

 A silylene-bridged Ir dimer in situ generated from [Ir(coe)2Cl]2 and Et2SiH2 was found to catalyze the hydrosilylation of N-heteroaromatics to furnish dearomatized azacyclic products with high activity (up to 1000 TONs), excellent selectivity, and good functional group tolerance.
A silylene-bridged Ir dimer in situ generated from [Ir(coe)2Cl]2 and Et2SiH2 was found to catalyze the hydrosilylation of N-heteroaromatics to furnish dearomatized azacyclic products with high activity (up to 1000 TONs), excellent selectivity, and good functional group tolerance.
Abstract
A silylene-bridged Ir dimer in situ generated from [Ir(coe)2Cl]2 and Et2SiH2 was found to catalyze the hydrosilylation of N-heteroaromatics to furnish dearomatized azacyclic products with high activity (up to 1000 TONs), excellent selectivity, and good functional group tolerance. The substrate scope was highly broad, including (iso)quinolines, substituted pyridines, pyrimidines, pyrazines, deazapurines, triazines, and benzimidazoles. Mechanistic studies such as a kinetic profile, rate-order assessment, and investigation of the electronic substituent effects on the initial rates were performed to access the detailed pathways. One pathway is proposed to involve an intramolecular insertion of the C N moiety of the substrates into the Ir–H bond of a resting species to form an Ir-amido silyl intermediate, followed by reductive elimination. The synthetic utility was proven by successful application to cinchona alkaloids, and facile post-synthetic transformations of the 1,2-dihydroquinoline products.
Introduction
Dearomatization of (hetero)arenes is a kinetically unfavorable process, thus necessitating the use of efficient catalytic systems to overcome the high resonance stabilization energies.1 Certain enzymes such as reductases or oxygenases are well known to catalyze dearomative transformations of arenes with excellent efficiency and selectivity,2 while selective dearomatization in synthetic chemistry has still been a great challenge.3 Compounds obtainable from the partial dearomatization of (hetero)aromatics are useful building units in synthesis, allowing for a variety of subsequent transformations. In particular, a partial reduction of pyridines can lead to 1,2- or 1,4-dihydropyridines,4 which are frequently utilized as a structural motif in the synthesis of alkaloids or pharmaceutically relevant molecules (Scheme 1).5 In this context, a list of elegant catalytic procedures has been developed for the reduction of pyridines to dihydropyridines with high efficiency and selectivity using H2, boranes, or silanes as reducing agents.6 For instance, Zhou and co-workers reported the 1,4-selective hydrogenation of Hantzsch pyridines to yield the corresponding dihydropyridines under a high pressure of H2.6a Catalytic hydroboration of pyridines was recently shown to operate giving rise to 1,4- or 1,2-dihydropyridines using Lewis acidic boranes6b or metal catalysts including Mg,6c Rh,6d or La.6e Similarly, hydrosilylation of pyridines catalyzed by cationic ruthenium species is known to produce N-silylated 1,4-dihydropyridines.6f–h In addition, Harder et al. developed a calcium complex-catalyzed 1,2-hydrosilylation of pyridines, albeit with limited scope.6i
Scheme 1. Partial reduction of pyridines and the use of dihydropyridines in alkaloid synthesis.

On the other hand, Brookhart et al. reported the preparation and reactivity of a series of chloro- or silylene-bridged dimeric (hydrido)iridium silyl complexes.7,8 Especially, a proposed silylene-bridged Ir dimer [Et2HSiIrH2(μ-SiEt2)]2, among those complexes, was revealed to promote the reductive hydrosilylation of carbonyls and imines.8,9 Given the facile in situ generation of this proposed complex and its notable reactivity particularly towards imines, we envisioned that this type of Ir species may catalyze the hydrosilylative dearomatization of N-heterocycles.
Herein, we report the use of an in situ generated silylene-bridged dinuclear Ir species as a catalyst for the hydrosilylative dearomatization of N-heteroaromatics with diethylsilane,10 furnishing a wide array of partially reduced azacyclic products with excellent selectivity (Scheme 2). This catalyst system shows broad substrate scope and high efficiency (∼1000 turnovers). Significantly, the present procedure does not require external ligands or additives to achieve such attractive features. Mechanistic investigations revealed details of a pathway involving C N insertion into an Ir–H bond of a bimetallic Ir resting species.
Scheme 2. This work: Ir-catalyzed selective hydrosilylation of N-heterocycles.

Results and discussion
Optimization study
At the outset, we presumed that the type of silane would be a crucial factor governing the catalytic activity for the dearomative hydrosilylation of N-heterocycles. When quinoline (2a) was treated with Et2SiH2 (1.5 equiv.) in the presence of 1.4 mol% of [Ir(coe)2Cl]2 in CDCl3, we were pleased to observe that 1-(diethylsilyl)-1,2-dihydroquinoline (3a) was formed in 62% NMR yield in 5 h at 25 °C (Table 1, entry 1). Prolonging the reaction for up to 17 h eventually led to a full conversion of the remaining 2a to 3a. Solvent effects were found to be significant for the catalytic performance: a reaction in THF-d8 gave 3a in 69% yield in 15 h (entry 2), whereas the use of nonpolar solvents such as C6D6 or hexane-d14 resulted in higher yields (85–99% in 4 h, entries 3 and 4). Notably, a reaction in neat Et2SiH2 (3 equiv.) led to a quantitative yield within 2 h (entry 5). Performing the hydrosilylation in a reaction vial resulted in a similar catalytic efficiency (compare entries 6 and 7). It was significant to see that even 0.1 mol% of the iridium precatalyst enabled quantitative 1,2-hydrosilylation of 2a at 55 °C in 60 h either in C6D6 or neat silane (3 equiv.) (entries 8 and 9). While tertiary or bulky secondary silanes were ineffective (entries 10–12), some sterically less demanding secondary (MePhSiH2) and primary silanes (PhSiH3) provided 3a with low efficiency (entries 13 and 14). Subtle structural variation of the iridium precursor also influenced the resulting catalytic activity to some extent (entries 15 and 16, and also see ESI† for details).
Table 1. Optimization of the 1,2-hydrosilylation of quinoline 2a a .
