

Abstract
BACKGROUND
The authors previously demonstrated that brief ischemia elicits cardiac troponin I (cTnl) release and myocyte apoptosis in the absence of necrosis. It remains uncertain whether other pathophysiological stresses can produce apoptosis and transient cTnI release without ischemia.
OBJECTIVES
The authors sought to determine whether a transient increase in left ventricular (LV) preload elicits cTnI release in the absence of ischemia.
METHODS
Propofol-anesthetized swine (N = 13) received intravenous phenylephrine (PE) (300 μg/min) for 1 h to increase left ventricular end-diastolic pressure (LVEDP) to ~30 mm Hg. Serial cTnI and echocardiographic function were assessed for 24 h, and myocardial tissue was analyzed for apoptosis and necrosis.
RESULTS
PE infusion increased systolic blood pressure from 137 ± 14 mm Hg to 192 ± 11 mm Hg (mean ± SD; p < 0.001) and increased LVEDP from 17 ± 2 mm Hg to 30 ± 5 mm Hg (p < 0.001). Myocardial flow measurements demonstrated no evidence of ischemia. Hemodynamics normalized rapidly after PE, but LV ejection fraction remained depressed (32 ± 21% vs. 58 ± 7%; p < 0.01) with normalization after 24 h (51 ± 16%; p = 0.31). Baseline transcoronary cTnI release was low (16 ± 20 ng/l) but increased to 856 ± 956 ng/l (p = 0.01) 1 h after LVEDP elevation. Circulating cTnI rose above the 99th percentile within 30 min and remained elevated at 24 h (1,462 ± 1,691 ng/l). Pathological analysis demonstrated myocyte apoptosis at 3 h (31.3 ± 11.9 myocytes/cm2 vs. 4.6 ± 3.7; p < 0.01), which normalized after 24 h (6.2 ± 5.6 myocytes/cm2; p = 0.46) without histological necrosis.
CONCLUSIONS
Transient elevations of LVEDP lead to cTnI release, apoptosis, and reversible stretch-induced stunning in the absence of ischemia. Thus, preload-induced myocyte injury may explain many cTnI elevations seen in the absence of clinical signs or symptoms of myocardial ischemia.
Keywords: cardiac troponin I, cardiomyocyte apoptosis, left ventricular preload, myocardial stretch, myocardial stunning, pressure overload
Serum cardiac troponin (cTn) concentrations are currently the preferred diagnostic biomarker for the noninvasive detection of myocardial injury and are commonly used in conjunction with clinical signs of ischemia to diagnose myocardial infarction (1). Nevertheless, it has become clear that transient elevations in cTn levels frequently occur in the absence of ischemia in patients, as well as following physiological stresses such as dobutamine stress testing and marathon running in normal hearts (2–5). The increased clinical utilization of cTn measurements and development of high-sensitivity cTn assays has led to a substantial increase in the prevalence of patients presenting with elevations in cTn reflecting myocardial injury not related to an acute coronary syndrome (ACS) (6). Indeed, in some studies, more than one-half of the patients with elevated serum cTn concentrations do not have clinical evidence of myocardial ischemia or evidence of an acute coronary syndrome (7–9). Although some cTn elevations are due to myocardial injury from myocarditis, the vast majority remain unexplained.
Excessive myocyte strain from elevated left ventricular (LV) filling pressures may be an important cause of irreversible myocyte injury in the absence of myocardial ischemia. This is supported by chronic elevations of cTn and B-type natriuretic peptide in heart failure and a variety of other fluid overload states (10). Previous in vitro studies have demonstrated that increased strain stimulates myocyte apoptosis, but whether this occurs in vivo is unclear (11). We previously demonstrated that elevating left ventricular end-diastolic pressure (LVEDP) to 25 mm Hg in the absence of ischemia leads to calpain-mediated cTnI degradation in the isolated rat heart (12). Although it is unclear whether apoptosis, cTnI release, and proteolytic cleavage of cTnI are causally related to one another, we recently demonstrated myocyte apoptosis and delayed cTnI release after 10 min of what had widely been held to be “reversible” ischemia (13). These findings raise the possibility that there may be other pathophysiological stimuli leading to apoptotic myocyte injury that explain cTn elevations in the many circumstances not associated with an ACS.
Accordingly, we increased LVEDP to 30 mm Hg for 1 h in closed-chest swine by increasing myocardial afterload with phenylephrine (PE). We assessed serial coronary venous as well as circulating cTnI levels, LV function, and myocyte apoptosis at times up to 24 h later. Because preload elevation can impede coronary flow (14,15), we excluded ischemia by assessing subendocardial flow with microspheres. Our results demonstrate that a transient 1-h elevation in preload is sufficient to stimulate myocyte apoptosis, elevate circulating cTnI for at least 24 h, and lead to stretch-induced LV systolic dysfunction or “stunning” in the absence of myocardial ischemia.
METHODS
All procedures and protocols conformed to institutional guidelines for the care and use of animals in research and were approved by the University at Buffalo Institutional Animal Care and Use Committee. A total of 13 Yorkshire swine (80 ± 10 kg) were enrolled in 1 of 2 protocols summarized in Figure 1 and described in detail in the following text. In each protocol, baseline hemodynamic measurements and echocardiography were performed before, during, and after a 60-min intravenous infusion of PE (300 μg/min) to elevate preload to ~30 mm Hg for 1 h. Detailed methodology is provided in the Online Appendix.
Figure 1. Experimental Protocol.
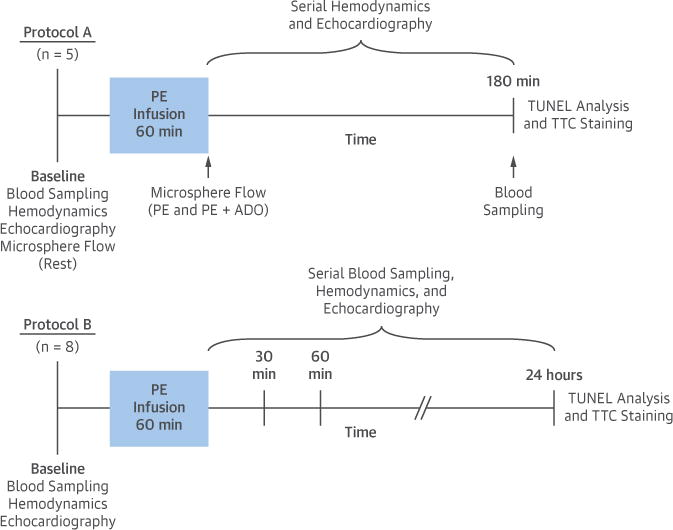
Following baseline data collection, swine were subjected to a 60-min intravenous infusion of PE (300 μg/min) and studied for 3 h (Protocol A; n = 5) or 24 h (Protocol B; n = 8). Serial blood sampling from the carotid artery and coronary sinus was performed at selected time points, along with assessment of hemodynamic parameters, left ventricular function, and myocardial perfusion. Following euthanasia, the heart was excised for gross assessment of infarction and histopathological quantification of myocyte apoptosis. ADO = adenosine; PE = phenylephrine; TTC = triphenyltetrazolium chloride; TUNEL = terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling.
