

Key Points
161533 TriKE–treated MDS NK cells proliferate and become activated to overcome tumor-induced NK cell dysfunction.
IL-15 induces the inhibitory checkpoint TIGIT on NK cells, but not when IL-15 is presented in the context of 161533 TriKE.
Abstract
Myelodysplastic syndrome (MDS) is a clonal heterogeneous stem cell disorder driven by multiple genetic and epigenetic alterations resulting in ineffective hematopoiesis. MDS has a high frequency of immune suppressors, including myeloid-derived suppressor cells (MDSCs), that collectively result in a poor immune response. MDSCs in MDS patients express CD155 that ligates the T-cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT) and delivers an inhibitory signal to natural killer (NK) cells. To mediate a productive immune response against MDS, negative regulatory checkpoints, like TIGIT, expressed on MDS NK cells must be overcome. NK cells can be directed to lyse MDS cells by bispecific killer engagers (BiKEs) that ligate CD16 on NK cells and CD33 on MDS cells. However, such CD16 × CD33 (1633) BiKEs do not induce the proliferative response in MDS NK cells needed to sustain their function. Here, we show that the addition of an NK stimulatory cytokine, interleukin-15 (IL-15), into the BiKE platform leads to productive IL-15 signaling without TIGIT upregulation on NK cells from MDS patients. Lower TIGIT expression allowed NK cells to resist MDSC inhibition. When compared with 1633 BiKE, 161533 trispecific killer engager (TriKE)–treated NK cells demonstrated superior killing kinetics associated with increased STAT5 phosphorylation. Furthermore, 161533 TriKE–treated MDS NK cells had higher proliferation and enhanced NK-cell function than 1633 BiKE–treated cells without the IL-15 linker. Collectively, our data demonstrate novel characteristics of the 161533 TriKE that support its application as an immunotherapeutic agent for MDS patients.
Visual Abstract
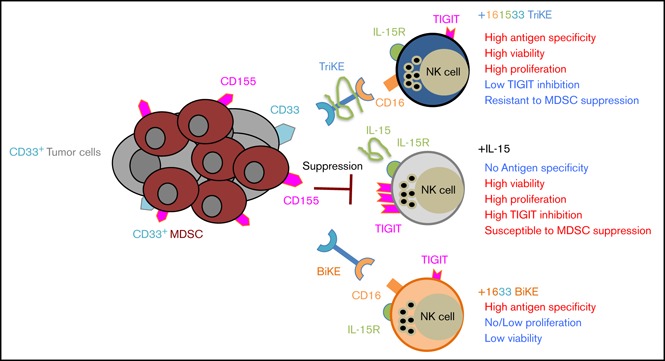
Introduction
The clonal disease of myelodysplastic syndrome (MDS) is characterized by morphological dysplasia, ineffective hematopoiesis leading to cytopenias, and risk of transformation to acute myeloid leukemia (AML).1,2 MDS incidence rates have dramatically increased in the population of the United States from 3.3 per 100 000 individuals from 2001-2004 to 70 per 100 000 annually3,4 and is especially prevalent in elderly patients (median age of 76 years at diagnosis).2 The median survival of patients with high-risk MDS is 7 months, as advanced age reduces eligibility for potentially curative allogeneic hematopoietic cell transplantation (allo-HCT).5 When allo-HCT is not an option, 3 chemotherapeutic agents have been approved by the US Food and Drug Administration for MDS. The hypomethylating agents azacitidine and decitabine reverse transcriptional inhibition of tumor-suppressor and DNA repair genes, whereas lenalidomide, an angiogenesis inhibitor, diminishes immunomodulation and anti-inflammatory changes.6 Given poor outcomes in patients who receive current drug therapies, more research is needed to develop and define novel therapeutic approaches.7
Natural killer (NK) cells are cytotoxic lymphocytes of the innate immune system that have been increasingly recognized in immune surveillance against cancer.8-10 Studies from our laboratory and others have shown the therapeutic potential of NK cells in the treatment of cancer. NK-cell function can be augmented by the use of monoclonal antibody therapies or through novel single-chain variable fragment (scFv) recombinant reagents termed bispecific and trispecific killer cell engagers (BiKEs and TriKEs), which target both the CD16 activating receptor expressed on mature NK cells and tumor antigens.11-13 We have shown that a CD16 × CD33 (1633) BiKE effectively activates blood and marrow MDS-NK cells to lyse CD33+ MDS cells.12 Due to its prominent role in NK cell development, homeostasis, proliferation, survival, and activation,14 a novel modified human interleukin-15 (IL-15) crosslinker was genetically engineered into the 1633 BiKE platform to improve NK-cell function in the tumor microenvironment.13 The modified IL-15 in the 161533 TriKE augmented healthy donor NK function and corrected posttransplant AML patient NK cell dysfunction. Additionally, 161533 TriKE improved in vivo NK cell expansion and tumor control in mice compared with the 1633 BiKE.13 Previously we have shown that soluble IL-15 and antibody engagement of CD16 increased MDS-NK inhibitory receptor T-cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT) expression, rendering canonical NK cells susceptible to myeloid-derived suppressor cell (MDSC)–mediated suppression15; however, how TriKE treatment affects TIGIT expression on NK cells remains unknown.