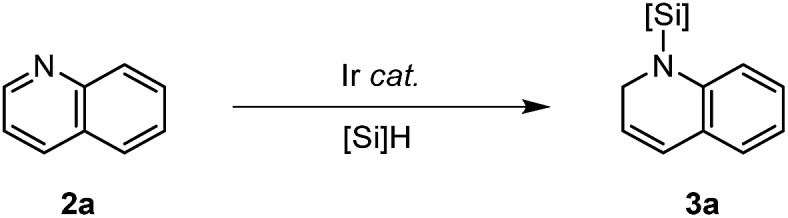
| ||||||
| Entry | Ir catalyst (mol%) | Silane (equiv.) | Solvent | T (°C) | Time (h) | Yield b (%) |
| 1 | [Ir(coe)2Cl]2 (1.4) | Et2SiH2 (1.5) | CDCl3 | 25 | 5 | 62 |
| 2 | [Ir(coe)2Cl]2 (1.4) | Et2SiH2 (1.5) | THF-d8 | 25 | 15 | 69 |
| 3 | [Ir(coe)2Cl]2 (1.4) | Et2SiH2 (1.5) | C6D6 | 25 | 4 | 85 |
| 4 | [Ir(coe)2Cl]2 (1.4) | Et2SiH2 (1.5) | Hexane-d14 | 25 | 4 | 99 |
| 5 | [Ir(coe)2Cl]2 (1.4) | Et2SiH2 (1.5) | Neat | 25 | 2 | 99 |
| 6 | [Ir(coe)2Cl]2 (1.4) | Et2SiH2 (1.5) | C6D6 | 55 | 1 | 99 |
| 7 c | [Ir(coe)2Cl]2 (1.4) | Et2SiH2 (1.5) | C6D6 | 55 | 1 | 99 |
| 8 | [Ir(coe)2Cl]2 (0.1) | Et2SiH2 (1.5) | C6D6 | 55 | 60 | 99 |
| 9 | [Ir(coe)2Cl]2 (0.1) | Et2SiH2 (3.0) | Neat | 55 | 60 | 99 |
| 10 | [Ir(coe)2Cl]2 (4.2) | Et3SiH (1.5) | C6D6 | 85 | 15 | <1 |
| 11 | [Ir(coe)2Cl]2 (4.2) | Me2PhSiH (1.5) | C6D6 | 55 | 15 | <1 |
| 12 | [Ir(coe)2Cl]2 (4.2) | Ph2SiH2 (1.5) | C6D6 | 55 | 15 | <1 |
| 13 | [Ir(coe)2Cl]2 (1.4) | PhMeSiH2 (1.5) | C6D6 | 25 | 4 | 40 |
| 14 | [Ir(coe)2Cl]2 (1.4) | PhSiH3 (1.5) | C6D6 | 25 | 15 | 29 d |
| 15 | [Ir(cod)OMe]2 (1.4) | Et2SiH2 (1.5) | C6D6 | 25 | 4 | 61 |
| 16 | [Ir(cod)Cl]2 (1.4) | Et2SiH2 (1.5) | C6D6 | 25 | 4 | 58 |
aCarried out in a J-Young NMR tube with 2a (0.35 mmol, 1 equiv.), Et2SiH2 (1.5–3.0 equiv.) and [Ir(coe)2Cl]2 (0.1–4.2 mol%), either neat or in solvent (0.35 mL), under an argon atmosphere.
bYield of 3a determined using 1H NMR.
cPerformed in a reaction vial.
dUnidentified side products were also formed.
1,2-Hydrosilylation of quinolines
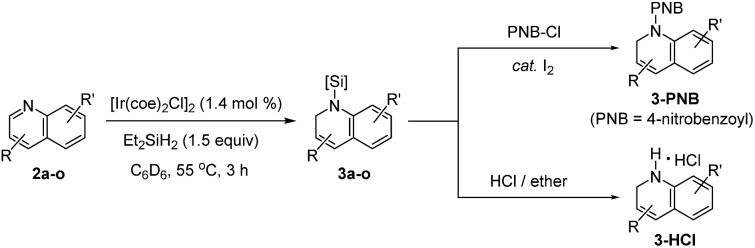
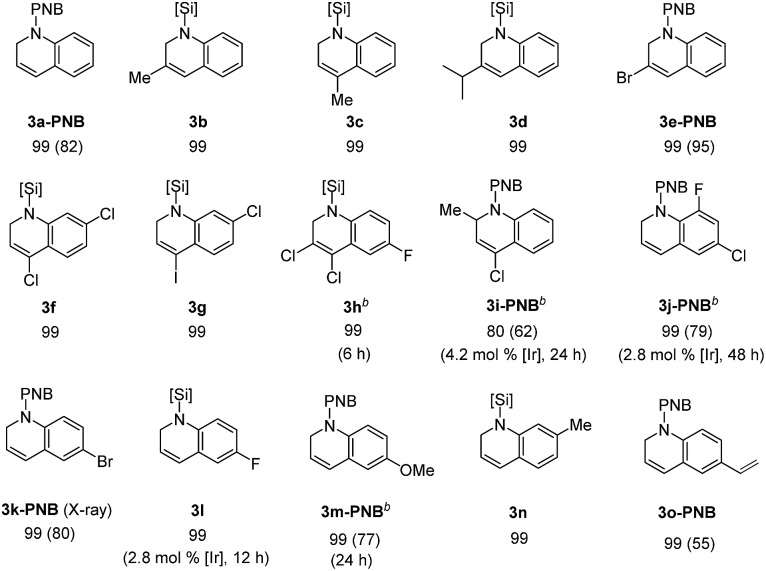
With the optimal conditions in hand (1.4 mol% [Ir(coe)2Cl]2, 1.5 equiv. Et2SiH2, C6D6, 55 °C), we investigated the scope for quinoline substrates (Table 2). An initial attempt to isolate 3a using silica column chromatography failed due to its instability.11 Thus, we conducted an N-benzoylation of 3a, that initially provided a 99% crude yield from 2a, to furnish 3a-PNB (82% isolated yield over two steps). Interestingly, 3a could also be converted to its hydrochloride salt (3a-HCl) in quantitative yield simply by treating the crude reaction mixture with an ether solution of HCl.12
Table 2. Scope of quinolines in the Ir-catalyzed 1,2-hydrosilylation a .
|
|
aSubstrate (0.35 mmol), Et2SiH2 (1.5 equiv.), and [Ir(coe)2Cl]2 (0.005 mmol, 1.4 mol%) in C6D6 (0.35 mL) at 55 °C; crude 1H NMR yields based on an internal standard (mesitylene or 1,4-dioxane); numbers in parentheses are isolated yields.
bWith 5 equiv. of Et2SiH2 (neat).
