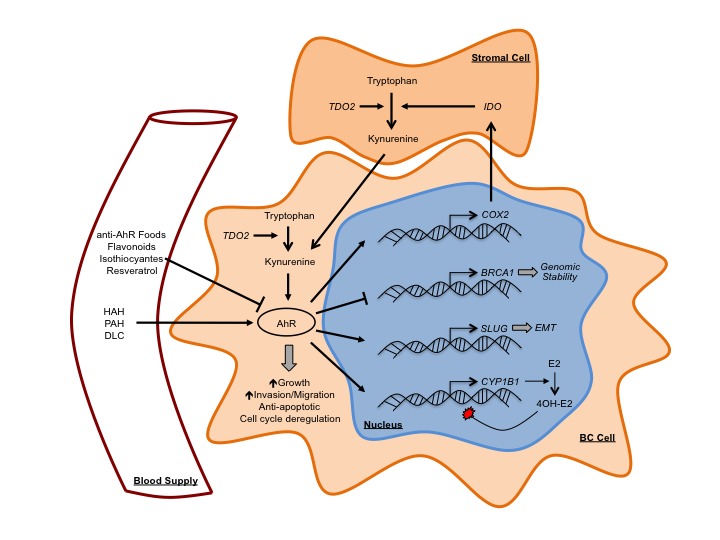
AhR in breast cancer and the role of dietary compounds. High levels of AhR induce malignant transformation in HMEC cells characterized by EMT, increased growth rates, abrogated cell cycle control, and increased migration and invasive potential. Exposure to exogenous AhR agonists such as halogenated aromatic hydrocarbons (HAH), polycyclic aromatic hydrocarbons (PAH), and other dioxin-like compounds (DLC) leads to activation of AhR in the mammary gland. The endogenous AhR agonist kynurenine is produced through metabolism of tryptophan by tryptophan-2,3-dioxygenase (TDO)2, which can be upregulated in the tumor stroma and intratumorally in TNBC; or Indoleamine 2,3-dioxygenase (IDO), which is induced in stromal cells by cancer cell upregulation of COX-II. Activated AhR induces expression of many key genes in BC including 1) COX-2, which enhances invasive capacity and metastatic potential of mammary tumors and may cause sustained hyperactivation of AhR through positive feedback by upregulating IDO in stromal cells leading to kynurenine accumulation; 2) CYP1B1, which metabolizes estradiol (E2) to the mutagenic 4OH-Estradiol metabolite; and 3) SLUG, a transcription factor largely involved in EMT. The activated AhR also mediates epigenetic silencing of BRCA1, which would otherwise function to maintain genomic stability through the stable homologous recombination pathway. Closed arrowheads represent stimulus. Blunted lines represent inhibition.
AhR Antagonism by Foods and BC Risk
An excellent starting point for considering the development of an anti-AhR Diet is a study by Amakura and colleagues [59] that utilized both receptor-binding and luciferase-based assays to determine the inhibitory effect of several fruit and vegetable extracts on AhR activation by TCDD. The receptor-binding assay (termed Ah-I) measures formation of TCDD-AhR complexes by ELISA in mammalian hepatic cytosol extracts. The luciferase-based assay (termed CALUX) utilizes a recombinant murine cell line containing the luciferase reporter gene under control of XRE and gives readout for AhR transactivation based on luciferase protein activity. In the Ah-I assay, cytosol was preincubated with freeze-dried food extracts at a concentration of 50 µM (or 50 µg/ml) for 10 min prior to treatment with TCDD (5 nM) for 2 h. For the CALUX assay, murine H1L1 hepatoma cells were cultured in 96-well plates and pre-treated with 25 µM (or 25 µg/ml) of food extracts for 10 min prior to a 20h incubation with TCDD (1nm). Green tea and its main bioactive constituent (-)-epigallocatechin gallate (EGCG) were used as positive controls for AhR inhibition based on previous studies indicating EGCG competes with TCDD for AhR binding [125]. Given our interest in whole foods as a mean to deliver AhR antagonists, we considered food extracts showing inhibitory effects similar to whole green tea in the Ah-I (~50 percent inhibition) and CALUX (~25 percent inhibition) assays. These foods included: broccoli, spinach, komatsuna (aka, Japanese mustard spinach), lettuce, shungiku (aka, Garland chrysanthemum), grapefruit, lemon, lime, and orange (Table 1). Although apple extract did not demonstrate inhibitory effects in the CALUX assay, a significant effect (~50 percent) was observed by the Ah-I detection method, so we decided to include apple intake in our review. Given the role of AhR in BC, we hypothesize that higher intakes of these foods decrease the risk of developing BC. To develop this working hypothesis, we sought to review studies investigating the effect of higher intakes of these foods on BC risk. Currently, clinical trials in humans analyzing BC development, patient outcomes, and biomarkers in response to dietary exposure to foods with AhR inhibitory properties are lacking, indicating an area open for investigation. In the following sections we review epidemiological/observational studies investigating the effect of higher intakes of these foods on BC risk. As a reference, we have provided information regarding the flavonoid content of these foods, based on a USDA Database of Flavonoid Content in selected foods [126]. This information is mentioned in the in the text for each food item where we list the major flavonoids and indicate their abundance in mg per 100g of food weight in parenthesis. To estimate the flavonoid dose from these foods in the studies that we reviewed (in mg/d), we used this database in conjunction with information from the USDA to convert volumetric measurements to weight measurements (https://ndb.nal.usda.gov/ndb/search/list). The estimates of flavonoid intake for each study are listed in Tables 2 to 5 under the column “Estimated flavonoid contribution (mg/d).” We were unable to identify studies investigating komatsuna and shungiku intake. Given the significant inhibitory effect these foods had on AhR activation this is an area open for investigation.
Table 1. Foods that inhibit AhR activation. From Amakura et al [59].
| Percent AhR Inhibition | |||
| Category | Food Item | Receptor Binding Assay (Ah-I) (against 5 nM TCDD) | Luciferase Assay (CALUX) (against 1 nM TCDD) |
| Whole Foods | |||
| Green tea | ~50% | ~25% | |
| Broccoli | ~75% | ~25% | |
| Komatsuna | ~90% | ~25% | |
| Lettuce | ~60% | ~25% | |
| Shungiku | ~55% | ~25% | |
| Spinach | ~75% | ~40% | |
| Grapefruit | ~70% | ~25% | |
| Lemon | ~60% | ~25% | |
| Lime | ~50% | ~50% | |
| Orange | ~80% | ~30% | |
| Apple | ~50% | n/a | |
| Asparagus | n/a | ~20% | |
| Carrot | ~15% | ~12.5% | |
| Cucumber | ~37.5% | n/a | |
| Eggplant | ~30% | n/a | |
| Green pepper | ~25% | n/a | |
| Pumpkin | ~25% | n/a | |
| Radish | ~20% | n/a | |
| American cherry | n/a | n/a | |
| Avocado | ~15% | n/a | |
| Kiwi fruit | ~30% | n/a | |
| Loquat | ~15% | n/a | |
| Papaya | ~37.5% | n/a | |
| Philippine mango | n/a | n/a | |
| Strawberry | ~37.5% | n/a | |
| Herbals | |||
| Basil | ~25% | ~10% | |
| Chinese sweet tea | ~50% | ~25% | |
| Cinnamon | ~25% | ~5% | |
| Clove | ~75% | ~25% | |
| Coffee | ~70% | ~5% | |
| Guava leaf | ~75% | ~37.5% | |
| Lavender | ~55% | ~20% | |
| Oolong tea | ~60% | ~25% | |
| Oregano | ~37.5% | n/a | |
| Peppermint | ~80% | ~25% | |
| Rosemary | ~60% | n/a | |
| Sage | ~85% | ~85% | |
| Tea | ~37.5% | ~25% | |
Broccoli
Broccoli is a cruciferous vegetable from the Brassica oleracea species. The most abundant flavanoids in broccoli include the flavonols kaempferol (~7.84 mg/100g) and quercetin (~3.26 mg/100g) [126]. It also contains lesser amounts of the flavone luteolin (0.8 mg/100g) and the flavonol myricetin (0.06 mg/100g). Broccoli, like most cruciferi, is also an abundant source of isothiocyanates [127], which are also inhibitory against AhR activation [65,66]. The relationship between cruciferous vegetable intake and cancer risk has been extensively investigated and meta-analyses have demonstrated that higher intakes of cruciferous vegetables may decrease the risk of overall BC (i.e., without regard for specific subtypes) by ~15 percent (RR= 0.85, 95% CI= 0.77-0.94) [128]. On the other hand, the association between specific cruciferous vegetables, such as broccoli, and BC risk has not been studied as intensely. We were unable to identify meta-analyses investigating broccoli intake and there was discrepancy among the observational studies identified in our searches (Table 2).
Table 2. Summary of studies investigating association between broccoli intake and BC risk.
| Author, year | Design | Cohort/Location | Menopausal status | Adjusted intake (g/d) | Estimated flavonoid contribution (mg/d) | Outcome | OR/HR/RR (95%CI) |
| Adebamowo, 2005 [129] | Cohort | NHS II | Pre | 29.3 – 35.2 | Luteolin, 0.23 – 0.28 | No association | RR= 0.99 (0.59-1.65) |
| Kaempferol, 2.30 – 2.76 | |||||||
| Myricetin, 0.018 – 0.021 | |||||||
| Quercetin, 0.95 – 1.14 | |||||||
| Ambrosone, 2004 [132] | Case-control | North East U.S.A. | Pre | ≥20.8 | Luteolin, ≥0.16 | Decreased risk | OR= 0.60 (0.40-1.00) |
| Kaempferol, ≥1.63 | |||||||
| Myricetin, ≥0.012 | |||||||
| Quercetin, ≥0.67 | |||||||
| Post | ≥26.7 | Luteolin, ≥0.21 | No association | OR= 1.00 (0.70-1.40) | |||
| Kaempferol, ≥2.09 | |||||||
| Myricetin, ≥0.016 | |||||||
| Quercetin, ≥0.87 | |||||||
| Boggs, 2010 [133] | Cohort | Black Women’s Health Study | All | 17.6 | Luteolin, 0.09 | No association | RR= 0.85 (0.67-1.09) |
| Pre | Kaempferol, 0.92 | No association | RR= 0.74 (0.51-1.07) | ||||
| Post | Myricetin, 0.007 | No association | RR= 0.91 (0.62-1.34) | ||||
| Quercetin, 0.38 | |||||||
| Farvid, 2016 [134] | Cohort | NHS | Pre | 11.7 | Luteolin, 0.09 | Adolescent intake; No association | HR= 1.06 (0.95-1.18) |
| Kaempferol, 0.92 | |||||||
| Myricetin, 0.007 | |||||||
| Quercetin, 0.38 | Early adult intake; No association | HR= 0.97 (0.92-1.02) | |||||
| Jung, 2013 [135] | Pooled cohort | Pooled analysis of 20 cohort studies | All | 78 | Luteolin, 0.62 | ER-; No association | RR= 0.93 (0.82-1.06) |
| Kaempferol, 6.11 | |||||||
| Myricetin, 0.047 | |||||||
| Quercetin, 2.54 | ER+; No association | RR= 1.06 (1.00-1.12) |
A study using data derived from the Nurses Health Study II (NHS II) cohort reported no effect of broccoli consumption (~29.3-35.2 g/d adjusted) on BC risk (RR= 0.99, 95% CI= 0.59-1.65) [129]. However, this study only looked at BC overall and did not stratify by menopausal status. Given that BC risk is elevated in postmenopausal women [130] and there are distinct risk factors for the different subtypes of BC [131], it is possible that stratifying by these factors modulates the observed effect.