MDSCs are a heterogeneous population of immature myeloid and granulocytic cells that acquire immunosuppressive properties. In humans, monocytic MDSCs are commonly identified by the expression of CD11b, CD33, and CD14 and lack or low expression levels of HLA-DR, whereas granulocytic MDSCs express CD33 and CD15/CD66b with low or no HLA-DR levels.16 MDSC expansion and activation have been associated with cancer and impaired immune effector cell function, including NK cells.17-21 In the current study, we evaluated the effects of an IL-15 linker within a TriKE (161533) that contained the engager moieties anti-CD16 and anti-CD33 to determine whether MDS-NK cell dysfunction could be overcome by this unique configuration.
Material and methods
Patients and healthy donors
Peripheral blood mononuclear cells (PBMCs) were obtained fresh or cryopreserved from MDS (myelodysplastic syndrome and myeloproliferative disease) patients (n = 16) or healthy donors (HDs) after Ficoll-Paque density gradient purification. Patient characteristics are listed in Table 1. Blood and patient samples were obtained from the National Marrow Donor Program/Center for International Blood and Marrow Transplant Research Repository and Memorial Blood Bank (Minneapolis, MN). All samples were deidentified before receipt, and use was approved by the University of Minnesota and National Marrow Donor Program institutional review boards in accordance with the Declaration of Helsinki.
Table 1.
Patient characteristics
| Patient number | Diagnosis | Age, y | Sex | Disease status |
|---|---|---|---|---|
| 1 | RAEB | 22 | Female | Advanced |
| 2 | RAEB | 53 | Female | Advanced |
| 3 | CIM | 63 | Female | Early |
| 4 | RA | 59 | Female | Early |
| 5 | RA | 46 | Male | Early |
| 6 | RAEB | 27 | Male | Advanced |
| 7 | Chronic MPS disorder | 42 | Male | Early |
| 8 | RA | 37 | Female | Early |
| 9 | RARBT | 24 | Male | Advanced |
| 10 | RAEB | 50 | Female | Advanced |
| 11 | CMML | 55 | Male | Advanced |
| 12 | OMM | 32 | Male | Early |
| 13 | RAEB | 57 | Male | Advanced |
| 14 | RARBT | 52 | Male | Advanced |
| 15 | RA | 41 | Female | Early |
CIM, chronic idiopathic myelofibrosis; CMML, chronic myelomonocytic leukemia; OMM, other myelodysplasia or myeloproliferative; RA, refractory anemia; RAEB, refractory anemia with excess blasts; RARBT, refractory anemia with excess blasts in transform.
Cell isolation
Peripheral blood CD33+ monocytes from HDs were isolated by magnetic positive selection (Miltenyi Biotec) and seeded at 2 × 106/mL in RPMI medium containing 10% heat-inactivated fetal bovine serum (FBS), IL-6 (10 ng/mL, Sigma-Aldrich), and granulocyte-macrophage colony-stimulating factor (10 ng/mL, R&D Systems) for 1 week and refreshed on day 3 of culture to generate MDSCs.22 NK cells were isolated from overnight-rested PBMCs by negative depletion (STEMCELL Technologies or Miltenyi Biotec). Control monocytes were isolated from overnight-rested PBMCs using anti-CD33 microbeads.
BiKEs and TriKEs
161533 TriKE molecule generation and protein production were described previously.13 In brief, a fragment encoding a G4S linker juxtaposed the VL and VH genes of the scFv. The 161533 NcoI/XhoI gene fragment encoded a start codon consisting, in sequential fashion, of an anti-human CD16 scFv,12 a 20-amino-acid flanking sequence (PSGQAGAAASESLFVSNHAY), human IL-15 with N72D substitution,23 a 7-amino-acid flanking sequence (EASGGPE), and an anti-CD33 scFv. Plasmid was transformed into the Escherichia coli strain BL21 (DE3) (EMD). Following 2-hour incubation, bacteria were harvested and cell pellets suspended and homogenized using a polytron homogenizer. After sonication and centrifugation, pellets were extracted and inclusion bodies extensively washed to remove endotoxin. Proteins were refolded using a sodium N-lauroyl-sarcosine air oxidation method.13 Refolded 161533 was purified by fast protein liquid ion exchange chromatography (Q Sepharose Fast Flow).13
Flow cytometry analysis
Cryopreserved PBMCs from MDS patients and HDs were cultured overnight in medium supplemented with 1633 BiKE (50 nM) or 161533 TriKE (50 nM) or left untreated. NK cell degranulation, interferon-γ (IFN-γ) production, and proliferation (Ki67) were evaluated after 6-hour stimulation with HL60 (20:1, adjusted to mean NK cell frequencies) prior to staining. Cells were stained with fluorochrome-conjugated antibodies as described in supplemental Table 1. Detection of CD107a (degranulation), Ki67 (proliferation), and IFN-γ production was performed after fixation and permeabilization (eBioscience) according to the manufacturer’s instructions. Cells were treated with the protein transport inhibitors Golgistop and Golgiplug 6 hours before staining. Alternatively, purified NK cells from HDs were cocultured with monocytes or MDSC for 5 days in the presence of IL-15 (10 nM, equivalent molar concentration to the IL-15 in TriKE), BiKE (50 nM), BiKE+IL-15, or TriKE (50 nM) and analyzed for NK-cell function. All cells were acquired by LSRII and analyzed by FlowJo 10.0.