Similarly, a range of N-silylated reduction products were in situ transformed to their isolable N-acyl (benzoyl or acetyl) amides or HCl salts. 3-Methylquinoline and 4-methylquinoline both reacted to give the desired products 3b and 3c, respectively, in quantitative yields in 3 h. 4-Chloro-2-methylquinoline (2i) and 6-chloro-8-fluoroquinoline (2j) were viable substrates for the 1,2-hydrosilylation, albeit under slightly more forcing conditions [2.8–4.2 mol% of Ir catalyst, neat silane (5 equiv.), and 24–48 h], affording 3i and 3j in 80% and 99% yield, respectively, which were subsequently converted to their benzoyl amides in good yield (3i-PNB; 62% and 3j-PNB; 79% over 2 steps). Quinoline substrates bearing halides at the C-6 position were also facile substrates for this hydrosilylation, providing 3k-PNB (80% over 2 steps) or 3l (99% crude yield). The structure of 3k-PNB was confirmed using X-ray crystallographic analysis.
Quinolines with electron-donating substituents at the C-6 or C-7 position were reduced to the desired N-silylated 1,2-dihydroquinolines (3m–n) in 99% NMR yields, and the former was subsequently isolated as an amide derivative (3m-PNB; 77%). It should be mentioned that a vinyl group remained intact during the hydrosilylation, as demonstrated by the quantitative formation of 3o (isolated as 3o-PNB).
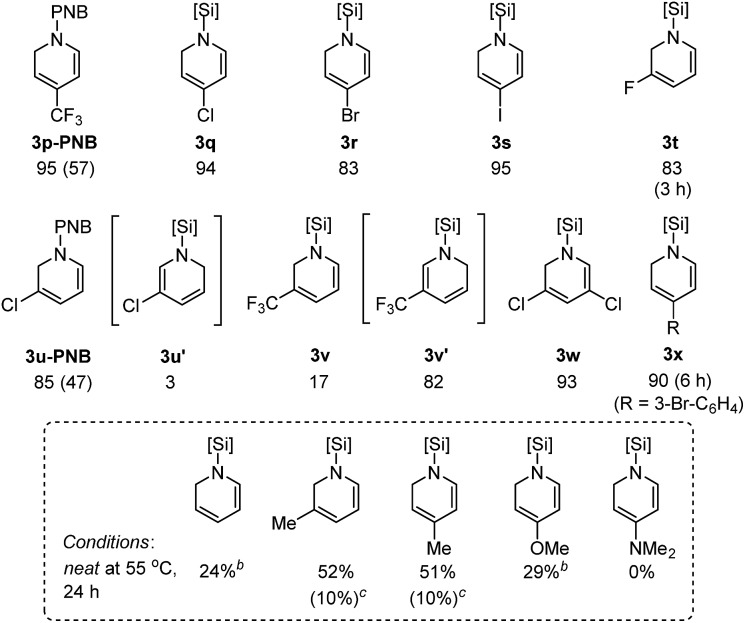
1,2-Hydrosilylation of substituted pyridines
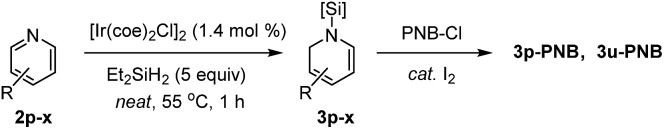
Next, we turned our attention to pyridine substrates which were presumed to be more challenging than quinolines for the present partial reduction (Table 3). With the newly optimized conditions for the hydrosilylation of 4-(trifluoromethyl)pyridine 2p in hand (see ESI† for detailed optimization study), a reaction of 2p in neat Et2SiH2 (5.0 equiv.) was carried out at 55 °C to provide the desired (4-trifluoromethyl)-1,2-dihydropyridine 3p in 95% yield in 1 h, which was subsequently converted to its N-benzoyl derivative (3p-PNB, 57% isolated yield). Similarly, 4-halopyridines were selectively converted to the corresponding N-silyl-1,2-dihydropyridines in high yield without reductive dehalogenation (3q–s). Likewise, 3-halopyridines underwent the dearomative hydrosilylation with high yields obtained (3t–v). Interestingly, the regioselectivity for the hydrosilylation of 3-halopyridines was altered depending on the C3-substituent: 3-fluoropyridine and 3-chloropyridine were 1,2-hydrosilylated selectively (83–85% crude yields) while 3-(trifluoromethyl) pyridine underwent 1,6-hydrosilylation to afford 3v′ in 82% crude yield with only 17% of the 1,2-dihydropyridine product (3v). Although it is still unclear what factors influence the product regioselectivity, comparing the observed catalytic selectivity for 3-fluoropyridine [99% of 1,2-selectivity] with 3-(trifluoromethyl)pyridine [83% of 1,6-selectivity + 17% of 1,2-selectivity], a steric factor seems to be involved in determining the regioselectivity. However, 3-chloropyridine was found to be converted selectively to the corresponding 1,2-dihydropyridine (97% of 1,2-selectivity). Since the steric bulkiness of a chloro group is believed to be comparable to that of a trifluoromethyl group, only steric effects of the 3-substituents cannot account for the regioselectivity observed in our study [e.g. 3-chloropyridine (97% of 1,2-selectivity) vs. 3-(trifluoromethyl)pyridine (17% of 1,2-selectivity)]. In a related study of a La-catalyzed hydroboration of pyridines,6e Marks et al. rationalized that appreciable bonding interactions between the C3-halogen and emerging C2–H2C might influence the dearomatization site selectivity for 3-substituted pyridines.
Table 3. Scope of pyridines a .
|
|
aSubstrate (0.35 mmol), Et2SiH2 (5 equiv.) and [Ir(coe)2Cl]2 (0.005 mmol, 1.4 mol%) at 55 °C; crude 1H NMR yields based on an internal standard (mesitylene or 1,4-dioxane); numbers in parentheses are isolated yields.
b4.2 mol% of [Ir(coe)2Cl]2.
cReaction in C6D6 (0.35 mL).
3,5-Dichloropyridine was reduced smoothly to the corresponding 1,2-dihydropyridine derivative (3w) with a 93% crude yield. A substrate with a 3-bromophenyl group at the C-4 position still displayed high reactivity to afford 3x in good yield. However, the hydrosilylation of pyridine and substituted pyridines bearing electron-donating groups was sluggish under the present conditions as shown in the dotted box (vide infra for possible explanations).
