

Abstract
Recombinant protein design allows modular protein domains with different functionalities and responsive behaviors to be easily combined. Inclusion of these protein domains can enable recombinant proteins to have complex responses to their environment (e.g., temperature-triggered aggregation followed by enzyme-mediated cleavage for drug delivery or pH-triggered conformational change and self-assembly leading to structural stabilization by adjacent complementary residues). These “smart” behaviors can be tuned by amino acid identity and sequence, chemical modifications, and addition of other components. A wide variety of domains and peptides have smart behavior. In this review, we will focus on protein designs for self-assembly or conformational changes due to stimuli such as shifts in temperature or pH.
Keywords: Biomaterials, Biopolymers, Hydrogels, Self-assembly, Stimuli-sensitive polymers
TOC image
Environmentally-responsive behavior in natural and recombinant proteins can be traced to specific amino acid sequences that can be used in modular protein design. These sequences can be used alone or in conjunction to create complex responses to multiple stimuli, including temperature, pH, and salt, and to cause structural changes and self-assembly.
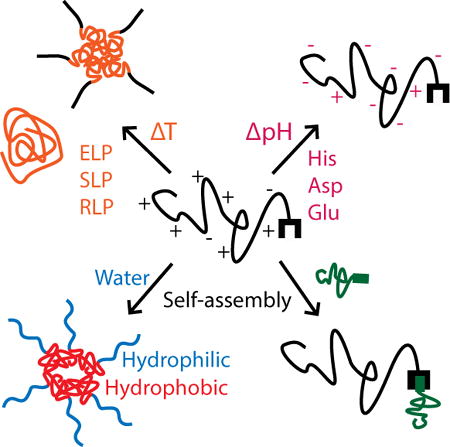
1. Introduction
Biomaterials with properties that change in response to environmental stimuli have drawn tremendous interest since the concept of smart materials was proposed. Progress in molecular biology has identified the structures and sequences of stimuli-responsive proteins and allowed them to be produced recombinantly. Because of the modularity inherent in recombinant protein design and production, these stimuli-responsive protein domains can be harnessed as building blocks for smart biomaterials. In this review, we categorized stimuli-responsive proteins into three categories based on the origin of their responsiveness.
1.1 Protein Sequences with Intrinsic Responsiveness
One of the commonly used modular domains in recombinant protein design is elastin-like polypeptides (ELPs). ELPs have a repetitive pentapeptide sequence with a guest residue position that allows flexibility in sequence design.[1] The guest residue can be used to tune the temperature-responsive behavior of ELPs based on the hydrophobicity or charge status of the residue.[1] Some amino acids can be used to confer additional environmental responses. Other protein sequences with temperature-responsiveness include resilin-like polypeptides (RLPs)[2] and silk-like polypeptides (SLPs),[3] and both RLPs and SLPs are composed of repetitive sequences.[2, 4] The temperature-responsive behavior of these recombinant proteins is also sensitive to environmental conditions including the ionic strength or pH of the system,[5] and thus these recombinant proteins are responsive to multiple stimuli. This multi-responsiveness can be harnessed for designing smart biomaterials with applications such as targeted drug delivery.[5]
1.2 Responsiveness from Charged Amino Acids
Including charged residues in recombinant protein design can create pH sensitivity. Histidine, aspartic acid, and glutamic acid are frequently used to confer responsiveness to pH values between 3 and 7 since the charge states of these residues change within this pH range. The changes in individual residues leads to a shift in overall charge status of the whole protein. At various pH values, these residues can induce intra-protein association[6] or bridge membranes and bilayers.[7] These charged residues can also provide chelation with metal groups[8] or pH-sensitivity in fluorescent markers.[9]
1.3 Responsiveness from Protein-protein Interaction
Some proteins can interact with other proteins in a controlled manner. For example, leucine zippers are helical domains that will form coiled coil structures only when the right partner is present in the system.[10] In addition to the formation of quaternary structures, some of these protein-protein interactions self-assemble into higher-order structures or networks. Surfactant-like proteins are one example in this category since they form micelle or vesicle structures upon self-assembling.[11] There are also split protein domains that form covalent bonds[12] or became fluorescent after reconstitution.[13]
In the following review, we discuss recombinant proteins and domains that are responsive to temperature, pH, and other proteins in the environment. The changes in protein structure and biochemical properties in response to the stimuli will be discussed, and the applications based on the responsiveness of these proteins will be summarized.
2. Temperature Sensitivity
Temperature sensitivity in proteins often causes shifts in protein structure and can lead to changes in hydrophobicity or solubility. Natural proteins such as elastin, resilin, and silk aggregate upon heating and form complex fibrous structures with elastomeric,[14, 15] resilient,[16] or rigid[17] mechanical properties, respectively. Repetitive polypeptide sequences derived from these proteins (i.e., ELPs, RLPs, and SLPs) also exhibit temperature sensitivity and maintain some of the mechanical properties of those native proteins. Upon heating or cooling, these temperature-sensitive polypeptides may phase separate,[18, 19] form aggregates,[20] or self-assemble into micelles or more complicated structures.[21]
2.1 Elastin-like Polypeptides
2.1.1 Elastin-like Polypeptide Structure and Lower Critical Solution Temperature
The self-assembly and mechanical properties of ELPs are similar to those of native elastin.[22] One of the most commonly used ELP sequences is based on the hydrophobic sequences of native elastin. The ELP is composed of a repeating pentapeptide sequence VPGXG, where X is a “guest residue” that can be any amino acid except for proline. ELPs can phase separate upon heating above their lower critical solution temperature (LCST). Below the LCST, ELPs are soluble and have a random structure.[19] Upon heating above the LCST, ELPs experience hydrophobic collapse into a compact structure containing β-spiral structures and eventually phase separate into two liquid phases – a protein-rich phase (“coacervate”) and a protein-poor phase. LCST behavior in ELPs can be exploited for a wide variety of applications, including protein purification, temperature-responsive drug delivery, and temperature-triggered self-assembly.[23, 24]
The LCST can be controlled through the sequence of the ELP. The identity of the guest residue can have a strong influence on LCST. Hydrophobic residues decrease the LCST, and hydrophilic residues increase the LCST.[1] The length of the ELP also influences the LCST; longer ELP sequences have lower LCSTs.[25] Information about sequence identity and length can be combined to predict the LCST of a de novo ELP design and assist in tuning the environmental responsiveness of the protein.[26] Environmental factors that affect the LCST include protein concentration, salts, and pH.
The LCST of ELP systems can also be influenced through chemical modifications. Although the LCST is highly tunable through ELP design, there are cases in which the actual LCST of the ELP may not be the same as the theoretical LCST or the inclusion of other functional or responsive groups may cause the LCST to be outside of the desired region.[25, 27] The LCST can be lowered by adding salts or raised by increasing concentration, but the application conditions may limit changes in salt or concentration. In these instances, the LCST can be changed by modifying the ELP directly. One such example is an ELP with methionine guest residues and an LCST of 27–33°C depending on concentration.[28] Methyl or benzyl groups were bound to the sulfur group of methionine and created a positively-charged sulfonium group. Benzylation increased the LCST by ~12°C, and the resulting coacervate had a lower opacity than unmodified ELP. This lowered opacity indicated that the ELP aggregates were at a lower density, and this result may be due to the hydrophobicity of the benzyl group or the electrostatic repulsions between sulfonium groups. The methylated ELP exhibited no LCST behavior between the testing range of 20°C and 80°C. The absence of detectable LCST behavior was most likely due to the strong hydrophilicity of the charged sulfonium groups. Thus, chemical modification provides a powerful tool for further altering the hydrophilicity of ELPs and tuning the LCST after the ELP has been designed.
2.1.2 Temperature-responsive Applications of ELPs
ELPs have been used for temperature-triggered drug release. In one such system created by the Raucher group, ELPs were conjugated to a chemotherapeutic drug, paclitaxel, which has poor water solubility and is currently delivered using an ethanol delivery system that can cause hypersensitive reactions.[29] The authors combined a cell-penetrating peptide, SynB1; a temperature-responsive ELP carrier; and an acid-sensitive prodrug of paclitaxel, 6-maleimidocaproyl hydrazine (Figure 1). A maleimide group on the drug allowed efficient coupling to a cysteine group on the C-terminus of the ELP. The drug was sensitive to acidic conditions due to a hydrazone bond between the ELP carrier and the paclitaxel drug. At the acidic pH of lysosomes and endosomes, this bond can be cleaved and can result in intracellular release of the drug in acidic cells and tissues.[30, 31] It was observed that, after three and six days, the ELP-drug fusion was 20 and 2 times less potent, respectively, than the free drug when tested in vitro.[29] The delayed effectiveness of the ELP-drug fusion may be due to its larger size, which causes it to enter cells more slowly than the free drug, whose small size and hydrophobicity allow it to quickly penetrate cell membranes. However, the ELP-drug fusion was designed to aggregate at temperatures above the LCST, and the cytotoxic effects of the drug increased when heated due to higher local drug concentrations. This temperature-sensitive behavior of the ELP-drug fusion allowed control over its release, whereas the free drug affected cells indiscriminately. In a paclitaxel-resistant cell line, free drug inhibited proliferation by ~80% at both 37°C and 42°C, whereas the ELP-drug fusion had no cytotoxicity at 37°C and inhibited proliferation by 50% at 42°C. Combining the temperature sensitivity of ELPs and the acid sensitivity of the paclitaxel prodrug allowed for targeted delivery and targeted release of a drug that would otherwise damage healthy tissues.
Figure 1.
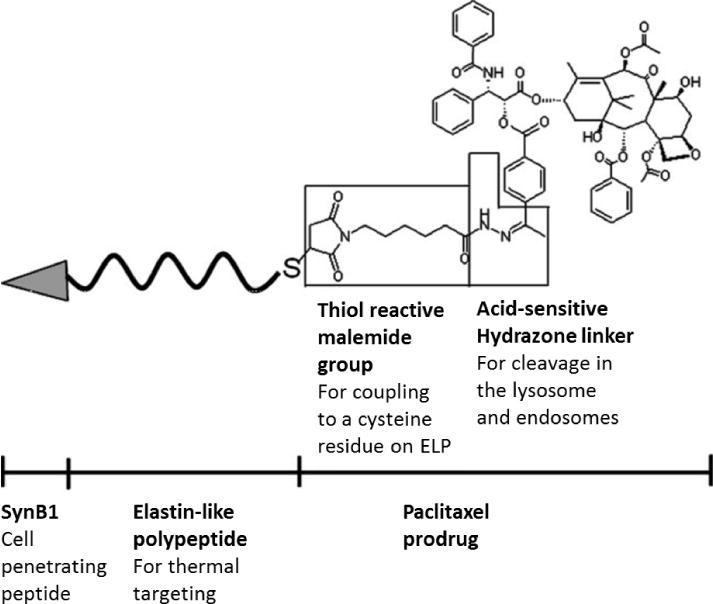
Schematic showing the structure of a fusion molecule consisting of a cell-penetrating peptide and a temperature-sensitive elastin-like polypeptide (ELP) domain bound to a paclitaxel prodrug through a thiol reaction to an acid-sensitive linker. Investigational New Drugs, A thermally responsive biopolymer conjugated to an acid-sensitive derivative of paclitaxel stabilizes microtubules, arrests cell cycle, and induces apoptosis, 30, 2012, 236, Shama Moktan, Claudia Ryppa, Feliz Kratz, Drazen Raucher, Copyright Springer Science+Business Media, LLC 2010, with permission of Springer
Another application of the temperature-responsiveness of ELPs is to combine it with other peptides to create micelles that self-assemble upon heating. One such micelle assembly was used for single-step protease detection[32] and consisted of ELP sequences (VPGVG) linked to a hydrophilic, charged domain by a matrix metalloproteinase (MMP)-sensitive peptide (VPMSMRGG) (Figure 2A). Upon heating above the critical micelle temperature (CMT), the ELP block underwent hydrophobic collapse and formed the micelle core while the hydrophilic block remained hydrated and formed the shell of the micelle. In the absence of protease, the fusion protein formed micelles with a hydrodynamic radius of 90 nm when heated above the CMT of 38°C. When protease was present, the MMP-sensitive linker was cleaved, the ELPs aggregated at 38°C to form a coacervate phase, and the hydrodynamic radius and turbidity of the mixture increased significantly (Figure 2B). Furthermore, at a fixed temperature, there was an increase in OD and hydrodynamic radius that depended on time. Increasing the protease concentration decreased the time required for turbidity to increase. Thus, the aggregation time was used to develop an assay for detection of protease concentration in both proteolysis buffer and a physiologically relevant buffer. The assay was accurate between 1 – 100 ng/mL, which is a range comparable to available assays for MMP. This method required only a single-step process and no sophisticated data analysis, whereas other assays such as precipitation-based or fluorescent assays require, respectively, multi-step purification methods or specialized equipment and analysis.