EXPERIMENTAL PROTOCOL
Pigs were sedated with a Telazol (tiletamine/zolazepam 100 mg/ml)/xylazine (100 mg/ml) mixture (0.04 ml/kg intramuscular), maintained on a continuous intravenous infusion of propofol (5 to 10 mg/kg/h), and mechanically ventilated with supplemental oxygen. A 6-F sheath was placed into the right carotid artery through which a 5-F catheter (Millar, Houston, Texas) was positioned in the LV for continuous pressure measurement and administration of fluorescent microspheres (Protocol A). The side port of the introducer was used to measure arterial pressure. A 6-F sheath was placed into the right jugular vein for intravenous administration of PE and also to advance a 5-F multipurpose catheter (Cordis, Milpitas, California) into the coronary sinus (CS) for coronary venous blood sampling (Protocol B). An additional 5-F multipurpose catheter (Cordis) was placed in the aorta through a 6-F sheath in the left carotid artery for arterial blood sampling (Protocol B).
After catheters were placed, animals were heparinized (100 U/kg intravenous) and hemodynamics allowed to equilibrate (~20 min) before data collection. Baseline hemodynamic parameters and function (echocardiography, GE Vivid 7; GE Healthcare, Little Chalfont, United Kingdom) were assessed as previously described (16,17). Briefly, the LV was imaged in the short-axis projection from a right parasternal approach to assess regional wall thickening, end-systolic and end-diastolic volumes, and LV ejection fraction using American Society of Echocardiography criteria. Following baseline data collection, intravenous PE infusion was initiated at 300 μg/min and titrated to maintain LVEDP at ~30 mm Hg for 60 min (range 200 to 700 μg/min). Hemodynamic parameters were continuously recorded, and echocardiography was performed every 15 min throughout the infusion period. After 60 min, the PE infusion was stopped, and hemodynamic and echocardiographic assessments were repeated during recovery as detailed for each protocol in the following text. At the end of the study, a final blood sample was collected, and the heart was arrested with intracardiac KCl under deep isoflurane anesthesia. Myocardial tissue was excised for histopathology, quantification of myocyte apoptosis, and 2,3,5-triphenyltetrazolium chloride (TTC) staining (18).
Protocol A: Short-term effects of elevated preload on LV function, cTnI release, and transmural myocardial perfusion (n = 5)
In the initial experiments, myocardial perfusion was assessed with fluorescent microspheres at baseline with resting flow measurements repeated during elevated preload at the conclusion of the PE infusion period. We then assessed flow reserve at elevated preload by infusing adenosine (0.9 mg/kg/min) to induce coronary vasodilation. After this, PE was stopped and preload returned to normal. Hemodynamic assessment and echocardiography were performed at selected time points for 3 h, at which time the animals were euthanized and tissue samples taken as outlined in the preceding text.
Protocol B: Transcoronary cTnI release, LV functional recovery, and myocyte apoptosis at 24 h (n = 8)
In the next series of experiments, serial blood samples were collected from the CS and aorta to assess transcoronary cTnI release at baseline as well as 30 min and 60 min after the PE infusion was stopped. Catheters were then removed, and the animals were recovered and returned to the animal facility. After 24 h, they were brought back to the laboratory under anesthesia, and echocardiography was repeated, after which the animals were euthanized and the heart was excised for tissue analyses as described in the preceding text.
STATISTICAL ANALYSIS
Data are expressed as mean ± SD. Hemodynamic data and echocardiographic data collected during and immediately after PE were pooled for the time points common to both protocols for statistical analysis. Temporal differences were assessed by repeated measures analysis of variance and the post hoc Holm-Sidak test. Because cTnI concentrations were not normally distributed, nonparametric testing (Friedman test with post hoc paired Wilcoxon signed rank test) was employed to assess differences in serum cTnI between sampling locations (i.e., CS vs. aorta) and time points. Post hoc tests were not adjusted for multiple comparisons. Statistical analysis was performed with IBM SPSS Statistics 23 (IBM, Armonk, New York), and the acceptable type 1 error rate was prospectively set at 5%.
RESULTS
EFFECTS OF A TRANSIENT INCREASE IN PRELOAD ON HEMODYNAMICS AND LV FUNCTION
Serial measurements of selected hemodynamic and echocardiographic parameters before, during, and after transient PE-mediated pressure overload for all animals are summarized in Figure 2 and Table 1. PE infusion increased LV systolic pressure from 133 ± 12 mm Hg to 187 ± 10 mm Hg (p < 0.01) and increased LVEDP from 17 ± 2 mm Hg to 30 ± 5 mm Hg (p < 0.01). There was no significant change in heart rate (from 97 ± 15 beats/min to 89 ± 20 beats/min; p = 0.29). Hemodynamic parameters normalized quickly after cessation of the 1-h PE infusion, but LV contractility as reflected by maximum rate of rise of left ventricular pressure (dP/dtmax) was depressed.
Figure 2. Hemodynamic Effects of PE.
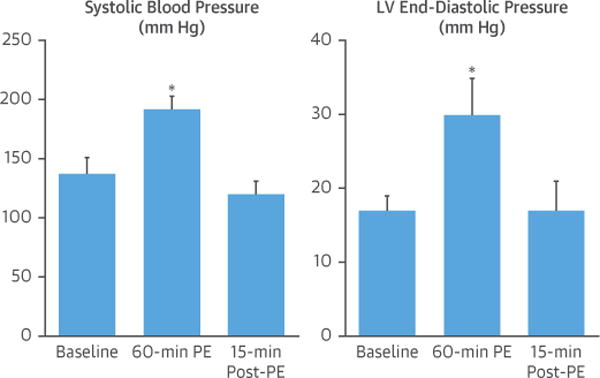
Intravenous PE elicited a significant increase in systolic blood pressure and LV end-diastolic pressure that persisted throughout the 60-min infusion period but normalized within 15 min after the end of the infusion period. Values are mean ± SD, n = 13. *p < 0.05 versus baseline. LV = left ventricular; PE = phenylephrine.
