

Abstract
According to the National Center of Health Statistics, cancer was the culprit of nearly 600,000 deaths in 2016 in the United States. It is by far one of the most heterogeneous diseases to treat. Treatment for metastasized cancers remains a challenge despite modern diagnostics and treatment regimens. For this reason, alternative approaches are needed. Chemoprevention using dietary phytochemicals such as triterpenoids, isothiocyanates, and curcumin in the prevention of initiation and/or progression of cancer poses a promising alternative strategy. However, significant challenges exist in the extrapolation of in vitro cell culture data to in vivo efficacy to animal models and particularly to human. In this review, the dose at which these phytochemicals elicit a response in vitro and in vivo of a multitude of cellular signaling pathways will be reviewed highlighting Nrf2-mediated anti-oxidative stress, anti-inflammation, epigenetics, cytoprotection, differentiation, and growth inhibition. The in vitro-in vivo dose response of phytochemicals can vary due in part to the cell line/animal model used, the assay system of the biomarker used for the readout, chemical structure of the functional analog of the phytochemical, and the source of compounds used for the treatment study. While the dose response varies across different experimental designs, the chemopreventive efficacy appears to remain and demonstrates the therapeutic potential of triterpenoids, isothiocyanates, and curcumin in cancer prevention and in health in general.
Keywords: triterpenoids, isothiocyanates, curcumin, chemoprevention, phytochemicals
I. Introduction
Cancer is now the leading cause of death in 21 states in the US (1). Cancer is a chronic disease that could be prevented (2, 3). Cancer development can take about 10–30 years to develop, from initiation, promotion to progression (Figure 1) (4). Thus, the slow development of the disease potentially allows the intervention in the progression of cancer into advanced stages and metastases (Figure 2). Recent evidence suggests epigenetic alterations precede genetic mutations during cancer development. As such, naturally occurring phytochemicals have been shown to have the ability to activate the anti-oxidative stress, Nrf-2 mediated pathway, antiinflammatory networks as well as others (5, 6), resulting in blocking cancer initiation, promotion and/or progression, in many in vitro and in vivo models (5). These pathways may be directly or indirectly regulated through epigenetic modulation by natural dietary phytochemicals. Among these, some of the most promising chemopreventive agents include ursolic acid (UA), sulforaphane (SFN), phenethylisothiocyanate (PEITC), and curcumin. The dose by which these phytochemicals produce their chemopreventive effects will be explored in this review.
Figure 1.
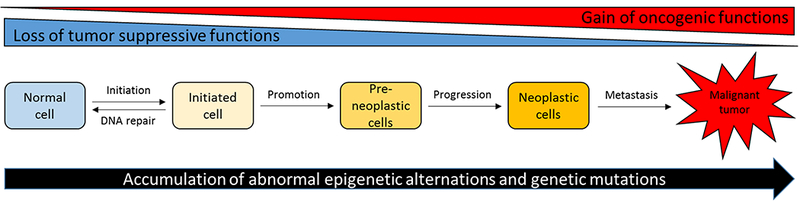
Development of carcinogenesis with aberrant epigenetics and genetics changes.
Figure 2.
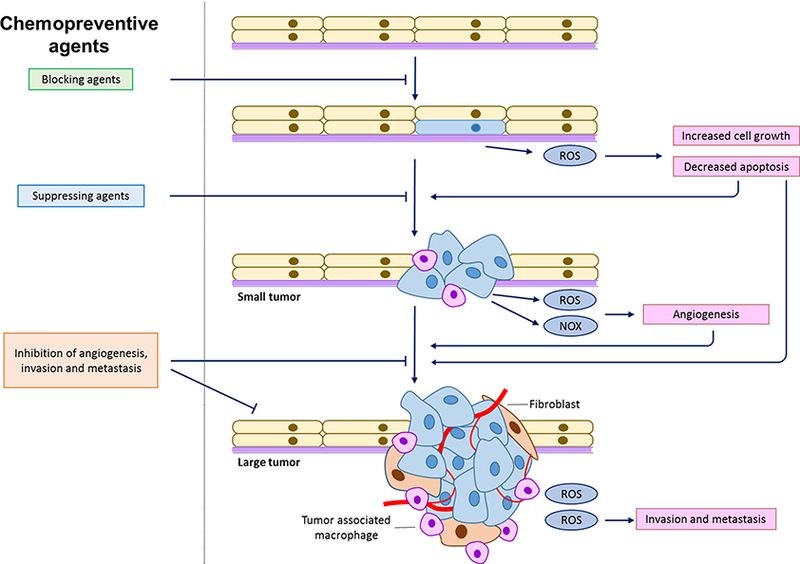
Illustration of chemopreventive agents on different carcinogenesis stages.