Figure 2.
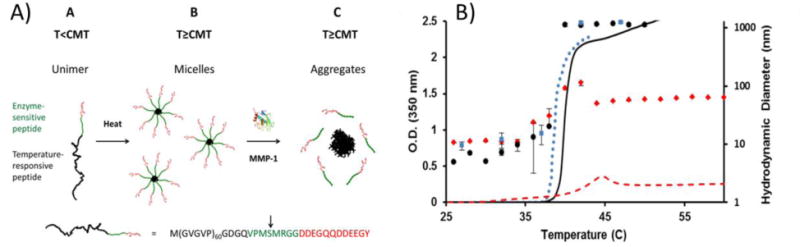
Micelle formation of a fusion protein consisting of elastin-like polypeptide (ELP) sequences, a matrix-metalloproteinase (MMP)-sensitive linker, and a hydrophilic domain. A) Upon heating, the protein formed micelles with an ELP core and a hydrophilic shell. When exposed to MMPs, the protein was cleaved and the ELP aggregated to form a coacervate phase. B) Change in aggregate size and solution opacity of ELP micelles. Optical density (lines) and hydrodynamic radius (individual data points) of MMP-sensitive ELP in the presence of MMP (black), MMP-sensitive ELP in the absence of MMP (red), and a negative control ELP without an MMP-sensitive linker or hydrophilic domain in the absence of MMP (blue). Reprinted (adapted) with permission from Analytical Chemistry, Simple Assay for Proteases Based on Aggregation of Stimulus-Responsive Polypeptides, 86, 2014, Ali Goorchian, Ashutosh Chilktoi, Gabriel P. Lopez. Copyright 2014 American Chemical Society.
A fusion protein consisting of an ELP fused to an SLP domain also formed micelles.[21] The more hydrophobic SLP domains formed the micelle core, and the more hydrophilic ELP blocks formed the hydrated shell (Figure 3A). Upon heating, the ELP domains aggregated and formed larger particles consisting of multiple micelles. The ability to modulate the temperature and process of this assembly was investigated by using three constructs with varying ratios of SLP to ELP – 1:8 in SE8Y, 2:8 in S2E8Y, and 4:8 in S4E8Y (Figure 3B). As expected, proteins with ratios of 1:8 or 2:8 formed particles upon heating, and the process was fully reversible. At the same concentration, the 2:8 protein solutions had a higher turbidity value than the 1:8 proteins, but the particles were the same size. These data thus suggested that the 2:8 protein formed more particles. Proteins with a 4:8 ratio formed micelles but had little particle formation upon heating to temperatures as high as 100°C. The high silk to elastin ratio may have interfered with the coacervation behavior of the ELP domains. This explanation was further supported by the observations that, at high temperatures, the 1:8 and 2:8 proteins had an increase in β-turn structure, which is characteristic of pure ELPs, whereas the 4:8 protein had β-sheet conformations, which are a feature often seen in SLPs. The SLP:ELP ratio also affected the nanostructure of the self-assembled aggregates. Upon heating and cooling, the 1:8 proteins had a uniform distribution of small particles, the 2:8 proteins had intermediate-sized aggregates composed of spherical particles, and the 4:8 proteins were composed of large aggregates (Figure 3C). In addition, the 2:8 and 4:8 proteins formed nanofibers. By controlling the relative amounts of hydrophobic and hydrophilic blocks, the temperature-triggered self-assembly of these ELP-SLP proteins was tuned, and a variety of nanostructures were created.
Figure 3.
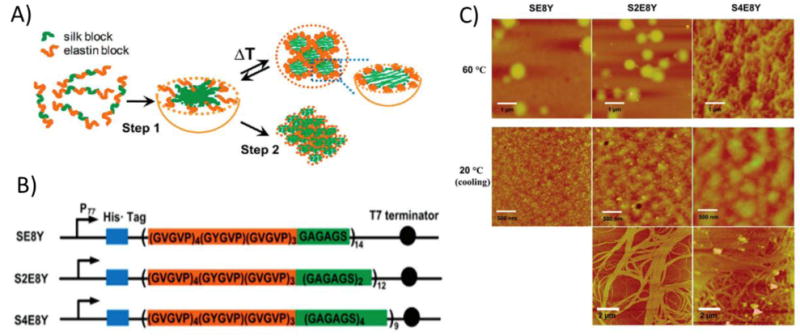
Silk-like and elastin-like polypeptide fusion proteins (SLP-ELP) exhibited a temperature-sensitive two step assembly process into coacervates, gels, and fibers. A) Scheme showing micelle assembly of SLP-ELP protein. Upon mixing, proteins self-assembled into micelles with hydrophobic SLPs in the core and ELPs in the shell. Upon heating above the lower critical solution temperature (LCST), the ELP block underwent hydrophobic collapse and aggregated with other ELP blocks in neighboring micelles. B) Sequence of ELP-SLP proteins. The frequency of the ELP guest residue was 1/8 tyrosine and 7/8 valine. The ratio of SLP to ELP varied between 1:8 (SE8Y), 2:8 (S2E8Y), and 4:8 (S4E8Y). C) Atomic force microscopy (AFM) images of aggregates and fibers formed by SLP-ELP proteins. When heated, the proteins formed small aggregates. Upon cooling, proteins with higher ratios of SLP to ELP formed fibrous networks. Reprinted (adapted) with permission from Biomacromolecules, Tunable Self-Assembly of Genetically Engineered Silk-Elastin-like Protein Polymers, 12, 2011, Xiao-Xia Xia, Qiaobing Xu, Xiao Hu, Guokui Qin, David L. Kaplan. Copyright 2011 American Chemical Society.
When designing ELP fusion proteins with temperature responsive behavior, the guest residues chosen and the relative location and length of the ELP can affect the thermal properties of the fusion protein as well as other protein functions. For example, protein structure, LCST behavior, and fluorescent activity were affected in a fusion of the fluorescent protein m-Cherry to different ELP sequences.[33] The two ELP sequences used were ELP0, which contained valine in all guest residue positions, and the more hydrophilic ELP1, which contained valine, glycine, and alanine guest residues. The proteins varied by the identity of the ELP sequence, the presence or absence of a histidine tag, the length of the ELP sequence, and the location of the ELP sequence at the C- or N-terminus of the protein. Protein structure was explored through small-angle X-ray scattering (SAXS). Proteins containing ELP0 had sharp, well-defined peaks that indicated ordered lamellar structure. Proteins containing ELP1 sequences had no peaks or broader peaks and were thus unstructured or less structured, respectively. This reduction in structure may be caused by the relatively lower hydrophobicity of ELP1. For both ELP0 and ELP1, longer ELP sequences in the fusion protein resulted in higher overall symmetry between the two protein domains and increased structure. Both increased mass and sequence symmetry are thought to promote ordered structures in block copolymers like this system.[34] The relative position of the ELP sequence in the fusion protein also affected LCST behavior. Fusion proteins with ELP0 at the N-terminus did not have a detectable LCST between 10 and 50 °C, whereas a protein with ELP0 at the C-terminus had an LCST of 32.5 °C. Fusion proteins with ELP1 did not have a detectable LCST within the tested range. It is presumed that the LCSTs of the ELP1 fusion proteins were all greater than 50 °C regardless of ELP sequence location, and these results could be due to the lower hydrophobicity of ELP1 compared to ELP0. It is possible that varying the location of ELP1 would have had an effect on LCST similar to that of the ELP0 fusion proteins, but the authors were not able to observe this phenomenon because they did not test for LCST values greater than 50 °C. Finally, in fusion proteins, it is important that an added domain does not limit the function of other sequences. In this instance, ELP location and length and the presence of a histidine tag affected m-Cherry fluorescence. Compared to m-Cherry alone, fusion proteins with short ELP sections located on the N-terminus and containing a His-tag had the best maintenance of fluorescent function as measured by absorbance at 586 nm (104% and 82% for ELP1 and ELP0, respectively). Overall, these results indicate that it is important to consider the location, length, and hydrophobicity of an ELP sequence when creating functional fusion proteins.
The temperature-responsiveness of ELPs can be used to trigger auto-crosslinking behavior by including molecular groups that bind when in close proximity. For example, oxidized cysteine residues included in ELPs crosslinked upon heating above the LCST because the hydrophobic collapse of ELPs decreased inter-protein chain distance.[35] These cysteine-containing ELPs gelled rapidly (~2.5 minutes) at physiological temperature and mildly oxidative conditions of 0.3 wt% hydrogen peroxide. This concentration of hydrogen peroxide is considered to be safe since 3% hydrogen peroxide is often used in clinical settings to clean wounds. Below the LCST, increasing the cysteine content of the ELP increased turbidity, and this result indicated that some intermolecular crosslinking occurred. Heating above the LCST accelerated the crosslinking process. This design was used for hydrogel-based drug delivery. Bovine serum albumin (BSA), which was used as a model protein, was encapsulated in the ELP gels and had first order release kinetics with an initial burst release followed by a slower release. The system was used with a radionuclide in an in vivo tumor system. The ELP-drug mixture was injected directly into the tumor and gelled in situ. The gel had a higher radionuclide retention in comparison to either a soluble ELP with no cysteines or a cysteine-containing ELP without hydrogen peroxide. It also distributed more homogenously in the tumor compared to the cysteine-containing ELP without hydrogen peroxide. In previous studies using ELPs for drug delivery to tumors, soluble ELPs had homogenous distributions but low retention times, and ELPs heated above their LCST had longer retention times but less homogenous distributions.[36] Thus, the combination of LCST behavior and crosslinking provided a superior ELP delivery method.[35] In addition, the crosslinkable cysteine-containing ELP can be used at low concentration and low viscosity; previous injectable ELP hydrogel systems required high concentrations and high viscosities and could consequently cause pain or discomfort to the patient upon injection.
2.2 Resilin-like Polypeptides
Resilin-like polypeptides also have temperature-sensitive behavior. It has been observed that as temperature increases, the proteins aggregate and shift from a random structure to increased β-sheet content.[20] Some RLPs also have an upper critical solution temperature (UCST), below which they form coacervate.[18, 37] RLPs can be constructed from a range of sequences from several insect species, and the variety of sequences leads to different temperature-responsive behaviors.[16] Because of the sequence diversity, the thermally-responsive behavior observed for specific RLP sequences cannot always be generalized to all RLP sequences, and function-oriented design of RLPs may need to be taken on a case-by-case basis. Depending on the RLP design, one or both temperature transitions may exist, and the structural and phase transitions may be reversible or irreversible.
For an RLP with both high- and low-temperature sensitivity, the temperatures of these transitions were tuned by mixing the RLP solution with proline or recombinant silk fibroin (RSF).[38] RLP and poly-proline are both considered hydrophilic when using the Kyte-Doolittle hydropathy scoring system. However, the rigid structure of poly-proline exposes nonpolar side groups, so the overall peptide is slightly hydrophobic. The RLP studied in this experiment had a UCST of 6°C and an LCST of 70°C, and polyproline had an LCST of 45°C. Upon heating, RLP and poly-proline co-assembled to form an aggregate with a proline core. The hydrophobicity and stable structure of proline reduced the LCST of the RLP-poly-proline mixture to 41°C. Next, RLP was combined with RSF, which has a very rigid structure of alternating hydrophobic and hydrophilic blocks. The data suggested that the mixture formed assemblies with an RSF core. The assemblies increased in size with heating and over time and resulted in irreversible gelation in the RLP-RSF mixture. Furthermore, rod-like structures were detected over time. As the RLP-RSF mixture aged, the LCST decreased, the β-sheet structure increased, and the size of the particles increased. These shifts in structure and particle behavior indicated that the RLP-RSF co-assembly rearranged with time. By simply mixing RLPs with hydrophobic proteins, the temperature at which LCST behavior occurred was modulated, and the behavior itself was expanded to include both temperature-responsive structural changes and self-assembly into higher order structures and networks.