Flow cytometry–based MDS target killing assay
Purified fresh HD NK cells and cryopreserved bone marrow (BM) samples from MDS patients were overnight rested in RPMI supplemented with 10% FBS for recovery. NK cells were added to CellTrace violet (5 μM, Invitrogen) fluorescently labeled MDS BM targets, and target killing was evaluated using Live/Dead dye (Invitrogen) after a 6-hour incubation at an effector to target (E:T) ratio of 2:1. Cultures were treated with IL-15 (30 nM), 1633 BiKE (30 nM), or 161533 TriKE (30 nM) or left untreated throughout the assay. MDS blast killing was assessed by gating on an intermediate CD45 and SSC low population, further gated to CD117+ and CD34+ cells within the CellTrace-positive population and assessed for the proportion of Live/Dead+ cells.
Phosflow
NK cells from HDs were stimulated with IL-15 (10 nM, equivalent molar concentration to the IL-15 in TriKE), BiKE (50 nM), or TriKE (50 nM) in the presence or absence of HL60 for 25 minutes before analysis of STAT5 phosphorylation. Cells were fixed and permeabilized (eBioscience) and stained for pSTAT5 (pY694) (BD Biosciences).
Live single-cell analysis
Cells
The HL60 cell line was maintained in RPMI 1640 supplemented with 10% FBS, 50 U/mL penicillin, and 50 mg/mL streptomycin. Human NK cells were freshly isolated from PBMCs of anonymous HDs by negative depletion (Miltenyi biotech). Isolated NK cells were cultured in RPMI 1640 supplemented with 10% FBS, 50 U/mL penicillin, and 50 mg/mL streptomycin for 12 to 24 hours before microwell imaging experiments.
Microwell assay and imaging
NK cells were stained with 0.5 μM CellTrace calcein red-orange AM and HL60 target cells with 1 μM CellTrace calcein green AM and 5 μM Far Red DDAO-SE or 2.5 μM CellTrace far red (all from Thermo Fisher Scientific) at 37°C for 10 to 20 minutes. Stained NK cells and stained target cells were seeded onto a silicon-glass microchip divided into 2 separate compartments containing 50 nM of either 161533 TriKE or 1633 BiKE. Each compartment covered at least 2500 microwells with dimensions of 50 × 50 × 300 μm3. Imaging of the microchip was performed using a Zeiss LSM 880 microscope equipped with an environmental chamber kept at 37°C and 5% CO2. Images were acquired using a 10× objective every 3 minutes for 9 hours.
Image analysis
The number of NK cells, as well as live and dead target cells in each well, was quantified automatically by a previously described software routine developed in MATLAB.24 Briefly, the microwells were segmented using the bright-field channel. Thresholds and a Gaussian filter were applied to the fluorescence channels. Target cells were detected using an algorithm based on the circular Hough transform.25 NK cells were detected by an algorithm for finding regional intensity peaks corresponding to single NK cells.25 NK cell–induced target cell death is accompanied by leakage of the calcein dye and loss of the green fluorescence signal. An automated algorithm detecting drops in the total calcein intensity from each well was used to find the time points of individual killing events. Tracking individual NK cells and adding up the distance migrated between each consecutive time point and dividing with the elapsed time calculated the average migration speed.
Live kinetic analysis of tumor cell killing
HL60 (acute promyelocytic leukemia) and MV-4-11 (biphenotypic B myelomonocytic leukemia, authenticated from ATCC, used within 3 months of the first passage) were labeled with red fluorescent CellTracker (5 μM, Invitrogen). For analysis of tumor cell killing, HL60 and MV-4-11 cells were plated at a concentration of 1 × 104 cells per well in 96-well flat bottom plates. Prior to analysis, NK cells were added at a 3:1 ratio onto the target cells. The number of killed target cells was monitored by hourly fluorescence imaging over 48 hours using an IncuCyte Live Cell Analysis System (Essen BioScience). Percent killing was quantified using IncuCyte Zoom software (Essen BioScience) and normalized to the number of spontaneous cells death in the target cell only control group according to the following calculation formula: (% killing = (overlap counts/red counts) × 100).
Statistical analysis
All data were first analyzed in the software mentioned above and summarized by Prism Version 6 software (GraphPad). All data were first tested for normal distribution. Thereafter, differences among groups were analyzed by a Student t test, 1- or 2-way analysis of variance (ANOVA), or nonparametric Mann-Whitney U tests (as indicated in the figure legends). Representative histograms or images were chosen based on the average values.
Results
161533 TriKE does not increase TIGIT expression in HD or MDS NK cells
We have previously shown that MDSCs are increased in MDS patients and suppress NK-cell function. Suppression of MDS-NK–cell function occurs as the result of high expression of the TIGIT ligand CD155 on MDS-MDSCs, which triggers inhibition of NK-cell function via TIGIT engagement.15 In the present study, we sought to examine whether 161533 TriKE would induce TIGIT expression in NK cells as seen with soluble recombinant IL-15. PBMCs from HDs and MDS patients were activated overnight with IL-15 or TriKE or left untreated alone or in the presence of HL60 target cells before analysis of TIGIT expression. In striking contrast to soluble IL-15 stimulation, IL-15 in the form of 161533 TriKE did not induce TIGIT expression on NK cells, as evidenced by significantly lower mean fluorescence intensity (MFI) in the absence of HL60 targets (731 ± 179 vs 250 ± 94, P = .04; Figure 1A, top and middle left panel). Similar findings were observed in MDS patient samples (MFI 790 ± 207 vs 188 ± 51, P = .02; Figure 1A, bottom left panel). Incubating PBMCs from HDs and MDS patients with HL60 target cells together with IL-15 induced significantly higher TIGIT expression compared with 161533 TriKE+HL60 in both HD (MFI [n = 6] 1017 ± 63 vs 342 ± 68, P < .0001; Figure 1B, top and middle right panel) and MDS NK cells (MFI [n = 11] 1105 ± 100 vs 200 ± 30, P < .0001; Figure 1B, bottom right panel). Stimulating PBMCs from MDS patients with 1633 BiKE did not induce TIGIT expression on NK cells (supplemental Figure 1A). In contrast, treating PBMCs with a combination of 1633 BiKE and exogenous soluble IL-15 drastically increased TIGIT expression on NK cells (supplemental Figure 1A). These data indicate that in the presence of tumor cells, 1633 BiKE or 161533 TriKE does not elevate NK cell TIGIT expression compared with soluble recombinant IL-15.
