

Abstract
Mitochondrial and lysosomal dysfunction have been implicated in substantia nigra dopaminergic neurodegeneration in Parkinson’s disease (PD), but how these pathways are linked in human neurons remains unclear. Here we studied dopaminergic neurons derived from patients with idiopathic and familial PD. We identified a time-dependent pathological cascade beginning with mitochondrial oxidant stress leading to oxidized dopamine accumulation and ultimately resulting in reduced glucocerebrosidase enzymatic activity, lysosomal dysfunction, and α-synuclein accumulation. This toxic cascade was observed in human, but not in mouse, PD neurons at least in part because of species-specific differences in dopamine metabolism. Increasing dopamine synthesis or α-synuclein amounts in mouse midbrain neurons recapitulated pathological phenotypes observed in human neurons. Thus, dopamine oxidation represents an important link between mitochondrial and lysosomal dysfunction in PD pathogenesis.
Parkinson’s disease (PD) is the second most common neurodegenerative disorder (1). Although several neuronal populations are affected in PD, substantia nigra pars compacta (SNc) dopaminergic neurons are among the first neurons to degenerate, leading to motor symptoms of PD (2). Familial genes in PD point to both mitochondrial and lysosomal mechanisms contributing to pathology (1). However, how mutations in components of two distinct essential cellular pathways both result in indistinguishable clinical and pathological phenotypes and finally cause the death of dopaminergic neurons in patients remains unclear. Moreover, because the majority of PD mouse models do not manifest nigral degeneration (3), it is unclear whether this interaction is specific to human neurons.
Loss of function mutations in DJ-1 cause early-onset autosomal recessive PD (4), and DJ-1 is known to orchestrate oxidant defenses, because its deletion leads to elevated mitochondrial oxidant stress (5). To examine whether elevated mitochondrial oxidant stress triggers downstream pathological phenotypes, fibroblasts from PD patients with a homozygous loss of function mutation in DJ-1 c.192G>C (6) were reprogrammed into induced pluripotent stem cells (iPSCs). These were then differentiated into dopaminergic neurons (hom 1 and hom 2), which were grown in long-term cultures (fig. S1, A to D, fig. S2, A and B). For comparison purposes, dopaminergic neurons were also generated from a healthy individual with a single copy of mutated DJ-1 (het), idiopathic PD patients without DJ-1 mutations and healthy individuals without PD. These neurons had anatomical and electrophysiological characteristics of SNc neurons (7) including robust, low-frequency autonomous pacemaking (fig. S2C) and prominent voltage sag upon brief injection of strong hyperpolarizing currents (fig. S2D). DJ-1 protein levels were decreased by ~50% in heterozygous and undetectable in homozygous DJ-1 mutant neurons (fig. S2E).
Neuromelanin and oxidized dopamine accumulate in dopaminergic neurons from genetic and sporadic PD patients
By day 50 (d50) in culture, mitochondrial oxidant stress was elevated in human homozygous DJ-1 mutant dopaminergic neurons (Fig. 1A). This increase was seen using both a genetically encoded, mitochondrial matrix redox sensor (mito-roGFP) (5) and by fluorescence measurements using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) at d50 and d70 (Fig. 1B). Increased mitochondrial oxidant stress was accompanied by a drop in basal respiration in mutant neurons at d50 (Fig. 1C), suggesting that the observed stress damaged mitochondria.
Figure 1. Neuromelanin and oxidized dopamine accumulate in PD patient dopaminergic neurons.