The phase transition temperatures of RLPs display similar behavior to those of ELPs and are sensitive to concentration and salt. A higher protein concentration lowers the transition temperature, and salts can increase or decrease the transition temperature.[20] RLPs were more soluble and had higher transition temperatures when exposed to increasing salt concentrations.[20] This observation is in contrast to ELPs, which generally experience lowered transition temperatures in the presence of salt.[39] At high salt concentrations, the RLP transition temperatures followed the Hofmeister anion series.[20] The transition temperature of the RLP was ~5°C higher in PBS and 10–14°C higher in 0.1M – 1M sodium chloride compared to pure water. These results suggest that salt ions interact with the charged groups of the RLP to increase solubility and that the identity of the salt ion affects the strength of this interaction.
Denaturants also affected the transition temperature by affecting hydrogen bonding or hydrophobic interactions.[20] Mixing the RLP with urea increased the transition temperature. This increase suggested that the aggregation process at the transition temperature relied on hydrogen bonding and that interfering with hydrogen bonds resulted in higher temperatures needed to induce aggregation. The presence of SDS removed any transition temperature aggregation behavior up to 85°C. SDS interacts with uncharged groups in proteins, and thus the elimination of transition temperature behavior implied that hydrophobic interactions are also important to the aggregation process.
The transition temperature of the RLPs in this study were also pH dependent.[20] The experimental isoelectric point of the RLP was 5.2, but the theoretical pI was 10.5. This disparity may be caused by conformational complexities in the protein as the proximity of charged residues in the protein chain can affect their individual pKa values and thus the overall pKa of the protein. The transition temperature of this RLP was lowest at pH 5, near the experimental pI, and increased by 25°C when the pH was increased to 10.
Because transition temperature behavior is dependent on hydrophilic and hydrophobic interactions, the temperature-responsive behavior of ELPs and RLPs is sensitive to pH, salts, and denaturants. If the protein will be applied in physiological conditions, saline conditions, or in harsh environments, the effect of these environments on the transition temperature will need to be considered.
2.3 Silk-like Polypeptides
Other temperature-responsive proteins include those with repetitive hydrophobic-hydrophilic blocks such as recombinant silk. One study investigated the temperature-triggered gelation at hot and cold temperatures of a recombinant silk derived from spider dragline silk.[3] Specifically, a recombinant protein composed of the C-terminal domain of Nephila clavipes major ampullate spidroin 1 was termed “NcCT.” When cooled to 2°C, the NcCT silk gelled reversibly due to hydrophobic interactions. When heated to 65°C, the NcCT protein gelled irreversibly. When NcCT was fused with short, repetitive sequences from the central region of the major ampullate spidroin (MaSp), the gelation of the fusion protein (NcCT-MaSp) only occurred at low temperatures. MaSp alone did not gel at high or low temperatures.
The structure of the NcCT gels formed at high or low temperatures varied. NcCT gels formed at high temperatures had sheet-like structures, whereas gels formed at low temperatures formed porous structures (Figure 4). Circular dichroism (CD) analysis suggested that structural changes may contribute to NcCT gelation at high temperatures. At low to intermediate temperatures (2–65°C), NcCT had an α-helix structure, and at temperatures above 65°C, the α-helix structure decreased, a result that indicated the α-helix structures loosened with increased heat. At higher temperatures, the NcCt-MaSp fusion protein had a similar decrease in α-helix structure but did not form gels. This result may indicate that the addition of the repetitive MaSp sequence interfered with the physical crosslinking of NcCT at higher temperatures.
Figure 4.

Temperature-triggered formation of gels by silk-like polypeptides at both low and high temperature. Scanning electron microscopy (SEM) images of hydrogels formed by proteins from the C-terminal domain of N. clavipes major ampullate spidroin 1 (NcCT) and the fusion proteins containing NcCT and repeated sequences from the central region of major ampullate spidroin (NcCT-MaSP). NcCT proteins, which do not contain repetitive sequences, formed irreversible hydrogels with sheet-like structures when crosslinked at high temperatures. Both proteins formed porous hydrogel networks when crosslinked at low temperatures. Reprinted (adapted) with permission from Biomacromolecules, Dual Thermosensitive Hydrogels Assembled from the Conserved C-Terminal Domain of Spider Dragline Silk, 16, 2015, Zhi-Gang Qian, Ming-Liang Zhou, Wen-Wen Song, Xiao-Xia Xia. Copyright 2015 American Chemical Society.
The crosslinking mechanisms of the NcCT and NcCT-MaSp proteins were further explored using 8-anilinonaphthalene-1-sulfonic acid (ANS) fluorescence. ANS fluoresces in hydrophobic conditions, and when mixed with aqueous protein solutions, will only fluoresce when it binds to exposed hydrophobic regions of proteins. The fluorescence of NcCT was significantly higher at 4°C and 65°C compared to 25°C. This increase indicated that NcCT exposed hydrophobic regions of the protein at low and high temperatures. The fusion NcCT-MaSp also had higher fluorescence at 4°C and 65°C compared to 25°C. Given that the fusion protein did not crosslink at high temperature, these results also suggest that the repetitive sequences of MaSp may interfere with the hydrophobic interactions that lead to crosslinking at high temperature. In all, these results contribute to the idea that irreversible gelation of NcCT at high temperatures was due to physical crosslinking of exposed hydrophobic regions and structural changes, whereas reversible gelation at low temperatures occurred due to hydrogen bonding and hydrophobic interactions without structural change. The dual-temperature hydrogel formation is conserved across recombinant silk sequences from several different spider sequences; this conservation may indicate that this gelation process is critical to web spinning in many spiders.
3. pH Sensitivity
In recombinant protein design, charged amino acids are often used to induce pH sensitivity by selecting side groups with pKa values near the desired pH range. Depending on the charge shift of the amino acid, the responsive behavior may occur above or below the pKa. In its charged state (either negatively or positively charged), an amino acid will be more hydrophilic and thus could result in increased interactions with water and decreased interactions with organic solvents. Oppositely charged amino acids may also interact with each other and shield solvent interactions. In their uncharged states, amino acids will be more hydrophobic, and there could be increased overall hydrophobicity of the protein or effects on the secondary structure in aqueous solvents.
3.1 pH-induced Delivery Systems
Histidine and glutamic acid are commonly used to create pH sensitivity in acidic conditions. The side group of histidine is protonated below its pKa of 6.0. The side group of glutamic acid becomes deprotonated above its pKa of 4.25. One study investigated a peptide containing both histidine and glutamic acid (HE) that was linked to an amphipathic peptide (MAP) and a cargo protein, glutathione S-transferase (GST), that models delivery of a protein-based drug in a tumorous system.[40] When tested in vitro, GST-MAP associated non-specifically with cells at neutral pH due to a large number of lysine groups in the MAP sequence. On the other hand, GST-MAP-HE had the positively charged lysine groups shielded at neutral pH by the negatively charged glutamic acid residues. At a slightly lower pH (6.0–6.5), the histidine groups of GST-MAP-HE began to protonate and interact with the glutamic acid groups, and the interaction between lysine and glutamic acid was disrupted. Thus, the lysine groups in the GST-MAP-HE sequence interacted with cells, and significantly higher cell association was observed at slightly acidic pH values compared to neutral pH (Figure 5A). pH sensitivity only occurred when the HE peptide and GST-MAP were linked; when free HE peptide and GST-MAP were mixed in equimolar amounts, cell association was not pH sensitive. In an in vivo tumor model, the fusion protein was enriched in the tumor, liver, and kidneys in less than 30 minutes and remained in the body for 24 hours (Figure 5B–C). The fusion protein GST-MAP-HE localized to the tumor site more efficiently than GST-MAP or GST-HE. Thus, this study showed that the simple addition of charged histidine and glutamic acid residues to the protein design induced pH-responsive drug delivery across a narrow pH range and reduced non-specific delivery at neutral pH.
Figure 5.
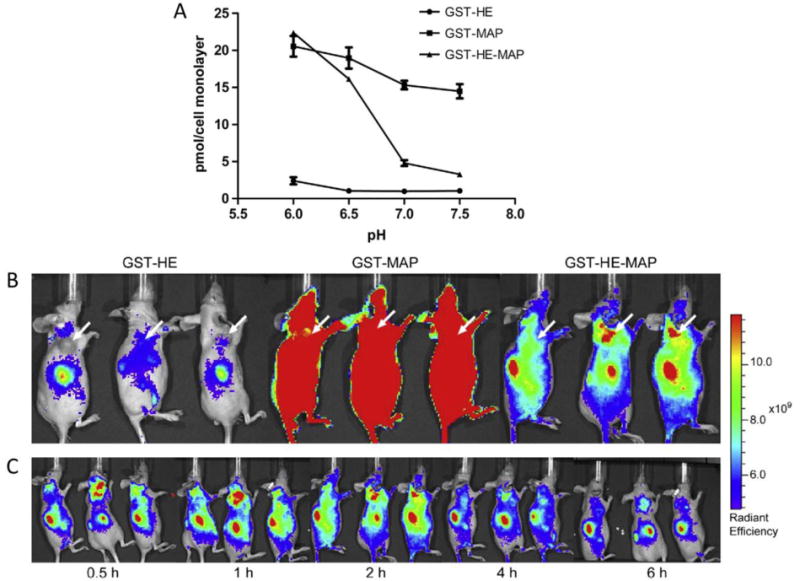
A cell penetrating peptide, named model amphipathic peptide (MAP), delivered a model drug, glutathione S-transferase (GST), in slightly acidic conditions in vivo because of its fusion with a histidine- and glutamic acid-containing peptide (HE). A) HeLa cell association of GST-carrying proteins. GST-HE-MAP was pH-sensitive and associated more strongly at pH <7, whereas GST-MAP or GST-HE were not pH sensitive. B) Fluorescently-labeled localization of GST-carrying proteins in a tumor model mouse system. GST-HE-MAP localized to tumor cells, whereas GST-MAP associated indiscriminately and GST-HE had very low association. C) Time monitoring of GST-MAP-HE protein in mouse after injection. GST-MAP-HE localized to the tumor site in less than 30 minutes. Reprinted from Biomaterials, 35/13, Likun Fei, Li-Peng Yap, Peter S. Conti, Wei-Chiang Shen, Jennica L. Zaro, Tumor targeting of a cell penetrating peptide by fusing with a pH-sensitive histidine-glutamate co-oligopeptide, 4082–4087, Copyright 2014, with permission from Elsevier.
pH-sensitive peptides can also be used to induce pH sensitivity in polymer systems. For example, pH-sensitive micelles that released a chemotherapeutic drug docetaxel (DTX) at low pH were created by conjugating poly(ethylene glycol) – poly(lactic acid) (PEG-PLA) molecules to either pH-sensitive histidine-glutamic acid peptides (HEO) or a cell-penetrating peptide composed of arginine and glycine (RGO) (Figure 6A).[41] RGO was not pH-sensitive in the pH region of interest. Thus, the ratio between RGO and HEO was used to control the pH response of the resulting micelle (Figure 6B). RGO/HEO mixtures were used to encapsulate DTX (DTX-PHPO) and were compared to PEG-PLA micelles with encapsulated DTX (DTX-PM). In the pH-sensitive DTX-PHPO system, larger percentages of DTX were released at pH 6.0–6.8 than at pH 7.4. At pH 6.8, which corresponds to the pH in tumors, the DTX-PHPO released 70% – 87% of the drug within 48 hours, whereas DTX-PM only released ~50%. Both micelle structures released ~50% of the drug within 48 hours at pH 7.4.
Figure 6.