Figure 1.

161533 TriKE does not increase TIGIT expression in HD or MDS NK cells. (A-B) Cryopreserved PBMCs from HDs (n = 6) and MDS patients (n = 5) were activated overnight with IL-15 or 161533 TriKE in the absence (A) or presence (B) of HL60 target cells and analyzed for NK cell TIGIT expression. One representative histogram (from HD) and pooled data are shown, and aggregate data are shown as mean ± standard error of the mean (SEM). Statistical analysis was performed using an unpaired Student t test.
161533 TriKE overcomes MDSC-mediated immune suppression, facilitating effective NK cell lysis of tumor cells
NK cells stimulated with 1633 BiKE are able to kill in vitro–generated CD33+ MDSCs as well as autologous CD33+ MDS targets in a 4-hour cytotoxicity assay.12 Given the low induction of TIGIT expression following 161533 TriKE treatment, we sought to investigate whether 161533 TriKE treatment of NK cells would overcome MDSC-mediated suppression in a longer-term 5-day assay.
Purified NK cells from HDs were cultured with monocytes or in vitro–generated MDSCs in the presence of IL-15, 1633 BiKE, or 161533 TriKE for 5 days prior to exposure to HL60 targets. NK cell degranulation, IFN-γ production, and proliferation were suppressed in the presence of MDSCs and IL-15 in contrast to monocyte cocultures (CD107a: 40% ± 2% vs 56 ± 3, P = .001; IFN-γ: 20 ± 3 vs 44% ± 5%, P = .005; Ki67: 29% ± 4% vs 51% ± 2%, P = .001; Figure 2A). Culture with 1633 BiKE alone without cytokine supplementation resulted in overall low NK cell activity as a result of poor NK cell survival (supplemental Figure 1B). In contrast, treating NK cells with 161533 TriKE maintained NK-cell function and proliferation in the presence of MDSCs (Figure 2A). Short-term overnight priming of NK cells with IL-15 or 161533 TriKE followed by washing and then coculture with MDSCs for 5 days did not rescue NK cells from MDSC suppression. The data show that 161533 TriKE is required throughout the duration of the 5-day culture to resist MDSC suppression, indicating a requirement for a sustained IL-15 survival signal (supplemental Figure 1C).
Figure 2.

161533 TriKE–treated NK cells overcome MDSC-induced immune suppression. (A) Purified HD-NK cells (n = 6) were cocultured with autologous monocytes or MDSCs for 5 days in the presence of IL-15 (equal molar concentration to the IL-15 in TriKE [50 nM], BiKE [50 nM], or TriKE [50 nM]. HL60 target cells were added 6 hours before staining and assessed for NK cell degranulation (CD107a), IFN-γ production, and proliferation (Ki67). Data are shown as mean ± SEM and statistical analyses were performed using paired Student t test. (B-C) NK cells (n = 4) were added at a 3:1 ratio onto red fluorescent CellTracker-labeled HL60 (B) and MV-4-11 (C) targets and then analyzed for tumor cell killing monitored by hourly fluorescence imaging over 48 hours using an IncuCyte Live Cell Analysis System. Percent killing was quantified using IncuCyte Zoom software (Essen BioScience) and normalized to the number of cells death in the target cell only control group. Statistical analyses of the slopes over time were assessed using a 2-way ANOVA. NS, not significant.
The ability of 161533 TriKE to provoke NK cell escape from suppression was evaluated further in a 30-hour live imaging assay using the IncuCyte Zoom platform (Figure 2B-C). Labeled malignant CD33+ myeloid cell lines that are intermediate (HL60) or highly (MV-4-11)26 resistant to resting NK cell killing were plated with NK cells in the presence of MDSCs or monocytes and the indicated drugs. TriKE alone was the only reagent treatment able to maintain normal NK cell cytotoxicity against HL60 and MV-4-11 targets in the presence of MDSCs (Figure 2B-C). Collectively, these data indicate that soluble IL-15 priming, even in the presence of BiKE, is insufficient to rescue NK cells from immune suppression, in contrast to IL-15 in the context of the TriKE reagent.