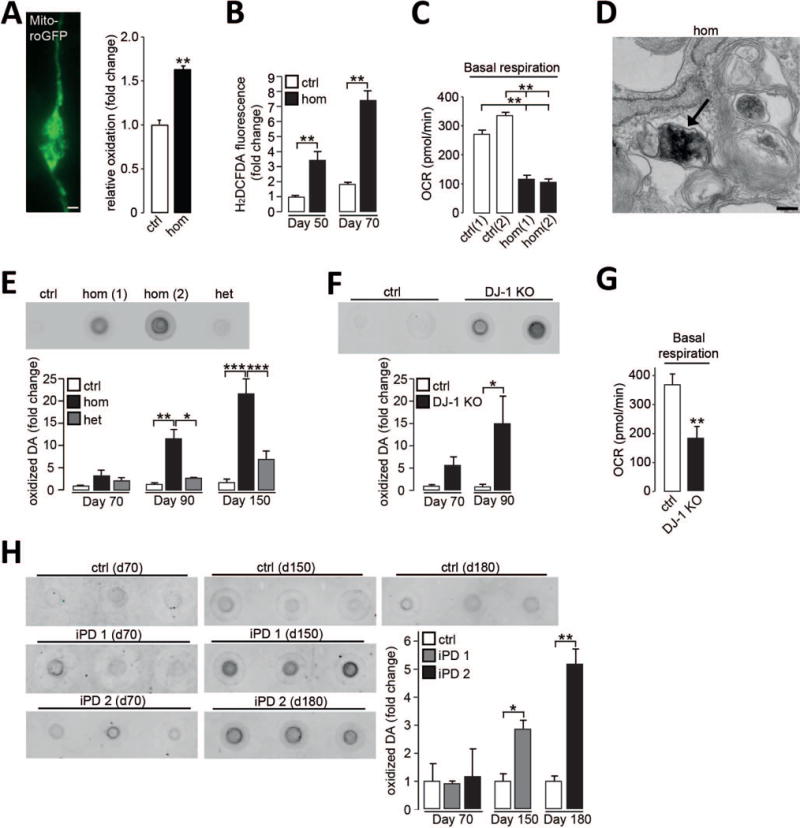
(A) Neuronal expression of mito-roGFP and quantification of relative oxidation in control (ctrl) and homozygous DJ-1 mutant (hom) neurons at d50 (n=3). Scale bar, 10μm. (B) H2DCFDA fluorescence in neurons at d50 and d70 (n=3). (C) Oxygen consumption rate (OCR) in neurons under basal conditions at d50 (n=3). (D) Electron microscopy (EM) image of neuromelanin deposition (arrow) in homozygous DJ-1 mutant neuron at d90. Scale bar 200nm. (E-F) Oxidized dopamine (DA) by nIRF at d90 in (E) homozygous DJ-1 mutant (hom(1) and hom(2)) neurons and controls, quantification at d70, d90, and d150 (n=3), het for heterozygous, or (F) DJ-1 KO and isogenic control neurons and quantification at d70 and d90 (n=3-4). (G) OCR in DJ-1 KO neurons and isogenic controls under basal conditions at d50 (n=12). (H) Oxidized dopamine in control and two idiopathic PD (iPD1; iPD2) neurons at d70, d150 and d180 (n=3). Quantification for each iPD line is normalized to control for each time point. Equal protein concentrations were used for nIRF assays. Error bars, mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, Student’s t test (A, B, F-H) or one-way analysis of variance (ANOVA) with Tukey post-hoc test (C, E).
Homozygous DJ-1 mutant neurons also accumulated membrane-bound, dense pigmented aggregates consistent with the appearance of neuromelanin (Fig. 1D, fig. S2F). Biochemical properties of these pigments, such as their insolubility to strong detergents, further indicated their similarity to neuromelanin extracted from human brain (8). Neuromelanin is a complex aggregate, including oxidized dopamine, proteins and lipids (9). To quantify the amount of oxidized dopamine in these neurons, a near infrared fluorescence (nIRF) assay was used (10). Oxidized dopamine progressively increased from d70 to d150 in homozygous DJ-1 mutant neurons, but not in controls (Fig. 1E). To determine whether the absence of DJ-1 protein was sufficient to induce this pathological phenotype, clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 was used to generate homozygous DJ-1 knock out (KO) iPSC-derived neurons (DJ-1 KO) from controls (fig. S3). These neurons recapitulated the phenotype of DJ-1 patient neurons, resulting in the progressive accumulation of oxidized dopamine from d70 to d90 (Fig. 1F), as well as decreased basal mitochondrial respiration (Fig. 1G).
Dopaminergic neurons from idiopathic PD patients (iPD1, (11); iPD2, fig. S1, E and F) also manifested a similar phenotype, including decreased basal respiration and oxidized dopamine accumulation, but at later time points (d150, d180) (Fig. 1H, fig. S4A). We observed partial inactivation of DJ-1 in iPD neurons (fig. S4B), suggesting that decreased DJ-1 function may contribute to these delayed pathological alterations in iPD neurons (12).
Dopamine-mediated modification of glucocerebrosidase (GCase) contributes to downstream lysosomal dysfunction
Because neuromelanin granules have been found in lysosomes (13), the accumulation of neuromelanin-like aggregates in mutant dopaminergic neurons suggested that lysosomal function might be impaired. While there was no detectable impairment in lysosomal proteolysis in homozygous DJ-1 mutant neurons at d70 (Fig. 2A), the activity of the lysosomal enzyme glucocerebrosidase (GCase), an important PD risk factor implicated in lysosomal dysfunction in PD (14), was decreased in lysosomal fractions (Fig. 2B). In contrast, there was no drop in the activity of another lysosomal enzyme, α-iduronate-2-sulfatase (α-i-2-sulf). However, lysosomal proteolysis was significantly diminished at a later time point (d180) (Fig. 2A, fig. S5), suggesting the contribution of accumulated oxidized dopamine to lysosomal dysfunction in homozygous DJ-1 mutant neurons. Idiopathic PD neurons exhibited a delayed decrease in GCase activity (d180) compared to DJ-1 mutant neurons (d70) (Fig. 2C). In order to determine if other forms of genetic PD might show similar alterations, we generated iPSC-derived neurons from PD patients with homozygous mutations in the parkin, PINK1, and LRRK2 (15, 16) genes and neurons carrying triplication of wildtype (WT) α-synuclein (17). We observed a similar increase in oxidized dopamine across all genotypes (fig. S4C, E to G) as well as decreased GCase activity in parkin mutant neurons (fig. S4D), further suggesting the presence of common phenotypes across both sporadic and familial forms of PD.