Conjugation of cell penetrating peptides with pH-sensitive peptides improves drug delivery in acidic environments. A) Scheme showing micelle structure and drug delivery mechanisms of PEG-PLA conjugated with a cell penetrating peptide composed of arginine and glycine (RGO) or charged peptides containing histidine and glutamic acid (HEO). PEG-PLA alone formed micelles that encapsulated the drug and delivered it passively to cells through diffusion (DTX-PM). RGO and HEO formed micelles with the drug encapsulated and the surface of the micelle decorated with peptides (DTX-PHPO). Upon lowering the pH, the micelles were endocytosed by the cells. B) Altering pH sensitivity of micelles by adjusting the ratio of charged peptides to uncharged peptides. RGO alone had a pH-insensitive zeta potential, whereas HEO was pH-sensitive. By increasing the ratio between HEO and RGO in PHPO, the point of zero charge shifted. A ratio of 45:55 HEO:RGO was selected for further testing since the point of zero charge was between the pH values of physiological tumor microenvironmental conditions. Reprinted from International Journal of Pharmaceutics, 466, Ammar Ouahab, Nihad Cheraga, Vitus Onoja, Yan Shen, Jiasheng Tu, Novel pH-sensitive charge-reversal cell penetrating peptide conjugated PEG-PLA micelles for docetaxel delivery: In vitro study. 233–245, Copyright 2014, with permission from Elsevier.
In vitro experiments showed that DTX-PHPO had higher rates of DTX release at pH 6.8 than pH 7.4, whereas DTX-PM had equal rates of release at both pH values.[41] The process of pH-sensitive cell penetration was also studied by encapsulating a fluorescent marker in the micelles. Both DTX-PM and DTX-PHPO were taken up by cells at pH 6.8 and 7.4. However, DTX-PHPO had a higher fluorescent signal at pH 6.8 than at pH 7.4. In addition, DTX-PHPO was twice as toxic to MCF-7 cells at pH 6.8 compared to pH 7.4. The increased drug release, cellular uptake, and cytotoxic efficiency of the pH-sensitive micelles at lower pH may be due to their structure. At neutral pH, the opposing charges of HEO and RGO cause the two peptides to interact with each other and may prevent RGO from acting as a cell-penetrating peptide (CPP). At lowered pH, the protonation of HEO reduces this interaction and allows RGO to act as a CPP, and, as a result, the cells can more readily endocytose the entire micelle. These results demonstrate that the combination of charged peptides with polymer-based delivery systems can be effective at delivering chemotherapeutic drugs in a targeted acidic pH environment.
Peptides containing histidine and glutamic acid can be conjugated to CPPs to deliver tumor-fighting enzymes directly into cells.[42] Arginine depletion has been shown to increase tumor cell death,[43] and thus a promising anti-cancer therapy was developed by creating a recombinant protein that combined arginine deiminase, which catalyzes arginine conversion to citrulline, with a histidine-glutamic acid oligopeptide and a CPP. Cells internalized 3.5 fold more protein at pH 6 compared to pH 7.5. MDA-MB-231 cells had 2.4 times more uptake of HE-coupled enzyme at hypoxic, low pH conditions compared to normal, neutral pH conditions. In contrast, pure enzyme and enzyme coupled with PEG had no statistical differences in uptake when comparing low pH to neutral conditions. The peptide-enzyme fusion had an in vivo tumor targeting profile similar to that of enzyme coupled with PEG. Thus, this combination of a pH sensitive peptide and CPP allowed for targeted delivery of a functional enzyme to acidic, hypoxic cells.
pH-sensitive cell penetrating peptides can also be combined with other functional proteins to create functional sites anchored in cell membranes. For example, this kind of protein could be used to target antibody binding to membranes that are in acidic, tumorous microenvironments. The diphtheria toxin transmembrane domain is soluble at physiological pH but, at pH values between 2 to 6, acts as a CPP and inserts into cell membranes. The transmembrane domain was combined in a fusion protein with the antibody-binding protein ZZ, which binds to the Fc region of IgG antibodies.[7] This fusion protein integrated into large unilamellar vesicles (LUV) at pH 5. By disrupting the membrane, the protein allowed hydrogen ions and small organic compounds through the membrane. The membrane interaction was not affected when the fusion protein was bound to antibodies. When small unilamellar vesicles (SUV) were treated with the fusion protein, antibody binding to the SUV was higher at pH 5 compared to physiological pH. This result was also observed in fibroblastic L cells and non-adherent FDC-P1 cells. Treating the cells with the fusion protein at pH 5 increased the amount of antibody binding to the cells compared to treating them at pH 7.4 and did not affect cell viability. Antibody binding to FDC-P1 cells treated with the fusion protein was lower when the fusion protein was pre-treated with an antibody that binds to the diphtheria toxin domain or when the cells treated with the fusion protein were incubated with rabbit serum before incubation with antibody. This reduction in binding indicated that the toxin section attached to the membrane and the ZZ section bound IgG antibodies. Thus, this study demonstrated the use of an acid-triggered CPP fused to an antibody-binding domain that can selectively bind to tumorous cells and act as a targeted functional site for therapeutic antibodies.
3.2 pH-induced Conformational Changes
Changes in protein charge can have strong effects on protein structure and conformation. Because the secondary structure of a protein is often driven by hydrogen bonding and hydrophobic or hydrophilic interactions, a shift in the charge of even a few amino acids can cause regions of the protein to attract or repulse itself and alter the overall structure. Thus, pH-sensitive amino acids can be used to create these conformational changes through protonation or deprotonation upon a shift in environmental pH.
Histidine rich protein II (HRPII) from Plasmodium falciparum has a large number of histidine residues and undergoes conformational changes and shifts in binding properties at pH values above 6.[8] HRPII is a candidate heme polymerase and binds to iron-containing hemes in the blood through histidine and aspartate side chains. At pH values above 6, uncharged histidine groups contribute to binding of heme iron centers. At pH values below 6, histidine becomes charged and has reduced binding contribution; instead, aspartate binds through carboxylate-metal ionic interactions. When mixed with heme at pH values lower than 6, HRPII was strongly unstructured with a small amount of α-helix structure, and there was no structural change when heme concentration was increased. At these lower pH values, interactions between positively charged histidine and negatively charged aspartate may increase the stability of the helices. Adding heme did not change the structure, presumably because the heme binds to aspartate at these pH values, and the aspartate residues are spaced 5–8 amino acids apart. This distance between aspartates prevents single hemes from binding to multiple aspartate groups. At pH values above 6, the protein alone had reduced α-helix structure, but increasing heme concentration increased the helicity of the protein. At higher pH, heme may stabilize the helix structure because a single heme bound to multiple adjacent histidines could exclude solvent molecules through multiple hemes on the protein surface. In conclusion, these charged residues had a strong effect on the protein’s overall structure and binding capabilities and resulted two different binding mechanisms depending on the pH of the environment. This shift in binding mechanisms may be useful to create pH-triggered selective binding sites for other metal ions or chemicals.
pH-sensitive conformational changes can also be designed in recombinant proteins using glutamic acid. Because protein structure relies partially on intra-protein charge interactions, shifts in H+ concentration can affect the protonation state, charge interactions, and secondary structure of proteins. Prothymosin A is composed of ~50% glutamic acid and aspartic acid.[6] At physiological conditions, the protein had a random coil secondary structure. The negative charge of glutamic acid and aspartic acid increased the hydrophilicity of the protein and promoted a random secondary structure. As pH was lowered, the structure of prothymosin A collapsed to a pre-molten globule because the negatively charged glutamic acid groups began to protonate and lowered the overall hydrophilicity of the protein. To study these shifts in hydrophobicity, ANS fluorescence was used. At pH values below 5, ANS fluorescence increased significantly, and these results indicated a decrease in hydrophilicity. Upon lowering the pH below 5, the helicity of the protein increased and the radius of gyration decreased. Thus, the protein became more compact and ordered. By combining charged residues that have varying pH responses, inter-residue interactions across a range of pH can cause large shifts in protein structure and morphology.
Adding pH-sensitive residues can also modulate conformational changes that are intrinsic to specific recombinant proteins. The LCST of ELPs is strongly influenced by charge since coacervate phase separation is driven by hydrophobic collapse. An ELP design that is more hydrophobic will generally have a lower LCST, and a more hydrophilic design will have a higher LCST. When pH-sensitive amino acids are used as guest residues in the ELP sequence, the overall charge contribution of the protein will shift as the pH of the solution changes and cause changes in the hydrophobicity and LCST of the ELP. For example, glutamic acids were included in one out of eight guest residues in a silk-ELP fusion sequence.[44] Glutamic acid becomes charged above pH 4 and thus increases overall protein charge and hydrophilicity. The LCST of this silk-ELP fusion increased from ~30°C to >100°C as the solution pH increased from pH 3 to pH 12. By simply incorporating charged residues as an ELP guest residue, the LCST became highly pH-sensitive. This pH-sensitivity may be useful for purification of salt- or temperature-sensitive proteins that are fused to a pH-sensitive ELP.
pH-triggered conformational changes are sometimes experienced by proteins in cellular pathways as they enter acidic organelles. Vesicular stomatitis virus uses glycoprotein G to fuse to endosomal membranes after endocytosis. Glycoprotein G undergoes a structural transition at low pH in order to pass through the acidic Golgi complex without negatively affecting its fusion ability.[45] After exiting the Golgi complex, the protein changes structure again at neutral pH to assist in fusion to membranes. Glycoprotein G has a large number of aspartic acid and glutamic acid residues, which may contribute to this conformational change.[46] At low pH, aspartic acid and glutamic acid are protonated, which increases hydrogen bonding and causes the residues to draw closer together. As the pH increases to 7, the residues deprotonate and repel each other. Individual glutamic and aspartic acid residues in glycoprotein G were mutated to asparagine, glutamine, or leucine. The aspartic acid group located at position 268, which is buried within the core of a six-helix structure at low pH, was found to be the key pH switch. When this aspartic acid was mutated, no conformational changes occurred at any pH. Mutating other aspartic acid residues had varying effects such as lowering the pH at which the conformational change occurred or resulting in less efficient fusion. The mutations used in this study illustrate the importance of individual charged residues on the overall structure of proteins.
pH-sensitive conformational changes can also cause the formation of larger protein aggregates. For example, α-synuclein is a natively unfolded protein that forms ordered fibrils at lower pH (Figure 7).[47] It has a highly acidic C-terminus and imperfectly repeated sequences (consensus KTKEGV) that are thought to form amphipathic helixes. α-synuclein has a low intrinsic hydrophobicity with high net charge at pH 7, and a low theoretical pI of 4.4.[48] Lowering the pH decreased the overall charge of α-synuclein and caused it to adopt a partially folded conformation with β-sheet structures.[47] The structural changes were reversible and independent of protein concentration. The radius of gyration also decreased at lower pH. α-synuclein was monomeric at pH 3, and thus, it was found that individual α-synuclein monomers homogenously increased in structure and became more compact at lower pH values. These structural changes were attributed to decreased charge repulsions at lowered pH and subsequent hydrophobic collapse. Incubating the proteins at low pH increased the kinetics of fibril formation, and thus the conformational changes correlated with increased rate of fibril formation. With a small decrease in pH and subsequent change in protein charge, this natively unfolded protein dramatically changed structure and formed higher order structures.
Figure 7.