161533 TriKE induces robust NK cell killing dynamics
To better understand the changes in NK-cell function mediated by TriKE vs BiKE, state-of-the-art microchip-based live-cell imaging was used to evaluate NK-cell function and target dynamics. Unstimulated NK cells and HL60 target cells were stained with distinct CellTracker dyes. The stained NK cells and target cells were then seeded onto a silicon-glass microchip, which was divided into 2 separate compartments containing either 161533 TriKE or 1633 BiKE and assessed by live imaging for 9 hours. Based on single-cell analysis, we found that 161533 TriKE had significantly higher NK cell cytotoxicity against HL60 target cells compared with 1633 BiKE (58% ± 3% vs 38% ± 2%, P = .009, Figure 3A). NK cells cultured in the presence of 161533 TriKE killed their target remarkably faster than 1633 BiKE incubation (time to first kill: 109 ± 6 vs 146 ± 6 minutes, P < .0001; Figure 3B-C, left). NK cells treated with 161533 TriKE were more mobile than NK cells treated with 1633 BiKE (average migration speed: 1.4 ± 0.01 vs 1.8 ± 0.01 μm/min, P < .0001; Figure 3C, right). Furthermore, 161533 TriKE treated NK cells were more likely than controls to be serial killers (Figure 3D; supplemental movie). Our data show that 161533 TriKE robustly improved NK cell killing dynamics, suggesting that IL-15 and CD16 signaling together contribute to the enhanced function of 161533 TriKE–treated NK cells.
Figure 3.

161533 TriKE induces robust NK cell killing dynamics. Purified NK cells by negative depletion were stained with 0.5 μM CellTrace calcein red-orange AM and HL60 target cells with distinct dyes for 10 to 20 minutes. The blue-stained NK cells and green-stained target cells were seeded onto a silicon-glass microchip divided into 2 separate compartments containing 161533 TriKE or 1633 BiKE. Imaging of the microchip was performed using a Zeiss LSM 880 microscope equipped with an environmental chamber kept at 37°C and 5% CO2. Images were acquired using a 10× objective every 3 minutes for 9 hours. (A-D) NK cell cytotoxicity (A), representative images of target cell killing (red cells) at different time points (B), time to first kill and migration (C), and serial killing (D) are shown from 5 independent experiments (n = 1031 NK cells for BiKE and n = 1202 NK cells for TriKE). Data were quantified by MATLAB, and statistical analyses were performed by paired Student t test (A-B) or Mann-Whitney U test (D).
161533 TriKE elicits strong STAT5 phosphorylation
A STAT5 phosflow assay was employed to investigate whether the IL-15 moiety in TriKE induces a similar type and magnitude of IL-15 receptor downstream signaling as soluble recombinant IL-15. Purified NK cells were cultured in medium, IL-15, BiKE, BiKE and IL-15, or TriKE with or without HL60 target cells for 25 minutes before analysis of STAT5 phosphorylation. IL-15 forms a complex with IL-15Rβ and the common γ-chain on the surface of NK cells to induce a signaling cascade resulting in the recruitment and activation of the transcription factor STAT5, which is required for a number of essential NK-cell functions.27 In the absence of targets, 161533 TriKE and soluble IL-15 activated STAT5 phosphorylation to a similar extent (pSTAT5 MFI 592 ± 25 vs 586 ± 25), whereas 1633 BiKE alone and media controls did not induce STAT5 phosphorylation (Figure 4; see supplemental Figure 2A for the gating strategy used). In contrast, in the presence of HL60 target cells, BiKE induced significant NK cell STAT5 phosphorylation compared with NK cells in the absence of target cells (MFI 595 ± 20 vs 513 ± 16, P = .001). However, target cells alone did not induce pSTAT5 in NK cells, indicating that CD16 ligation can induce of STAT5 phosphorylation when crosslinked to targets. As further proof of this, ligating CD16 with an agonistic anti-CD16 on purified NK cells augmented STAT5 phosphorylation compared with the medium control (supplemental Figure 2). 161533 TriKE induced higher levels of STAT5 phosphorylation in NK cells in the presence of target cells than exogenous IL-15 and target cells (MFI 670 ± 30 vs 600 ± 32, P = .03). In both settings (with or without targets), 161533 TriKE and 1633 BiKE plus IL-15 induced similar STAT5 phosphorylation. Collectively, the data demonstrate STAT5 phosphorylation synergy mediated by the TriKE or IL-15 plus BiKE when targets cells are present to induce an immune synapse that triggers CD16 in conjunction with IL-15.
Figure 4.

Crosslinked CD16 enhances the IL-15–induced phosphor-STAT5 signal. Isolated NK cells (n = 9; by negative depletion) from HDs were stimulated with IL-15 (equal molar concentration to the IL-15 in TriKE [50 nM], BiKE [50 nM], BiKE + IL-15 [both 50 nM], or TriKE [50 nM]) with or without HL60 targets for 25 minutes before fixation and STAT5 phosphorylation analysis. Symbols represent distinct individuals, and statistical analysis was performed using a paired Student t test.
161533 TriKE restores NK-cell function in patients with MDS
Studies from our group have previously shown that 1633 BiKE enhances NK cell responses to HL60 cells and endogenous CD33+ MDS targets12 and that 161533 TriKE can stimulate NK cell proliferation in post-HCT samples without the addition of exogenous soluble IL-15.13 Having shown robust killing dynamics and maintenance of normal NK cell activity in the presence of immune-suppressive cells through low expression of the inhibitory checkpoint TIGIT in TriKE-treated NK cells compared with soluble recombinant IL-15, the ability of 161533 TriKE to restore NK cell proliferation and function in samples from MDS patients with active disease was investigated next.