Dearomative hydrosilylation of additional N-heteroaromatics
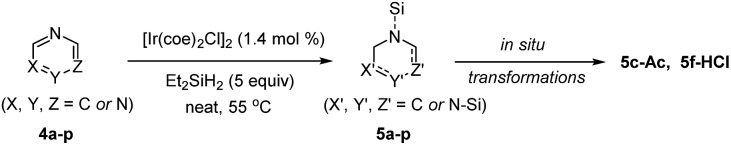
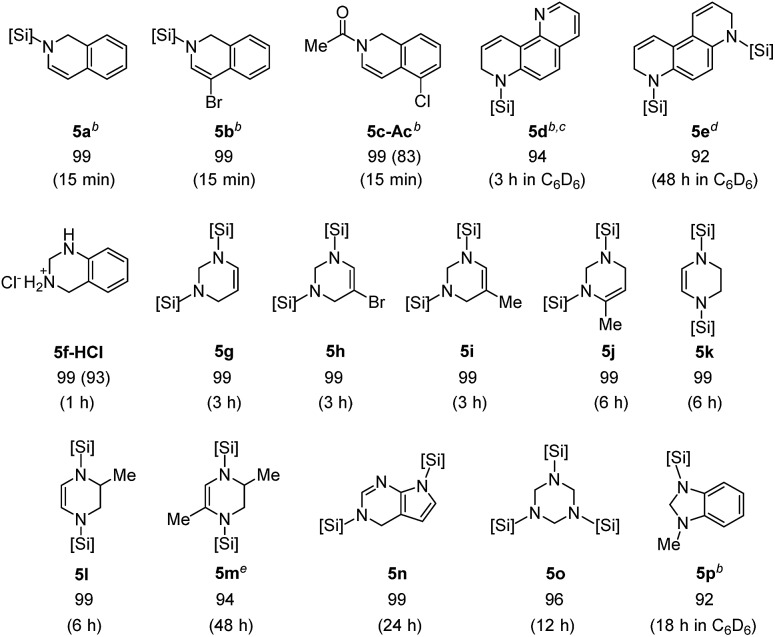
Based on the high reactivity observed for the selective hydrosilylation of quinolines and substituted pyridines, the present iridium catalytic system was next applied to additional types of aromatic N-heterocycles (Table 4). Isoquinolines rapidly reacted with Et2SiH2 (1.5 equiv.) in the presence of [Ir(coe)2Cl]2 (1.4 mol%) to provide N-silyl-1,2-dihydroisoquinolines in quantitative yields within 15 min at 55 °C (5a–c).
Table 4. Substrate scope using N-heteroaromatics a .
|
|
aSubstrate (0.35 mmol), Et2SiH2 (5 equiv.) and [Ir(coe)2Cl]2 (1.4 mol%) at 55 °C; crude 1H NMR yields based on an internal standard (mesitylene or 1,4-dioxane); numbers in parentheses are isolated yields of 5c-Ac and 5f-HCl.
bWith 1.5 equiv. of Et2SiH2.
c2.8 mol% of [Ir(coe)2Cl]2.
dMono-silylated product (5e′, 6%) was also observed.
e4.2 mol% of [Ir(coe)2Cl]2.
An N-silylated dihydroisoquinoline 5c was readily transformed to its N-acetyl amide (5c-Ac, 83% isolated yield), thus demonstrating its high potential utility in synthetic chemistry. The degree of hydrosilylation was found to be controllable in the case of phenanthrolines: 1,7-phenathroline underwent mono-hydrosilylation at the sterically more accessible C N bond with a 94% crude yield by employing 1.5 equiv. of Et2SiH2 (5d), whereas 4,7-phenanthroline was doubly hydrosilylated to give 5e in high yield with 5 equiv. of Et2SiH2. We were pleased to observe that a series of pyrimidines was also selectively reduced with silane to give 1,2,3,4-tetrahydropyrimidines in excellent yields (1–6 h at 55 °C).
It is notable that the reaction of 5-bromopyrimidine was chemoselective without dehalogenation (5h). Similarly, pyrazines underwent double hydrosilylation with excellent selectivity to afford 1,2,3,4-tetrahydropyrazines (5k–m). Interestingly, a reaction of 2-methylpyrazine was regioselective leading to 5l. The current catalytic system was also efficient for the selective hydrosilylation of 7-deazapurine to furnish a partial reduction product 5n in quantitative yield in 24 h, while leaving another C N moiety intact. Gratifyingly, triazine underwent triple hydrosilylation with the use of excess silane (5 equiv.) to give a saturated product 5o in excellent yield in 12 h.13 When 1-methyl-1H-benzo[d]imidazole was subjected to the present conditions, the corresponding hydrosilylated product (5p) was formed in high yield.
Mechanistic studies
(A). Kinetic studies of the Ir-catalyzed 1,2-hydrosilylation
At the outset of the mechanistic investigations, we set two distinctive working hypotheses (Scheme 3). One process was proposed to involve the transfer of a silylium ion from a plausible silyl iridium hydride species (1) to a N-heteroaromatic substrate (Het) to form [(Et2SiH)(Het)]+, followed by hydride attack via an outer-sphere route at the C-2 or C-4 position to give the N-silylated 1,2- or 1,4-dihydropyridine [Scheme 3 (left)]. Another conceivable scenario is an inner-sphere process involving C N insertion of Het into the Ir–H bond of 1 upon prior N-coordination via [Het-1] to form an amido silyliridium complex [N-Ir-Si] that eventually releases the N-silylated 1,2-reduction product through reductive elimination [Scheme 3 (right)].
Scheme 3. Two working hypotheses for the present hydrosilylation.

In order to assess the rate-order of [Ir(coe)2Cl]2, Et2SiH2, and 2a in the 1,2-hydrosilylation, the initial-rate (vi) kinetics of the individual components was studied using 1H NMR spectroscopy (Scheme 4). A plot of vi for the formation of 3avs. a range of initial concentrations of [Ir(coe)2Cl]2 showed that the reaction was first-order in [Ir(coe)2Cl]2, while the initial rate plot across a range of concentrations of Et2SiH2 indicated zero-order for [Et2SiH2]. It is notable that the reaction was fractional-order in terms of the quinoline substrate (2a): zero-order dependence at high concentration (above ∼0.5 M), but approaching to first-order at concentrations of [2a] below ∼0.5 M (see ESI† for details). At present, however, the exact reason for the observed rate-order alteration for 2a is not clear, and requires further rationalization.14
Scheme 4. Observed rate order for each component based on the initial-rate kinetics.