In line with this hypothesis, a case-control study investigating broccoli intake in women from North Eastern US reported higher intakes of broccoli (≥ 20.8 g/d) to be inversely associated with BC risk in premenopausal women (OR= 0.60, 95% CI= 0.40-1.00) [132]. However, there was no effect of higher broccoli intakes (≥ 26.7 g/d) among postmenopausal women (OR= 1.0, 95% CI= 0.70-1.40). On the other hand, prospective analysis using data from the Black Women’s Health Study (BWHS) cohort reported no effect of broccoli consumption (17.6 g/d) on BC risk among all (RR= 0.85, 95% CI= 0.67-1.09) premenopausal (RR= 0.74, 95% CI= 0.51-1.07), and postmenopausal (RR= 0.91, 95%CI= 0.62-1.34) women [133]. A potential explanation for the lack of observed effect in the BWHS is that the amount of broccoli consumed by the group with the highest intake was inadequate to elicit an effect. For example, in the case-control study from the US [132] intakes of at least 20.8 g/d were needed to observe a significant inverse association.
To determine if the period of exposure mediates the effect of broccoli consumption on risk in premenopausal women, we looked at a study by Farvid and colleagues [134], which used NHS II data to analyze the association between BC risk and the intake of various fruits and vegetables during adolescence (high school) and early adulthood. Although authors reported no effect of broccoli consumption during either period, the amount considered (11.7 g/d) was again likely inadequate to elicit an effect.
Given the association between AhR and HR-/TNBC, we turned to an analysis of 20 cohort studies that sought to investigate the effect of fruit and vegetable intake on BC risk stratified by ER status [135]. When determining the relative risk for consumption of 78 g/d, authors found no effect on either ER- or ER+ BC. An overlooked factor in all of these studies investigating broccoli intake, which likely contributes significant confounding effects, is the lack of consideration for whether broccoli was consumed as raw or cooked by study participants. Given that various cooking methods can drastically impact bioactive contents of foods in different ways [136], these are variables that need to be considered for analysis and when reporting data. Additionally, there are other factors that may influence the bioavailability of certain compounds from food. For example, Liu and colleagues showed that the gut microbiome played an integral role in hydrolysis of glucoraphanin to the bioactive isothiocyanate sulforaphane [137]. These authors also demonstrated that frequent broccoli consumption was required to ensure sustained microbial hydrolysis of glucoraphanin to sulforaphane. S-(−)7-hydroxy-3-(4-hydroxyphenyl)-chroman, known more commonly as S-(−)equol, is a bioactive formed through microbial metabolism of the soy isoflavone daidzein that has demonstrated some anti-tumorigenic properties including selective affinity for ERβ [138,139], anti-androgenic activity [140], and demethylation of the BRCA1/2 promoter [141]. A study [139] found the capacity to detect urinary S-(−)equol in human infants was related to early dietary exposure to soy or cow milk formula and that breast-fed infants had inferior S-(−)equol production. These data lend credence to the idea that chronic exposure to certain foods (perhaps over the lifetime) may be needed in order to derive maximal benefits from the bioactive constituents.
Lettuce
Lettuce belongs to the asteraceae family of flowering plants. The flavonoid content of lettuce is highly dependent on the variety. The main flavonoid found in all varieties of lettuce is the flavonol quercetin (1.42-7.61 mg/100g) [126]. However, the other flavonols such as kaempferol (0.01-0.15 mg/100g) and myricetin (0-0.07 mg/100g) and the flavones apigenin (0-0.13 mg/100g) and luteolin (0-0.95 mg/100g) are also found. Table 3 summarizes data from studies investigating the association between lettuce intake and BC risk. A case-control study conducted in Athens, Greece reported lettuce consumption (12 g/d) to be associated with a decreased risk of BC (OR= 0.75, 95%CI= 0.59-0.95) among all women without regard for molecular subtype or menopausal status [142]. On the other hand, a case-control study out of Nagoya, Japan failed to identify the same observation in their subjects with higher intakes of lettuce (≥ 14.4 g/d), regardless of whether or not family history was considered as a variable [143]. A likely source of disagreement between these two studies is the limited adjustment for potential confounding variables in the multivariate analysis from the Japanese study. For example, risk factors that were controlled for in the Greek study [142] but not in the Japanese study [143] included: age of menarche, menopausal status, parity, duration of lactation, ponderal index, and education status among others.
Table 3. Summary of studies investigating association between lettuce and spinach intake and BC risk.
| Food item | Author, year | Design | Cohort/Location | Menopausal status | Adjusted intake (g/d) | Estimated flavonoid contribution (mg/d) | Outcome | OR/HR/RR (95%CI) |
| Lettucea | ||||||||
| Farvid, 2016 [134] | Cohort | NHS | Pre | 9.6 | Apigenin, 0 – 0.012 | Adolescent intake; No association | OR= 0.99 (0.95-1.04) | |
| Luteolin, 0 – 0.091 | ||||||||
| Kaempferol, 0.001 – 0.014 | ||||||||
| Myricetin, 0 – 0.006 | Early adult intake; No association | OR= 1.00 (0.98-1.01) | ||||||
| Quercetin, 0.14 – 0.73 | ||||||||
| Jung, 2013 [135] | Pooled cohort | Pooled analysis of 20 cohort studies | All | 56 | Apigenin, 0 – 0.073 | ER-; Decreased risk | RR= 0.91 (0.84-0.98) | |
| Luteolin, 0 – 0.532 | ||||||||
| Kaempferol, 0.006 – 0.084 | ER+; No association | RR= 1.01 (0.97-1.05) | ||||||
| Myricetin, 0 – 0.039 | ||||||||
| Quercetin, 0.80 – 4.26 | ||||||||
| Katsouyanni, 1986 [142] | Case-control | Athens, Greece | All | 12 | Apigenin, 0 – 0.016 | Decreased risk | OR= 0.75 (0.59-0.95) | |
| Luteolin, 0 – 0.114 | ||||||||
| Kaempferol, 0.001 – 0.018 | ||||||||
| Myricetin, 0 – 0.008 | ||||||||
| Quercetin, 0.17 – 0.91 | ||||||||
| Huang, 2004 [143] | Case-control | Nagoya, Japan | All | ≥14.4 | Apigenin, 0 – 0.018 | Fam Hx-; No Association | OR= 0.98 (0.88-1.09) | |
| Luteolin, 0 – 0.137 | ||||||||
| Kaempferol, 0.001 – 0.022 | ||||||||
| Myricetin, 0 – 0.01 | Fam Hx+; No association | OR= 0.98 (0.62-1.53) | ||||||
| Quercetin, 0.20 – 1.10 | ||||||||
| Spinach | ||||||||
| Boggs, 2010 [133] | Cohort | Black Women’s Health Study | All | 12 | Luteolin, 0.089 | No association | RR= 0.79 90.62-1.00) | |
| Pre | Kaempferol, 0.766 | No association | RR= 0.73 (0.51-1.05) | |||||
| Post | Myricetin, 0.042 | No association | RR= 0.84 (0.59-1.19) | |||||
| Quercetin, 0.476 | ||||||||
| Farvid, 2016 [134] | Cohort | NHS | Pre | 8 | Luteolin, 0.059 | Adolescent intake; No association | HR= 1.09 (0.98-1.21) | |
| Kaempferol, 0.51 | ||||||||
| Myricetin, 0.028 | ||||||||
| Quercetin, 0.318 | Early adult intake; No association | HR= 0.99 (0.92-1.06) | ||||||
| Jung, 2013 [135] | Pooled cohort | Pooled analysis of 20 cohort studies | All | 15 | Luteolin, 0.111 | ER-; No association | RR= 0.91 (0.80-1.04) | |
| Kaempferol, 0.957 | ||||||||
| Myricetin, 0.052 | ||||||||
| Quercetin, 0.596 | ER+; No association | RR= 1.00 (0.90-1.11) | ||||||
| Longnecker, 1997 [144] | Case-control | North East U.S.A. | All | ≥8.75 | Luteolin, ≥0.065 | Decreased risk | RR= 0.64 (0.43-0.94) | |
| Kaempferol, ≥0.56 | ||||||||
| Myricetin, ≥0.031 | ||||||||
| Quercetin, ≥0.347 |
aRanges for lettuce flavonoid load due to differences in content between butterhead, romaine, green leaf, red leaf, and iceberg varieties.
Neither of the aforementioned studies on lettuce consumption considered whether stratification by menopausal status or BC molecular subtype modulated the observed effect. Prospective analysis of NHS II cohort data indicated that lettuce consumption (9.6 g/d) during both adolescence (OR= 0.99, 95%CI= 0.95-1.04) and early adulthood (OR= 0.98, 95%CI= 0.62-1.53) had no effect on premenopausal BC risk [134]. However, the amount of lettuce intake used to calculate OR in this study seems low compared to the amount needed to elicit an effect in the case-control study from Athens, Greece [142]. In line with the hypothesis that there is a dose-dependency is the result observed by Jung and colleagues [135] that intake of 56 g/d was associated with a decreased risk of ER- BC (RR= 0.91, 95%CI= 0.84-0.98). Interestingly, authors found no association with ER+ BC, lending credence to the potential of a molecular subtype dependency. There is a need for more investigation here, given the limited number of studies investigating the effect of lettuce consumption on BC risk in general and the lack of studies attempting to parse out mediating factors (i.e., menopausal status and molecular subtype). In addition, there are several varieties of lettuce, which vary substantially in their flavonoid content. Given that no studies take into consideration the type of lettuce consumed, this variable likely confounds the available data and needs to be explored.