A dramatic decrease of circulating NK cell frequencies in MDS patient peripheral blood samples, when compared with HD samples, was noted in this cohort (1.2% ± 0.3% [n = 14] vs 13% ± 2% [n = 7], P < .0001; Figure 5A). This is consistent with our earlier study in MDS patients.12 Thus, strategies are needed to support NK cell proliferation in these patients. Therefore, PBMCs from HDs and MDS patients were cultured overnight in medium alone or in the presence of 1633 BiKE or 161533 TriKE. HL60 target cells were then added and adjusted to a 20:1 E:T ratio 6 hours before analysis. The gating strategy is shown in supplemental Figure 3. As previously reported, MDS-NK cell degranulation and IFN-γ production were enhanced by 1633 BiKE compared with cells cultured with target cells alone12 (CD107a 29% ± 2% vs 15.5% ± 3%, P < .0001; IFN-γ 17% ± 4% vs 11% ± 3%, P = .002; Figure 5B). However, 1633 BiKE did not enhance proliferation of MDS or HD NK cells (Figure 5B-C). In marked contrast, 161533 TriKE has a superior effect on MDS-NK cell responses compared with 1633 BiKE (CD107a 37% ± 3% vs 29% ± 2%, P = .001; IFN-γ; 27% ± 1% vs 17% ± 4%, P = .0001; Figure 5B). In addition, 161533 TriKE significantly enhanced both MDS (Ki67 21% ± 3% vs 11% ± 2% vs 12% ± 3%, P ≤ .025) and HD-NK cell proliferation (Ki67; 21% ± 2% vs 12% ± 2% vs 16% ± 2%, P ≤ .03) compared with PBMCs plus target cells alone or PBMCs plus targets and 1633 BiKE (Figure 5B-C). Treating HD PBMCs with 161533 TriKE and exogenous IL-15 had no synergistic effect on NK-cell function, suggesting that IL-15 signaling with the TriKE was already saturated (supplemental Figure 4). Allogeneic killing of patient BM MDS blasts was evaluated using flow cytometric cytotoxicity assays. There was a significant increase in NK cell killing of primary MDS blasts when treated with 161533 TriKE compared with no treatment or exogenous IL-15 alone (Figure 6). Our data suggest that incorporation of the IL-15 molecule into TriKE enhances NK cell antitumor activity and proliferation in MDS patient samples.
Figure 5.

161533 TriKE is superior to 1633 BiKE to restore NK-cell function and proliferation in patients with MDS. (A) Cryopreserved PBMCs from MDS patients (n = 14) and HDs (n = 7) were rested overnight and stained, and NK cell frequencies were determined by flow cytometry. (B-C) Cryopreserved PBMCs from MDS patients (B) or HDs (C) were activated overnight with IL-15 (equal molar concentration to the IL-15 in TriKE [50 nM], BiKE [50 nM], or TriKE [50 nM]). Target cells were then added 6 hours prior to staining. NK cell degranulation (CD107a), IFN-γ production, and proliferation (Ki67) were evaluated. Data are shown as mean ± SEM, and statistical analyses were performed using paired Student t test.
Figure 6.

Improved MDS-NK cell cytotoxicity following treatment with 161533 TriKE. Purified HD-NK cells were cultured with CellTrace dye–labeled MDS BM targets (E:T ratio 2:1) for 6 hours in the presence of IL-15 (30 nM), 1633 BiKE (30 nM), 161533 TriKE (30 nM), or no treatment. MDS blast killing was assessed by gating on an intermediate CD45 and SSC low population, further gated to CD117+ and CD34+ cells within the CellTrace-positive population, and assessed for the proportion of dead cells using Live/Dead dye. Gating strategy and pooled data (mean ± SEM) are shown, and statistical analyses were performed using 1-way ANOVA.
Discussion
MDS is characterized by multiple genetic and epigenetic alterations that result in a morphologic heterogeneous disease. A diverse set of antigens helps the immune system distinguish tumor cells from their normal counterparts. However, tumor cells can escape the immune response through recruitment of immune suppressive cells and increased expression of immune checkpoints. Earlier studies have shown that NK cells play an important role in the immune surveillance against tumors,8,28 yet tumor-induced immune suppression may dampen the efficacy of NK cell therapies against hematological malignancies.29
Several reports have shown the role of MDSCs in immune suppression in the tumor microenvironment and association with poor immune effector antitumor responses.20,30,31 Therefore, targeting MDSCs or strategies to resist their suppression mechanisms are desired. In this study, we show a unique feature of 161533 TriKE molecules; besides improving NK cell killing dynamics, these molecules induce NK cell resistance to MDSC-mediated suppression through a mechanism that is different than CD33-targeted killing of MDSC directly. 161533 TriKE, unlike soluble recombinant IL-15, 1633 BiKE plus IL-15, and tumor target cell stimulation, does not induce the expression of the inhibitory immune checkpoint TIGIT on NK cells, which at least partially explains the NK cell resistance to MDSC suppression. We have previously shown that blocking TIGIT from the interaction with CD155 on MDS-MDSCs rescues NK-cell function.15 The lack of induced TIGIT expression could be explained by local and directed delivery of IL-15 instead of systematic activation of NK cells in the case of soluble IL-15. This suggests that IL-15 in the context of the TriKE is being presented differently. This concept has been suggested by others. A study by Romano et al has shown that IL-15 crosspresentation to T cells breaks the tolerance against tumors expressing self-antigens compared with exogenous soluble IL-15.32 TIGIT expression is associated with poor T cell antitumor responses and MDSC-mediated suppression of NK cells.15,33,34
The main rationale for using IL-15 is that it is the natural homeostatic factor known to induce differentiation, enhance NK cell survival, and stimulate activation and proliferation. While other cytokine linkers might be considered, they may be less optimal. For example, IL-2 increases regulatory T cells in cancer patients that correlate with worse prognosis. Other cytokines, such as IL-12/IL-18, can certainly increase cytokine production by NK cells but exhibit less proliferative potential and have higher safety concerns for human therapy. Lastly, the clinical development of IL-15 supports the in vivo activity on human NK cells and was the basis for our choice as the linker in the TriKE molecule.35-37
NK cells are dramatically decreased in patients with MDS, making them insufficient to control established disease. Thus, novel strategies to manipulate NK cell proliferation, persistence, and functional maintenance are needed in the MDS setting. Allo-HCT can be curative for MDS patients. Nevertheless, many patients are ineligible for transplantation because of the risks associated with allo-HCT, limiting its availability to the majority of the patients.38 Although alternate therapies exist, the clonal heterogeneity in MDS leads to high relapse rates and risk of mortality as well as progression to AML.39,40 Therefore, new therapeutic approaches are highly sought. In this study, we show that 161533 TriKE can restore NK-cell function, including proliferation and targeting of primary MDS blasts.