Figure 2. Dopamine-mediated modification of GCase and lysosomal dysfunction in PD patient neurons.
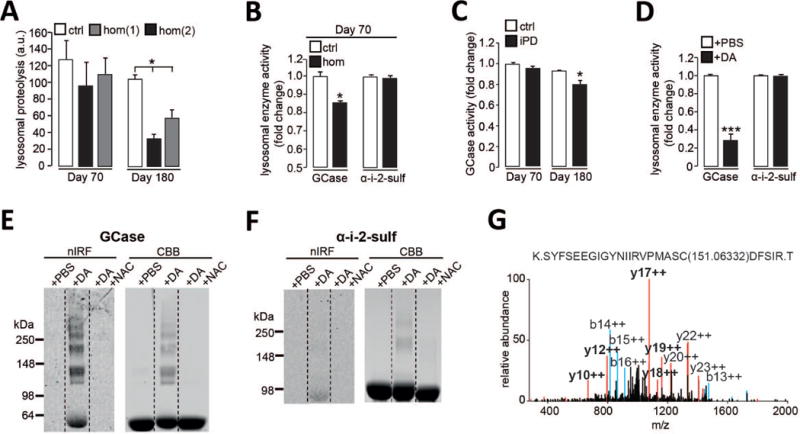
(A) Lysosomal proteolysis in control (ctrl) and homozygous DJ-1 mutant (hom) neurons at d70 and d180 (n=3). (B) GCase and a-i-2-sulf activity in lysosomal fractions from control and homozygous DJ-1 mutant neurons at d70 (n=3). (C) GCase activity in lysosomal fractions from control and idiopathic PD (iPD) neurons at d70 and d180 (n=3). (D) Recombinant GCase and α-i-2-sulf activity after incubation with DA or phosphate-buffered saline (PBS) (n=5). (E-F) Oxidized DA by in-gel nIRF of (E) recombinant GCase or (F) α-i-2-sulf after incubation with DA, DA+NAC or PBS. Coomassie brilliant blue (CBB) was used to visualize total protein. (G) MS/MS spectrum of GCase treated with DA. Modified cysteine is indicated with mass adduct in parentheses. Prominent b (blue) and y (red) ions and fragments containing the additional mass (bold) are indicated. Error bars, mean ± SEM. *P<0.05, ***P<0.001, one-way ANOVA with Tukey post-hoc test (A) or Student’s t test (B-D).
Because elevated oxidant stress and oxidized dopamine were detected concomitantly with decreased GCase activity (Fig. 1 and 2, A to C), we next sought to determine if reduced GCase function could have resulted from oxidation of the protein. Indeed, the activity of recombinant GCase, but not α-i-2-sulf, was reduced by incubation with dopamine (Fig. 2D). Using in-gel nIRF, dopamine incubation was found to modify GCase compared to α-i-2-sulf; and these changes were blocked upon antioxidant treatment with n-acetyl cysteine (NAC) (Fig. 2, E and F). Using tandem mass spectrometry (MS/MS), we identified cysteine residues in the catalytic site of GCase that were modified by dopamine quinones, reactive oxidized forms of dopamine (Fig. 2G). These results suggested that oxidant stress directly modifies and disrupts GCase enzymatic activity and contributes to lysosomal dysfunction in PD neurons.
Mitochondrial antioxidants and calcium modulators attenuate the toxic cascade in DJ-1 mutant dopaminergic neurons
We found that long-term treatment with either the mitochondrial-targeted antioxidant mito-TEMPO or NAC blunted the accumulation of oxidized dopamine and improved lysosomal GCase activity and proteolysis (Fig. 3, A to D). Furthermore, because increased Ca2+ entry through Cav1 voltage-gated calcium channels enhances mitochondrial oxidant stress and dopamine synthesis (5, 18), we tested whether Cav1 channel antagonist isradipine (19) or the calcium-dependent serine/threonine phosphatase calcineurin inhibitor FK506 (20) could also regulate dopamine oxidation. Both isradipine and FK506 significantly diminished the accumulation of oxidized dopamine (Fig. 3E), highlighting the importance of Ca2+ in regulating dopamine oxidation in DJ-1 mutant neurons.