Spinach
The main flavonoids found in spinach include the flavonols kaempferol (6.38 mg/100g) and quercetin (3.97 mg/100g) with minor amounts of myricetin (0.35 mg/100g) and the flavone luteolin (0.74 mg/100g) [126]. A summary of data from studies investigating the association between spinach intake and BC is in Table 3. For spinach, we encountered the same issue we did with broccoli regarding whether or not the authors considered cooked vs. raw in their analysis. There was only one study that analyzed the effects of cooked and raw spinach separately, the results from which demonstrate the necessity to consider such variables [144]. In a case-control analysis conducted across several U.S. states, higher intake of raw spinach (≥ 8.75 g/d) was reported as inversely associated with overall BC risk (RR= 0.64, 95% CI= 0.43-0.94). However, higher consumption of cooked spinach (≥ 26.25 g/d) had no statistically significant association (RR= 0.88, 95% CI= 0.62-1.42) [144]. This clearly demonstrates a difference in effect between raw and cooked spinach and perhaps why studies that do not differentiate between the two have failed to identify significant associations. For example, there was no association between spinach consumption (12 g/d) and overall BC among all (RR= 0.79, 95% CI= 0.62-1.00), premenopausal (RR= 0.73, 95% CI= 0.51-1.05) or postmenopausal (RR= 0.84, 95% CI= 0.59-1.19) women in a prospective analysis of the BWHS [133]. In addition, Farvid and colleagues [134] reported no effect on premenopausal BC risk associated with either adolescent (HR= 1.09, 95% CI= 0.98-1.21) or early adulthood (HR= 0.99, 95% CI= 0.92-1.06) spinach consumption (8 g/d). Finally, in analysis of data from 20 prospective cohort studies, authors reported no effect of spinach consumption (15 g/d) on either ER- (RR= 0.91, 95% CI= 0.80-1.04) or ER+ (RR= 1.00, 95% CI= 0.90-1.11) BC [135].
Citrus Fruits: Grapefruit, Lemons, Limes, Oranges
The category of citrus fruits demonstrates the necessity to perform more detailed studies that analyze items separately on the basis of individual food rather than food group. Several studies have investigated the effects of general citrus fruit consumption on BC risk [145-149]. However, although we found studies that investigated the individual effects of grapefruit (n=5) [133,135,150-152] and orange (n=3) [133-135] consumption on BC risk, we were unable to identify any looking at lime or lemon intake. This is unfortunate, given that both of these citrus fruits demonstrated significant and consistent inhibitory effects against AhR activation in vitro [59]. Thus, this clearly represents an area of nutritional epidemiology that is largely open for investigation.
Meta-analyses indicate higher consumption of citrus fruit is inversely associated with BC risk [146]. Interestingly, analysis of data from the Shanghai Breast Cancer Study suggests this effect may be specific to BC cases that are either completely HR+ (i.e., both ER+ and PR+; OR= 0.78, 95% CI= 0.63–0.96) or completely HR- (i.e., both ER- and PR-; OR= 0.71, 95% CI= 0.54–0.93) [145]. Despite meta-analytical evidence regarding the protective effect of citrus fruit consumption, there is discrepancy in the observational literature. Some reports suggest there is an inverse association [145,148], while others report no association [147,149]. A potential explanation for this divergence in evidence is the likelihood that different individual fruits represent the majority of fruits consumed under the citrus fruit category among the different studies. This is conceivable given the differential effects observed for grapefruits and oranges.
The most abundant flavonoid in grapefruit is the flavanone naringenin (32.64 mg/100g), with trace amounts of hesperetin (0.35 mg/100g), the flavone luteolin (0.6 md/100g), and the flavonols kaempferol (0.01 mg/100g), myricetin (0.01 mg/100g), and quercetin (0.33 mg/100g) [126]. Table 4 provides a summary of data from studies investigating the association between citrus fruits and BC risk. None of the studies we reviewed indicated an inverse association between grapefruit consumption and BC risk. On the other hand, analysis of data from the Multi Ethnic Cohort (MEC) study indicates a potential adverse effect associated with higher consumption (≥ 60 g/d) of grapefruit (RR= 1.30, 95% CI= 1.06-1.58) in postmenopausal women [150]. Authors speculated this could be due changes in estrogen metabolism since elevation of serum estrogens is a significant risk factor for postmenopausal BC [153] and grapefruit is an inhibitor of cytochrome P450 3A4 (CYP3A4) [154], which metabolizes E2 to 2OH-estradiol [155]. However, although concomitant intake of grapefruit during oral administration of exogenous estradiols leads to their elevation in serum [156,157], there does not appear to be an elevating effect on endogenous E2 or estrone levels [158].
Table 4. Summary of studies investigating association between citrus fruit intake and BC risk.
| Citrus Fruit | Author, year | Design | Cohort/Location | Menopausal status | Adjusted intake (g/d)a | Estimated flavonoid contribution (mg/d) | Outcome | OR/HR/RR (95%CI) |
| Grapefruit | ||||||||
| Boggs, 2010 [133] | Cohort | Black Women’s Health Study | All | 20-32 | Hesperetin, 0.07 – 0.112 | No association | RR= 0.94 (0.78-1.17) | |
| Pre | Naringenin, 6.53 – 10.44 | No association | RR= 1.17 (0.84-1.61) | |||||
| Post | Luteolin, 0.12 – 0.19 | No association | RR= 0.81 (0.61-1.09) | |||||
| Kaempferol, 0.002 – 0.003 | ||||||||
| Myricetin, 0.002 – 0.003 | ||||||||
| Quercetin, 0.066 – 0.106 | ||||||||
| Jung, 2013 [135] | Pooled cohort | Pooled analysis of 20 cohort studies | All | 120 | Hesperetin, 0.42 | ER-; Noassociation | RR= 1.06 (0.98-1.16) | |
| Naringenin, 39.17 | ||||||||
| Luteolin, 0.72 | ||||||||
| Kaempferol, 0.012 | ER+; No association | RR= 1.00 (0.92-1.09) | ||||||
| Myricetin, 0.012 | ||||||||
| Quercetin, 0.396 | ||||||||
| Monroe, 2007 [150] | Cohort | MEC | Post | ≥60 | Hesperetin, 0.21 | Increased risk | RR= 1.30 (1.06-1.58) | |
| Naringenin, 19.58 | ||||||||
| Luteolin, 0.36 | ||||||||
| Kaempferol, 0.006 | ||||||||
| Myricetin, 0.006 | ||||||||
| Quercetin, 0.198 | ||||||||
| Kim, 2008 [151] | Cohort | NHS | All | 25-40 | Hesperetin, 0.09 – 0.14 | No association | RR= 1.00 (0.86-1.15) | |
| Post | Naringenin, 8.16 – 13.06 | No association | RR= 0.97 (0.83-1.14) | |||||
| Luteolin, 0.15 – 0.24 | ||||||||
| Kaempferol, 0.003 – 0.004 | ||||||||
| Myricetin, 0.003 – 0.004 | ||||||||
| Quercetin, 0.083 – 0.132 | ||||||||
| Spencer, 2009 [152] | Cohort | EPIC | All | ≥60 | Hesperetin, 0.21 | No association | HR= 0.93 (0.77-1.13) | |
| Pre | Naringenin, 19.58 | No association | HR= 0.97 (0.75-1.27) | |||||
| Post | Luteolin, 0.36 | No association | HR= 1.19 (0.81-1.75) | |||||
| Kaempferol, 0.006 | ||||||||
| Myricetin, 0.006 | ||||||||
| Quercetin, 0.198 | ||||||||
| Oranges | ||||||||
| Boggs, 2010 [133] | Cohort | Black Women’s Health Study | All | 38.4-73.6 | Hesperetin, 8.40 – 16.10 | No association | RR= 1.03 (0.87-1.23) | |
| Pre | >Naringenin, 2.73 – 5.23 | No association | RR= 1.09 (0.83-1.42) | |||||
| Post | Luteolin, 0.23 – 0.52 | No association | RR= 1.01 (0.79-1.31) | |||||
| Kaempferol, 0.004 – 0.007 | ||||||||
| Myricetin, 0.004 – 0.007 | ||||||||
| Quercetin, 0.077 – 0.147 | ||||||||
| Farvid, 2016 [134] | Cohort | NHS | Pre | 25.6-49.1 | Hesperetin, 5.60 – 10.74 | Adolescent intake; No association | HR= 1.01 (0.94-1.07) | |
| Naringenin, 1.82 – 3.49 | ||||||||
| Luteolin, 0.180 – 0.344 | ||||||||
| Kaempferol, 0.003 – 0.005 | ||||||||
| Myricetin, 0.003 – 0.005 | Early adult intake; Decreased risk | HR= 0.93 (0.88-0.99) | ||||||
| Quercetin, 0.051 – 0.098 | ||||||||
| Jung, 2013 [135] | Pooled cohort | Pooled analysis of 20 cohort studies | All | 131 | Hesperetin, 28.65 | ER-; No association | RR= 0.93 (0.83-1.04) | |
| Hesperetin, 28.65 | ||||||||
| Naringenin, 9.3 | ||||||||
| Luteolin, 0.917 | ||||||||
| Kaempferol, 0.013 | ER+; No association | RR= 1.01 (0.95-1.05) | ||||||
| Myricetin, 0.013 | ||||||||
| Quercetin, 0.262 |
aRange b/c FFQ failed to differentiate Large (160g) vs. Medium (128g) and Small (100g) grapefruit.
Despite the potential adverse effect of grapefruit intake on postmenopausal BC risk identified in the MEC study [150], a follow up prospective analysis of NHS data failed to confirm this observation in postmenopausal women (RR= 1.00, 95% CI= 0.86-1.15) with the highest intakes of grapefruit (25-40 g/d) [151]. Authors additionally reported no effect of grapefruit consumption on BC risk among all women (RR= 0.97, 95% CI= 0.83-1.14) regardless of menopausal status [151]. Similarly, another study reported no effect of grapefruit consumption (20-32 g/d) on BC risk among all (RR= 0.94, 95% CI= 0.78-1.17), premenopausal (RR= 1.17, 95% CI= 0.84-1.61) and postmenopausal (RR= 0.81, 95% CI= 0.61-1.09) women among the BWHS [133].
A potential explanation for the disparity observed between these two studies and the MEC study, which reported an elevated risk, is the difference in the amount of grapefruit consumed by the groups with the highest intakes in these studies. It is likely that consumption among these groups was inadequate to reach a potential threshold that was achieved (≥ 60 g/d) in the MEC study [150]. However, this explanation is challenged by a study reporting no effect of grapefruit consumption (≥ 60 g/d) on BC risk among all (HR= 0.93, 95% CI= 0.77-1.13), premenopausal (HR= 0.97, 95% CI= 0.75-1.27), and postmenopausal (HR= 1.19, 95% CI= 0.81-1.75) women from the EPIC cohort [152]. This could potentially be the result of population differences between the MEC and EPIC cohorts.