II. The importance of dose in the in vitro and in vivo chemopreventive effects of phytochemicals
A. Triterpenoids
Triterpenoids are a natural class of compounds produced through the cyclization of squalene widely used in Asian medicine. Approximately 20,000 sources of triterpenoids exist in nature (7). This class of compounds has been shown to have anti-inflammatory and anti-cancer properties. Specifically, the triterpenoids, oleanolic acid (OA, 3B-hydroxyolean-12-en-28oic acid) (8) and ursolic acid (UA, 3B-hydroxy-urs-12-en-28-oic-acid), an isomer of OA, have shown great promise in these areas. The anti-cancer, anti-inflammatory, and chemopreventive effects of these compounds vary with dose and will be explored in this review.
UA is found in blackberries, blueberries, holy basil, thyme, lavender, catnip, peppermint leaves, olive oil, rosemary and apple peels (7)). It has been shown to modulate a number of pathways implicated in the progression and the survival of cancer. The same can be said of OA, a triterpenoid found in ginseng root and the olive plant, bearberries, heather, three leaved caper, reishi, Chinese elder, and Sodom’s apple (9). UA and OA are often found in combination and share many of their pharmacological properties (10). In addition to its anti-inflammatory and anti-cancer effects, OA possesses a wide range of pharmacological activities such as anti-viral, anti-microbial, anti-parasitic, anti-diabetic, and anti-analgesic (9) effects.
A multitude of experiments have shown UA is able inhibit proliferation and induce apoptosis of a variety of cell lines and in various animal models. Triterpenoids have been shown to exert their anti-inflammatory properties through the modulation of ROS and the attenuation of iNOS, COX-2 and NF-κB, a key factor in controlling transcription of DNA, cytokine production and cell survival. In the T lymphoma Hut-78 cells, UA inhibited proliferation and induced early apoptosis at 10–80 μM, with the highest effect achieved at 80 μM. This was noted in the downregulation of NF-κB p65, and p50 proteins and the upregulation of caspase-8, caspase-3, and caspase-9. In addition, COX-2 mRNA also decreased in the presence of UA (11). UA and OA have been shown to inhibit the proliferation of non-small cell lung cancer A549 cells in a nude mouse model at low and high doses of 50 and 100 mg/kg bw, respectively. At 100 mg/kg bw UA significantly inhibited the growth of the cells noted in tumor weight. Further investigations, noted UA and OA increased expression of Bid and decreased the protein levels of MMP-2, Ki-67, and CD34 (12). Additionally, UA has been shown to inhibit proliferation and induce apoptosis in MTC-SK cells, a medullary thyroid carcinoma cell line) at 10 μM and 20 μM in vitro (13). Additionally, UA and OA have been shown to induce apoptosis through a multitude of pathways in prostate cancer. These include the activation of JNK and inhibition of Akt pathways in PC-3 cells at 80 μM (14), and the down regulation of Bcl-2 in PC-3 and LNCaP prostate cancer cells at 55 μM and 45 μM respectively (15).