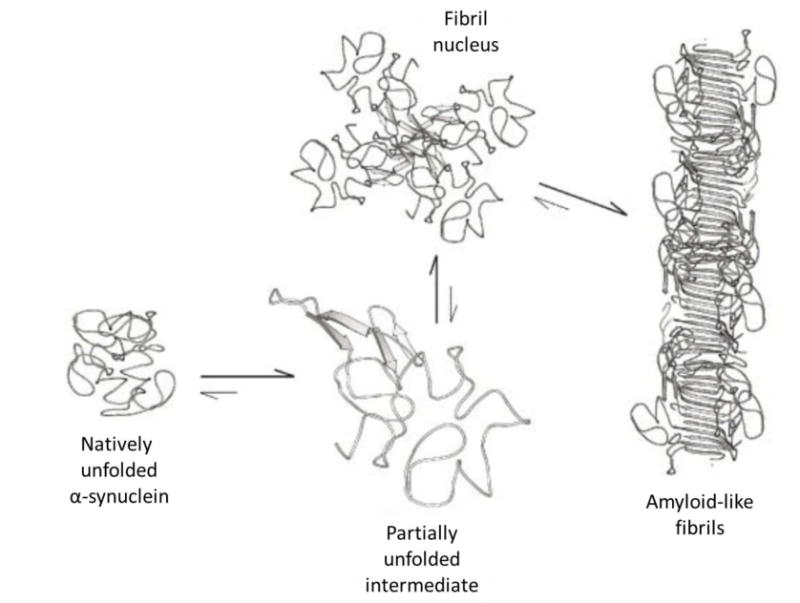
Scheme showing self-assembly model of α-synuclein. In native conditions, the protein is unstructured. As pH was lowered, hydrophobic regions were exposed and associated with the hydrophobic regions on another α-synuclein. This association created small fibril nuclei that further assembled into fibrils. Reprinted with permission from The Journal of Biological Chemistry, Evidence for a Partially Folded Intermediate in α-Synuclein Fibril Formation, 276, 2001, Vladimir N. Uversky, Jie Li, Anthony L. Fink. Copyright 2001 The American Society for Biochemistry and Molecular Biology.
3.3 pH-sensitive Fluorescence
pH-sensitive fluorescent proteins are also dependent on changes in secondary structure conformation, and any disruptions to the structure will alter the fluorescence. One such protein is EosFP, which emits green light at 516 nm in its native tetrameric state.[49] Irradiation at ~390 nm causes an EosFP monomer to cleave into two smaller proteins, one of which contains the fluorophore that emits red light at 581 nm. This conversion process relies on histidine residues in an HYG tripeptide in the chromophore and is more effective at pH values below 7. Once converted, the red light emission is pH sensitive and decreases in intensity when the pH is lowered from pH 7 to 5.5. The green light emission is not pH sensitive. The protein was used to transfect bacteria to monitor the process of phagocytosis.[50] After brief irradiation with 400 nm light, the transfected bacteria were excited with 490 nm light, and both red and green light were detected. When mixed with blood, the bacteria were phagocytosed by granulocytes. Any phagocytosed bacteria that were fused with lysosomes within the cell were exposed to the acidic environment of the lysosome. This lower pH reduced red light emission but did not affect green light emission. Thus, the processes of phagocytosis and lyososome fusion were monitored by the kinetics of red light emission; a faster reduction in red light indicated more rapid or efficient phagocytosis. Furthermore, by comparing the signals of pH-insensitive green fluorescence and pH-sensitive red fluorescence, this protein can be used to locate and accurately measure acidic organelles.
Another pH-sensitive fluorescent protein, enhanced Cyan fluorescence protein (ECFP), can be used for precise pH measurements both in vitro and in vivo. The fluorescent lifetime of ECFP decreased by 40% at pH 5.5 in comparison to pH 7, and the emission spectrum changed.[51] This change in intensity and lifetime was fully reversible when the pH returned to 7. In cells, the fluorescent lifetime decreased by 25% when cells were incubated in a buffer at pH 5.8, and this decrease was fully reversible. At physiological pH, ECFP fluoresces through tryptophan groups. These tryptophan groups are flanked by many charged residues, including histidine, tyrosine, and aspartic acid, but whether these residues are responsible for the fluorescent quenching is not clear, and the exact mechanism of the fluorescent quenching of ECFP at lowered pH is not well understood. When ECFP was fused with a tumor marker, chromagranin A, it was able to target acidic secretory granules, whereas LysoTracker, an organic dye commonly used for visualizing acidic organelles, appeared to be in larger organelles.[52] This pH-sensitive fusion protein was used to make pH measurements accurate up to 0.2 pH units by measuring the fluorescence lifetime of the protein in a cell and comparing this value to a calibration curve. The pH of secretory granules was measured to be 5.5 ± 0.05, which falls within the reported range of 5.4 to 5.8. In all, this reversibly pH-sensitive fluorescent protein can be used to make precise measurements of acidic pH values within cells.
One of the most commonly used fluorescent proteins, green fluorescent protein (GFP), was altered to respond to pH.[9] GFP has several key residues that control the conformation of its fluorescent chromophore. Through site-directed mutagenesis, histidine was introduced in flanking positions. One promising mutant showed a small amount of pH sensitivity since a shift from pH 7.4 to 6.0 caused a decrease in excitation at 395 nm and a 26% increase in excitation at 475 nm. From this mutant, a library of GFP mutants was screened for pH sensitivity. From this library, the authors selected two designs: one protein with an inverse ratio between the excitation peaks of 410 and 470 nm that changed between pH 7.5 and 5.5 and another protein with concurrent reductions in the excitation peaks as pH was lowered. The exact mechanism of these behaviors is unknown, but the authors created pH-sensitive GFP fusion proteins that were targeted to vesicles. They measured the pH of the local environment through fluorescent imaging of the change in excitation ratio (Figure 8). Thus, by mutating this fluorescent protein to obtain a pH-sensitive version, the pH of cellular compartments was easily measured.
Figure 8.
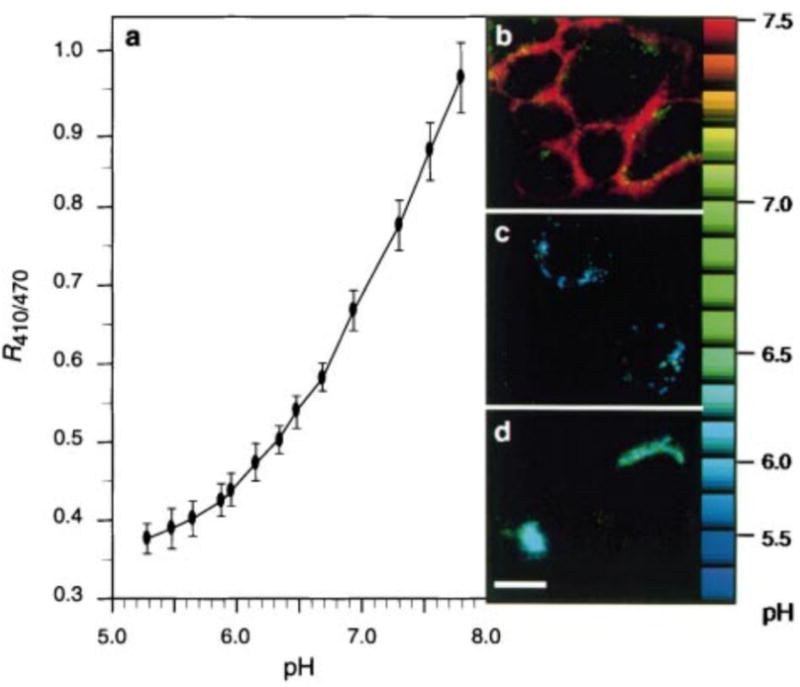
A pH-sensitive fluorescent protein derived from green fluorescent protein was used as a pH indicator. A) Standard curve for pH measurement created by expressing the fluorescent protein on cell surfaces and culturing the cells in buffers with varying pH. The fluorescent protein was used for pH measurement of cell components by fusing with organelle-targeting proteins including B) glycophosphatidylinositol for cell surfaces, C) cellubrevin for endosomes, and D) TGN38 for the trans-Golgi network. The relative fluorescence localized to the organelles was measured and compared to the standard curve to determine the pH. Scale bar represents 10 μm. Reprinted by permission from Macmillan Publisher Ltd: Nature, Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins, Gero Miesenbock, Dino A. De Angelis, James E. Rothman, 394, 1998, Copyright 1998.
4. Self-assembly
Protein structures can be categorized into four levels: primary and secondary structures that involve peptide bonds and local interactions, tertiary structures that involve interactions among domains, and quaternary structures that are defined by interactions among proteins. An increasing number of protein domains responsible for tertiary and quaternary structures have been identified and harnessed to make recombinant proteins that are capable of assembly into complicated supramolecular structures. For example, ELPs discussed in section 2.1 are known to self-assemble into micelles or vesicles at specific conditions (e.g., temperature and pH).
4.1 Oleosin: An Example of Amphiphilic Protein Self-assembly
Surfactant-like proteins are also known to undergo self-assembly spontaneously because of their amphiphilic nature. One actively studied surfactant-like protein is oleosin, which is found in plant seeds and stabilizes oil bodies. The Hammer group constructed a series of recombinant oleosin variants and demonstrated that they could self-assemble into various structures under different conditions.[11] Wild type oleosin is composed of three segments – an N-terminal hydrophilic block, a central hydrophobic block, and a C-terminal hydrophilic block. Recombinant variants were created with a shortened central hydrophobic block and by varying the lengths of the hydrophilic blocks on both ends.
4.1.1 Ionic Strength-dependent Self-assembly
Concentration-dependent self-assembly was observed with recombinant oleosin variants using emulsion preparation; the resulting structures were dependent on the type of variant in the organic phase and the ionic strength in the aqueous phase (Figure 9).[11] At low ionic strength (e.g., deionized water), either lamellar or fibril structures were observed, depending on the relative lengths of the hydrophilic blocks to the hydrophobic block. Variants with higher hydrophilic portions formed fibers whereas variants with lower hydrophilic fractions formed sheets. As the ionic strength increased, variants with higher hydrophilic portions started to form vesicle structures. A mixture of fibers and vesicles was observed at moderate ionic strength (35 mM), and vesicle structures dominated at high ionic strength (70 and 140 mM). On the other hand, variants with lower hydrophilic fractions formed dominantly lamellar structures in solutions up to 70 mM in ionic strength. Coexistence of sheets and vesicles was observed at 140 mM. These results suggested that the ratio between hydrophilic and hydrophobic blocks had a significant effect on self-assembly structures. Larger hydrophilic segments prevent structures with smaller curvatures, and variants with smaller hydrophilic blocks were more likely to assemble into lower-curvature structures (e.g., sheets) because the small hydrophilic blocks occupied less space.
Figure 9.
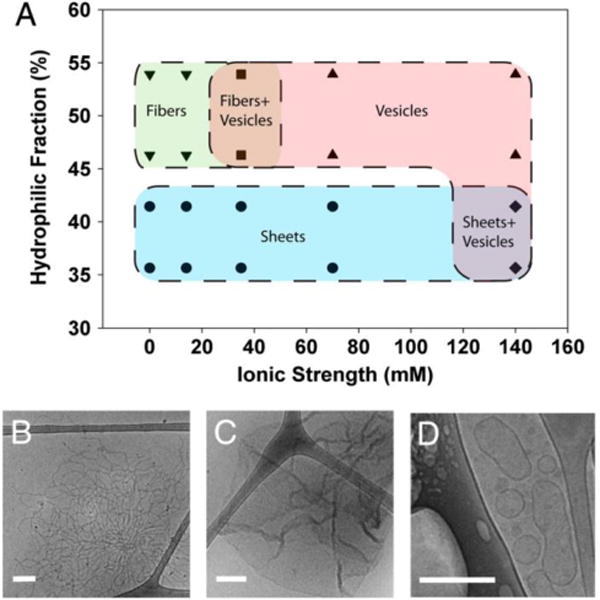
Self-assembly structures determined by protein hydrophilicity and ionic strength in solution. A) Structures of self-assembled oleosins. Proteins with a lower hydrophilic fraction favored structures with smaller curvature (e.g., sheets). For proteins with the same hydrophilic fraction, higher ionic strength in the solution favored vesicle formation. Data points represent actual experimental observations. B-D) Representative cryo-TEM images of fibers, sheets, and vesicles, respectively. Reprinted with permission from Proceedings of the National Academy of Sciences of the United States of America, Self-assembly of tunable protein suprastructures from recombinant oleosin, 109, 2012, Kevin B. Vargo, Ranganath Parthasarathy, Daniel A. Hammer. Copyright 2012, the National Academy of Sciences of the United States of America.