We and others have reported the efficacy of BiKEs and bispecific antibodies in augmenting antitumor activity.12,41-43 Here, we show that 161533 TriKE provokes unique killing kinetics in NK cells, highlighted by an increase in serial killing, higher target cytotoxicity, and faster NK cell migration dynamics compared with NK cells treated with 1633 BiKE. These findings are explained by the addition of the modified IL-15 linker. These properties support the use of TriKE as an immunotherapeutic agent with several advantages, including targeting and reduced TIGIT upregulation, over IL-15 monotherapy in the MDS setting. As a cautionary note, similar to chimeric antigen receptor T cells, depletion of myeloid cells has to be considered when targeting CD33, which is also present at lower levels on normal myeloid progenitors. In addition, CD16 is expressed on other immune cells such as neutrophils, monocytes, and macrophages, and the signaling effect on those cells by 161533 TriKE needs to be considered. The expected short half-life of the TriKE will likely make off-target problems more manageable. These issues will become clear after testing of the TriKE clinically, which is projected to start in quarter 3/4 of 2018. Collectively, 161533 TriKE shows therapeutic promise in the MDS setting, with unique CD16/IL-15 signaling to enhance NK cell killing dynamics without provoking inhibitory checkpoint expression.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors would like to acknowledge the Translational Therapy Shared Resource and the Flow cytometry core at the University of Minnesota for their services.
This work was supported by the National Institutes of Health, National Cancer Institute (grants P01 CA111412 [D.J.W., S.R.S., S.C., and J.S.M.], P01 CA65493 [B.R.B., S.C., and J.S.M.], and R35 CA197292 [M.F., S.C., and J.S.M.]), the National Heart, Lung, and Blood Institute (grants R01 HL122216 and R01 HL56067) (B.R.B.), the National Institute of Allergy and Infectious Diseases (R37 AI 34495) (B.R.B.), the US Department of Defense (grant CA150085) (M.F.), and the Swedish Foundation for Strategic Research (grant SBE13-0092) (D.S.).
The sponsors of this study are public or nonprofit had no role in gathering, analyzing, or interpreting the data. The content of this publication does not necessarily reflect the views or polices of the National Institutes of Health, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Authorship
Contribution: D.S., L.B., K.G., T.L., and P.H. performed experiments and analyzed data; J.C., E.W., S.R.S., D.J.W., and S.C. provided patient samples and clinical data and interpretation; and D.S., M.F., B.R.B., D.A.V., B.Ö., and J.S.M. designed and performed experiments, interpreted data, and wrote the paper.
Conflict-of-interest disclosure: J.S.M. and D.A.V. serve on the Scientific Advisory Board of GT Biopharma and have received research funds and clinical trial support from these relationships. These relationships have been reviewed and managed by the University of Minnesota in accordance with its conflict of interest polices. M.F. has received research funds from GT Biopharma. The remaining authors declare no competing financial interests.
Correspondence: Jeffrey S. Miller, University of Minnesota, 420 Delaware St SE, Minneapolis, MN 55455; e-mail: mille011@umn.edu.