The reactions of a series of substituted quinolines with Et2SiH2 were monitored using 1H NMR to compare their relative rates of hydrosilylation (Scheme 5). The observed initial rates at the early stage (up to 15% conversion of the quinoline substrate) revealed a relevance of the substituents present: 3-Br ∼ 3-I > 4-I > 4-Br > 4-Cl > 4-H > 4-Me (see ESI† for more details).15 Although the relationship between the electron-withdrawing ability of the substituents and the observed initial rates still needs to be further rationalized, the present results at this stage suggest that: (i) softer halide substituents at the para position lead to a higher reaction efficiency than harder ones at the same position; (ii) electron-withdrawing ability partially contributes to the rate enhancement.
Scheme 5. Relative initial rates for substituted quinolines.

(B). Stoichiometric reaction of [Ir(coe)2Cl]2, 3-methyl-pyridine, and Et2SiH2
A reaction of [Ir(coe)2Cl]2 with 15 equiv. of Et2SiH2 in toluene-d8 resulted in the formation of an unsymmetrical structure of two isomeric silylene-bridged Ir dimers [(C8H14)(Et2SiH)Ir(H)2(μ-SiEt2)2Ir(H)3] (I/I′) in a ratio of 3 : 2 (Fig. 1A).161H–1H COSY analysis suggested selective interactions between the olefinic protons of the cyclooctene and the dihydrides at one Ir centre of the bimetallic species (see ESI† for details of the NMR spectroscopic data). It is noteworthy that Brookhart previously proposed a symmetrical structure of a silylene-bridged Ir dimer [(Et2SiH)Ir(H)2(μ-SiEt2)]2, which was assumed to be generated in situ under similar conditions.8 Subsequent addition of 3-methylpyridine (5 equiv.) as a model substrate17 into a solution containing I/I′ and excessive silane led to quantitative conversion of I/I′ into a new bimetallic iridium species (6a) within 10 min at 25 °C (Fig. 1B). A set of signals assignable to the coordinated 3-methylpyridine at δ 8.85 (Hb), 8.36 (Hc), 6.79 (Hd), 6.59 (He), and 1.80 (methyl) appeared accordingly in the 1H NMR spectrum. The Ir-hydride signals at δ –13.01 and –13.20 are assignable to trihydrides and dihydrides, respectively.
Fig. 1. The reaction of [Ir(coe)2Cl]2 (1 equiv.) with Et2SiH2 (15 equiv.) at 25 °C for 10 min in toluene-d8 ((A) blue); 1H and 29Si{1H}NMR spectra obtained 10 min ((B) red) and 18 h ((C) green) after the addition of 3-methylpyridine (5 equiv.) at 25 °C.

As can be seen in Fig. 1B, and since the relative intensity of the 1H peaks at δ 6.59 (He), –13.01, and –13.20 was determined to be 1 : 3 : 2, one molecule of 3-methylpyridine was proposed to coordinate to the bimetallic Ir species forming a pyridine adduct (6a), by assuming that 6a was a pentahydride bimetallic species.18 In addition, a 2D-NOESY (nuclear Overhauser effect spectroscopy) experiment for 6a at –70 °C revealed that one 3-methylpyridine molecule coordinates exclusively to one Ir centre with dihydrides and a terminal silyl group, while the other metal centre does not coordinate to the excess pyridine substrates (see ESI† for the NOESY experiment). For the 29Si spectrum, two peaks at δ 130.4 and 44.5 were assigned to the bridged silylene and terminal silyl moieties, respectively, based on the literature.7,8,19 A peak at δ 78.7 is attributable to a silicon with an η2 coordination mode, leading to the assignment of 6a to a structure that contains a substructure of “–Ir–(Et2)Si–H(η2)···Ir–”. Indeed, a number of binuclear metal complexes with η2 metal–H–Si interactions have been reported,20 among which a silylene-bridged dinuclear titanocene complex [Cp2Ti-(η2-H-SiHPh)(μ-H)TiCp2] is known to have a 29Si resonance at ca. 87 ppm due to the η2-silane moiety.20j Furthermore, regarding the Si–H coupling constant [1JSi–H(η2)] for 6a, we observed two small peaks with a J value of 44 Hz at δ –13.14 and –13.25 (see ESI† for the expanded Ir-hydride region). Fast atom bombardment mass spectrometry (FAB-MS) analysis of this stoichiometric reaction mixture showed a peak that corresponds to a fragment ion of [6a + H]+ (see ESI† for the mass analysis).
(C). Observation of resting species in the hydrosilylation turnover
With a prolonged reaction time, 3-methylpyridine was observed to undergo the 1,2-hydrosilylation process to afford the corresponding 3-methyl-N-silyl-1,2-dihydropyridine in 9% NMR yield in 18 h at 25 °C (Fig. 1C). It is notable that a constant amount of 6a was observed throughout the reaction course. Similarly, a reaction of [Ir(coe)2Cl]2, Et2SiH2, and 3-fluoropyridine 2t (1 : 35 : 35) at 25 °C selectively produced 3-fluoro-N-silyl-1,2-dihydropyridine 3t (36% conversion in 12 h). During this conversion, an analogous resting species of a 2t adduct was also observed, showing a peak at δ –12.97 and multiple peaks over δ –13.2 to –13.5 (1H NMR) in addition to silicon peaks at δ 130.5, 87.2, and 44.4 (see ESI† for details). These results strongly indicate that N-heteroaromatic adducts of a bimetallic Ir species, such as 6a, exist in the catalytic resting state.21
(D). Mechanistic proposal
Based on our mechanistic studies, in combination with the precedent literature,6c–e,i we propose a catalytic cycle for the Ir-catalyzed 1,2-hydrosilylation of N-heteroaromatics (Scheme 6). The initiation step involves the generation of two isomeric iridium olefin adducts I/I′via a reaction of [Ir(coe)2Cl]2 with excess Et2SiH2. The in situ generated I/I′ adducts undergo a facile ligand exchange with the heteroarene substrate to form a substrate-bound bimetallic species (II) as the dominant catalyst resting species.
Scheme 6. Proposed mechanistic pathway for the Ir-catalyzed dearomative 1,2-hydrosilylation of heteroaromatics.

We propose that the key hydride insertion takes place within the coordinated heteroaromatic adduct species. The intramolecular C N insertion into an Ir–H bond of II giving rise to III is believed to be turnover-limiting at high concentrations of substrates. We assume that this step is influenced by the electronic nature of the substrates, leading to higher rates with heteroarenes bearing electron-withdrawing substituents. Intermediate III will subsequently deliver the partially reduced N-silyl product Pvia reductive elimination,6d,22 with the concomitant generation of an unsaturated Ir species IV. Although σ-bond metathesis at the Ir-amido moiety of III6c,6e,23 with excess Et2SiH2 would be an alternative product releasing path, we rather favor the reductive elimination route which would be entropically more beneficial over the metathesis process, particularly in our catalytic system. An inhibitory off-cycle process involving the coordination of P to II leading to V is assumed to be a minor occurrence in the overall process.