Table 1.
Selected Hemodynamic and Echocardiographic Parameters Before, During, and After Intravenous PE Infusion: Protocol A & B Combined (n = 13)
| Baseline (n = 13) |
60-Min PE (n = 13) |
60-Min Post-PE (n = 13) |
|
|---|---|---|---|
| Heart rate, beats/min | 97 ± 15 | 89 ± 20 | 91 ± 17 |
| LV peak systolic pressure, mm Hg | 133 ± 12 | 187 ±10* | 117 ± 12* |
| LV end-diastolic pressure, mm Hg | 17 ± 2 | 30 ± 5* | 17 ± 3 |
| LV dP/dtmax, mm Hg/s | 2178 ± 330 | 2037 ±349* | 1316 ± 232* |
| LV end-diastolic volume, ml | 115 ± 19 | 147 ± 27* | 145 ± 37* |
| LV end-systolic volume, ml | 46 ± 13 | 91 ± 26* | 96 ± 44* |
| Stroke volume, ml | 69 ± 12 | 56 ± 11* | 48 ± 16* |
| LV ejection fraction, % | 61 ± 8 | 39 ± 9* | 37 ± 19* |
Values are mean ± SD.
p <0.05 versus baseline.
dP/dtmax = maximum rate of rise of left ventricular pressure; LV = left ventricular; PE = phenylephrine.
Serial echocardiography (Table 1) demonstrated that the PE-induced increases in preload and afterload increased LV end-diastolic volume (from 115 ± 22 ml to 145 ± 28 ml; p = 0.01) and end-systolic volume (from 46 ± 15 ml to 90 ± 23 ml; p < 0.01), reflecting a reduced LV stroke volume (from 69 ± 13 ml to 54 ± 9 ml; p = 0.03). As a result, LV ejection fraction decreased from 60 ± 8% to 38 ± 6% during PE (p < 0.01). Interestingly, despite normalization of LV systolic and end-diastolic pressure after cessation of PE, the increases in LV end-diastolic volume (144 ± 33 ml; p < 0.01 vs. baseline) and end-systolic volume (97 ± 40 ml; p < 0.01 vs. baseline) as well as the reduction in ejection fraction (35 ± 15%; p < 0.01 vs. baseline) persisted.
Animals in protocol B (n = 8) were followed for 24 h, and serial echocardiographic measurements evaluating late recovery are summarized in Figure 3 and Table 2. After 24 h (n = 8), paired analysis demonstrated normalization of LV end-diastolic volume (106 ± 47 ml vs. 122 ± 18 ml at baseline; p = 0.38) and end-systolic volume (52 ± 26 ml vs. 52 ± 12 ml at baseline; p = 0.96). As a result, ejection fraction increased and was not significantly different than baseline values (51 ± 16% vs. 58 ± 7% at baseline; p = 0.31). Thus, the LV dilatation and systolic dysfunction elicited by a 1-h episode of pressure overload was reversible.
Figure 3. A Transient Increase in LV Preload Produces Reversible LV Dilatation and Systolic Dysfunction.
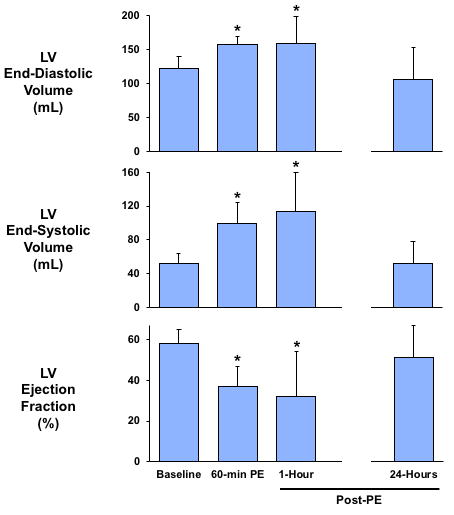
The acute increase in arterial blood pressure and LV preload induced by intravenous phenylephrine resulted in a significant increase in LV end-diastolic and end-systolic volume, as well as an impairment in LV ejection fraction that persisted upon normalization of blood pressure but returned to baseline levels 1 day later. Values are mean ± SD. *p < 0.05 versus baseline. Abbreviations as in Figure 2.
Table 2.
Serial Echocardiographic Parameters Before, During, and Up to 24 h After Intravenous PE Infusion: Protocol B Only (n = 8)
| Baseline (n = 8) |
60-Min PE (n = 8) |
60-Min Post-PE (n = 8) |
24-h Post-PE (n = 8) |
|
|---|---|---|---|---|
| LV end-diastolic volume, ml | 122 ± 18 | 157 ± 21* | 159 ± 39* | 106 ± 47† |
| LV end-systolic volume, ml | 52 ± 12 | 99 ± 25* | 114 ± 46* | 52 ± 26† |
| Stroke volume, ml | 70 ± 13 | 58 ± 12* | 45 ± 20* | 54 ± 26 |
| LV ejection fraction, % | 58 ± 7 | 37 ± 10* | 32 ± 22* | 51 ± 16† |
Values are mean ± SD.
p < 0.05 vs. baseline.
p < 0.05 vs. 60-min post-PE.
Abbreviations as in Table 1.
ABSENCE OF SUBENDOCARDIAL ISCHEMIA DURING ELEVATED PRELOAD (N = 5)
Despite the marked increase in LV end-diastolic pressure produced by PE, there was no evidence of myocardial ischemia (Figure 4). Resting subendocardial flow increased from 0.9 ± 0.3 ml/min/g to 1.0 ± 0.2 ml/min/g (p = 0.64) following PE with no change in the sub-endocardial/subepicardial flow ratio (1.2 ± 0.3 ml/min/g vs. 1.2 ± 0.3 ml/min/g at baseline; p = 0.96). Furthermore, there was considerable adenosine flow reserve during preload elevation with myocardial flow increasing in each transmural layer as well as on > a full-thickness basis (0.9 ± 0.2 ml/min/g at rest to 4.9 ± 1.2 ml/min/g following adenosine; p < 0.01). All of these results indicate that elevated preload following PE did not lead to subendocardial ischemia.
Figure 4. Transmural Myocardial Perfusion and Flow Reserve Is Preserved During PE-Induced Preload Elevation.