While the cumulative evidence suggests grapefruit intake is not protective against BC and may actually increase the risk, the available evidence regarding the effect of oranges is limited. Oranges have a very similar flavonoid content to grapefruits, however the major compound in oranges is the flavanone hesperetin (21.87 mg/100g) with smaller amounts of naringenin (7.1 mg/100g), luteolin (0.7 mg/100g), kaempferol (0.01 mg/100g), myricetin (0.01 mg/100g), and quercetin (0.2 mg/100g) [126]. Analysis of data from the Black Women’s Health study indicates higher consumption of oranges (~38.4-73.6 g/d, depending on the size of fruit used for serving reference) has no effect on BC risk among all (RR= 1.03, 95% CI= 0.87-1.23), premenopausal (RR= 1.09, 95% CI= 0.83-1.42), and postmenopausal (RR= 1.01, 95% CI= 0.79-1.31) women [133]. On the contrary, Farvid and colleagues [134] report that early adulthood intake of oranges (~25.6-49.1 g/d) is inversely associated with premenopausal BC risk (HR= 0.93, 95% CI= 0.88-0.99) in their analysis of NHS II data. It is difficult to compare conclusions from these two studies because the exact amount of orange intake in these subjects is unknown. In the BWHS [133] subjects with higher intakes of oranges were reported to consume ≥ three servings/week, whereas calculations in the NHS II study [134] were based on two servings/week intake. In both studies a serving was considered one fruit, however the size was not specified and could be interpreted to be large (~184g), medium (~131g), or small (~96g). Hence, why we provided ranges for the estimated daily consumption above. This suggests that this lack of specification may be driving confounding among the reported data. Despite the discrepancy between the BWHS [133] and the NHS II [134], pooled prospective analysis of several studies indicates no effect of the intake of oranges (131 g/d) on either ER- (RR= 0.93, 95% CI= 0.83-1.04) or ER+ (RR= 1.01, 95% CI= 0.95-1.05) BC among all women [135].
It is clear that more investigations into the effect of specific citrus fruits on BC risk are needed. Analysis of citrus fruit consumption in general suggests a protective effect [145,149], however in the case of both grapefruit and orange consumption, the evidence is less convincing. Furthermore, to our knowledge, there are no studies investigating the individual effects of lemon and lime intake on BC risk. Given that these two citrus fruits are among the most potent and consistent inhibitors of AhR activation in vitro [59], these studies are especially warranted to determine their contribution to the protective effect associated with citrus fruit intake.
Apple
Although apple extract did not produce an inhibitory effect on AhR activation in the CALUX assay, it had a significant effect in the Ah-I assay (~50 percent inhibition) and is a significant source of quercetin (up to 4 mg/100g) [126], which is one of the more potent flavonoid AhR antagonists (IC50= ~1.5 µM, against 1 nm TCDD activation) [67]. We encountered two potential sources of confounding when analyzing studies that investigated apple intake and BC risk. The first issue was similar to that observed with lettuce in that the type of apples consumed were not specified (i.e., Fuji, Gala, Golden Delicious, Granny Smith, etc.), which is significant given that the flavonoid content can vary depending on the variety. For example, Fuji apples have ~2.35 mg/100g of quercetin, whereas Red Delicious apples have ~3.86 mg/100g [126]. The second issue was similar to that observed with the studies on oranges, which did not specify the size of the fruit used as reference for serving size. Thus, we were unable to determine if these studies calculated intake of large (~223g), medium (~182g), or small (~149g) apples. As a result, for studies that did not specify, we calculated estimated daily intakes as ranges.
Table 5 summarizes data from observational studies investigating the effect of apple intake on BC risk. Meta analyses suggest higher intake of apples is inversely associated with BC risk (OR=0.79, 95% CI= 0.73-0.87, P < 0.001) [159]. However, a case-control study in Shanghai, China found no effect of apple consumption (57 g/d) on BC risk among all women (OR= 0.86, 95% CI= 0.66-1.11) [148]. Similar intake levels (59.6-89.2 g/d) were shown to have no effect on risk among all (RR= 1.02, 95% CI= 0.83-1.25), premenopausal (RR= 1.04, 95% CI= 0.76-1.41) and postmenopausal (RR= 0.99, 95% CI= 0.72-1.36) women in the BWHS [133]. In contrast, an Italian case-control study reported a protective effect (OR= 0.82, 95% CI= 0.73-0.92) associated with apple intake (149-223 g/d) among all women [160]. A likely explanation for this difference is that the estimated daily intake of subjects in this study was much higher than subjects in the Chinese study, although prospective analysis of NHS II data suggests this level of intake (149-223 g/d) does not have an effect in premenopausal women (RR= 1.16, 95% CI= 0.77-1.76) [129]. Interestingly, intake of apples during adolescence (39.7-59.5 g/d) appears to be slightly protective against premenopausal BC (RR= 0.93, 95% CI= 0.87-0.99) [134]. Finally, the pooled analysis by Jung and colleagues [135] suggests this protection may be specific to ER- forms of BC (RR= 0.92, 95% CI= 0.85-0.99), as no effect of apple intake (138 g/d) on ER+ BC risk was observed (RR= 0.98, 95% CI= 0.94-1.02) [135].
Table 5. Summary of studies investigating association between apple intake and BC risk.
| Author, year | Design | Cohort/Location | Menopausal status | Adjusted intake (g/d) | Estimated flavonoid contribution (mg/d)a | Outcome | OR/HR/RR (95%CI) |
| Adebamowo, 2005 [129] | Cohort | NHS II | Pre | 149-223 | E3G, 0 – 0.022 | No association | RR= 1.16 (0.77-1.76) |
| EGCG, 0.17 – 4.30 | |||||||
| Luteolin, 0 – 0.022 | |||||||
| Myricetin, 0 – 0.022 | |||||||
| Quercetin, 3.5 – 8.6 | |||||||
| Boggs, 2010 [133] | Cohort | Black Women’s Health Study | All | 59.6-89.2 | E3G, 0 – 0.009 | No association | RR= 1.02 (0.83-1.25) |
| Pre | EGCG, 0.07 – 1.72 | No association | RR= 1.04 (0.76-1.41) | ||||
| Post | Luteolin, 0 – 0.009 | No association | RR= 0.99 (0.72-1.36) | ||||
| Myricetin, 0 – 0.009 | |||||||
| Quercetin, 1.40 – 3.44 | |||||||
| Farvid, 2016 [134] | Cohort | NHS | Pre | 39.7-59.5 | E3G, 0 – 0.006 | Adolescent intake; Decreased risk | RR= 0.93 (0.87-0.99) |
| EGCG, 0.04 – 1.15 | |||||||
| Luteolin, 0 – 0.006 | |||||||
| Myricetin, 0 – 0.006 | Early adult intake; No association | RR= 0.99 (0.87-0.99) | |||||
| Quercetin, 0.932 – 2.30 | |||||||
| Jung, 2013 [135] | Pooled cohort | Pooled analysis of 20 cohort studies | All | 138 | E3G, 0 – 0.014 | ER-; Decreased risk | RR= 0.92 (0.85-0.99) |
| EGCG, 0.15 – 2.66 | |||||||
| Luteolin, 0 – 0.014 | ER+; No association | RR= 0.98 (0.94-1.02) | |||||
| Myricetin, 0 – 0.014 | |||||||
| Quercetin, 3.24 – 5.33 | |||||||
| Malin, 2003 [148] | Case-control | Shanghai, China | All | 57 | E3G, 0 – 0.006 | No association | OR= 0.86 (0.66-1.11) |
| EGCG, 0.63 – 1.10 | |||||||
| Luteolin, 0 – 0.006 | |||||||
| Myricetin, 0 – 0.006 | |||||||
| Quercetin, 1.34 – 2.20 | |||||||
| Gallus, 2005 [160] | Case-control | Italy | All | 149-223 | E3G, 0 – 0.022 | Decreased risk | OR= 0.82 (0.73-0.92) |
| EGCG, 0.17 – 4.30 | |||||||
| Luteolin, 0 – 0.022 | |||||||
| Myricetin, 0 – 0.022 | |||||||
| Quercetin, 3.5 – 8.6 |
aRanges b/c FFQ failed to differentiate Large (223g) vs. Medium (182g) and Small (149g) apples and due to differences in content between Fuji, Gala, Golden Delicious, Granny Smith, Red Delicious varieties; E3G, (-)-Epicatechin 3-gallate; EGCG, (-)-Epigallocatechin 3-gallate.
Blueberry
Although blueberries were not tested against AhR activation [59], it is likely they exert an inhibitory effect, due to their significant flavonoid load. Thus, it is fair to assume that higher intakes of blueberries are inversely associated with BC risk. Analysis of NHS II study revealed that blueberry intake (two to four serv/wk) had no effect on BC risk in premenopausal women [129]. In contrast, Fung and colleagues [161] demonstrated that higher intakes of blueberries (> one serving/week) conferred ~30 percent reduction in ER- BC risk among postmenopausal women (RR= 0.69, 95% CI= 0.50-0.95). Based on this limited data there may be a protective effect of blueberry intake specifically against postmenopausal BC, however given that studies investigating this association are limited further investigation is required to parse out any potential effects.
Two varieties of blueberry (rabbiteye and high bush) have published data regarding their flavonoid content. Rabbiteye blueberries contain a significant amount of the catechins (-)-epicatechin (25.66 mg/100g) and (+)-catechin (98.47 mg/100g) and flavonols kaempferol (2.36 mg/100g), myricetin (2.92 mg/100g), and quercetin (14.42 mg/100g) [126]. On the other hand, high bush blueberries have significantly less catechins (0.62 mg/100g, (-)-epicatechin; 5.29 mg/100g (+)-catechin) and flavonols [kaempferol (1.66 mg/100g), myricetin (1.3 mg/100g), and quercetin (7.67 mg/100g)] than rabbiteye varieties. Therefore, future studies that seek to investigate the effect of blueberry consumption on BC risk should take this variable into consideration when assessing intake levels.
Discussion and Future Research
The global prevalence of BC is increasing [2]. Moreover, there are no targeted therapies for TNBC and the systemic cytotoxic therapeutics used on these patients often fail to slow tumor progression [15,16]. This highlights the need to continue to 1) develop effective preventive approaches; and 2) interrogate potential novel molecular targets. Overexpression of the AhR is detected in human [46,99] and rodent [37,100] mammary tumors and associates with TNBC in cell lines and clinical samples [37,117]. Expressing high levels of AhR protein is sufficient to induce malignant transformation in non-tumorigenic cell lines [45], and depletion of AhR attenuates tumorigenic properties of TNBC cells [47]. The activated AhR is also responsible for coordinating a transcriptionally repressive chromatin state at the BRCA1 promoterthrough epigenetic silencing [29,32,33]. Exogenous activating ligands of the AhR are ubiquitous [43] and high levels of endogenous agonists can be produced intratumorally [86] and in the inflammatory tumor microenvironment [54]. Moreover, constitutively active AhR has been detected in multiple malignancies [97,98] including BC [46]. This collective information suggests exposure to compounds that antagonize activated AhR may be preventive and therapeutic in BC. Specific foods [59] and food bioactives, namely flavonoids, [32-34,60-66] have inhibitory effects on AhR activation. Thus, we hypothesize that a diet heavy in these constituents would provide preventive effects against BC.