Synthetic applications
To prove the practicability of the present catalytic procedure, the 1,2-hydrosilylation of quinolines was examined on a large scale (Scheme 7). Pleasingly, the reaction of quinoline was readily amenable to gram scale synthesis, using a neat reaction mixture with only 0.1 mol% of the iridium precursor [Ir(coe)2Cl]2 at 55 °C, to give 3a-Ac in 90% yield over 2 steps. In a similar manner, the hydrosilylation of 4-methylquinoline was easily scaled up using 1.5 equiv. of Et2SiH2 to afford the N-acetyl amide product (3c-Ac) in good yield. Interestingly, a gram scale reaction of 3-bromoquinoline occurred smoothly at room temperature to give 3e-PNB in 86% yield.
Scheme 7. Synthetic applications.

Epoxidation of isolated compounds 3a-PNB and 3c-PNB was successfully achieved using an in situ generated oxidant, 3-methyl-3-(trifluoromethyl)dioxirane, under mild conditions,24 thus demonstrating the synthetic utility of the partially reduced products obtained through the present catalytic procedure. Cinchona alkaloids are representative compounds with biological activity bearing a quinoline moiety and, therefore, facile transformations of them are important in synthetic and pharmaceutical chemistry.25 When hydroquinine (7a) was subjected to the dearomative hydrosilylation conditions in neat Et2SiH2 (5 equiv.), the desired N-silyl-1,2-reduction product (8a) was formed with a 95% crude yield, which was subsequently transformed to the corresponding ammonium salt, 8a-2HCl.
Conclusions
We have developed a highly efficient, convenient, and scalable procedure for the Ir-catalyzed selective hydrosilylation of a wide array of N-heteroaromatics, including (iso)quinolines, substituted pyridines, pyrimidines, pyrazines, deazapurines, triazines, and benzimidazoles. The reaction occurs smoothly under mild conditions to provide diverse dearomatized azacyclic products in satisfactory yields. The catalyst system requires a [Ir(coe)2Cl]2 precursor only, with an absence of any external ligands or additives. Mechanistic considerations of the kinetics, rate law and NMR studies led us to propose a hydrosilylation path involving intramolecular insertion of a C N unit into an Ir–H bond of a key silylene-bridged Ir dimer. Spectroscopic analysis under various reaction conditions revealed a bimetallic structure of the catalytic resting species. The synthetic utility of the present scalable catalytic procedure was demonstrated through the facile 1,2-hydrosilylation of cinchona alkaloids and successful post-synthetic transformations (e.g. epoxidation) of the partially reduced azacycle products. Considering the attractive features of the present procedure, it is anticipated to find wide application in synthetic and medicinal chemistry. More detailed mechanistic studies, including DFT calculations, on the present catalytic system are now underway and the results will be communicated in due course.
Supplementary Material
Acknowledgments
We thank Prof. Kiyoung Park (KAIST) for valuable discussions. This research was supported by the Institute for Basic Science (IBS-R010-D1) in Korea.
Footnotes
†Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization of new compounds, and X-ray analysis. CCDC 1457941. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc01037g
References
- (a) Krygowski T. M., Cryaňski M. K. Chem. Rev. 2001;101:1385. doi: 10.1021/cr990326u. [DOI] [PubMed] [Google Scholar]; (b) Balaban A. T., Oniciu D. C., Katritzky A. R. Chem. Rev. 2004;104:2777. doi: 10.1021/cr0306790. [DOI] [PubMed] [Google Scholar]
- (a) Boll M. J. Microbiol. Biotechnol. 2005;10:132. doi: 10.1159/000091560. [DOI] [PubMed] [Google Scholar]; (b) Hudlicky T., Gonzalez D., Gibson D. T. Aldrichimica Acta. 1999;32:35. [Google Scholar]; (c) Hudlicky T., Reed J. W. Synlett. 2009:685. [Google Scholar]; (d) Kung J. W., Baumann S., von Bergen M., Müller M., Hagedoorn P.-L., Hagen W. R., Boll M. J. Am. Chem. Soc. 2010;132:9850. doi: 10.1021/ja103448u. [DOI] [PubMed] [Google Scholar]
- (a) Pape A. R., Kaliappan K. P., Kündig E. P. Chem. Rev. 2000;100:2917. doi: 10.1021/cr9902852. [DOI] [PubMed] [Google Scholar]; (b) Roche S. P., Porco J. A. Angew. Chem., Int. Ed. 2011;50:4068. doi: 10.1002/anie.201006017. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bull J. A., J Mousseau J., Pelletier G., Charette A. B. Chem. Rev. 2012;112:2642. doi: 10.1021/cr200251d. [DOI] [PubMed] [Google Scholar]
- (a) Eisner U., Kuthan J. Chem. Rev. 1972;72:1. [Google Scholar]; (b) Sliwa W. Heterocycles. 1986;24:181. [Google Scholar]; (c) Wan J.-P., Liu Y. RSC Adv. 2012;2:9763. [Google Scholar]
- (a) Stout D. M., Meyers A. I. Chem. Rev. 1982;82:223. [Google Scholar]; (b) Lavilla R. J. Chem. Soc., Perkin Trans. 1. 2002:1141. [Google Scholar]; (c) Edraki N., Mehdipour A. R., Khohneviszadeh M., Miri R. Drug Discovery Today. 2009;14:1058. doi: 10.1016/j.drudis.2009.08.004. [DOI] [PubMed] [Google Scholar]; (d) Wender P. A., Schaus J. M., White A. W. J. Am. Chem. Soc. 1980;102:6157. [Google Scholar]; (e) Satoh N., Akiba T., Yokoshima S., Fukuyama T. Angew. Chem., Int. Ed. 2007;46:5734. doi: 10.1002/anie.200701754. [DOI] [PubMed] [Google Scholar]; (f) Mizoguchi H., Oikawa H., Oguri H. Nat. Chem. 2014;6:57. doi: 10.1038/nchem.1798. [DOI] [PubMed] [Google Scholar]; (g) Duttwyler S., Chen S., Lu C., Mercado B. Q., Bergman R. G., Ellman J. A. Angew. Chem., Int. Ed. 2014;53:3877. doi: 10.1002/anie.201310517. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zhao G., Deo U. C., Ganem B. Org. Lett. 2001;3:201. doi: 10.1021/ol006810x. [DOI] [PubMed] [Google Scholar]; (i) Barbe G., Charette A. B. J. Am. Chem. Soc. 2008;130:13873. doi: 10.1021/ja8068215. [DOI] [PubMed] [Google Scholar]