4.1.2 Sequence Charge-dependent Self-assembly
Because of its large hydrophobic domain, native oleosin and the recombinant variants described above had limited solubility in aqueous solvents.[11] The low solubility in aqueous environments hampers the potential biological applications because organic solvents often have low cytocompatibility. Thus, the Hammer group created a water-soluble oleosin variant (Oleosin-30) and verified its self-assembly in a pure aqueous phase.[53] Solubility in water was increased by deleting 65% of the hydrophobic segment, and this change potentially reduced the secondary structure predicted in native oleosin. To further study the effect of protein structure on self-assembly, five glycine residues were inserted into the remaining hydrophobic segment (Oleosin-30G) to promote random coil structure. In both Oleosin-30 and Oleosin-30G, the hydrophilic portions were uncharged. To study the influence of charge states on self-assembly, two more variants were constructed. In Oleosin-30G(+) and Oleosin-30G(−), residues were mutated such that all charged residues were positively or negatively charged, respectively.
Oleosin-30 showed improved solubility in phosphate buffer and was able to self-assemble into micelles. A two-stage assembly was observed with Oleosin-30G. It was proposed that at a concentration lower than the critical micelle concentration (CMC), Oleosin-30G assembled into a loose structure, which was not dense enough to completely expel water from the core. As the concentration reached the CMC, more proteins participated in the assembly process and resulted in micelles that had densely packed structures. The charged variants, Oleosin-30G(+) and Oleosin-30G(-), exhibited a similar two-stage self-assembly of micelles. However, compared to Oleosin-30G, Oleosin-30G(+) had a much higher CMC, whereas Oleosin-30G(-) had a lower CMC. Circular dichroism (CD) revealed concentration-dependent changes in secondary structures for Oleosin-30G, which adopted a random coil structure at low concentrations and displayed restricted secondary structures only at high concentrations. Conversely, over the concentration range examined, Oleosin-30G(-) had a restricted structure, which decreased the penalty in conformational entropy upon self-assembly and made the assembly energetically more favorable. This result suggested that protein charge affected protein self-assembly, not only through electrostatic interactions but also by affecting secondary structures.
The development of water-soluble Oleosin variants has provided insights into controlling self-assembly structures by either external factors (e.g., ionic strength in the solution) or internal factors (e.g., protein sequence and charge). To utilize these lessons in a biological application, the Hammer group functionalized Oleosin-30 with the cell-binding domain RGDS. In this case, the RGDS domain was fused to the C-terminus of Oleosin-30 and did not change the self-assembly behavior of the protein.[53] In the future, additional functionalized self-assembling materials can be developed for various applications, but the effect of additional domains on self-assembly should be taken into consideration.
4.2 Self-assembly to Form Hydrogels
Some protein-protein interactions have been utilized to physically crosslink protein chains. For example, the Heilshorn group developed Mixing-Induced Two-Component Hydrogels (MITCHs) based on the interaction between WW domains and proline-rich sequences.[54] The MITCH system is composed of two recombinant proteins with WW domains exclusively in one protein and proline-rich sequences exclusively in the other one. Gelation was not observed with solutions of each individual protein. However, when the two components were mixed together, WW domains in one protein bound to proline-rich sequences in the other protein and resulted in hydrogel formation. The gelation kinetics and the mechanical properties of the hydrogel were tuned by using WW domains with different binding affinities for proline-rich sequences and by incorporating different numbers of WW domains and proline-rich sequences in the MITCH components. The gelation time of MITCH was as short as ~30 seconds. This short gelation time allowed homogeneous cell encapsulation throughout the hydrogels. By taking advantage of the modularity in recombinant protein design, bioactive peptide sequences can be incorporated easily into the backbone of the MITCH components forming the network or can be fused with short proline-rich tags that crosslink into the MITCH network.[55] It is also possible to encapsulate free growth factor within the MITCH hydrogels.[55] By controlling the affinity of the proline-rich tags for the WW domains within the MITCH network, bioactive peptides can be released from the network with tunable kinetics.
Leucine zippers are small coiled-coil domains that can self-assemble into superhelical structures. The sequence of a leucine zipper is composed of heptad repeats, and the identity of the amino acids comprising the heptads can modulate the self-assembly conditions, helix stability, and oligomeric state of the superhelix.
Leucine zippers have been used as physical crosslinking domains to make hydrogels. The Tirrell group first designed a recombinant protein composed of a hydrophilic polyelectrolyte domain and two pH-sensitive leucine zippers on the N- and C-termini.[56] The sequences of the leucine zippers were rich in glutamic acid, and thus the stability of the leucine zipper self-assembly depended on pH. At basic pH, the glutamic acids in the leucine zippers became charged and resulted in increased charge repulsion and decreased stability of the self-assembly. As a result, no gelation was observed due to the lack of interactions between leucine zippers. At near-neutral pH, the glutamic acids in the sequence were neutralized and resulted in decreased charge repulsion between leucine zippers and increased stability of the assembly. The stabilized self-assembly of leucine zippers led to gelation of the solution over time. In addition to being sensitive to pH, the stability of the leucine zipper self-assembly could be altered at elevated temperature. The solution exhibited gelation behavior over time at 23 °C but remained a viscous liquid when heated to 55 °C. However, the gelation behavior was restored when the solution was allowed to cool down to 23 °C, and this result suggested that the denaturation of the leucine zippers upon heating was reversible. The dual-sensitivity of the leucine zipper self-assembly makes them an attractive material because of the expanded flexibility in designing and controlling their self-assembly.
In hydrogels crosslinked by interactions between proteins, the resulting stability not only depends on the strength of the interactions, but also on the arrangement of the interacting domains in the recombinant protein. Shen and coworkers studied the effect of domain arrangement on hydrogel stability by designing a series of leucine zipper-fusion proteins.[57] In this work, two different leucine zippers were used as the interacting domains. Specifically, one leucine zipper (denoted as A) associates as a tetramer, and the other leucine zipper (designated as P) forms a pentamer. However, A and P do not interact with each other. Three recombinant proteins were designed: AC10A had two A leucine zipper domains flanking a polyelectrolyte domain C10, PC10P had P instead of A as the flanking domains, and PC10A had both P and A domains in one protein. All three proteins formed hydrogels with different storage moduli (G’). The lower G’ of the AC10A hydrogels compared to PC10P hydrogels could be due to the different oligomeric states of the P and A domains. Meanwhile, PC10A hydrogels showed a slightly higher G’ than PC10P hydrogels, and the erosion rate of PC10A hydrogels was reduced by up to two orders of magnitude when compared to those of AC10A and PC10P hydrogels. These results confirmed the hypothesis that more interchain interactions were involved in PC10A hydrogels because P and A domains on one protein chain had to interact with their counterparts on other chains. On the other hand, two A or two P domains on the same protein chain can interact with each other to from intrachain interactions in AC10A or PC10P hydrogels, respectively, and result in less stable hydrogels. This system provides insights into the effects of self-assembled structures on the stability of a higher-order network.
Because self-interacting protein domains usually interact through non-covalent, transient associations, the resulting hydrogels often have weak or moderate mechanical properties and are not very stable over time. However, it is possible to reinforce the transient interactions by introducing covalent bonds, such as disulfide bonds between cysteines, into the network to improve hydrogel mechanical properties and stability. For example, Huang and coworkers introduced cysteine residues into a leucine zipper-based recombinant protein such that they would be in close proximity when the two leucine zippers associated.[55] The protein solution at pH 7.4 spontaneously gelled within three hours at 37 °C. The hydrogels were treated with a dehydrothermal process and reconstituted before testing for stability. The dehydrothermal treatment facilitated the formation of disulfide bonds between cysteines. More than 60% of the original weight remained after a 30-day incubation. On the other hand, proteins without the additional dehydrothermal treatment were able to form hydrogels as well but with much lower stability because the network was not secured by disulfide bonds. A similar strategy was applied to temperature-triggered gelation of alanine-rich ELP hydrogels[58] that were toughened by introducing cysteines at the ELP chain ends.[59] These results demonstrated the utilization of external covalent bonds to reinforce structures after self-assembly.
Covalent bonds can also be introduced within the self-assembly domains. In a work conducted by Fernández-Colino and coworkers, a leucine zipper sequence with intrinsic cysteine residues was utilized to make self-assembled hydrogels.[60] Leucine zippers were fused on two ends of an ELP domain (EI-ZC) with an LCST at 16 °C. Two control constructs were designed. To investigate the contribution of disulfide bond formation on hydrogel stability, EI-ZL was constructed with the cysteine residues in the leucine zippers being mutated to leucine residues. To study the contribution from interactions between leucine zippers, EI was constructed with only the ELP domain. Gelation of concentrated protein solutions occurred when the temperature was above the LCST of the ELP domain. However, when hydrogels were shaken in excess PBS, the EI hydrogel dissolved immediately whereas both EI-ZC and EI-ZL hydrogels remained structurally intact. The disulfide bonds in EI-ZC hydrogels resulted in 2% erosion, whereas the EI-ZL hydrogels that lacked cysteine residues had 30% erosion. This result suggested that although the gelation was induced by coacervation of the ELP domain, the interactions between leucine zippers reinforced the network. The EI-ZC gels had a stable complex modulus at 1300 Pa. On the other hand, the complex modulus of EI-ZL continuously decreased over time until it reached 130 Pa. The above work demonstrated that weak self-assembled structures (e.g., coacervation of the ELP domain) can be strengthened by stronger protein-protein interactions (e.g., leucine zipper interactions) and that covalent bonds (e.g., disulfide bonds between cysteines) can secure the transient self-assembly and increase the stability of the network.
Recombinant self-assembly domains have also been used to make hydrogels of composite materials. One example is the dual-component Dock-and-Lock (DnL) self-assembly mechanism developed by the Burdick group to make recombinant protein-synthetic polymer hydrogels.[61, 62] The DnL system is composed of a recombinant dimeric-docking domain (rDDD) derived from cAMP-dependent protein kinase A and an anchoring domain (AD) from A-kinase anchoring protein, which was conjugated to each end of a 4-arm PEG to comprise the synthetic polymer component. Gelation did not occur with only one component but did occur spontaneously when both components were mixed.[61] Gelation was initiated with the dimerization of the docking domains and was further secured by the interaction between the dimerized docking domains and the anchoring domains. The DnL hydrogels were shear-thinning and self-healing, and they were used to encapsulate human mesenchymal stem cells (hMSCs) in an injectable delivery system.[61] The DnL hydrogels were also reinforced by post-gelation photo-crosslinking.[62] Specifically, photoreactive methacrylate groups were introduced to the ends of the anchoring component to allow photo-crosslinking by UV radiation. Photo-crosslinked DnL hydrogels showed higher storage moduli and slower erosion rates than the DnL hydrogels without photo-crosslinking. Encapsulated hMSCs showed comparable viability after three days of culture in photo-crosslinked gels and gels without photo-crosslinking.[62] The DnL system demonstrates that recombinant self-assembly domains can also be harnessed to make hybrid polymer hydrogels.
4.3 Complementation of Protein Fragments
Some recombinant protein domains are fragments derived from a single protein and have a tendency to reconstitute with their counterparts. One such example is the SpyTag and SpyCatcher system, which are split domains derived from the autocatalytic isopeptide bond-forming subunit from Streptococcus pyogenes.[63, 64] When both domains are present, an isopeptide bond is formed between a specific aspartic acid on SpyTag and a specific lysine on SpyCatcher. Zhang et al. demonstrated that by strategically arranging the locations of SpyTag and SpyCatcher within a recombinant protein chain, the resulting topology of the protein chain can be controlled, as illustrated in Figure 10.[65] These two domains have also been harnessed to form covalently crosslinked ELP hydrogels.[66] The stability of the hydrogels was controlled by the number of SpyTag and SpyCatcher domains and the arrangement of them within the ELP-fusion protein chains. Unlike other networks that are based on transient self-assembly (e.g., the MITCH system described in section 4.2), SpyTag and SpyCatcher directly result in covalent networks with good stability.
Figure 10.