Figure 3. Mitochondrial antioxidants and calcium modulators attenuate the toxic cascade in DJ-1 mutant dopaminergic neurons.
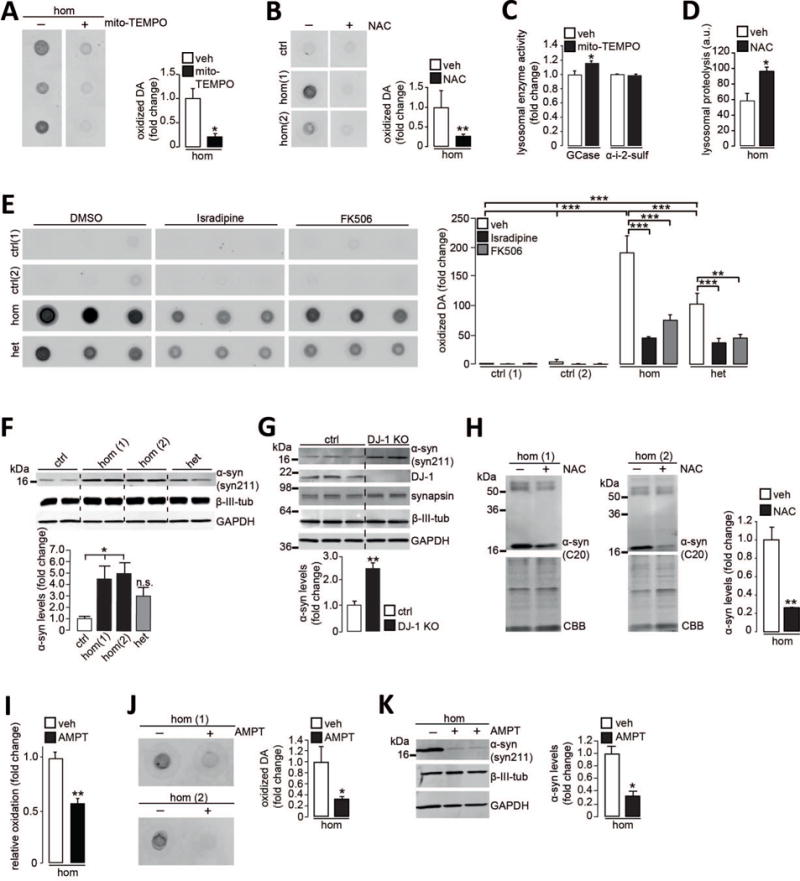
(A-B) Oxidized dopamine (DA) in homozygous DJ-1 mutant neurons (hom) treated with (A) mito-TEMPO or (B) NAC compared to vehicle (veh) at d70 (n=3). (C) GCase and α-i-2-sulf activity in lysosomal fractions from homozygous DJ-1 mutant neurons treated with mito-TEMPO or vehicle at d70 (n=3). (D) Lysosomal proteolysis in homozygous DJ-1 mutant neurons treated with NAC or vehicle at d180 (n=3). a.u., arbitrary units. (E) Oxidized DA in homozygous DJ-1 mutant, heterozygous DJ-1 carrier (het) and control neurons treated with isradipine, FK506 or vehicle (DMSO, dimethyl sulfoxide) at d90 (n=3-6). (F-G) Immunoblot analysis of α-synuclein (syn211 antibody) at d70 in Triton X-100 (T)-soluble neuronal lysates from (F) control, heterozygous DJ-1 carrier and two homozygous DJ-1 mutant lines (n=4) or (G) a gene-edited DJ-1 KO line (n=3 or 4). β-III-tubulin, synapsin, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were used as loading controls. (H) T-insoluble α-synuclein (C20 antibody) at d70 in homozygous DJ-1 mutant neurons treated with NAC or vehicle. CBB used as loading control (n=4). (I) Homozygous DJ-1 mutant neurons expressing mito-roGFP treated with AMPT at d50 (n=3). (J-K) Homozygous DJ-1 mutant neurons treated with AMPT and analyzed at d70 for (J) oxidized DA by nIRF (n=3) or (K) T-soluble α-synuclein (syn211 antibody). GAPDH and β-III-tubulin loading controls (n=3). Treatment applied for 30 days (A-C, E, H-K) or 140 days (D). Error bars, mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, Student’s t test (A-D, G-K) or one-way ANOVA with Tukey post-hoc test (E-F).