In this review we conducted an initial development of this working hypothesis by performing a literature search of observational studies investigating the association between consumption of foods demonstrating AhR inhibitory capacities and BC risk. The foods included broccoli, lettuce, spinach, grapefruit, orange, and apple. Although discrepancy between studies for these foods makes the currently available evidence unconvincing, there are several differences between studies likely owing to this disparity and confounding the ability to draw precise conclusions. Perhaps the most logical drivers of this heterogeneity are population differences and the difference in intake levels between the highest consumers from the different studies. An example of this latter driver exists in our analysis of lettuce. The Greek case-control study [142] and the pooled prospective analysis [135] found protective effects among individuals consuming 12 g/d and 56 g/d, respectively, but in contrast ~9.6 g/d had no effect on risk in the NHS II cohort [134]. Another example is in the case of broccoli. Among women from North Eastern U.S.A, intakes ≥ 20.8 g/d were inversely associated with BC risk [132], whereas 11.7 g/d was not protective among the NHS II cohort [134]. Future analyses should seek to determine dose-dependent effects based on standardized absolute intake values (as opposed to quantiles of intake). This may be imperative, for example, in the case of grapefruit, where higher consumption may have an adverse impact on risk [150].
Another issue that should be addressed in future investigations is the type/variety of food item in question. In this review, this was most relevant in the case of lettuce and apples. Both foods come in multiple varieties, which differ in their bioactive content. For example, red leaf lettuce carries ~7.61 mg of quercetin per 100g of food weight, whereas iceberg lettuce has ~1.42 mg/100g [126]. Unless investigators factor this into their data collection, study subjects consuming red leaf and iceberg lettuce in the same amount would be grouped together for multivariate analysis despite the large difference in flavonoid dose. A similar confounding effect is brought when investigators do not differentiate the specific size of the fruit used for the serving size in data collection. For example, the FFQ employed in the NHSII has a serving size of one fruit for apples, oranges, grapefruits, etc. Unless this is taken into consideration subjects consuming three small apples are grouped with those consuming three large. Again, despite a considerable difference in bioactive load.
Perhaps the most important unexplored variable in analyses that consider the association between the intake of a particular food item and cancer risk is the effect of cooking and cooking method. This was raised as a concern in our analysis of broccoli and this concern was validated by a study investigating spinach intake and cancer risk. In a case-control study conducted in the US, higher consumption of raw, but not cooked, spinach was found to be inversely associated with BC risk [144]. This was the only study we reviewed that investigated this difference in spinach, and none of the studies considered this for broccoli. Given that cooking can drastically alter bioactive contents of food [136], this too needs to be considered in future nutritional epidemiology investigations.
Conclusions
This review served as an initial exploration of our working hypothesis that an anti-AhR diet is protective against BC. The evidence to support this hypothesis among currently published observational studies is lacking, however this is likely owing to the lack of standardization between studies and the confounding variables discussed above. Although a comprehensive overview was out of the scope of this work, preclinical studies have demonstrated that dietary AhR antagonists and similar compounds have therapeutic promise in BC [35,162]. It is likely that future studies that take more rigorous approaches to identify and account for confounding variables (i.e., dose, variety, cooking method) that impact pertinent biological activities (i.e., the AhR pathway) may derive more accurate conclusions in regard to the biological endpoint (i.e., BC) in question.
Acknowledgments
This work was supported by a grant from the US Department of Defense Breast Cancer Program (DAMD-15- 1-0387).
Glossary
- AhR
aryl hydrocarbon receptor
- ARNT
aryl hydrocarbon receptor nuclear translocator
- BC
breast cancer
- BWHS
Black Women’s Health Study
- COX
cyclooxygenase
- DMBA
7,12-Dimethyl-benz[a]anthracene
- DNMT
DNA methyltransferase
- EMT
epithelial-to-mesenchymal transition
- EPIC
European Prospective Investigation into Cancer and Nutrition
- ER
estrogen receptor
- FFQ
food frequency questionnaire
- HAH
halogenated aromatic hydrocarbons
- HER
human epidermal growth factor receptor
- HR
hormone receptor
- IDO
Indoleamine 2,3-dioxygenase
- MBD
methyl binding domain
- MEC
Multi Ethnic Cohort
- NHS
nurses health study
- PAH
polycyclic aromatic hydrocarbons
- PR
progesterone receptor
- SERM
selective estrogen receptor modulator
- TCDD
2,3,7,8-Tetrachlorodibenzo-p-dioxin
- TDO
tryptophan-2,3-dioxygenase
- TNBC
triple negative breast cancer
- XRE
xenobiotic response element
Author Contributions
MD, OS, and DR contributed to the conceptual development, organization, and review of the manuscript; MD and DR were responsible for review of literature, collecting and cataloging published data, and writing of the manuscript. DR was responsible for the content of the manuscript.
References
- Torre LA, Islami F, Siegel RL, Ward EM, Jemal A. Global cancer in women: burden and trends. Cancer Epidemiol Biomarkers Prev. 2017 [DOI] [PubMed] [Google Scholar]
- Tao Z, Shi A, Lu C, Song T, Zhang Z, Zhao J. Breast Cancer: epidemiology and Etiology. Cell Biochem Biophys. 2015;72(2):333–8. [DOI] [PubMed] [Google Scholar]
- Duncan JA, Reeves JR, Cooke TG. BRCA1 and BRCA2 proteins: roles in health and disease. Mol Pathol. 1998;51(5):237–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolou P, Fostira F. Hereditary breast cancer: the era of new susceptibility genes. BioMed Res Int. 2013;2013:747318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi RE, Pericleous M, Mandair D, Whyand T, Caplin ME. The role of dietary factors in prevention and progression of breast cancer. Anticancer Res. 2014;34(12):6861–75. [PubMed] [Google Scholar]
- World Cancer Research Fund International/American Institute for Cancer Research Continuous Update Project Report: Diet, Nutrition, Physical Activity and Breast Cancer. WCRF International, Bedford Square, London;2017. [Google Scholar]
- Toss A, Cristofanilli M. Molecular characterization and targeted therapeutic approaches in breast cancer. Breast Cancer Res. 2015;17:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn SG, Kim SJ, Kim C, Jeong J. Molecular Classification of Triple-Negative Breast Cancer. J Breast Cancer. 2016;19(3):223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A, Pineda E, Adamo B, Galvan P, Fernandez A, Gaba L, et al. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast. 2015;24 Suppl 2:S26–35. [DOI] [PubMed] [Google Scholar]
- Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 2011;5(1):5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr AE, Margenthaler JA. Molecular profiling of breast cancer. Surg Oncol Clin N Am. 2014;23(3):451–62. [DOI] [PubMed] [Google Scholar]
- Hwang KT, Kim EK, Jung SH, Lee ES, Kim SI, Lee S, et al. Tamoxifen therapy improves overall survival in luminal A subtype of ductal carcinoma in situ: a study based on nationwide Korean Breast Cancer Registry database. Breast Cancer Res Treat. 2018; 169(2):311-322. [DOI] [PubMed] [Google Scholar]
- Dowsett M, Forbes JF, Bradley R, Ingle J, Aihara T, Bliss J, et al. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet. 2015;386(10001):1341–52. [DOI] [PubMed] [Google Scholar]
- O’Sullivan CC, Bradbury I, Campbell C, Spielmann M, Perez EA, Joensuu H, et al. Efficacy of Adjuvant Trastuzumab for Patients With Human Epidermal Growth Factor Receptor 2–Positive Early Breast Cancer and Tumors ≤ 2 cm: A Meta-Analysis of the Randomized Trastuzumab Trials. J Clin Oncol. 2015;33(24):2600–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirshfield KM, Ganesan S. Triple-negative breast cancer: molecular subtypes and targeted therapy. Curr Opin Obstet Gynecol. 2014;26(1):34–40. [DOI] [PubMed] [Google Scholar]
- Schmadeka R, Harmon BE, Singh M. Triple-negative breast carcinoma: current and emerging concepts. Am J Clin Pathol. 2014;141(4):462–77. [DOI] [PubMed] [Google Scholar]
- Savage KI, Harkin DP. BRCA1, a ‘complex’ protein involved in the maintenance of genomic stability. FEBS J. 2015;282(4):630–46. [DOI] [PubMed] [Google Scholar]
- Eisen A, Lubinski J, Gronwald J, Moller P, Lynch HT, Klijn J, et al. Hormone Therapy and the Risk of Breast Cancer in BRCA1 Mutation Carriers. J Natl Cancer Inst. 2008;100(19):1361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosse SA, van Beers EH, Tielen IH, Horlings H, Peterse JL, Hoogerbrugge N, et al. Prediction of BRCA1-association in hereditary non-BRCA1/2 breast carcinomas with array-CGH. Breast Cancer Res Treat. 2009;116(3):479–89. [DOI] [PubMed] [Google Scholar]
- Lakhani SR, Van De Vijver MJ, Jacquemier J, Anderson TJ, Osin PP, McGuffog L, et al. The pathology of familial breast cancer: predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J Clin Oncol. 2002;20(9):2310–8. [DOI] [PubMed] [Google Scholar]
- Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J Clin Oncol. 2010;28(7):1145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner NC, Reis-Filho JS, Russell AM, Springall RJ, Ryder K, Steele D, et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene. 2007;26(14):2126–32. [DOI] [PubMed] [Google Scholar]
- Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100(14):8418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–9. [DOI] [PubMed] [Google Scholar]
- Li M, Zhang J, Ouyang T, Li J, Wang T, Fan Z, et al. Incidence of BRCA1 somatic mutations and response to neoadjuvant chemotherapy in Chinese women with triple-negative breast cancer. Gene. 2016;584(1):26–30. [DOI] [PubMed] [Google Scholar]