References
- 1.Arber DA, Orazi A, Hasserjian R, et al. . The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. [DOI] [PubMed] [Google Scholar]
- 2.Ma X, Does M, Raza A, Mayne ST. Myelodysplastic syndromes: incidence and survival in the United States. Cancer. 2007;109(8):1536-1542. [DOI] [PubMed] [Google Scholar]
- 3.Cogle CR. Incidence and burden of the myelodysplastic syndromes. Curr Hematol Malig Rep. 2015;10(3):272-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cogle CR, Kurtin SE, Bentley TG, et al. . The incidence and health care resource burden of the myelodysplastic syndromes in patients in whom first-line hypomethylating agents fail. Oncologist. 2017;22(4):379-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gangat N, Patnaik MM, Begna K, et al. . Survival trends in primary myelodysplastic syndromes: a comparative analysis of 1000 patients by year of diagnosis and treatment. Blood Cancer J. 2016;6(4):e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeidan AM, Linhares Y, Gore SD. Current therapy of myelodysplastic syndromes. Blood Rev. 2013;27(5):243-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenberg PL, Stone RM, Al-Kali A, et al. . Myelodysplastic Syndromes, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2017;15(1):60-87. [DOI] [PubMed] [Google Scholar]
- 8.Miller JS, Soignier Y, Panoskaltsis-Mortari A, et al. . Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):3051-3057. [DOI] [PubMed] [Google Scholar]
- 9.Ruggeri L, Capanni M, Casucci M, et al. . Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood. 1999;94(1):333-339. [PubMed] [Google Scholar]
- 10.Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene. 2008;27(45):5932-5943. [DOI] [PubMed] [Google Scholar]
- 11.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12(4):278-287. [DOI] [PubMed] [Google Scholar]
- 12.Gleason MK, Ross JA, Warlick ED, et al. . CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood. 2014;123(19):3016-3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vallera DA, Felices M, McElmurry R, et al. . IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res. 2016;22(14):3440-3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huntington ND, Legrand N, Alves NL, et al. . IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J Exp Med. 2009;206(1):25-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarhan D, Cichocki F, Zhang B, et al. . Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res. 2016;76(19):5696-5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bronte V, Brandau S, Chen SH, et al. . Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brimnes MK, Vangsted AJ, Knudsen LM, et al. . Increased level of both CD4+FOXP3+ regulatory T cells and CD14+HLA-DR−/low myeloid-derived suppressor cells and decreased level of dendritic cells in patients with multiple myeloma. Scand J Immunol. 2010;72(6):540-547. [DOI] [PubMed] [Google Scholar]
- 18.Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015;66:97-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. 2009;182(1):240-249. [DOI] [PubMed] [Google Scholar]
- 20.Hoechst B, Voigtlaender T, Ormandy L, et al. . Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50(3):799-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res. 2014;20(15):4096-4106. [DOI] [PubMed] [Google Scholar]
- 22.Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. 2010;185(4):2273-2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu X, Marcus WD, Xu W, et al. . Novel human interleukin-15 agonists. J Immunol. 2009;183(6):3598-3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guldevall K, Brandt L, Forslund E, et al. . Microchip screening platform for single cell assessment of NK cell cytotoxicity. Front Immunol. 2016;7:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MathWorks. MATLAB and Imaging Toolbox. Natick, MA: The MathWorks; 2015.
- 26.Chan WK, Kung Sutherland M, Li Y, Zalevsky J, Schell S, Leung W. Antibody-dependent cell-mediated cytotoxicity overcomes NK cell resistance in MLL-rearranged leukemia expressing inhibitory KIR ligands but not activating ligands. Clin Cancer Res. 2012;18(22):6296-6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17(9):1025-1036. [DOI] [PubMed] [Google Scholar]
- 28.Cooley S, Weisdorf DJ, Guethlein LA, et al. . Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood. 2010;116(14):2411-2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gras Navarro A, Björklund AT, Chekenya M. Therapeutic potential and challenges of natural killer cells in treatment of solid tumors. Front Immunol. 2015;6:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu T, Ramakrishnan R, Altiok S, et al. . Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest. 2011;121(10):4015-4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romano E, Cotari JW, Barreira da Silva R, et al. . Human Langerhans cells use an IL-15R-α/IL-15/pSTAT5-dependent mechanism to break T-cell tolerance against the self-differentiation tumor antigen WT1. Blood. 2012;119(22):5182-5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kong Y, Zhu L, Schell TD, et al. . T-cell immunoglobulin and ITIM domain (TIGIT) associates with CD8+ T-cell exhaustion and poor clinical outcome in AML patients. Clin Cancer Res. 2016;22(12):3057-3066. [DOI] [PubMed] [Google Scholar]
- 34.Blake SJ, Dougall WC, Miles JJ, Teng MW, Smyth MJ. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188. [DOI] [PubMed] [Google Scholar]
- 35.Steel JC, Waldmann TA, Morris JC. Interleukin-15 biology and its therapeutic implications in cancer. Trends Pharmacol Sci. 2012;33(1):35-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Romee R, Leong JW, Fehniger TA. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica (Cairo). 2014;2014:205796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Romee R, Cooley S, Berrien-Elliott MM, et al. . First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood. 2018;blood-2017-12-823757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Welniak LA, Blazar BR, Murphy WJ. Immunobiology of allogeneic hematopoietic stem cell transplantation. Annu Rev Immunol. 2007;25(1):139-170. [DOI] [PubMed] [Google Scholar]
- 39.Greenberg P, Cox C, LeBeau MM, et al. . International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89(6):2079-2088. [PubMed] [Google Scholar]
- 40.Greenberg PL, Tuechler H, Schanz J, et al. . Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiernik A, Foley B, Zhang B, et al. . Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin Cancer Res. 2013;19(14):3844-3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vallera DA, Zhang B, Gleason MK, et al. . Heterodimeric bispecific single-chain variable-fragment antibodies against EpCAM and CD16 induce effective antibody-dependent cellular cytotoxicity against human carcinoma cells. Cancer Biother Radiopharm. 2013;28(4):274-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singer H, Kellner C, Lanig H, et al. . Effective elimination of acute myeloid leukemic cells by recombinant bispecific antibody derivatives directed against CD33 and CD16. J Immunother. 2010;33(6):599-608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.