Oxidant stress has also been found to drive α-synuclein aggregation, the primary structural component of Lewy bodies and a pathological hallmark of PD (21). Furthermore, dopamine adducts can modify α-synuclein and promote its aggregation and a recent study reports that loss of DJ-1 function is associated with Lewy body pathology (22). Soluble α-synuclein was elevated at d70 in both homozygous DJ-1 mutant and CRISPR/Cas9-generated DJ-1 KO neurons (Fig. 3, F and G). We observed a time-dependent increase in oxidized, insoluble α-synuclein from d70 to d100 in mutant neurons (fig. S6, A and B), but not in heterozygous DJ-1 neurons or controls. NAC treatment also decreased α-synuclein accumulation in DJ-1 mutant neurons (Fig. 3H, fig. S6, C and D). To determine whether dopamine contributed to this phenotype, neurons were treated with α-methyl-p-tyrosine (AMPT), which competitively inhibits tyrosine hydroxylase and prevents dopamine synthesis. AMPT treatment diminished mitochondrial oxidant stress as well as oxidized dopamine (Fig. 3, I and J) and reduced soluble α-synuclein levels in DJ-1 mutant neurons (Fig. 3K), suggesting that mitochondrial oxidant stress and oxidized dopamine contribute to α-synuclein accumulation in PD neurons.
Increasing dopamine or α-synuclein in mouse neurons recapitulates pathology in human neurons
We next examined whether this toxic cascade observed in human PD neurons also occurred in SNc dopaminergic neurons from PD mouse models. In contrast to human neurons, SNc from DJ-1 KO mice showed negligible amounts of oxidized dopamine at 3 and 12 months of age (Fig. 4, A and B). In addition, soluble and insoluble α-synuclein levels were normal in SNc from DJ-1 KO mice (Fig. 4C, fig. S7, A to B) and lysosomal GCase activity was not altered (Fig. 4G) compared to WT mice. Moreover, tyrosine hydroxylase (TH) immunoreactivity in the SNc (Fig. 4I) was not decreased in DJ-1 KO mice, indicating lack of nigral pathology.
Figure 4. Increasing dopamine synthesis or amounts of α-synuclein in mouse midbrain neurons recapitulates pathological phenotypes observed in human neurons.
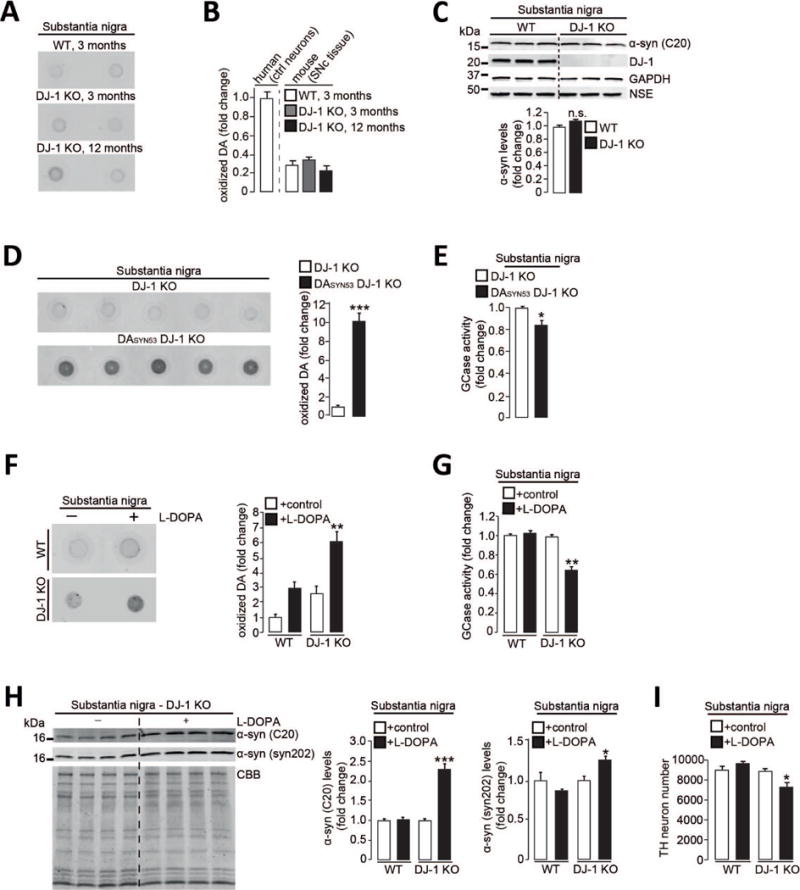
(A-B) Oxidized DA in substantia nigra (SN) from DJ-1 KO and WT mice at 3 and 12 months compared to human control neurons at d70. (C) T-soluble α-synuclein (C20 antibody) in SNc of WT and DJ-1 KO mice. GAPDH and NSE *neural-specific enolase) loading controls (n=3 per group). (D-E) SN from DJ-1 KO and DASYN53 x DJ-1 KO mice analyzed at 8 months of age for (D) oxidized DA and (E) GCase activity (n=5 per group). (F-I) WT and DJ-1 KO mice were fed L-DOPA-supplemented or vehicle-treated chow for 6 months, substantia nigra analyzed at 14 months for (F) oxidized DA (n=3-4 per group), (G) GCase activity (n=3-4 per group), (H) T-insoluble α-synuclein (C20 and syn202 antibodies). CBB used as loading control (n=3-4 per group), and (I) number of DAB (3,3′-diaminobenzidine)-stained TH-positive neurons (n=3 per group). Equal protein concentrations were used for nIRF assays. Error bars, mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, Student’s t test (C-E, H) or one-way ANOVA with Tukey post-hoc test (B, F-G, I,). n.s. = not significant.