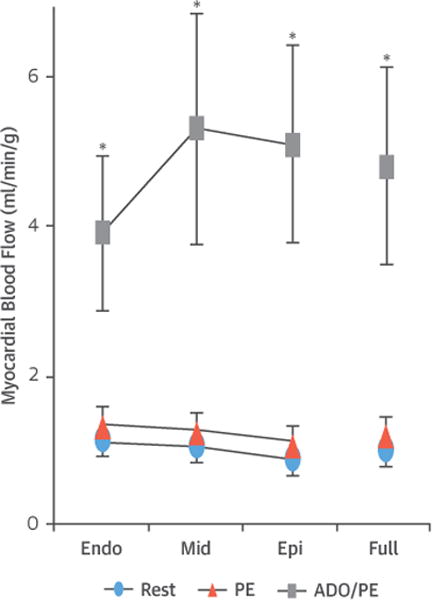
Subendocardial blood flow was similar at baseline and at the end of the 60-min PE infusion period, with administration of ADO demonstrating preservation ofsubendocardial flow reserve throughout the LV during intravenous PE infusion. The sub-endocardial/subepicardial blood flow ratio was not affected by intravenous PE. These argue against the presence of subendocardial ischemia during PE-induced pressure overload. Values are mean ± SD and represent average values from the anterior and posterior LV wall. *p < 0.05 versus baseline; †p < 0.05 versus 60-min PE. ADO = adenosine; endo = subendocardial; epi = subepicardial; mid = mid-myocardial; other abbreviations as in Figure 2.
POST-MORTEM HISTOPATHOLOGICAL ANALYSIS
The absence of infarction was confirmed in hearts excised 3 h (n = 5) and 24 h (n = 5) after transient preload elevation by TTC staining. Likewise, hematoxylin and eosin-stained myocardial tissue sections collected 3 h after preload elevation showed no myocyte nuclear loss or inflammatory cell infiltration (Figure 5A). Contraction band necrosis was not observed. Although myocyte necrosis was absent, hearts excised 3 h after preload elevation exhibited apoptosis with an ~6-fold increase in terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)-positive cardiomyocytes (Figure 5B) in the anterior (30.5 ± 9.6 myocytes/cm2) and posterior (32.2 ± 14.9 myocytes/cm2) regions of the LV (both p < 0.01 vs. normal controls, which averaged 4.6 ± 3.7 myocytes/cm2, n = 9). In tissue excised 24 h after PE, myocyte apoptosis returned to control levels (anterior wall 6.5 ± 4.4 myocytes/cm2; p = 0.40 vs. normal control; posterior wall 6.0 ± 7.2 myocytes/cm2; p = 0.64 vs. normal control). Activated caspase-3, an alternative marker of apoptosis, was not detectable in any myocytes from normal hearts (n = 4) (13), but increased to 12.8 ± 18.2 myocytes/cm2 3 h after transient preload elevation (Figure 5C). Although myocyte TUNEL staining normalized after 24 h, there was a persistent increase in myocyte caspase-3 activity (15.2 ± 9.1 myocytes/cm2).
Figure 5. An Acute Elevation in LV Preload Causes a Transient Increase in Cardiomyocyte Apoptosis.
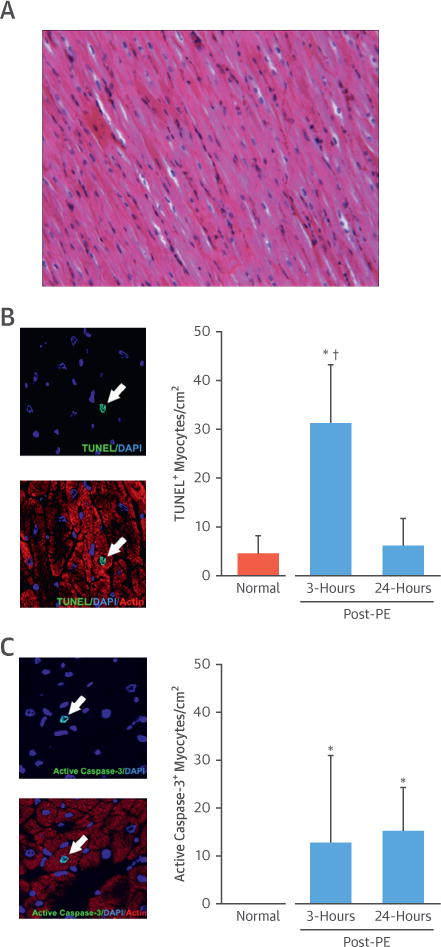
(A) Representative hematoxylin and eosin-stained myocardial tissue section demonstrating absence of contraction band necrosis, nuclear loss, and inflammatory cell infiltration. (B) Example images of a TUNEL+ myocyte 3 h after the cessation of intravenous PE are shown in the left panel (arrow). There was a significant, ~6-fold increase in myocyte apoptosis throughout the LV 3 h after intravenous PE that returned toward normal values 24 h later. (C) Example images of an active caspase-3+ myocyte 3 h after the cessation of intravenous PE (left). Activated caspase-3 was not detectable in any myocytes from normal hearts (n = 4), but increased 3 h and 24 h after transient preload elevation. Values are mean ± SD. *p < 0.05 versus normal control; †p < 0.05 versus 24-h post-PE. DAPI = 4′,6-diamidino-2-phenylindole; other abbreviations as in Figures 1 and 2.
EFFECTS OF TRANSIENT PRELOAD ELEVATION ON MYOCARDIAL Tnl RELEASE
Western blot analysis demonstrated a faint 26-kDa cTnI degradation band in myocardial tissue samples collected 3 h after PE (0.22 ± 0.14 vs. 0.01 ± 0.02 densitometric units in normal controls; p < 0.01) (Figure 6). There was no change in the intensity of the primary 31-kDa cTnI band. Serum cTnI concentrations at baseline averaged 5.2 ± 5.8 ng/l (n = 13) and were below the 99th percentile upper reference limit that we have established for this assay (38 ng/l, n = 39 normal swine [13]). Analysis of peripheral blood from animals in protocol A (n = 5) demonstrated a significant rise in circulating cTnI concentrations 3 h after cessation of the PE infusion (392 ± 390 ng/l; p < 0.01 vs. baseline). In protocol B (n = 8), transcoronary cTnI release (ΔcTnI = cTnI[coronary sinus] _ cTnI[aorta]) was low at baseline (16 ± 20 ng/l) but increased to 57 ± 60 ng/l (p = 0.12) 30 min after cessation of preload elevation. Transcoronary cTnI became significantly elevated after 1 h (856 ± 956 ng/l; p = 0.01 vs. baseline). The cumulative cTnI release arising after preload elevation led to peripheral circulating cTnI rising above baseline values at 30 min (127 ± 54 ng/l; p < 0.01 vs. baseline) and increasing further at 1 h (509 ± 401 ng/l; p < 0.01 vs. baseline) (Figure 7). Although circulating cTnI levels remained elevated at 24 h (1,462 ± 1,691 ng/l; p = 0.04), there was no longer ongoing myocardial release because cTnI levels in the aorta (1,462 ± 1,691 ng/l) and CS (1,454 ± 1,741 ng/l) were similar (p = 0.98). Collectively, these results indicate that stretch-induced LV stunning elicited by a transient elevation in preload is accompanied by measurable cTnI release within the first hour after cessation of pressure overload that remains elevated for up to 24 h.