- Yamashita N, Tokunaga E, Kitao H, Hitchins M, Inoue Y, Tanaka K, et al. Epigenetic Inactivation of BRCA1 Through Promoter Hypermethylation and Its Clinical Importance in Triple-Negative Breast Cancer. Clin Breast Cancer. 2015;15(6):498–504. [DOI] [PubMed] [Google Scholar]
- Ignatov T, Poehlmann A, Ignatov A, Schinlauer A, Costa SD, Roessner A, et al. BRCA1 promoter methylation is a marker of better response to anthracycline-based therapy in sporadic TNBC. Breast Cancer Res Treat. 2013;141(2):205–12. [DOI] [PubMed] [Google Scholar]
- Lips EH, Mulder L, Oonk A, van der Kolk LE, Hogervorst FB, Imholz AL, et al. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br J Cancer. 2013;108(10):2172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockings JK, Thorne PA, Kemp MQ, Morgan SS, Selmin O, Romagnolo DF. The ligand status of the aromatic hydrocarbon receptor modulates transcriptional activation of BRCA-1 promoter by estrogen. Cancer Res. 2006;66(4):2224–32. [DOI] [PubMed] [Google Scholar]
- Jeffy BD, Chirnomas RB, Chen EJ, Gudas JM, Romagnolo DF. Activation of the aromatic hydrocarbon receptor pathway is not sufficient for transcriptional repression of BRCA-1: requirements for metabolism of benzo[a]pyrene to 7r,8t-dihydroxy-9t,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene. Cancer Res. 2002;62(1):113–21. [PubMed] [Google Scholar]
- Jeffy BD, Schultz EU, Selmin O, Gudas JM, Bowden GT, Romagnolo D. Inhibition of BRCA-1 expression by benzo[a]pyrene and its diol epoxide. Mol Carcinog. 1999;26(2):100–18. [DOI] [PubMed] [Google Scholar]
- Papoutsis AJ, Borg JL, Selmin OI, Romagnolo DF. BRCA-1 promoter hypermethylation and silencing induced by the aromatic hydrocarbon receptor-ligand TCDD are prevented by resveratrol in MCF-7 cells. J Nutr Biochem. 2012;23(10):1324–32. [DOI] [PubMed] [Google Scholar]
- Papoutsis AJ, Lamore SD, Wondrak GT, Selmin OI, Romagnolo DF. Resveratrol prevents epigenetic silencing of BRCA-1 by the aromatic hydrocarbon receptor in human breast cancer cells. J Nutr. 2010;140(9):1607–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papoutsis AJ, Selmin OI, Borg JL, Romagnolo DF. Gestational exposure to the AhR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin induces BRCA-1 promoter hypermethylation and reduces BRCA-1 expression in mammary tissue of rat offspring: preventive effects of resveratrol. Mol Carcinog. 2015;54(4):261–9. [DOI] [PubMed] [Google Scholar]
- Romagnolo DF, Daniels KD, Grunwald JT, Ramos SA, Propper CR, Selmin OI. Epigenetics of breast cancer: modifying role of environmental and bioactive food compounds. Mol Nutr Food Res. 2016;60(6):1310–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnolo DF, Donovan MG, Papoutsis AJ, Doetschman TC, Selmin OI. Genistein prevents BRCA1 CpG methylation and proliferation in human breast cancer cells with activated aromatic hydrocarbon receptor. Curr Dev Nutr. 2017;1(6):e000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnolo DF, Papoutsis AJ, Laukaitis C, Selmin OI. Constitutive expression of AhR and BRCA-1 promoter CpG hypermethylation as biomarkers of ERα-negative breast tumorigenesis. BMC Cancer. 2015:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel J, Haarmann-Stemmann T. An introduction to the molecular basics of aryl hydrocarbon receptor biology. Biol Chem. 2010;391(11):1235–48. [DOI] [PubMed] [Google Scholar]
- Tian J, Feng Y, Fu H, Xie HQ, Jiang JX, Zhao B. The Aryl Hydrocarbon Receptor: A Key Bridging Molecule of External and Internal Chemical Signals. Environ Sci Technol. 2015;49(16):9518–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushmore TH, Pickett CB. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J Biol Chem. 1990;265(24):14648–53. [PubMed] [Google Scholar]
- Munzel PA, Lehmkoster T, Bruck M, Ritter JK, Bock KW. Aryl hydrocarbon receptor-inducible or constitutive expression of human UDP glucuronosyltransferase UGT1A6. Arch Biochem Biophys. 1998;350(1):72–8. [DOI] [PubMed] [Google Scholar]
- Yueh MF, Huang YH, Hiller A, Chen S, Nguyen N, Tukey RH. Involvement of the xenobiotic response element (XRE) in Ah receptor-mediated induction of human UDP-glucuronosyltransferase 1A1. J Biol Chem. 2003;278(17):15001–6. [DOI] [PubMed] [Google Scholar]
- Murray IA, Patterson AD, Perdew GH. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer. 2014;14(12):801–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safe S, Lee SO, Jin UH. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicol Sci. 2013;135(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks J, Eltom SE. Malignant transformation of mammary epithelial cells by ectopic overexpression of the aryl hydrocarbon receptor. Curr Cancer Drug Targets. 2011;11(5):654–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltom SE, Gasmelseed AA, Saudoudi-Guentri D. The aryl hydrocarbon receptor is over-expressed and constitutively activated in advanced breast carcinoma. Cancer Res. 2006;66(8 supplement):408. [Google Scholar]
- Goode G, Ballard BR, Manning HC, Freeman ML, Kang Y, Eltom SE. Knockdown of aberrantly upregulated aryl hydrocarbon receptor reduces tumor growth and metastasis of MDA-MB-231 human breast cancer cell line. Int J Cancer. 2013;133(12):2769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JB, Goode GD, Eltom SE. The Aryl Hydrocarbon Receptor: A Target for Breast Cancer Therapy. J Cancer Ther. 2013;4(7):1177–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNatale BC, Murray IA, Schroeder JC, Flaveny CA, Lahoti TS, Laurenzana EM, et al. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol Sci. 2010;115(1):89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185(6):3190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberg M, Bergander L, Hakansson H, Rannug U, Rannug A. Identification of the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole, in cell culture medium, as a factor that controls the background aryl hydrocarbon receptor activity. Toxicol Sci. 2005;85(2):935–43. [DOI] [PubMed] [Google Scholar]
- Phelan D, Winter GM, Rogers WJ, Lam JC, Denison MS. Activation of the Ah receptor signal transduction pathway by bilirubin and biliverdin. Arch Biochem Biophys. 1998;357(1):155–63. [DOI] [PubMed] [Google Scholar]
- Schroeder JC, Dinatale BC, Murray IA, Flaveny CA, Liu Q, Laurenzana EM, et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry. 2010;49(2):393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478(7368):197–203. [DOI] [PubMed] [Google Scholar]
- Pilotte L, Larrieu P, Stroobant V, Colau D, Dolusic E, Frederick R, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci USA. 2012;109(7):2497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller AJ, Sharma MD, Chandler PR, Duhadaway JB, Everhart ME, Johnson BA, 3rd, et al. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc Natl Acad Sci USA. 2008;105(44):17073–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson P, McGuire J, Rubio C, Gradin K, Whitelaw ML, Pettersson S, et al. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc Natl Acad Sci USA. 2002;99(15):9990–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moennikes O, Loeppen S, Buchmann A, Andersson P, Ittrich C, Poellinger L, et al. A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res. 2004;64(14):4707–10. [DOI] [PubMed] [Google Scholar]
- Amakura Y, Tsutsumi T, Nakamura M, Kitagawa H, Fujino J, Sasaki K, et al. Preliminary Screening of the Inhibitory Effect of Food Extracts on Activation of the Aryl Hydrocarbon Receptor Induced by 2,3,7,8-Tetrachlorodibenzo-p-dioxin. Biol Pharm Bull. 2002;25(2):272–4. [DOI] [PubMed] [Google Scholar]
- Degner SC, Papoutsis AJ, Selmin O, Romagnolo DF. Targeting of aryl hydrocarbon receptor-mediated activation of cyclooxygenase-2 expression by the indole-3-carbinol metabolite 3,3′-diindolylmethane in breast cancer cells. J Nutr. 2009;139(1):26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner SC, Kemp MQ, Hockings JK, Romagnolo DF. Cyclooxygenase-2 promoter activation by the aromatic hydrocarbon receptor in breast cancer mcf-7 cells: repressive effects of conjugated linoleic acid. Nutr Cancer. 2007;59(2):248–57. [DOI] [PubMed] [Google Scholar]
- Miller ME, Holloway AC, Foster WG. Benzo-[a]-pyrene increases invasion in MDA-MB-231 breast cancer cells via increased COX-II expression and prostaglandin E2 (PGE2) output. Clin Exp Metastasis. 2005;22(2):149–56. [DOI] [PubMed] [Google Scholar]
- Zhang S, Qin C, Safe SH. Flavonoids as aryl hydrocarbon receptor agonists/antagonists: effects of structure and cell context. Environ Health Perspect. 2003;111(16):1877–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciolino HP, Yeh GC. The flavonoid galangin is an inhibitor of CYP1A1 activity and an agonist/antagonist of the aryl hydrocarbon receptor. Br J Cancer. 1999;79(9-10):1340–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdull Razis AF, Hanlon N, Soltys E, Krizova V, Iori R, Plant KE, et al. The naturally occurring aliphatic isothiocyanates sulforaphane and erucin are weak agonists but potent non-competitive antagonists of the aryl hydrocarbon receptor. Arch Toxicol. 2012;86(10):1505–14. [DOI] [PubMed] [Google Scholar]
- Abdull Razis AF, Konsue N, Dervetzoglou M, Plant KE, Plant N, Ioannides C. Phenethyl isothiocyanate, a naturally occurring phytochemical, is an antagonist of the aryl hydrocarbon receptor. Mol Nutr Food Res. 2012;56(3):425–34. [DOI] [PubMed] [Google Scholar]
- Ashida H, Fukuda I, Yamashita T, Kanazawa K. Flavones and flavonols at dietary levels inhibit a transformation of aryl hydrocarbon receptor induced by dioxin. FEBS Lett. 2000;476(3):213–7. [DOI] [PubMed] [Google Scholar]
- Beedanagari SR, Bebenek I, Bui P, Hankinson O. Resveratrol Inhibits Dioxin-Induced Expression of Human CYP1A1 and CYP1B1 by Inhibiting Recruitment of the Aryl Hydrocarbon Receptor Complex and RNA Polymerase II to the Regulatory Regions of the Corresponding Genes. Toxicol Sci. 2009;110(1):61–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hestermann EV, Brown M. Agonist and chemopreventative ligands induce differential transcriptional cofactor recruitment by aryl hydrocarbon receptor. Mol Cell Biol. 2003;23(21):7920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai R, Shirai Y, Saito N, Fukuda I, Nishiumi S, Yoshida K, et al. Suppression mechanisms of flavonoids on aryl hydrocarbon receptor-mediated signal transduction. Arch Biochem Biophys. 2010;501(1):134–41. [DOI] [PubMed] [Google Scholar]