Because mutations in α-synuclein that result in its accumulation lead to familial PD, we then asked whether increasing levels of mutant α-synuclein in vivo might drive this toxic cascade in DJ-1 KO mice. We generated mice with both dopamine neuron-specific overexpression of human α-synuclein A53T (23) and constitutive DJ-1 deficiency (DASYN53 x DJ-1 KO) (fig. S8A). These mice showed elevated levels of oxidized dopamine in nigral neurons (Fig. 4D), and decreased lysosomal GCase activity (Fig. 4E) compared to DJ-1 KO mice. Heterozygous expression of DJ-1 in DASYN53 mice was sufficient to ameliorate pathological phenotypes observed in DASYN53 x DJ-1 KO mice (fig. S8, B to D) suggesting that a combination of a-synuclein accumulation and loss of DJ-1 contributes to nigral degeneration in mice.
We next investigated whether increasing dopamine levels in vivo might also trigger pathology in mice. Chronic feeding of WT or DJ-1 KO mice with levodopa (L-DOPA)-supplemented chow led to oxidized dopamine accumulation in the nigra (Fig. 4F, fig. S7C). L-DOPA treatment in DJ-1 KO mice but not in WT mice was sufficient to disrupt SNc GCase enzymatic activity (Fig. 4G), increase α-synuclein accumulation (Fig. 4H, fig. S7D), and trigger the loss of dopaminergic neurons (Fig. 4I and fig. S7E). L-DOPA treatment had no effect on homocysteine levels (fig. S7F).
Altered calcium and dopamine metabolism contribute to differences between human and mouse neurons
To directly compare human neurons with mouse neurons, we extended these experiments to mouse iPSC-derived dopaminergic neurons generated from DJ-1 KO and WT fibroblasts (fig. S9A). Consistent with the in vivo results, DJ-1 KO mouse neurons did not manifest significant levels of oxidized dopamine at d40, d70 or d90 (Fig. 5A). However, treatment of these neurons with L-DOPA resulted in increased oxidized dopamine (Fig. 5B), disrupted lysosomal GCase activity (Fig. 5C) and decreased neuronal survival (fig. S9B). There were no pathological changes observed in L-DOPA-treated mouse WT neurons (Fig. 5, B and C), further suggesting that both mitochondrial oxidant stress and increased oxidized dopamine contribute to neurotoxicity of dopaminergic neurons.
Figure 5. Alterations in calcium homeostasis and dopamine metabolism contribute to intrinsic differences between human and mouse dopaminergic neurons.
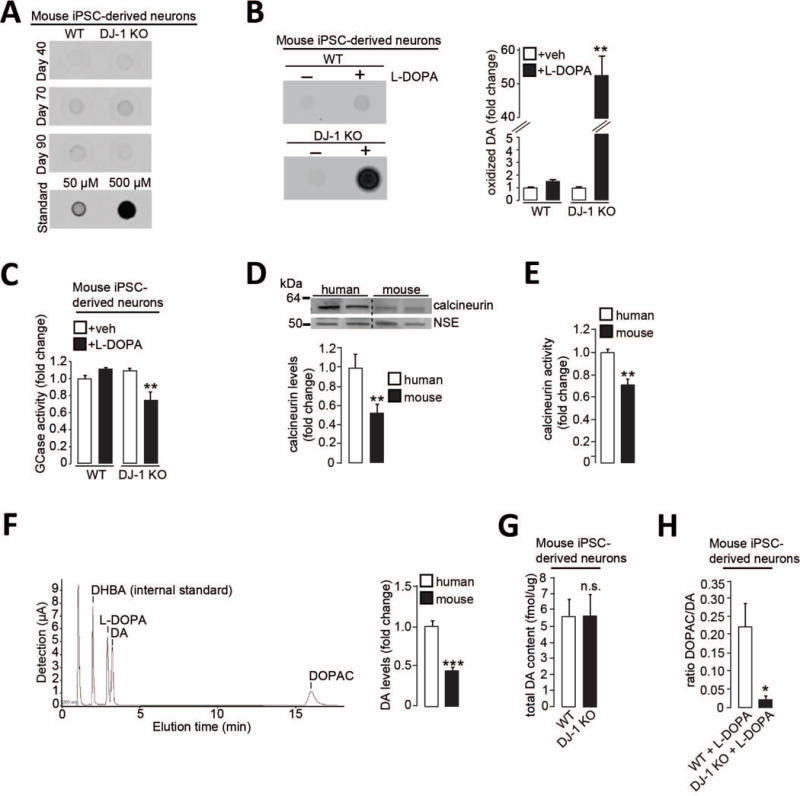
(A-C) WT and DJ-1 KO mouse iPSC-derived dopaminergic neurons were analyzed for (A) oxidized DA at d40, d70 and d90 (50μM and 500μM standards shown), or treated with L-DOPA or vehicle and analyzed at d55 for (B) oxidized DA (n=3) and (C) lysosomal GCase activity (n=3). (D-F) Control human and WT mouse iPSC-derived dopaminergic neurons analyzed for (D) T-soluble calcineurin (n=5-6), (E) calcineurin activity (n=5), and (F) DA amounts by HPLC (n=5 (mouse); n=10 (human)). HPLC chromatogram in a 4 μM standard sample is shown. Dihydroxybenzylamine (DHBA) was used as internal standard. (G) WT and DJ-1 KO mouse iPSC-derived dopaminergic neurons were analyzed for total DA content at d55 (n=5). (H) The ratio of DOPAC/DA for WT and DJ-1 KO mouse iPSC-derived dopaminergic neurons treated with L-DOPA (n=8). Equal protein concentration was used in nIRF assays. Error bars, mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, Student’s t test (D-H) or one-way ANOVA with Tukey post-hoc test (B-C). n.s. = not significant.