Figure 6. cTnI Degradation Following an Acute Elevation in LV Preload.
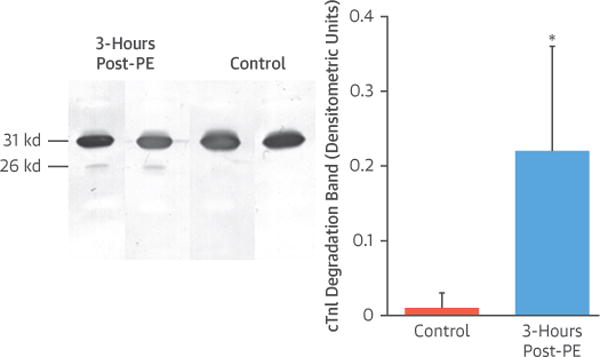
Western Blot images show the presence of a cTnl degradation band (26 kDa) in myocardial tissue collected 3 h after cessation of intravenous PE (left). Densitometric measurements revealed that elevating preload in the absence of ischemia increased cTnI degradation compared with control animals, as indicated by a significant increase in the integrated density of the 26 kDa cTnI degradation band (right). Values are mean ± SD. *p < 0.05 versus control. cTnI = cardiac troponin I; PE = phenylephrine.
Figure 7. A Transient Increase in LV Preload Elicits cTnI Release.
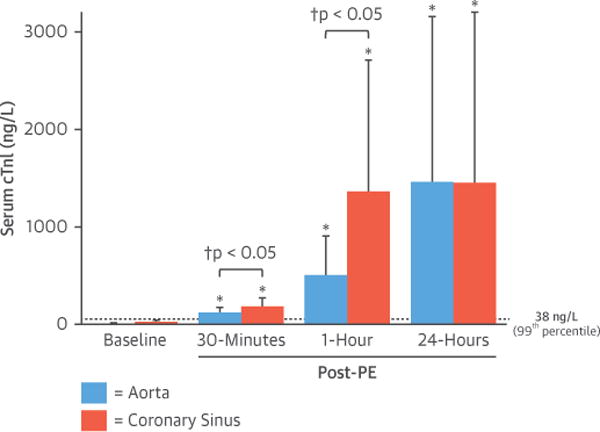
Serial blood sampling revealed a significant increase in serum cTnI in blood obtained from the aorta and coronary sinus 30 min and 1 h after cessation of intravenous PE, with a concomitant elevation in the transcoronary cTnI gradient indicative of active myocardial cTnI release. The next day, circulating levels remained elevated, but there was no longer a significant gradient between the aorta and coronary sinus. Values are mean ± SD. *p < 0.05 versus baseline. †p < 0.05 coronary sinus versus aorta. Dashed line = 99th percentile of normal animals (38 ng/l [13]). Abbreviations as in Figures 1 and 2.
DISCUSSION
The present study provides several new and important findings that improve our understanding of the pathophysiology and consequences of mechanical stretch-induced cardiomyocyte injury (Central Illustration). Using a transient increase in arterial blood pressure to raise LV preload in normal swine, we observed marked LV chamber dilatation and a reduction in ejection fraction that persisted despite normalization of arterial blood pressure and LVEDP indicative of strain-induced myocardial injury. Function returned to normal 24 h later, consistent with myocardial stunning. Importantly, microsphere perfusion data demonstrated that subendocardial blood flow and flow reserve remained normal, confirming that reversible LV dysfunction was not secondary to myocardial ischemia, but rather reflected “stretch-induced stunning.” This transient dysfunction was accompanied by evidence of myocyte injury as reflected by elevations in serum cTnI and a transient increase in myocyte apoptosis in the absence of necrosis. Collectively, our results demonstrate that acute hemodynamic overload can provoke transient LV dysfunction resulting in myocyte injury in the absence of ischemia or necrosis. The rise in serum cTnI following stretch-induced myocyte injury and apoptosis in the absence of ischemia may explain many of the cTn elevations identified in fluid overload states including congestive heart failure, renal failure as well as others.
CENTRAL ILLUSTRATION. cTnl Release and Reversible LV Dysfunction Following Transient Preload Elevation.
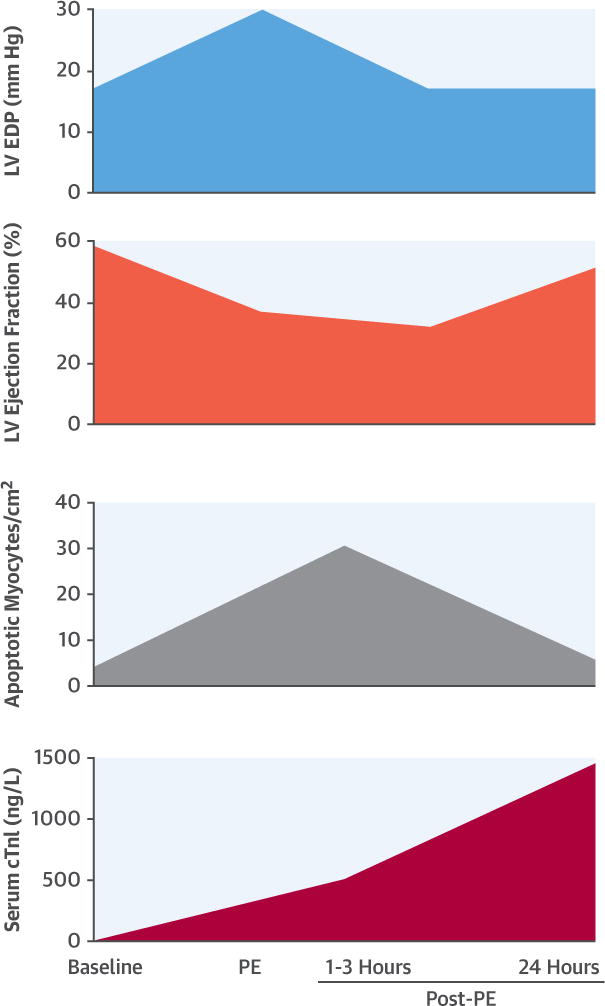
In a porcine model oftransient preload elevation elicited by intravenous administration of PE, we demonstrated reversible systolic dysfunction that was accompanied by a temporary 6-fold increase in myocyte apoptosis in the absence of myocardial ischemia. Although histopathological analyses confirmed the absence of necrosis or infarction, we observed a gradualrise of serum cardiac troponin I (cTnI) that exceeded the 99th percentile of normal animals 30 min after preload elevation and rose to readily detectable levels 24 h later. Collectively, these results demonstrate that a transient elevation in preload produces mechanical stretch-induced myocyte injuryand measurable cTnI release in the absence of ischemia or necrosis that is associated with reversible contractile dysfunction and myocyteapoptosis. This may explain why cTnI elevations are frequently observed in patients without clinical signs or symptoms of myocardial ischemia. EDP = end-diastolic pressure; LV = left ventricular; PE = phenylephrine.