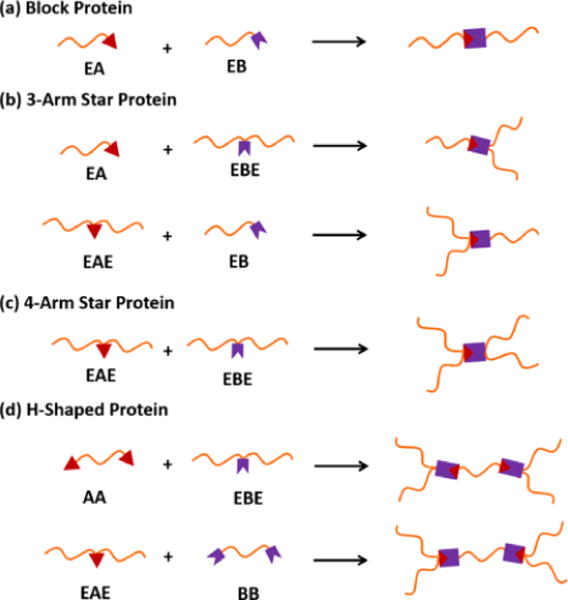
Illustration of protein chain topologies that can be formed by different arrangements of SpyTag and SpyCatcher within protein chains. A: SpyTag, B: SpyCatcher, and E: ELP. Reprinted with permission from Journal of the American Chemical Society, Controlling Macromolecular Topology with Genetically Encoded SpyTag-SpyCatcher Chemistry, 135, 2013, Wen-Bin Zhang, Fei Sun, David A. Tirrell, Frances H. Arnold. Copyright 2013, American Chemical Society.
Fluorescent proteins have been split into N- and C-terminal domains that reconstitute the intact protein. Because these split domains do not fluoresce until reconstituted, they have been utilized as a tool for bimolecular fluorescence complementation (BiFC) applications to study protein-protein interactions. In most studies, a pair of split fluorescent domains is fused to a pair of protein domains whose interactions are being investigated. When the protein pair associates, the split fluorescent domains fused to them will be drawn into close proximity and result in fluorescence. On the other hand, if the protein pair does not interact, the split domains will remain separated, and reconstitution and subsequent fluorescence will not occur efficiently. BiFC pairs are widely used for detecting protein-protein interactions in live cells. Comprehensive reviews on using split fluorescent domains for BiFC are available.[13, 67] Recently, Sjöhamn et al. utilized a BiFC pair to stabilize the interaction between human aquaporin 0 (hAQP0) and calmodulin (CaM).[68] hAQP0 is a membrane protein and is difficult to purify as part of a complex with its regulator CaM. When both hAQP0 and CaM fused with BiFC domains were co-expressed in yeast, a fluorescent signal was observed, and these results suggested that the fusion with BiFC domains did not alter the interaction between hAQP0 and CaM. Moreover, the complexes of hAQP0 and CaM remained intact after Ni-affinity chromatography followed by size exclusion chromatography. In contrast, independently expressed hAQP0 and CaM formed complexes that dissociated during size exclusion chromatography. This result suggested that the weak interactions between hAQP0 and CaM were augmented by the interaction of the BiFC domains. Thus, this study demonstrated a new application of BiFC in purification of protein complexes with weak interactions.
5. Conclusions
Temperature- and pH-responsive behavior can be included in proteins by careful selection of amino acid sequences. These responsive behaviors can cause conformational changes that trigger self-assembly into higher order structures.
The temperature-responsive proteins explored here are generally unstructured and experience changes in hydrophobicity and structure when the environmental temperature is varied. Fusing these temperature-responsive peptides with other peptides of varying hydrophobicity or rigidity results in proteins that can form higher order structures, such as micelles or fibers, upon heating. These fusion proteins can be used for temperature-triggered drug delivery or gelation.
Including charged residues such as histidine, aspartic acid, and glutamic acid in recombinant protein design can create pH-sensitivity. At varying pH values, these residues can induce intra-protein association, bind to metal groups, bridge membranes and bilayers, and change protein conformation. pH-responsive recombinant proteins can be used for targeted drug delivery to acidic organelles within cells or to tumors, where extracellular pH is lower than physiological pH. pH-induced conformational changes can also be used to trigger fluorescence in acidic environments or to create large fibrous aggregates.
Environmentally-responsive proteins can be designed to self-assemble by alternating hydrophobic and hydrophilic blocks within the protein. The length and order of the blocks can be used to tune self-assembly behaviors such as induction of gelation upon mixing or by an environmental trigger. Complementary binding sequences on two different proteins can be used for two-component gelation in which the two separate solutions will not gel alone but will gel quickly upon mixing. This characteristic is desirable for storage stability and medical applications. Combination of proteins with other environmentally-sensitive or functional peptides can create multiple responses, including pH-triggered fluorescent changes, chemical bond formation after self-assembly, and thermal gelation with structure-stabilizing auto-crosslinking.
Through careful sequence selection, proteins can be created that have smart behavior in response to multiple environmental triggers. These behaviors can be combined to create highly selective delivery systems, complex changes in higher-order structure, and reversible changes in phases and mechanical properties. Many of these behaviors are not fully understood and will require detailed study to uncover the mechanisms that drive the responsive behavior.
There are a wide range of other environmental triggers that have yet to be used for smart behavior in protein-based systems. Polymeric materials that have light-triggered smart behavior have been used for rapid photo-crosslinking. These UV light-crosslinking mechanisms have begun to be developed for recombinant protein-based materials[69–71] and have promise for further biomedical applications since near infrared and UV light can penetrate up to 40 cm into skin.[69–72]
Tissue engineering materials that can react to changes in mechanical forces or electrical stimulus may help to grow electrically- or mechanically-sensitive tissues in vitro for organ replacement or in vivo for tissue regeneration. Mechanical stimulation may be used as an environmental trigger since some proteins undergo structural changes in response to mechanical forces. For example, proteins from egg capsules of marine snails[73] and from hagfish slime threads[73] undergo strain-induced transitions in their secondary structures from α-helices to β-sheets. Electrical stimulation may affect cardiac cell growth,[74] and using conductive scaffolds to grow electrically sensitive cells may improve their growth.[75] Electrically conductive polymers[76] have been used for neural cell growth[75] and have been successfully combined with proteins.[77] Electrically conductive proteins do exist. For example, crystallized bovine hemoglobin can act as a semiconductor,[78] some bacteria use redox proteins on their outer membrane to transfer electrons from the surface of iron oxides,[79] and photosynthetic protein complexes convert light into chemical energy through electron transfer.[80, 81] However, electrically-sensitive proteins have not been well developed for biological applications.
In addition, some organisms have a magnetosensing ability that allows them to sense magnetic fields. The biological mechanism behind this behavior is not well understood. Recently, Qin et al. reported a possible magnetic complex in fruit flies that could contribute to magnetosensing at a molecular level.[82] Essential components of the complex were recombinantly produced, and the assembled recombinant complex showed a tendency to align with a magnetic field in vitro. In the future, protein-based smart materials could be created that have sophisticated, multi-layered responses to their surroundings by developing and combining protein sequences that self-assemble or are responsive towards temperature, pH, mechanical, electrical, light, or magnetic triggers.
Acknowledgments
This work was supported by the National Science Foundation (Award DMR-1309787 and CBET-1512285 to J.C.L) and the National Institutes of Health (NIDCR R03DE021755 and NIAMS R21AR065644 to J.C. L.).
Biographies
Sydney Hollingshead received her BS in chemical engineering from the University of California at Santa Barbara in 2013. She is currently working towards her PhD in chemical engineering at Purdue University. Her research focuses on creating adhesive recombinant proteins for soft tissue medical applications.

Charng-Yu Lin obtained his BS and MS in chemical engineering from National Taiwan University, Taiwan R.O.C. in 2010 and 2013, respectively. He is pursuing his PhD from the School of Chemical Engineering at Purdue University. He specializes in constructing functional biomaterials based on modular recombinant proteins. He focuses on developing materials for stem cell differentiation in tissue engineering and recombinant protein-based probes for epigenetic markers in cell differentiation.

Julie C. Liu received a B.S.E. in chemical engineering from Princeton University. She obtained her Ph.D. in chemical engineering at the California Institute of Technology in 2006 and was an NIH postdoctoral fellow in the department of cell biology at the University of Massachusetts Medical School. She is an associate professor of chemical engineering and biomedical engineering at Purdue University. Her research interests include biomimetic materials, tissue engineering, stem cell differentiation, surgical adhesives, and detection of epigenetic modifications. Her research has been funded by the NSF, NIH, DoD, American Heart Association, and a 3M Nontenured Faculty Award.

References
- 1.Urry DW, Gowda D, Parker TM, Luan CH, Reid MC, Harris CM, Pattanaik A, Harris RD. Biopolymers. 1992;32:1243. doi: 10.1002/bip.360320913. [DOI] [PubMed] [Google Scholar]
- 2.Su RSC, Kim Y, Liu JC. Acta Biomater. 2014;10:1601. doi: 10.1016/j.actbio.2013.06.038. [DOI] [PubMed] [Google Scholar]
- 3.Qian ZG, Zhou ML, Song WW, Xia XX. Biomacromolecules. 2015;16:3704. doi: 10.1021/acs.biomac.5b01231. [DOI] [PubMed] [Google Scholar]
- 4.Dinjaski N, Kaplan DL. Curr Opin Biotechnol. 2016;39:1. doi: 10.1016/j.copbio.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Smits F, Buddingh BC, van Eldijk MB, van Hest J. Macromol Biosci. 2015;15:36. doi: 10.1002/mabi.201400419. [DOI] [PubMed] [Google Scholar]
- 6.Uversky VN, Gillespie JR, Millett IS, Khodyakova AV, Vasiliev AM, Chernovskaya TV, Vasilenko RN, Kozlovskaya GD, Dolgikh DA, Fink AL. Biochemistry. 1999;38:15009. doi: 10.1021/bi990752+. [DOI] [PubMed] [Google Scholar]
- 7.Nizard P, Liger D, Gaillard C, Gillet D. FEBS Lett. 1998;433:83. doi: 10.1016/s0014-5793(98)00890-4. [DOI] [PubMed] [Google Scholar]
- 8.Lynn A, Chandra S, Malhotra P, Chauhan VS. FEBS Lett. 1999;459:267. doi: 10.1016/s0014-5793(99)01260-0. [DOI] [PubMed] [Google Scholar]
- 9.Miesenböck G, De Angelis DA, Rothman JE. Nature. 1998;394:192. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 10.Apostolovic B, Danial M, Klok HA. Chem Soc Rev. 2010;39:3541. doi: 10.1039/b914339b. [DOI] [PubMed] [Google Scholar]