Using high-performance liquid chromatography (HPLC) for detection of dopamine and its metabolites, we found that human control neurons showed elevated amounts of dopamine compared to mouse WT neurons (Fig. 5F), suggesting a difference in dopamine metabolism between species. Consistent with this notion is the observation that L-DOPA treatment was sufficient to increase oxidized dopamine in human control (fig. S10), but not mouse WT neurons (Fig. 5B). Dopamine can be either converted into non-toxic downstream metabolites including 3,4-dihydroxyphenylacetic acid (DOPAC), or form toxic dopamine quinones and aminochrome that are normally sequestered in neuromelanin (9). We hypothesized that increased oxidative stress in DJ-1 KO mouse neurons may preferentially result in oxidized dopamine and neuromelanin upon treatment with L-DOPA. Although the baseline amounts of total dopamine were comparable between mouse WT and DJ-1 mutant neurons (Fig. 5G), mutant neurons showed a dramatically decreased DOPAC/dopamine ratio when treated with L-DOPA compared to WT neurons (Fig. 5H). Thus, decreased dopamine degradation into non-toxic metabolites may be responsible for the accumulation of oxidized dopamine.
Because the calcineurin inhibitor FK506 diminished the accumulation of oxidized dopamine (Fig. 3E), we examined if calcineurin might be altered between mice and humans. Control human neurons showed both increased protein levels and activity of calcineurin (Fig. 5, D and E) compared to mouse WT neurons, further suggesting that alterations in calcium homeostasis may also contribute to inter-species differences in dopamine metabolism.
Discussion
Here we have found that elevated mitochondrial oxidant stress in human SNc dopaminergic neurons triggers a dopamine-dependent toxic cascade leading to lysosomal dysfunction and α-synuclein accumulation – causally linking three major pathological features of PD. This discovery complements previous work demonstrating that lysosomal dysfunction leads to deficits in mitophagy and the buildup of dysfunctional mitochondria (24), and raises the possibility of a pathogenic positive feedback between these two organelles. The pathogenic cascade from mitochondria to lysosomes was only evident in human – and not mouse – dopaminergic neurons. This species difference was inferred to be a consequence of higher levels of dopamine in human neurons based upon several lines of evidence, including the selective accumulation of neuromelanin in human neurons and the ability of levodopa, the synthetic precursor of dopamine, to induce the pathological cascade in mouse dopaminergic neurons modeling PD. In addition, our data also suggest that increased cytosolic dopamine contributes to elevated mitochondrial oxidant stress, suggesting a vicious cycle of dopamine and mitochondrial oxidation in human midbrain neurons.
Although increased α-synuclein inhibits GCase trafficking and lysosomal function in various cell types (19), our data suggest that increased oxidant stress and dopamine adduct formation modified the catalytic site of GCase and lowered its activity preferentially in midbrain dopaminergic neurons. Because similar alterations in oxidized dopamine and GCase were seen in neurons obtained from patients with idiopathic and familial PD, we propose that this mechanism applies across various forms of PD. In support of this notion, we showed that elevation of α-synuclein in the substantia nigra of DJ-1 KO mice also resulted in increased oxidized dopamine, diminished activity of GCase and neurodegeneration.