Although elevated serum cTn has traditionally been considered the gold-standard biomarker of acute myocardial necrosis, the present findings add to a growing body of evidence that cTn release can occur without necrotic myocyte death (6). This is supported by the increasing prevalence of low levels of cTn using high-sensitivity assays in normal subjects. Potential causes of cTn release in the absence of ischemia include normal myocyte protein turnover, the low baseline rate of apoptotic myocyte cell death, release of proteolytic degradation products of cTn, increased cellular membrane permeability, and the formation and release of membranous blebs (19). Regardless of its cause, an elevated cTn is associated with a relatively worse prognosis in patients with established cardiac disease (20,21), noncardiac disease (22) as well as in asymptomatic cross-sectional populations (23). Although it is not possible to distinguish specific mechanisms among patients, it is clear that increases in serum cTn reflect myocyte injury. This has fueled the notion that cTn is an “organ-specific,” not “disease-specific,” biomarker (24), which complicates the diagnostic use of cTn assays in patients presenting without symptoms or signs of ischemia that are typical of an ACS.
Among potential nonischemic causes of myocyte injury, myocardial stretch has been implicated as an important mechanism, in large part based on clinical observations of elevated cTn concentrations in patients with high filling pressures in the setting of chronic heart failure. For example, Logeart et al. (10) observed a significant correlation between circulating cTnI and B-type natriuretic peptide levels in patients with nonischemic heart failure. Because the latter is known to positively correlate with LV filling pressures, it was suggested that preload-induced stretch could elicit myocyte injury and cTn release in these patients. Furthermore, Takashio et al. (25) reported transcoronary cTnT release in nonischemic heart failure patients undergoing cardiac catheterization and found that LVEDP was an independent determinant of cTnT release. A caveat was that patients in both studies exhibited increased myocardial wall thickness and/or LV mass, raising the possibility that hypertrophy-induced subendocardial ischemia was responsible for cTn release. The use of otherwise normal hearts in our animal model, along with the microsphere perfusion measurements to exclude subendocardial ischemia, provides important validation for the concept that even short-term preload-induced mechanical deformation and diastolic myocyte strain can provoke significant myocyte injury, apoptosis, and cTn release.
Our findings fill an important void in the existing knowledge of stretch-induced myocyte injury and cTn release by bridging the gap between clinical data and prior in vitro studies. The observation that an acute increase in LV preload elicits myocyte apoptosis and reversible LV contractile dysfunction builds on previous in vitro data from Cheng etal. (11), who observed an ~20-fold increase in apoptotic myocyte death and impairment in myocardial force development following excessive stretch of isolated papillary muscles. Furthermore, the stretch-induced cTnI degradation we demonstrate in the present study is consistent with the in vitro work of Feng et al. (12), where elevated preload in the isolated rat heart led to calpain-mediated cTnI proteolysis in the absence of ischemia. Our results demonstrate the in vivo relevance of these prior studies to show that preload-induced stretch elicits tissue cTnI proteolysis and myocyte apoptosis that ultimately leads to cTnI release in the absence of ischemia. Because we could not assess the specific form of circulating cTn, it is unclear whether cTnI released from myocytes has undergone proteolytic modification or is released in an intact form from reversibly injured myocytes. The notion that cTn could be released from viable myocytes following mechanical stretch is supported by the findings of Hessel et al. (26), who demonstrated release of intact cTn from viable myocytes in cell culture via stimulation of stretch-responsive integrins. Nevertheless, this has been challenged because the magnitude of cell permeability change required to allow large molecular weight proteins such as cTnI (~ 31 kDa) to escape would be unlikely to occur without concomitant alterations in intracellular calcium flux which would ultimately lead to hypercontracture and calcium-mediated cell death (24). Thus, cTn release from irreversibly injured myocytes undergoing apoptosis (and possibly transitioning to secondary necrosis [27]) remains the most likely source of elevated serum cTn concentrations following elevated preload.
STUDY LIMITATIONS
There are several experimental considerations that merit discussion. First, because our study design did not include serial collection of blood samples between 3 and 24 h after myocardial stretch, we could not determine time course or the peak magnitude of circulating concentrations of cTnI. Given the ~2-h half-life of cTnI in the blood (28) and our finding that circulating cTnI concentrations were significantly elevated at 24 h without active transcoronary release, it is likely that peak cTnI values occur between 3 and 24 h after acute stretch. Likewise, although myocyte apoptosis returned to normal values after 24 h, we cannot exclude the possibility that it may have been higher between 3 and 24 h following cessation of PE. Second, echocardiographic measurement of LV volumes and ejection fraction indicate that systolic function normalized 24 h after acute preload elevation consistent with reversible stretch-induced “stunning.” Potentially more sensitive indices of contractile function such as myocardial strain or dP/dt were not assessed at this time point and may have revealed subtle, persistent abnormalities in systolic dysfunction, although it should be noted that LV ejection fraction has been shown to be superior to dP/dt in assessing LV systolic function in certain large animal models (29). Third, although TUNEL positivity is consistent with the onset of apoptosis, it has been suggested that it may also reflect myocyte oncosis in experimental models of myocardial infarction (30). Alternatively, as we previously discussed in the context of brief ischemia-induced myocyte injury (13,27), TUNEL positivity could occur secondary to focal necrosis, with DNA fragmentation arising after rupture of the outer mitochondrial membrane resulting in cytochrome c release, and caspase activation (31). Fourth, a porcine-specific cTnI assay without the sensitivity of current high-sensitivity human assays was used to measure circulating cTnI concentrations, which may have affected our ability to detect very low levels of baseline transcoronary cTnI release or mild elevations early after stretch-induced myocyte injury. Finally, we only evaluated cTnI, and whether similar findings apply to troponin T measurements during preload elevation remains to be established.