- Han EH, Kim JY, Jeong HG. Effect of biochanin A on the aryl hydrocarbon receptor and cytochrome P450 1A1 in MCF-7 human breast carcinoma cells. Arch Pharm Res. 2006;29(7):570–6. [DOI] [PubMed] [Google Scholar]
- Beischlag TV, Luis Morales J, Hollingshead BD, Perdew GH. The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr. 2008;18(3):207–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. Exactly the Same but Different: Promiscuity and Diversity in the Molecular Mechanisms of Action of the Aryl Hydrocarbon (Dioxin) Receptor. Toxicol Sci. 2011;124(1):1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PM, Wang D, Wang Y, Li Y, Uchizono JA, Chan WK. p23 co-chaperone protects the aryl hydrocarbon receptor from degradation in mouse and human cell lines. Biochem Pharmacol. 2012;84(6):838–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji N, Fukuda K, Nagata Y, Okada H, Haga A, Hatakeyama S, et al. The activation mechanism of the aryl hydrocarbon receptor (AhR) by molecular chaperone HSP90. FEBS Open Bio. 2014;4:796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endler A, Chen L, Shibasaki F. Coactivator Recruitment of AhR/ARNT1. Int J Mol Sci. 2014;15(6):11100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankinson O. Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch Biochem Biophys. 2005;433(2):379–86. [DOI] [PubMed] [Google Scholar]
- Elferink CJ, Whitlock JP., Jr 2,3,7,8-Tetrachlorodibenzo-p-dioxin-inducible, Ah receptor-mediated bending of enhancer DNA. J Biol Chem. 1990;265(10):5718–21. [PubMed] [Google Scholar]
- Wu L, Whitlock JP., Jr Mechanism of dioxin action: ah receptor-mediated increase in promoter accessibility in vivo. Proc Natl Acad Sci USA. 1992;89(11):4811–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TA, Hoivik D, Lee JE, Safe S. Interactions of nuclear receptor coactivator/corepressor proteins with the aryl hydrocarbon receptor complex. Arch Biochem Biophys. 1999;367(2):250–7. [DOI] [PubMed] [Google Scholar]
- Soshilov AA, Denison MS. Ligand promiscuity of aryl hydrocarbon receptor agonists and antagonists revealed by site-directed mutagenesis. Mol Cell Biol. 2014;34(9):1707–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry EC, Kende AS, Rucci G, Totleben MJ, Willey JJ, Dertinger SD, et al. Flavone antagonists bind competitively with 2,3,7, 8-tetrachlorodibenzo-p-dioxin (TCDD) to the aryl hydrocarbon receptor but inhibit nuclear uptake and transformation. Mol Pharmacol. 1999;55(4):716–25. [PubMed] [Google Scholar]
- Stejskalova L, Dvorak Z, Pavek P. Endogenous and exogenous ligands of aryl hydrocarbon receptor: current state of art. Curr Drug Metab. 2011;12(2):198–212. [DOI] [PubMed] [Google Scholar]
- Chiaro CR, Patel RD, Perdew GH. 12(R)-Hydroxy-5(Z),8(Z),10(E),14(Z)-eicosatetraenoic acid [12(R)-HETE], an arachidonic acid derivative, is an activator of the aryl hydrocarbon receptor. Mol Pharmacol. 2008;74(6):1649–56. [DOI] [PubMed] [Google Scholar]
- D’Amato NC, Rogers TJ, Gordon MA, Greene LI, Cochrane DR, Spoelstra NS, et al. A TDO2-AhR Signaling Axis Facilitates Anoikis Resistance and Metastasis in Triple-Negative Breast Cancer. Cancer Res. 2015;75(21):4651–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikov O, Wang Z, Stanford EA, Parks AJ, Ramirez-Cardenas A, Landesman E, et al. An Aryl Hydrocarbon Receptor-Mediated Amplification Loop That Enforces Cell Migration in ER(−)/PR(−)/Her2(−) Human Breast Cancer Cells. Mol Pharmacol. 2016;90(5):674–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Li CF, Kuo CC, Tsai KK, Hou MF, Hung WC. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res. 2014:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen NT, Nakahama T, Le DH, Van Son L, Chu HH, Kishimoto T. Aryl Hydrocarbon Receptor and Kynurenine: Recent Advances in Autoimmune Disease Research. Front Immunol. 2014:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds GP, Pearson SJ. Increased brain 3-hydroxykynurenine in Huntington’s disease. Lancet. 1989;2(8669):979–80. [DOI] [PubMed] [Google Scholar]
- Ogawa T, Matson WR, Beal MF, Myers RH, Bird ED, Milbury P, et al. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology. 1992;42(9):1702–6. [DOI] [PubMed] [Google Scholar]
- Jaronen M, Quintana FJ. Immunological Relevance of the Coevolution of IDO1 and AHR. Front Immunol. 2014:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandi Y, Vecsei L. The kynurenine system and immunoregulation. J Neural Transm (Vienna). 2012;119(2):197–209. [DOI] [PubMed] [Google Scholar]
- Vogel CF, Goth SR, Dong B, Pessah IN, Matsumura F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem Biophys Res Commun. 2008;375(3):331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai S, Kikuchi H. The inhibitory mechanisms of the tyrosine kinase inhibitors herbimycin a, genistein, and tyrphostin B48 with regard to the function of the aryl hydrocarbon receptor in Caco-2 cells. Biosci Biotechnol Biochem. 2010;74(1):36–43. [DOI] [PubMed] [Google Scholar]
- Ciolino HP, Daschner PJ, Yeh GC. Dietary flavonols quercetin and kaempferol are ligands of the aryl hydrocarbon receptor that affect CYP1A1 transcription differentially. Biochem J. 1999;340(Pt 3):715–22. [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Degroot DE, Hayashi A, He G, Denison MS. CH223191 is a ligand-selective antagonist of the Ah (Dioxin) receptor. Toxicol Sci. 2010;117(2):393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P, Chang H, Ho WL, Wu MH, Su JM. Association of aryl hydrocarbon receptor and cytochrome P4501B1 expressions in human non-small cell lung cancers. Lung Cancer. 2003;42(3):255–61. [DOI] [PubMed] [Google Scholar]
- Tran C, Richmond O, Aaron L, Powell JB. Inhibition of constitutive aryl hydrocarbon receptor (AhR) signaling attenuates androgen independent signaling and growth in (C4-2) prostate cancer cells. Biochem Pharmacol. 2013;85(6):753–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Solomon S, Fraser LR, Trombino AF, Liu D, Sonenshein GE, et al. Constitutive regulation of CYP1B1 by the aryl hydrocarbon receptor (AhR) in pre-malignant and malignant mammary tissue. J Cell Biochem. 2008;104(2):402–17. [DOI] [PubMed] [Google Scholar]
- Trombino AF, Near RI, Matulka RA, Yang S, Hafer LJ, Toselli PA, et al. Expression of the aryl hydrocarbon receptor/transcription factor (AhR) and AhR-regulated CYP1 gene transcripts in a rat model of mammary tumorigenesis. Breast Cancer Res Treat. 2000;63(2):117–31. [DOI] [PubMed] [Google Scholar]
- Ikuta T, Kawajiri K. Zinc finger transcription factor Slug is a novel target gene of aryl hydrocarbon receptor. Exp Cell Res. 2006;312(18):3585–94. [DOI] [PubMed] [Google Scholar]
- Beaver LM, Yu TW, Sokolowski EI, Williams DE, Dashwood RH, Ho E. 3,3′-Diindolylmethane, but not indole-3-carbinol, inhibits histone deacetylase activity in prostate cancer cells. Toxicol Appl Pharmacol. 2012;263(3):345–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higdon JV, Delage B, Williams DE, Dashwood RH. Cruciferous vegetables and human cancer risk: epidemiologic evidence and mechanistic basis. Pharmacol Res. 2007;55(3):224–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorey LE, Hagman AM, Williams DE, Ho E, Dashwood RH, Benninghoff AD. 3,3′-Diindolylmethane induces G1 arrest and apoptosis in human acute T-cell lymphoblastic leukemia cells. PLoS One. 2012;7(4):e34975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Dashwood RH, Bjeldanes LF, Williams DE, Bailey GS. Mechanisms of indole-3-carbinol (I3C) anticarcinogenesis: inhibition of aflatoxin B1-DNA adduction and mutagenesis by I3C acid condensation products. Food Chem Toxicol. 1995;33(10):851–7. [DOI] [PubMed] [Google Scholar]
- Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–34. [DOI] [PubMed] [Google Scholar]
- Benson JM, Shepherd DM. Dietary ligands of the aryl hydrocarbon receptor induce anti-inflammatory and immunoregulatory effects on murine dendritic cells. Toxicol Sci. 2011;124(2):327–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortelboer HM, van der Linden EC, de Kruif CA, Noordhoek J, Blaauboer BJ, van Bladeren PJ, et al. Effects of indole-3-carbinol on biotransformation enzymes in the rat: in vivo changes in liver and small intestinal mucosa in comparison with primary hepatocyte cultures. Food Chem Toxicol. 1992;30(7):589–99. [DOI] [PubMed] [Google Scholar]
- Bjeldanes LF, Kim JY, Grose KR, Bartholomew JC, Bradfield CA. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc Natl Acad Sci USA. 1991;88(21):9543–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinck PH, Forkert PG, Riddick DS, Okey AB, Michnovicz JJ, Bradlow HL. Ah receptor binding properties of indole carbinols and induction of hepatic estradiol hydroxylation. Biochem Pharmacol. 1993;45(5):1129–36. [DOI] [PubMed] [Google Scholar]
- Meng Q, Qi M, Chen DZ, Yuan R, Goldberg ID, Rosen EM, et al. Suppression of breast cancer invasion and migration by indole-3-carbinol: associated with up-regulation of BRCA1 and E-cadherin/catenin complexes. J Mol Med (Berl). 2000;78(3):155–65. [DOI] [PubMed] [Google Scholar]
- Wattenberg LW, Loub WD. Inhibition of polycyclic aromatic hydrocarbon-induced neoplasia by naturally occurring indoles. Cancer Res. 1978;38(5):1410–3. [PubMed] [Google Scholar]
- Chen I, McDougal A, Wang F, Safe S. Aryl hydrocarbon receptor-mediated antiestrogenic and antitumorigenic activity of diindolylmethane. Carcinogenesis. 1998;19(9):1631–9. [DOI] [PubMed] [Google Scholar]
- Okino ST, Pookot D, Basak S, Dahiya R. Toxic and chemopreventive ligands preferentially activate distinct aryl hydrocarbon receptor pathways: implications for cancer prevention. Cancer Prev Res (Phila). 2009;2(3):251–6. [DOI] [PubMed] [Google Scholar]
- Thomson CA, Ho E, Strom MB. Chemopreventive properties of 3,3′-diindolylmethane in breast cancer: evidence from experimental and human studies. Nutr Rev. 2016;74(7):432–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen CJ, Wu LX, Fu LJ, Shen DY, Zhang X, Zhang YW, et al. Preferential induction of CYP1A1 over CYP1B1 in human breast cancer MCF-7 cells after exposure to berberine. Asian Pac J Cancer Prev. 2014;15(1):495–9. [DOI] [PubMed] [Google Scholar]