- 11.Vargo KB, Parthasarathy R, Hammer DA. Proc Natl Acad Sci USA. 2012;109:11657. doi: 10.1073/pnas.1205426109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shah NH, Muir TW. Chem Sci. 2014;5:446. doi: 10.1039/C3SC52951G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kodama Y, Hu CD. Biotechniques. 2012;53:285. doi: 10.2144/000113943. [DOI] [PubMed] [Google Scholar]
- 14.Wise SG, Yeo GC, Hiob MA, Rnjak-Kovacina J, Kaplan DL, Ng MK, Weiss AS. Acta Biomater. 2014;10:1532. doi: 10.1016/j.actbio.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waterhouse A, Wise SG, Ng MK, Weiss AS. Tissue Eng Part B: Reviews. 2011;17:93. doi: 10.1089/ten.TEB.2010.0432. [DOI] [PubMed] [Google Scholar]
- 16.Lyons RE, Wong DC, Kim M, Lekieffre N, Huson MG, Vuocolo T, Merritt DJ, Nairn KM, Dudek DM, Colgrave ML. Insect Biochem Mol Biol. 2011;41:881. doi: 10.1016/j.ibmb.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Gosline J, Guerette P, Ortlepp C, Savage K. J Exp Biol. 1999;202:3295. doi: 10.1242/jeb.202.23.3295. [DOI] [PubMed] [Google Scholar]
- 18.Dutta NK, Truong MY, Mayavan S, Roy Choudhury N, Elvin CM, Kim M, Knott R, Nairn KM, Hill AJ. Angew Chem Int Ed. 2011;50:4428. doi: 10.1002/anie.201007920. [DOI] [PubMed] [Google Scholar]
- 19.Urry D, Shaw R, Prasad K. Biochem Biophys Res Commun. 1985;130:50. doi: 10.1016/0006-291x(85)90380-8. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Luo T, Kiick KL. Macromol Rapid Commun. 2015;36:90. doi: 10.1002/marc.201400521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xia XX, Xu Q, Hu X, Qin G, Kaplan DL. Biomacromolecules. 2011;12:3844. doi: 10.1021/bm201165h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keeley FW, Bellingham CM, Woodhouse KA. Phil Trans R Soc B. 2002;357:185. doi: 10.1098/rstb.2001.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hassouneh W, MacEwan SR, Chilkoti A. Methods Enzymol. 2012;502:215. doi: 10.1016/B978-0-12-416039-2.00024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Kong N, Laver B, Liu J. Small. 2016;12:973. doi: 10.1002/smll.201502429. [DOI] [PubMed] [Google Scholar]
- 25.Meyer DE, Chilkoti A. Biomacromolecules. 2004;5:846. doi: 10.1021/bm034215n. [DOI] [PubMed] [Google Scholar]
- 26.McDaniel JR, Radford DC, Chilkoti A. Biomacromolecules. 2013;14:2866. doi: 10.1021/bm4007166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christensen T, Hassouneh W, Trabbic-Carlson K, Chilkoti A. Biomacromolecules. 2013;14:1514. doi: 10.1021/bm400167h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kramer JR, Petitdemange R, Bataille L, Bathany K, Wirotius AL, Garbay B, Deming TJ, Garanger E, Lecommandoux S. ACS Macro Lett. 2015;4:1283. doi: 10.1021/acsmacrolett.5b00651. [DOI] [PubMed] [Google Scholar]
- 29.Moktan S, Ryppa C, Kratz F, Raucher D. Invest New Drugs. 2012;30:236. doi: 10.1007/s10637-010-9560-x. [DOI] [PubMed] [Google Scholar]
- 30.Rodrigues PC, Scheuermann K, Stockmar C, Maier G, Fiebig HH, Unger C, Mülhaupt R, Kratz F. Bioorg Med Chem Lett. 2003;13:355. doi: 10.1016/s0960-894x(02)01002-8. [DOI] [PubMed] [Google Scholar]
- 31.Kratz F, Beyer U, Schutte MT. Crit Rev Ther Drug Carrier Syst. 1999;16:245. doi: 10.1615/critrevtherdrugcarriersyst.v16.i3.10. [DOI] [PubMed] [Google Scholar]
- 32.Ghoorchian A, Chilkoti A, López GP. Anal Chem. 2014;86:6103. doi: 10.1021/ac5012574. [DOI] [PubMed] [Google Scholar]
- 33.Qin G, Perez PM, Mills CE, Olsen BD. Biomacromolecules. 2016;17:928. doi: 10.1021/acs.biomac.5b01604. [DOI] [PubMed] [Google Scholar]
- 34.Bates FS, Fredrickson GH. Phys Today. 1999;52:32. [Google Scholar]
- 35.Asai D, Xu D, Liu W, Quiroz FG, Callahan DJ, Zalutsky MR, Craig SL, Chilkoti A. Biomaterials. 2012;33:5451. doi: 10.1016/j.biomaterials.2012.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu W, MacKay JA, Dreher MR, Chen M, McDaniel JR, Simnick AJ, Callahan DJ, Zalutsky MR, Chilkoti A. J Control Release. 2010;144:2. doi: 10.1016/j.jconrel.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lyons RE, Elvin CM, Taylor K, Lekieffre N, Ramshaw JA. Biotechnol Bioeng. 2012;109:2947. doi: 10.1002/bit.24565. [DOI] [PubMed] [Google Scholar]
- 38.Whittaker JL, Dutta NK, Knott R, McPhee G, Voelcker NH, Elvin C, Hill A, Choudhury NR. Langmuir. 2015;31:8882. doi: 10.1021/acs.langmuir.5b01014. [DOI] [PubMed] [Google Scholar]
- 39.Cho Y, Zhang Y, Christensen T, Sagle LB, Chilkoti A, Cremer PS. J Phys Chem B. 2008;112:13765. doi: 10.1021/jp8062977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fei L, Yap LP, Conti PS, Shen WC, Zaro JL. Biomaterials. 2014;35:4082. doi: 10.1016/j.biomaterials.2014.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ouahab A, Cheraga N, Onoja V, Shen Y, Tu J. Int J Pharm. 2014;466:233. doi: 10.1016/j.ijpharm.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 42.Yeh TH, Chen YR, Chen SY, Shen WC, Ann DK, Zaro JL, Shen LJ. Mol Pharm. 2015;13:262. doi: 10.1021/acs.molpharmaceut.5b00706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phillips MM, Sheaff MT, Szlosarek PW. Cancer Res Treat. 2013;45:251. doi: 10.4143/crt.2013.45.4.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagarsekar A, Crissman J, Crissman M, Ferrari F, Cappello J, Ghandehari H. J Biomed Mater Res. 2002;62:195. doi: 10.1002/jbm.10272. [DOI] [PubMed] [Google Scholar]
- 45.Gaudin Y, Tuffereau C, Durrer P, Flamand A, Ruigrok R. J Virol. 1995;69:5528. doi: 10.1128/jvi.69.9.5528-5534.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferlin A, Raux H, Baquero E, Lepault J, Gaudin Y. J Virol. 2014;88:13396. doi: 10.1128/JVI.01962-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uversky VN, Li J, Fink AL. J Biol Chem. 2001;276:10737. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 48.Uversky VN, Gillespie JR, Fink AL. Proteins: Struct, Funct, Genet. 2000;41:415. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 49.Wiedenmann J, Ivanchenko S, Oswald F, Schmitt F, Röcker C, Salih A, Spindler KD, Nienhaus GU. Proc Natl Acad Sci USA. 2004;101:15905. doi: 10.1073/pnas.0403668101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schreiner L, Huber-Lang M, Weiss ME, Hohmann H, Schmolz M, Schneider EM. J Cell Commun Signal. 2011;5:135. doi: 10.1007/s12079-010-0112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Villoing A, Ridhoir M, Cinquin B, Erard M, Alvarez L, Vallverdu G, Pernot P, Grailhe R, Mérola F, Pasquier Hln. Biochemistry. 2008;47:12483. doi: 10.1021/bi801400d. [DOI] [PubMed] [Google Scholar]
- 52.Poëa-Guyon S, Pasquier H, Mérola F, Morel N, Erard M. Anal Bioanal Chem. 2013;405:3983. doi: 10.1007/s00216-013-6860-y. [DOI] [PubMed] [Google Scholar]
- 53.Vargo KB, Sood N, Moeller TD, Heiney PA, Hammer DA. Langmuir. 2014;30:11292. doi: 10.1021/la502664e. [DOI] [PubMed] [Google Scholar]
- 54.Foo CTWP, Lee JS, Mulyasasmita W, Parisi-Amon A, Heilshorn SC. Proc Natl Acad Sci USA. 2009;106:22067. doi: 10.1073/pnas.0904851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mulyasasmita W, Cai L, Dewi RE, Jha A, Ullmann SD, Luong RH, Huang NF, Heilshorn SC. J Control Release. 2014;191:71. doi: 10.1016/j.jconrel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Science. 1998;281:389. doi: 10.1126/science.281.5375.389. [DOI] [PubMed] [Google Scholar]
- 57.Shen W, Zhang K, Kornfield JA, Tirrell DA. Nat Mater. 2006;5:153. doi: 10.1038/nmat1573. [DOI] [PubMed] [Google Scholar]
- 58.Glassman MJ, Olsen BD. Biomacromolecules. 2015;16:3762. doi: 10.1021/acs.biomac.5b01026. [DOI] [PubMed] [Google Scholar]
- 59.Glassman MJ, Avery RK, Khademhosseini A, Olsen BD. Biomacromolecules. 2016;17:415. doi: 10.1021/acs.biomac.5b01210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fernández-Colino A, Arias FJ, Alonso M, Rodríguez-Cabello JC. Biomacromolecules. 2015;16:3389. doi: 10.1021/acs.biomac.5b01103. [DOI] [PubMed] [Google Scholar]
- 61.Lu HD, Charati MB, Kim IL, Burdick JA. Biomaterials. 2012;33:2145. doi: 10.1016/j.biomaterials.2011.11.076. [DOI] [PubMed] [Google Scholar]
- 62.Lu HD, Soranno DE, Rodell CB, Kim IL, Burdick JA. Adv Healthc Mater. 2013;2:1028. doi: 10.1002/adhm.201200343. [DOI] [PubMed] [Google Scholar]
- 63.Zakeri B, Howarth M. J Am Chem Soc. 2010;132:4526. doi: 10.1021/ja910795a. [DOI] [PubMed] [Google Scholar]
- 64.Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, Howarth M. Proc Natl Acad Sci USA. 2012;109:E690. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang WB, Sun F, Tirrell DA, Arnold FH. J Am Chem Soc. 2013;135:13988. doi: 10.1021/ja4076452. [DOI] [PubMed] [Google Scholar]
- 66.Sun F, Zhang WB, Mahdavi A, Arnold FH, Tirrell DA. Proc Natl Acad Sci USA. 2014;111:11269. doi: 10.1073/pnas.1401291111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miller KE, Kim Y, Huh WK, Park HO. J Mol Biol. 2015;427:2039. doi: 10.1016/j.jmb.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sjöhamn J, Båth P, Neutze R, Hedfalk K. Protein Sci. 2016;25:2196. doi: 10.1002/pro.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McGann CL, Dumm RE, Jurusik AK, Sidhu I, Kiick KL. Macromol Biosci. 2016;16:129. doi: 10.1002/mabi.201500305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carrico IS, Maskarinec SA, Heilshorn SC, Mock ML, Liu JC, Nowatzki PJ, Franck C, Ravichandran G, Tirrell DA. J Am Chem Soc. 2007;129:4874. doi: 10.1021/ja070200b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nagapudi K, Brinkman WT, Leisen JE, Huang L, McMillan RA, Apkarian RP, Conticello VP, Chaikof EL. Macromolecules. 2002;35:1730. [Google Scholar]
- 72.Frangioni JV. Curr Opin Chem Biol. 2003;7:626. doi: 10.1016/j.cbpa.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 73.Fu J, Guerette PA, Miserez A. Biomacromolecules. 2015;16:2327. doi: 10.1021/acs.biomac.5b00552. [DOI] [PubMed] [Google Scholar]
- 74.Radisic M, Park H, Shing H, Consi T, Schoen FJ, Langer R, Freed LE, Vunjak-Novakovic G. Proc Natl Acad Sci USA. 2004;101:18129. doi: 10.1073/pnas.0407817101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schmidt CE, Shastri VR, Vacanti JP, Langer R. Proc Natl Acad Sci USA. 1997;94:8948. doi: 10.1073/pnas.94.17.8948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Balint R, Cassidy NJ, Cartmell SH. Acta Biomater. 2014;10:2341. doi: 10.1016/j.actbio.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 77.Zhang H, Wang K, Xing Y, Yu Q. Mater Sci Eng, C. 2015;56:564. doi: 10.1016/j.msec.2015.06.024. [DOI] [PubMed] [Google Scholar]
- 78.Rosenberg B. J Chem Phys. 1962;36:816. [Google Scholar]
- 79.Nakamura R, Kai F, Okamoto A, Newton GJ, Hashimoto K. Angew Chem Int Ed. 2009;48:508. doi: 10.1002/anie.200804750. [DOI] [PubMed] [Google Scholar]
- 80.Le RK, Raeeszadeh-Sarmazdeh M, Boder ET, Frymier PD. Langmuir. 2015;31:1180. doi: 10.1021/la5031284. [DOI] [PubMed] [Google Scholar]
- 81.Iwuchukwu IJ, Vaughn M, Myers N, O’Neill H, Frymier P, Bruce BD. Nat Nanotechnol. 2010;5:73. doi: 10.1038/nnano.2009.315. [DOI] [PubMed] [Google Scholar]
- 82.Qin S, Yin H, Yang C, Dou Y, Liu Z, Zhang P, Yu H, Huang Y, Feng J, Hao J. Nat Mater. 2016;15:217. doi: 10.1038/nmat4484. [DOI] [PubMed] [Google Scholar]