- (a) Chen Q.-A., Chen M. W., Yu C.-B., Shi L. S., Wang D.-S., Yang Y., Zhou Y. G. J. Am. Chem. Soc. 2011;133:16432. doi: 10.1021/ja208073w. [DOI] [PubMed] [Google Scholar]; (b) Fan X., Zheng J., Li Z. H., Wang H. J. Am. Chem. Soc. 2015;137:4916. doi: 10.1021/jacs.5b03147. [DOI] [PubMed] [Google Scholar]; (c) Arrowsmith M., Hill M. S., Hadlington T., Kociok-Köhn G., Weetman C. Organometallics. 2011;30:5556. [Google Scholar]; (d) Oshima K., Ohmura T., Suginome M. J. Am. Chem. Soc. 2012;134:3699. doi: 10.1021/ja3002953. [DOI] [PubMed] [Google Scholar]; (e) Dudnik A. S., Weidner V. L., Motta A., Delferro M., Marks T. J. Nat. Chem. 2014;6:1100. doi: 10.1038/nchem.2087. [DOI] [PubMed] [Google Scholar]; (f) Gutsulyak D. V., Est A. V. D., Nikonov G. I. Angew. Chem., Int. Ed. 2011;50:1384. doi: 10.1002/anie.201006135. [DOI] [PubMed] [Google Scholar]; (g) Lee S.-H., Gutsulyak D. V., Nikonov G. I. Organometallics. 2013;32:4457. [Google Scholar]; (h) Königs C. D. F., Klare H. F. T., Oestreich M. Angew. Chem., Int. Ed. 2013;52:10076. doi: 10.1002/anie.201305028. [DOI] [PubMed] [Google Scholar]; (i) Intemann J., Bauer H., Pahl J., Maron L., Harder S. Chem.–Eur. J. 2015;21:11452. doi: 10.1002/chem.201501072. [DOI] [PubMed] [Google Scholar]
- Park S., Kim B. G., Göttker-Schnetmann I., Brookhart M. ACS Catal. 2012;2:307. [Google Scholar]
- Cheng C., Brookhart M. J. Am. Chem. Soc. 2012;134:11304. doi: 10.1021/ja304547s. [DOI] [PubMed] [Google Scholar]
- Cheng C., Brookhart M. Angew. Chem., Int. Ed. 2012;51:9422. doi: 10.1002/anie.201205154. [DOI] [PubMed] [Google Scholar]
- For our recent reports in the relevant reactions, see: ; (a) Gandhamsetty N., Joung S., Park S.-W., Park S., Chang S. J. Am. Chem. Soc. 2014;136:16780. doi: 10.1021/ja510674u. [DOI] [PubMed] [Google Scholar]; (b) Gandhamsetty N., Park J., Jeong J., Park S.-W., Park S., Chang S. Angew. Chem., Int. Ed. 2015;54:6832. doi: 10.1002/anie.201502366. [DOI] [PubMed] [Google Scholar]; (c) Gandhamsetty N., Park S., Chang S. J. Am. Chem. Soc. 2015;137:15176. doi: 10.1021/jacs.5b09209. [DOI] [PubMed] [Google Scholar]
- Forrest T. P., Dauphinee G. A., Deraniyagala S. A. Can. J. Chem. 1985;63:412. [Google Scholar]
- Although the quinolinium salt 3a-HCl slowly underwent rearomatization over time in DMSO-d6 at 25 °C, returning to quinoline 2a, solid 3a-HCl was stable enough to be stored at –30 °C for over 1 week without being rearomatized
- Treatment of 4f with 1.1 equiv. of Et2SiH2 under otherwise identical conditions gave a mixture of isomeric mono-hydrosilylated products, while the reaction of 4l led to a mixture of doubly reduced product 5l and the remaining starting material 4l (see ESI for the crude NMR spectra)
- Initially, we assumed an inhibition effect via coordination of 3a and/or cyclooctene, which might lead to the alteration of the rate order from zero to first. However, no inhibition effect was found in terms of the reaction rate, as it was evident that hydrosilylations of 2a with Et2SiH2 in the presence of a significant amount of 3a or cyclooctene showed a catalytic activity similar to that obtained under the standard conditions (see ESI for details)
- A similar reactivity trend was observed for the La-catalyzed 1,2-hydroboration of substituted pyridines, in which the turnover frequency (TOF) increased in the order of 3-I > 3-Cl > 3-Br > 3-F ∼ 3-H, while 3-halopyridines were more reactive than the same type of 4-halopyridines (ref. 6e)
- Two types of hydride resonance at –12.5 and –12.8 ppm, assignable to Ir-(H)3 and Ir-(H)2 respectively for both I/I′, were clearly separate from each other in 1H NMR studies over a range of –70 to 25 °C without coalescence, suggesting that the hydrides at each Ir center are fluxional even at –70 °C, but do not undergo cross-exchange between the two Ir centers at least at 25 °C (see ESI for this variable temperature experiment)
- In order to observe the resting state(s) of the present process, we preliminarily carried out the reaction of quinoline 2a with Et2SiH2 under various conditions. It turned out that multiple Ir-hydride species were formed as the catalytic turnover increased. This rendered the resting species difficult to characterize. For this reason, 3-methylpyridine which had displayed lower reactivity under the catalytic conditions was chosen as a model substrate