- Vacher S, Castagnet P, Chemlali W, Lallemand F, Meseure D, Pocard M, et al. High AHR expression in breast tumors correlates with expression of genes from several signaling pathways namely inflammation and endogenous tryptophan metabolism. PLoS One. 2018;13(1):e0190619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safe SW, Porter W, Duan R, McDougal A. Ah receptor agonists as endocrine disruptors: antiestrogenic activity and mechanisms. Toxicol Lett. 1998;28(102-103):343–7. [DOI] [PubMed] [Google Scholar]
- Simon TW, Budinsky RA, Rowlands JC. A model for aryl hydrocarbon receptor-activated gene expression shows potency and efficacy changes and predicts squelching due to competition for transcription co-activators. PLoS One. 2015;10(6):e0127952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H, et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature. 2007;446(7135):562–6. [DOI] [PubMed] [Google Scholar]
- Wormke M, Stoner M, Saville B, Walker K, Abdelrahim M, Burghardt R, et al. The Aryl Hydrocarbon Receptor Mediates Degradation of Estrogen Receptor α through Activation of Proteasomes. Mol Cell Biol. 2003;23(6):1843–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S, Valen E, Sandelin A, Matthews J. Dioxin Increases the Interaction Between Aryl Hydrocarbon Receptor and Estrogen Receptor Alpha at Human Promoters. Toxicol Sci. 2009;111(2):254–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mense SM, Chhabra J, Bhat HK. Preferential induction of cytochrome P450 1A1 over cytochrome P450 1B1 in human breast epithelial cells following exposure to quercetin. J Steroid Biochem Mol Biol. 2008;110(1-2):157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozic JG, Chakraborty C, Lala PK. Cyclooxygenase inhibitors retard murine mammary tumor progression by reducing tumor cell migration, invasiveness and angiogenesis. Int J Cancer. 2001;93(4):497–506. [DOI] [PubMed] [Google Scholar]
- Williams SN, Shih H, Guenette DK, Brackney W, Denison MS, Pickwell GV, et al. Comparative studies on the effects of green tea extracts and individual tea catechins on human CYP1A gene expression. Chem Biol Interact. 2000;128(3):211–29. [DOI] [PubMed] [Google Scholar]
- USDA Database for the Flavonoid Content of Selected Foods Release 3.1 [Internet]. U.S. Department of Agriculture, Agricultural Research Service. 2014. Available from: Nutrient Data Laboratory Home Page: http://www.ars.usda.gov/nutrientdata/flav
- Oliviero T, Verkerk R, Vermeulen M, Dekker M. In vivo formation and bioavailability of isothiocyanates from glucosinolates in broccoli as affected by processing conditions. Mol Nutr Food Res. 2014;58(7):1447–56. [DOI] [PubMed] [Google Scholar]
- Liu X, Lv K. Cruciferous vegetables intake is inversely associated with risk of breast cancer: a meta-analysis. Breast. 2013;22(3):309–13. [DOI] [PubMed] [Google Scholar]
- Adebamowo CA, Cho E, Sampson L, Katan MB, Spiegelman D, Willett WC, et al. Dietary flavonols and flavonol-rich foods intake and the risk of breast cancer. Int J Cancer. 2005;114(4):628–33. [DOI] [PubMed] [Google Scholar]
- Surakasula A, Nagarjunapu GC, Raghavaiah KV. A comparative study of pre- and post-menopausal breast cancer: risk factors, presentation, characteristics and management. J Res Pharm Pract. 2014;3(1):12–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm J, Eriksson L, Ploner A, Eriksson M, Rantalainen M, Li J, et al. Assessment of Breast Cancer Risk Factors Reveals Subtype Heterogeneity. Cancer Res. 2017;77(13):3708–17. [DOI] [PubMed] [Google Scholar]
- Ambrosone CB, McCann SE, Freudenheim JL, Marshall JR, Zhang Y, Shields PG. Breast cancer risk in premenopausal women is inversely associated with consumption of broccoli, a source of isothiocyanates, but is not modified by GST genotype. J Nutr. 2004;134(5):1134–8. [DOI] [PubMed] [Google Scholar]
- Boggs DA, Palmer JR, Wise LA, Spiegelman D, Stampfer MJ, Adams-Campbell LL, et al. Fruit and Vegetable Intake in Relation to Risk of Breast Cancer in the Black Women’s Health Study. Am J Epidemiol. 2010;172(11):1268–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farvid MS, Chen WY, Michels KB, Cho E, Willett WC, Eliassen AH. Fruit and vegetable consumption in adolescence and early adulthood and risk of breast cancer: population based cohort study. BMJ. 2016;•••:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Spiegelman D, Baglietto L, Bernstein L, Boggs DA, van den Brandt PA, et al. Fruit and vegetable intake and risk of breast cancer by hormone receptor status. J Natl Cancer Inst. 2013;105(3):219–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Reis LC, de Oliveira VR, Hagen ME, Jablonski A, Flores SH, de Oliveira Rios A. Effect of cooking on the concentration of bioactive compounds in broccoli (Brassica oleracea var. Avenger) and cauliflower (Brassica oleracea var. Alphina F1) grown in an organic system. Food Chem. 2015;172:770–7. [DOI] [PubMed] [Google Scholar]
- Liu X, Wang Y, Hoeflinger JL, Neme BP, Jeffery EH, Miller MJ. Dietary Broccoli Alters Rat Cecal Microbiota to Improve Glucoraphanin Hydrolysis to Bioactive Isothiocyanates. Nutrients. 2017;9(3):pii: E262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morito K, Hirose T, Kinjo J, Hirakawa T, Okawa M, Nohara T, et al. Interaction of phytoestrogens with estrogen receptors alpha and beta. Biol Pharm Bull. 2001;24(4):351–6. [DOI] [PubMed] [Google Scholar]
- Brown NM, Galandi SL, Summer SS, Zhao X, Heubi JE, King EC, et al. S-(–)equol production is developmentally regulated and related to early diet composition. Nutr Res. 2014;34(5):401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund TD, Munson DJ, Haldy ME, Setchell KD, Lephart ED, Handa RJ. Equol is a novel anti-androgen that inhibits prostate growth and hormone feedback. Biol Reprod. 2004;70(4):1188–95. [DOI] [PubMed] [Google Scholar]
- Bosviel R, Durif J, Dechelotte P, Bignon YJ, Bernard-Gallon D. Epigenetic modulation of BRCA1 and BRCA2 gene expression by equol in breast cancer cell lines. Br J Nutr. 2012;108(7):1187–93. [DOI] [PubMed] [Google Scholar]
- Katsouyanni K, Trichopoulos D, Boyle P, Xirouchaki E, Trichopoulou A, Lisseos B, et al. Diet and breast cancer: a case-control study in Greece. Int J Cancer. 1986;38(6):815–20. [DOI] [PubMed] [Google Scholar]
- Huang XE, Hirose K, Wakai K, Matsuo K, Ito H, Xiang J, et al. Comparison of lifestyle risk factors by family history for gastric, breast, lung and colorectal cancer. Asian Pac J Cancer Prev. 2004;5(4):419–27. [PubMed] [Google Scholar]
- Longnecker MP, Newcomb PA, Mittendorf R, Greenberg ER, Willett WC. Intake of carrots, spinach, and supplements containing vitamin A in relation to risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 1997;6(11):887–92. [PubMed] [Google Scholar]
- Bao PP, Shu XO, Zheng Y, Cai H, Ruan ZX, Gu K, et al. Fruit, vegetable, and animal food intake and breast cancer risk by hormone receptor status. Nutr Cancer. 2012;64(6):806–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JK, Bae JM. Citrus fruit intake and breast cancer risk: a quantitative systematic review. J Breast Cancer. 2013;16(1):72–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foschi R, Pelucchi C, Dal Maso L, Rossi M, Levi F, Talamini R, et al. Citrus fruit and cancer risk in a network of case-control studies. Cancer Causes Control. 2010;21(2):237–42. [DOI] [PubMed] [Google Scholar]
- Malin AS, Qi D, Shu XO, Gao YT, Friedmann JM, Jin F, et al. Intake of fruits, vegetables and selected micronutrients in relation to the risk of breast cancer. Int J Cancer. 2003;105(3):413–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, Iwasaki M, Hara A, Inoue M, Sasazuki S, Sawada N, et al. Fruit and vegetable intake and breast cancer risk defined by estrogen and progesterone receptor status: the Japan Public Health Center-based Prospective Study. Cancer Causes Control. 2013;24(12):2117–28. [DOI] [PubMed] [Google Scholar]
- Monroe KR, Murphy SP, Kolonel LN, Pike MC. Prospective study of grapefruit intake and risk of breast cancer in postmenopausal women: the Multiethnic Cohort Study. Br J Cancer. 2007;97(3):440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EH, Hankinson SE, Eliassen AH, Willett WC. A prospective study of grapefruit and grapefruit juice intake and breast cancer risk. Br J Cancer. 2008;98(1):240–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer EA, Key TJ, Appleby PN, van Gils CH, Olsen A, Tjonneland A, et al. Prospective study of the association between grapefruit intake and risk of breast cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC). Cancer Causes Control. 2009;20(6):803–9. [DOI] [PubMed] [Google Scholar]
- Key T, Appleby P, Barnes I, Reeves G. Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst. 2002;94(8):606–16. [DOI] [PubMed] [Google Scholar]
- Ameer B, Weintraub RA. Drug interactions with grapefruit juice. Clin Pharmacokinet. 1997;33(2):103–21. [DOI] [PubMed] [Google Scholar]
- Tsuchiya Y, Nakajima M, Yokoi T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005;227(2):115–24. [DOI] [PubMed] [Google Scholar]
- Schubert W, Cullberg G, Edgar B, Hedner T. Inhibition of 17 beta-estradiol metabolism by grapefruit juice in ovariectomized women. Maturitas. 1994;20(2-3):155–63. [DOI] [PubMed] [Google Scholar]
- Weber A, Jager R, Borner A, Klinger G, Vollanth R, Matthey K, et al. Can grapefruit juice influence ethinylestradiol bioavailability? Contraception. 1996;53(1):41–7. [DOI] [PubMed] [Google Scholar]
- Monroe KR, Stanczyk FZ, Besinque KH, Pike MC. The effect of grapefruit intake on endogenous serum estrogen levels in postmenopausal women. Nutr Cancer. 2013;65(5):644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiani R, Minelli L, Rosignoli P. Apple intake and cancer risk: a systematic review and meta-analysis of observational studies. Public Health Nutr. 2016;19(14):2603–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallus S, Talamini R, Giacosa A, Montella M, Ramazzotti V, Franceschi S, et al. Does an apple a day keep the oncologist away? Ann Oncol. 2005;16(11):1841–4. [DOI] [PubMed] [Google Scholar]
- Fung TT, Chiuve SE, Willett WC, Hankinson SE, Hu FB, Holmes MD. Intake of specific fruits and vegetables in relation to risk of estrogen receptor-negative breast cancer among postmenopausal women. Breast Cancer Res Treat. 2013;138(3):925–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magne Nde CB, Zingue S, Winter E, Creczynski-Pasa TB, Michel T, Fernandez X, et al. Flavonoids, Breast Cancer Chemopreventive and/or Chemotherapeutic Agents. Curr Med Chem. 2015;22(30):3434–46. [PubMed] [Google Scholar]