CONCLUSIONS
Although the prognostic significance of elevated serum cTn concentrations appears to be independent of its underlying cause, it is increasingly recognized that various forms of nonischemic myocyte injury underlie many of the clinical cTnI elevations detected in the absence of underlying ischemic heart disease. Myocyte apoptosis arising from preload-induced mechanical stretch (or other causes [13,32]) may therefore explain elevations in cTn in pathologic conditions where an ACS is not present such as heart failure, pulmonary embolism, or renal failure to name a few. This reinforces the concept that cTn is an “organ-specific,” but not “disease-specific,” biomarker (24) and supports use of the term “myocardial injury” to describe cTn elevations in patients without ACS or supply-demand imbalance resulting in “reversible” ischemia distal to a stable coronary stenosis. Finally, like chronic regional apoptosis occurring in conjunction with reversible ischemia in hibernating myocardium, the impact of repetitive periods of preload elevation on myocyte loss in nonischemic myocardium may be considerable. Although speculative, this could lead to cellular hypertrophy and myocardial dysfunction in the absence of ischemia. Preload-induced myocyte loss could also contribute to adverse LV remodeling of remote normally-perfused regions after myocardial infarction. Further studies evaluating chronic repetitive preload elevation will be required to determine whether these acute responses impact cardiac pathophysiology in the chronic state (11,33).
PERSPECTIVES.
COMPETENCYIN MEDICAL KNOWLEDGE
Serum troponin I (cTnI) levels can rise in conditions associated with elevated left ventricular preload even in the absence of clinically recognized myocardial ischemia.
TRANSLATIONAL OUTLOOK
Further studies are needed to clarify the role of myocardial distension in patients with heart failure, renal failure, sepsis, pulmonary embolism, and other conditions associated with cardiac troponin elevations and their relationship to myocyte apoptosis and ventricular remodeling.
Acknowledgments
These studies could not have been completed without the assistance of Elaine Granica, Beth Palka, Anne Coe, and Marsha Barber. The authors also thank Dr. Saraswati Pokharel for assistance related to the histopathological evaluation of myocardial tissue specimens.
Funded by the National Heart Lung and Blood Institute (HL-055324, HL-061610, and F32HL-114335), the American Heart Association (17SDG33660200), the National Center for Advancing Translational Sciences (UL1TR001412), the Department of Veterans Affairs (1IO1BX002659), and the Albert and Elizabeth Rekate Fund in Cardiovascular Medicine. Dr. Iyer has been a proctor for Edwards Lifesciences, Boston Scientific, and Medtronic. Dr. Canty has been a consultant for Lantheus Medical Imaging. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
ABBREVIATIONS AND ACRONYMS
- ACS
acute coronary syndrome
- CS
coronary sinus
- cTn
cardiac troponin
- dP/dt
rate of rise of left ventricular pressure
- LV
left ventricle/ventricular
- LVEDP
left ventricular end-diastolic pressure
- PE
phenylephrine
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- TTC
triphenyltetrazolium chloride
APPENDIX
For an expanded Methods section, please see the online version of this paper.
References
- 1.Thygesen K, ALpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Circulation. 2012;126:2020–35. doi: 10.1161/CIR.0b013e31826e1058. [DOI] [PubMed] [Google Scholar]
- 2.Goodman SG, Steg PG, Eagle KA, et al. The diagnostic and prognostic impact of the redefinition of acute myocardial infarction: lessons from the Global Registry of Acute Coronary Events (GRACE) Am Heart J. 2006;151:654–60. doi: 10.1016/j.ahj.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 3.Turer AT, Addo TA, Martin JL, et al. Myocardial ischemia induced by rapid atrial pacing causes troponin T release detectable by a highly sensitive assay: insights from a coronary sinus sampling study. J Am Coll Cardiol. 2011;57:2398–405. doi: 10.1016/j.jacc.2010.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siriwardena M, Campbell V, Richards AM, Pemberton CJ. Cardiac biomarker responses to dobutamine stress echocardiography in healthy volunteers and patients with coronary artery disease. Clin Chem. 2012;58:1492–4. doi: 10.1373/clinchem.2012.187682. [DOI] [PubMed] [Google Scholar]
- 5.Predel HG. Marathon run: cardiovascular adaptation and cardiovascular risk. Eur Heart J. 2014;35:3091–8. doi: 10.1093/eurheartj/eht502. [DOI] [PubMed] [Google Scholar]
- 6.Giannitsis E, Katus HA. Cardiac troponin level elevations not related to acute coronary syndromes. Nat Rev Cardiol. 2013;10:623–34. doi: 10.1038/nrcardio.2013.129. [DOI] [PubMed] [Google Scholar]
- 7.Hamm CW, Giannitsis E, Katus HA. Cardiac troponin elevations in patients without acute coronary syndrome. Circulation. 2002;106:2871–2. doi: 10.1161/01.cir.0000044342.50593.63. [DOI] [PubMed] [Google Scholar]
- 8.Jeremias A, Gibson CM. Narrative review: alternative causes for elevated cardiac troponin levels when acute coronary syndromes are excluded. Ann Intern Med. 2005;142:786–91. doi: 10.7326/0003-4819-142-9-200505030-00015. [DOI] [PubMed] [Google Scholar]
- 9.Agewall S, Giannitsis E, Jernberg T, Katus H. Troponin elevation in coronary vs. non-coronary disease. Eur Heart J. 2011;32:404–11. doi: 10.1093/eurheartj/ehq456. [DOI] [PubMed] [Google Scholar]
- 10.Logeart D, Beyne P, Cusson C, et al. Evidence of cardiac myolysis in severe nonischemic heart failure and the potential role of increased wall strain. Am Heart J. 2001;141:247–53. doi: 10.1067/mhj.2001.111767. [DOI] [PubMed] [Google Scholar]