Our long-term midbrain cultures demonstrated that early treatment with mitochondrial antioxidants reduced downstream accumulation of oxidized dopamine and α-synuclein, and rescued lysosomal dysfunction, highlighting the importance of early therapeutic intervention in the pathogenic cascade. The inherent differences between human and mouse dopaminergic neuronal vulnerability emphasize the value of studies in human neurons to identify pathways and targets for therapeutic development in PD and related synucleinopathies.
Supplementary Material
One Sentence Summary.
Dopamine oxidation links mitochondrial oxidant stress to lysosomal dysfunction in human, but not mouse, dopaminergic PD neurons.
Acknowledgments
We are grateful to D. Sulzer, E.V. Mosharov, J.W. Shim, G. Caraveo, and J. Zheng for helpful discussions and suggestions. We thank the Northwestern Stem Cell Core Facility for the generation of mouse iPSC lines (supported by P30 NS081774). We are grateful to K. Hamada as well as M.K.V. Suresh for technical assistance and C. Leong for help with the assessment of off-target gene editing in the CRISPR-edited lines. The mito-GFP construct was a gift from P. Schumacker (Northwestern University). This work was supported by NIH (grants R01NS076054 to D.K., NS047085 to D.J.S, R01NS092823 to J.R.M, R00DC013805-02 to J.N.S., and T32NS041234 and F32NS101778 to Y.C.W.); The JPB Foundation and The Michael J. Fox and IDP Foundations to D.J.S; German Academic Exchange Service to L.F.B.; Les Turner ALS Foundation, Target ALS, and MDA to E.K.; and the German Research Council grant KR2119/8-1 and the Fond National de Recherche PEARL program grant FNR/P13/6682797 to R.K. All data necessary to support this paper’s conclusions are available in the supplementary materials.
Footnotes
SUPPLEMENTARY MATERIALS
Supplementary information: Entire western blots from main figures
References
References and Notes
- 1.Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386:896–912. doi: 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- 2.Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nature reviews Neuroscience. 2017;18:101–113. doi: 10.1038/nrn.2016.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson’s disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonifati V, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 5.Guzman JN, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krebiehl G, et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PloS one. 2010;5:e9367. doi: 10.1371/journal.pone.0009367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zecca L, et al. New melanic pigments in the human brain that accumulate in aging and block environmental toxic metals. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17567–17572. doi: 10.1073/pnas.0808768105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sulzer D, et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:11869–11874. doi: 10.1073/pnas.97.22.11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazzulli JR, Burbulla LF, Krainc D, Ischiropoulos H. Detection of Free and Protein-Bound ortho-Quinones by Near-Infrared Fluorescence. Analytical chemistry. 2016;88:2399–2405. doi: 10.1021/acs.analchem.5b04420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazzulli JR, et al. Activation of beta-Glucocerebrosidase Reduces Pathological alpha-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2016;36:7693–7706. doi: 10.1523/JNEUROSCI.0628-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi J, et al. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. The Journal of biological chemistry. 2006;281:10816–10824. doi: 10.1074/jbc.M509079200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plum S, et al. Proteomic characterization of neuromelanin granules isolated from human substantia nigra by laser-microdissection. Scientific reports. 2016;6:37139. doi: 10.1038/srep37139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mazzulli JR, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller JD, et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell stem cell. 2013;13:691–705. doi: 10.1016/j.stem.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper O, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Science translational medicine. 2012;4:141ra190. doi: 10.1126/scitranslmed.3003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. Alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:1931–1936. doi: 10.1073/pnas.1520335113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mosharov EV, et al. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simuni T, et al. Phase II safety, tolerability, and dose selection study of isradipine as a potential disease-modifying intervention in early Parkinson’s disease (STEADY-PD) Movement disorders : official journal of the Movement Disorder Society. 2013;13:1823–1831. doi: 10.1002/mds.25639. [DOI] [PubMed] [Google Scholar]
- 20.Caraveo G, et al. Calcineurin determines toxic versus beneficial responses to alpha-synuclein. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E3544–3552. doi: 10.1073/pnas.1413201111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong YC, Krainc D. Alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nature medicine. 2017;23:1–13. doi: 10.1038/nm.4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taipa R, et al. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain : a journal of neurology. 2016;139:1680–1687. doi: 10.1093/brain/aww080. [DOI] [PubMed] [Google Scholar]
- 23.Chen L, Xie Z, Turkson S, Zhuang X. A53T human alpha-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35:890–905. doi: 10.1523/JNEUROSCI.0089-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gegg ME, Schapira AH. Mitochondrial dysfunction associated with glucocerebrosidase deficiency. Neurobiology of disease. 2016;90:43–50. doi: 10.1016/j.nbd.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.