The anti-inflammatory capabilities of UA have been shown to be expansive in a number of experimental models. UA has been shown to inhibit tumor promotion by TPA in a two-stage skin carcinogenesis ICR mouse model at 2 μmol applied topically prior to application of TPA. UA reduced TPA-induced inflammation and decreased the gene expression of IL-1, IL-22, and Cox-2 inflammatory genes. In addition, UA reduced binding of NF-Kβ, Egr-1, and AP-1 (12). Furthermore UA has recently been shown to inhibit cell growth and proliferation of pancreatic cell lines AsPC-1, MIA, PaCa-2, and Panc-28 cells at 5–20 μM in vitro (16). UA suppressed NF-Kβ activation and was able to suppress its target genes in Panc-28 cells in vitro. UA’s anti-cancer effects were further confirmed in an orthotopically implanted pancreatic cancer model in which UA inhibited pancreatic cancer at a dose of 250 mg/kg bw given orally daily. Moreover, UA and OA have been shown to prevent ROS-induced hepatocellular carcinoma in vivo in a male Wistar rat model at an oral dosage of 20 mg/kg bw (17) and skin cancer through the attenuation of chemically induced ROS and protect against DNA damage induced hydrogen peroxide at concentrations 5 and 10 μM in murine keratinocyte Ca3/7 cells (18). Furthermore, UA has been shown to induce anti-inflammatory activity at 5 μM through the suppression of NF-Kβ in activated T cells, B cells and macrophages (19). UA’s anti-inflammatory role was further solidified in a study demonstrating UA was able to reduce NF-KB activation and the release of cytokines at 10 μM and 50 μM in human colon cancer COLO 205 cells (20). The study extended their findings in a (DSS)-induced acute murine colitis treated model. When induced and treated with either UA 10 mg/kg or 20 mg/kg disease activity decreased (20).
UA has demonstrated anti-oxidative activity through the modulation of several pathways. When colorectal cancer Caco-2 cells were treated with UA it resulted in the normalization of antioxidant levels and protection against oxidative damage at 5 μM and 10 μM and (21). Furthermore, UA and OA have attenuated H2O2 and in neuroblastic PC12 cells at 20 μM and 40 μM (22).
A recent study demonstrates UA’s role in epigenetic modulation in which UA increased phosphorylation of SAPK/JNK pathway in human non-small cell lung cancer H1299 and A549 cells in vitro at a concentration of 30 μM. Further investigations demonstrated UA was able to decrease the expression of SP1 and in turn regulate DNMT1 and EZH2 expression in H1299 and A549 cells (23). UA has also been reported to increase the acetylation of histone H3 and inhibit HDAC activity in vitro (24).
UA and OA have been shown to promote the differentiation of glioma, melanoma, and thyroid cancer cell lines, A375, U87, and ARO cell lines respectively, through the inhibition of endogenous reverse transcriptase (RT) at 10, 15, and 20 μM (25). Furthermore, UA has been shown to induce the differentiation of HL60, U-937, and THP-1 leukemic cells at 10, 20 and 30 μM via the activation of the ERK1/2 MAPK pathway (26). UA inhibits proliferation and induces apoptosis of ovarian epithelial cancer SKOV sphere cells at 12.5–50 μg/mL. In addition, UA downregulates the expression of EMT markers including Snail, Slug, Twist, vimentin, N-cadherin and fibronectin. These effects translated in vivo in a SKOV3 sphere cell xenograft athymic nude BALB/c-nu mouse model at 60 mg/kg bw (27).
Triterpenoids demonstrate their anti-cancer activities at a concentration range of 5 μM to 80 μM in vitro and 10–250 mg/kg in vivo. The variations in concentrations can be attributed to pharmacological effects related to cell line, assay system, animal model and source of compounds. While there are less than a handful of clinical studies evaluating UA in humans, similar doses to those evaluated in in vivo models have shown efficacy in human. A clinical study evaluating the effect of 150 mg of UA given orally once a day for 12 weeks on metabolic syndrome, insulin sensitivity, and inflammation lead to a transient remission in 50% of patients (28). Another study evaluating UA at 50.94 mg for use in sarcopenia demonstrated a significant increase in the right-handgrip of female subjects in comparison to the control group (29). Overall, triterpenoids hold great promise in the area of chemoprevention and as such are being synthetically modified in order to increase potency in vivo and will be the topic of discussion in a future review article.