- Each hydride signal at δ –13.01 and –13.20 remained sharp over a range of –70 to 60 °C without broadening and/or coalescence (see ESI for this variable temperature experiment). This observation suggests that the hydrides of 6a (or 6a′) do not undergo scrambling between the two bridged Ir centers. For relevant discussions on this issue, see: ; (a) Taw F. L., Bergman R. G., Brookhart M. Organometallics. 2004;23:886. [Google Scholar]; (b) Liang X., Crabtree R. H. J. Am. Chem. Soc. 1989;111:2527. [Google Scholar]; (c) Duckett S. B., Perutz R. N. J. Chem. Soc., Chem. Commun. 1991:28–31. [Google Scholar]; (d) Tanke R. S., Crabtree R. H. Organometallics. 1991;10:415. [Google Scholar]; (e) Delpech F., Sabo-Etienne S., Chaudret B., Daran J.-C. J. Am. Chem. Soc. 1997;119:3167. [Google Scholar]; (f) Park S., Brookhart M. J. Am. Chem. Soc. 2012;134:640. doi: 10.1021/ja209567m. [DOI] [PubMed] [Google Scholar]; (g) Metsänen T. T., Hrobárik P., Klare H. F. T., Kaupp M., Oestreich M. J. Am. Chem. Soc. 2014;136:6912. doi: 10.1021/ja503254f. [DOI] [PubMed] [Google Scholar]
- (a) Calimano E., Tilley T. D. Dalton Trans. 2010;39:9250. doi: 10.1039/b925856f. [DOI] [PubMed] [Google Scholar]; (b) Calimano E., Tilley T. D. J. Am. Chem. Soc. 2009;131:11161. doi: 10.1021/ja903737j. [DOI] [PubMed] [Google Scholar]
- For selected reviews on transition metal–silane σ-complexes, see: For examples of dinuclear transition metal complexes with M–H–Si interactions in an η2-mode, see: ; (a) Nikonov G. Adv. Organomet. Chem. 2005;53:217. [Google Scholar]; (b) Corey J. Y. Chem. Rev. 2011;111:863. doi: 10.1021/cr900359c. [DOI] [PubMed] [Google Scholar]; (c) Schubert U. Adv. Organomet. Chem. 1990;30:151. [Google Scholar]; (d) Lin Z. Chem. Soc. Rev. 2002;31:239. doi: 10.1039/b106620j. [DOI] [PubMed] [Google Scholar]; (e) Schneider J. I. Angew. Chem., Int. Ed. Engl. 1996;108:1132. [Google Scholar]; (f) Crabtree R. H. Angew. Chem., Int. Ed. Engl. 1993;32:789. [Google Scholar]; (g) Lachaize S., Sabo-Etienne S. Eur. J. Inorg. Chem. 2006:2115–2127. [Google Scholar]; (h) Bennett M. J., Simpson K. A. J. Am. Chem. Soc. 1971;93:7156. [Google Scholar]; (i) Ciriano M., Green M., Howard J. A. K., Proud J., Spencer J. L., Stone F. G. A., Tsipis C. A. J. Chem. Soc., Dalton Trans. 1978:801. [Google Scholar]; (j) Aitken C. T., Harrod J. F., Samuel E. J. Am. Chem. Soc. 1986;108:4059. [Google Scholar]; (k) Fryzuk M. D., Rosenberg L., Rettig S. J. Organometallics. 1991;10:2537. [Google Scholar]; (l) Suzuki H., Takao T., Tanaka M., Moro-oka Y. J. Chem. Soc., Chem. Commun. 1992:476. [Google Scholar]; (m) Takao T., Yoshida S., Suzuki H. Organometallics. 2005;24:521. [Google Scholar]
- For examples of binuclear metal complexes in catalysis, see: ; (a) Van den Beuken E. K., Feringa B. L. Tetrahedron. 1998;54:12985. [Google Scholar]; (b) Li H., Marks T. J. Proc. Natl. Acad. Sci. U. S. A. 2006;103:15295. doi: 10.1073/pnas.0603396103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Steinhagen H., Helmchen G. Angew. Chem., Int. Ed. 1996;35:2339. [Google Scholar]; (d) Broussard M. E., Juma B., Train S. G., Peng W.-J., Laneman S. A., Stanley G. G. Science. 1993;260:1784. doi: 10.1126/science.260.5115.1784. [DOI] [PubMed] [Google Scholar]; (e) McKenzie C. J., Robson R. Chem. Commun. 1988:112. [Google Scholar]; (f) Hostetler M. J., Bergman R. G. J. Am. Chem. Soc. 1990;112:8621. [Google Scholar]; (g) Hostetler M. J., Butts M. D., Bergman R. G. J. Am. Chem. Soc. 1993;115:2743. [Google Scholar]; (h) Guo N., Stern C. L., Marks T. J. J. Am. Chem. Soc. 2008;130:2246. doi: 10.1021/ja076407m. [DOI] [PubMed] [Google Scholar]; (i) Nagashima H., Suzuki A., Iura T., Ryu K., Matsubara K. Organometallics. 2000;19:3579. [Google Scholar]; (j) Hashimoto H., Aratani I., Kabuto C., Kira M. Organometallics. 2003;22:2199. [Google Scholar]
- (a) Hopmann K. H., Bayer A. Coord. Chem. Rev. 2014;268:59. [Google Scholar]; (b) Herrera V., Muñoz B., Landaeta V., Canudas N. J. Mol. Catal. A: Chem. 2001;174:141. [Google Scholar]; (c) Nakatani N., Hasegawa J.-Y., Sunada Y., Nagashima H. Dalton Trans. 2015;44:19344. doi: 10.1039/c5dt02767e. [DOI] [PubMed] [Google Scholar]; (d) Brown M., Cooley N. A. Chem. Rev. 1988;88:1031. [Google Scholar]; (e) Driver M. S., Hartwig J. F. J. Am. Chem. Soc. 1995;117:4708. [Google Scholar]
- Hao L., Harrod J. F., Lebuis A.-M., Mu Y., Shu R., Samuel E., Woo H.-G. Angew. Chem., Int. Ed. 1998;37:3126. doi: 10.1002/(SICI)1521-3773(19981204)37:22<3126::AID-ANIE3126>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Yang D., Wong M.-K., Yip Y.-C. J. Org. Chem. 1995;60:3887. [Google Scholar]
- (a) Kolb H. C., VanNieuwenhze M. S., Sharpless K. B. Chem. Rev. 1994;94:2483. [Google Scholar]; (b) Johnson R. A. and Sharpless K. B., in Catalytic Asymmetric Synthesis, ed. I. S. Ojima, John Wiley & Sons, Inc., New York, 2000. [Google Scholar]; (c) Maruoka K., in Asymmetric Phase Transfer Catalysis, Wiley-VCH Verlag GmbH, Weinheim, 2008. [Google Scholar]; (d) Houk K. N., List B. Acc. Chem. Res. 2004;37:487. doi: 10.1021/ar0300571. [DOI] [PubMed] [Google Scholar]; (e) Doyle A. G., Jacobsen E. N. Chem. Rev. 2007;107:5713. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]; (f) Hashimoto T., Maruoka K. Chem. Rev. 2007;107:5656. doi: 10.1021/cr068368n. [DOI] [PubMed] [Google Scholar]; (g) Song C.-E., in Cinchona Alkaloids in Synthesis & Catalysis: Ligands, Immobilization and Organocatalysis, Wiley-VCH Verlag GmbH, Weinheim, 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.