- 11.Cheng W, Li B, Kajstura J, et al. Stretch-induced programmed myocyte cell death. J Clin Invest. 1995;96:2247–59. doi: 10.1172/JCI118280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng J, Schaus BJ, Fallavollita JA, Lee TC, Canty JM., Jr Preload induces troponin I degradation independently of myocardial ischemia. Circulation. 2001;103:2035–7. doi: 10.1161/01.cir.103.16.2035. [DOI] [PubMed] [Google Scholar]
- 13.Weil BR, Young RF, Shen X, et al. Brief myocardial ischemia produces cardiac troponin I release and focal myocyte apoptosis in the absence of pathological infarction in swine. J Am Coll Cardiol Basic Trans Science. 2017;2:105–14. doi: 10.1016/j.jacbts.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ellis AK, Klocke FJ. Effects of preload on the transmural distribution of perfusion and pressure-flow relationships in the canine coronary vascular bed. Circ Res. 1979;46:68–77. doi: 10.1161/01.res.46.1.68. [DOI] [PubMed] [Google Scholar]
- 15.Aversano T, Klocke FJ, Mates RE, Canty JM., Jr Preload-induced alterations in capacitance-free diastolic pressure-flow relations. Am J Physiol. 1984;246:H410–7. doi: 10.1152/ajpheart.1984.246.3.H410. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki G, Iyer V, Lee TC, Canty JM., Jr Autologous mesenchymal stem cells mobilize cKit+ and CD133+ bone marrow progenitor cells and improve regional function in hibernating myocardium. Circ Res. 2011;109:1044–54. doi: 10.1161/CIRCRESAHA.111.245969. [DOI] [PubMed] [Google Scholar]
- 17.Weil BR, Suzuki G, Leiker MM, Fallavollita JA, Canty JM., Jr Comparative efficacy of intracoronary allogeneic mesenchymal stem cells and cardiosphere-derived cells in swine with hibernating myocardium. Circ Res. 2015;117:634–44. doi: 10.1161/CIRCRESAHA.115.306850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fallavollita JA, Canty JM., Jr Ischemic cardiomyopathy in pigs with two-vessel occlusion and viable, chronically dysfunctional myocardium. Am J Physiol Heart Circ Physiol. 2002;282:H1370–9. doi: 10.1152/ajpheart.00138.2001. [DOI] [PubMed] [Google Scholar]
- 19.White HD. PathobioLogy of troponin elevations: do elevations occur with myocardial ischemia as well as necrosis? J Am Coll Cardiol. 2011;57:2406–8. doi: 10.1016/j.jacc.2011.01.029. [DOI] [PubMed] [Google Scholar]
- 20.Sarkisian L, Saaby L, PouLsen TS, et al. Prognostic impact of myocardiaL injury reLated to various cardiac and noncardiac conditions. Am J Med. 2016;129:506–14.e1. doi: 10.1016/j.amjmed.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 21.Sarkisian L, Saaby L, PouLsen TS, et al. Clinical characteristics and outcomes of patients with myocardial infarction, myocardial injury, and nonelevated troponins. Am J Med. 2016;129:446.e5–21. doi: 10.1016/j.amjmed.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 22.Guest TM, Ramanathan AV, Tuteur PG, Schechtman KB, Ladenson JH, Jaffe AS. Myocardial injury in critically ill patients. A frequently unrecognized complication. JAMA. 1995;273:1945–9. [PubMed] [Google Scholar]
- 23.McEvoy JW, Chen Y, Ndumele CE, et al. Six-year change in high-sensitivity cardiac troponin t and risk of subsequent coronary heart disease, heart failure, and death. JAMA Cardiol. 2016;1:519–28. doi: 10.1001/jamacardio.2016.0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park KC, Gaze DC, Collinson PO, Marber MS. Cardiac troponins: from myocardial infarction to chronic disease. Cardiovasc Res. 2017;113:1708–18. doi: 10.1093/cvr/cvx183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takashio S, Yamamuro M, Izumiya Y, et al. Coronary microvascular dysfunction and diastolic load correlate with cardiac troponin T release measured by a highly sensitive assay in patients with nonischemic heart failure. J Am Coll Cardiol. 2013;62:632–40. doi: 10.1016/j.jacc.2013.03.065. [DOI] [PubMed] [Google Scholar]
- 26.Hessel MH, Atsma DE, van der Valk EJ, Bax WH, Schalij MJ, van der Laarse A. Release of cardiac troponin I from viabLe cardiomyocytes is mediated by integrin stimulation. Pflugers Arch. 2008;455:979–86. doi: 10.1007/s00424-007-0354-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amgalan D, Pekson R, Kitsis RN. Troponin release following brief myocardial ischemia: apoptosis versus necrosis. J Am Coll CardioL Basic Trans Science. 2017;2:118–21. doi: 10.1016/j.jacbts.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerhardt W, Katus H, Ravkilde J, et al. S-troponin T in suspected ischemic myocardial injury compared with mass and cataloytic concentrations of S-creatine kinase isoenzyme MB. Clin Chem. 1991;37:1405–11. [PubMed] [Google Scholar]
- 29.Ishikawa K, Chemaly ER, Tilemann L, et al. Assessing left ventricular systolic dysfunction after myocardial infarction: are ejection fraction and dP/dt(max) complementary or redundant? Am J Physiol Heart Circ Physiol. 2012;302:H1423–8. doi: 10.1152/ajpheart.01211.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohno M, Takemura G, Ohno A, et al. “Apoptotic” myocytes in infarct area in rabbit hearts may be oncotic myocytes with DNA fragmentation: analysis by immunogold electron microscopy combined with In situ nick end-labeling. Circulation. 1998;98:1422–30. doi: 10.1161/01.cir.98.14.1422. [DOI] [PubMed] [Google Scholar]
- 31.Konstantinidis K, Whelan RS, Kitsis RN. Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol. 2012;32:1552–62. doi: 10.1161/ATVBAHA.111.224915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh K, Communal C, Sawyer DB, Colucci WS. Adrenergic regulation of myocardial apoptosis. Cardiovasc Res. 2000;45:713–9. doi: 10.1016/s0008-6363(99)00370-3. [DOI] [PubMed] [Google Scholar]
- 33.Leri A, Claudio PP, Li Q, et al. Stretch-mediated release of angiotensin II induces myocyte apoptosis by activating p53 that enhances the local renin-angiotensin system and decreases the Bcl-2-to-Bax protein ratio in the cell. J Clin Invest. 1998;101:1326–42. doi: 10.1172/JCI316. [DOI] [PMC free article] [PubMed] [Google Scholar]