B. Isothiocyanates (ITCs)
Numerous epidemiological and pharmacological studies suggest a correlation between the consumption of cruciferous vegetables and a reduced cancer risk in humans (30, 31). Over 200 naturally-occurring glucosinolates are found in cruciferous vegetables (32), which consist of a β-D-thioglucose group, a sulfonated oxime group, and a side chain derived from methionine, phenylalanie, tryptophane, or branch-chained amino acids (33). Interestingly, the chemopreventive effects are mostly attributed to the isothiocyanate (ITC)-containing compounds rather than their glucosinolate precursors. ITCs, converted by myrosinase mediated hydrolysis from glucosinolate, are characterized by the sulfur containing N=C=S functional group with a wide structural diversity. Ally isothiocyanate (AITC) from cabbage, mustard, and horseradish; benzyl isothiocyanate (BITC) and phenethyl isothiocyanate (PEITC) from watercress and garden cress; and sulforaphane (SFN) from broccoli, cauliflower, and brassicas have been mostly studied against a variety of human malignancies (34). In cell culture models, micromolar concentrations of ITCs have shown potent anti-cancer effects through different mechanisms in vitro (35, 36). Several pharmacokinetic studies have provided evidence that the concentration range is achievable in vivo. For example, in a pharmacokinetics study of PEITC in rats, it demonstrated that plasma concentration of PEITC could reach 9.2 and 42.1 μM after an oral dose of 10 and 100 μmol/kg body weight in rats (37). Interestingly, it was also found that PEITC was highly bound to serum protein bound in the rats with the protein binding ratio around 98.1% and was not concentration-dependent. The high plasma concentration was due to the high oral bioavailability, which was 115 and 93% at doses of 10 and 100 μmol/kg (37). Compared to PEITC, SFN is relative less associated with protein binding and the binding ratio did not increase with time (38). In an in vitro study, the initial protein binding by PEITC was almost 3-fold higher than that of SFN. Four hours after incubation, cellular protein binding of PEITC became 6-fold higher than that of SFN (38). In an in vitro setting, PEITC also modified bovine serum albumin (BSA) covalently to a greater extent than SFN occurring exclusively at cysteine residue (38). Oral administration of 50 μmol SFN in rats resulted in a peak plasma concentration of 20 μM at 4 h (39). In a chemopreventive study using the ApcMin/+ mouse model, SFN inhibited adenoma formation with a steady-state concentration of 3–13 nmol/g (roughly equivalent to 3–10 μM) in the gastrointestinal tract (40).
In clinical studies, there are several reports showing that the ITCs could potentially impact in the prevention of cancer. After receiving 1 week of PEITC treatment (10 mg in 1 mL of olive oil, 4 times per day), tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) metabolic activation ratio was reduced by 7.7% in a clinical trial containing 82 smokers, which suggests that PEITC could be a potent inhibitor of lung carcinogenesis in smokers (41). In a double-blinded, randomized, placebo-controlled clinical trial, 78 senior patients with increasing PSA levels after radical prostatectomy were given 60 mg sulforaphane 3 times daily for 6 months, a much lower plasma PSA level were found in the sulforaphane treated group, which potentially suggests a promising treatment in recurrence of prostate cancer after prostatectomy (42). A randomized controlled clinical study consisting of 54 women subjects revealed a mean 81.7 g/d intake of cruciferous vegetable, enriching of SFN, for over 4 years (August 2009 to December 2013) was associated with a lower level of Ki-67, a cellular marker for proliferation, in breast ductal carcinoma in situ tissue, which strengthen the correlation of cruciferous vegetable consumption and lowering breast cancer risk (43).
Next, we will discuss the potential molecular targets for ITCs mediated anti-cancer activity. The chemopreventive effect of ITCs is considered to be associated with their ability to induce the expression of phase II drug metabolism/detoxifying enzymes. It has been extensively documented that SFN exerts potent activation of phase II/antioxidative gene expression in both in vitro and in vivo studies (44). In rats, 40 μmol/kg/day SFN treatments were found to increase GST and NQO1 activities in the duodenum, forestomach, and bladder tissues (45). In hepatocytes, SFN induced UGT1A1 and GSTA1 mRNA expression and protected cells against the 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP, commonly found in cooked meat and considered as risk factors for cancer)-DNA adduct formation (46). Similarly, PEITC was found to induce hepatic phase II enzymes, resulting in decreased PhIP-DNA adduct levels in rat tissues (47). In mice, 12 h after an oral dose of PEITC, upregulation of several GST isozymes in the liver were identified using a microarray approach (48). Markedly, a number of studies on ITCs suggest that the induction of phase II/antioxidant enzymes is NF-E2-related factor-2 (Nrf2) dependent (48–50).
Mechanistic studies demonstrated that ITCs activate the Nrf2 pathway by modifying Nrf2-Keap1 interactions. Using a liquid-tandem mass spectrometry approach, Hong et al. provided evidence that SFN can directly react with the thiol groups of Keap1. The formation of SFN-Keap1 thionoacyl adducts releases Nrf2 from the Nrf2-Keap1-Cul3 degradation complex; this stabilization of cellular Nrf2 consequently results in Nrf2 nuclear translocation and activation (51). On the other hand, PEITC may induce the Nrf2/ARE signal through a different mechanism potentially mediated by mitogen-activated protein kinases (MAPKs). It has been reported that PEITC induces ARE activity through the attenuation of c-Jun N-terminal kinase-1 (JNK1) and extracellular signal-regulated kinase (ERK) inhibitors (50). In the same study, in vitro kinase assays showed that JNK1 and ERK2 directly phosphorylate Nrf2 protein. Collectively, PEITC increased the phosphorylation of ERK1/2 and JNK1/2 in cells, which, in turn, caused phosphorylation of Nrf2 and subsequent release from Keap1 binding, and resulted in translocation activation of the Nrf2/ARE pathway. To note, Nrf2-deficient mice have shown increased susceptibility in carcinogenesis models and less effective towards preventive treatment (52–54). Therefore, transcriptional induction of Nrf2/ARE mediated phase II enzymes would be considered as an important mechanism for the chemopreventive effects of ITCs.
Inactivation of the NF-κB pathway by ITCs is another important mechanism that can contribute to their anti-cancer activities. Experimental evidence suggests that ITCs stabilize IκB by inhibiting its phosphorylation and degradation, resulting in a reduction in nuclear translocation of p65 (a subunit of NF-κB) and NF-κB activation. In PC-3 cells, both SFN (20 and 30 μM) and PEITC (5 and 7.5 μM) strongly inhibited nuclear translocation of p65, with the concomitant decreased expression of NF-κB regulated genes such as Bcl-XL, cyclin D1, and vascular endothelial growth factor (VEGF) (55). Correspondingly, PEITC and SFN were found to inhibit lipopolysaccharide (LPS)-induced NFκB luciferase activity in human colorectal cancer HT-29 cells, which was also mediated through the inhibition of IκKβ phosphorylation (56). In addition, SFN was proposed to interact with glutathione and other redox regulators like Ref-1 and thioredoxin, which in turn indirectly impairs the NFκB-DNA binding ability (57). Another study by Heiss et al suggested SFN directly interacts with Cys residues of NFκB subunits by forming dithiocarbamate, which results in decreased DNA binding abilities (58). Collectively, these findings indicate that redox modulation and thiol reactivity play certain roles in regulating NFκB-dependent transcription by SFN. Interestingly, studies on the crosstalk between Nrf2 and NFκB signaling have shown that Nrf2 downstream targets may inhibit of NFκB nuclear translocation (59, 60). Accordingly, pre-treatment of SFN (25 mg/kg per day) mitigated dextran sodium sulphate (DSS)-induced acute colitis in vivo, while increased expression of Nrf2-dependent genes and reduced expression of inflammatory were observed in colon tissues (61). Similarly, SFN restored the number of sunburn cells to basal levels in Nrf2 WT but not Nrf2 knockout (KO) mice after UV irradiation. The inflammatory markers were lower in SFN treated Nrf2 WT tissues compared to Nrf2 KO tissues (62). These results suggest activation of Nrf2 by SFN can, in part, contribute to the suppression of proinflammatory signaling pathways.
Given that epigenetics lies on the molecular interface between genetics and environmental factors, there is a growing interest in evaluating the potential of dietary phytochemicals to block or reverse epigenetic abnormalities in cancer development. In a recent study, Wong et al. reported the effects of SFN on promoter DNA methylation profiles in prostate epithelial cells (PrEC), androgen-dependent (LNCaP) and androgen-independent (PC-3) prostate cancer cells (63). SFN treatment was found to decrease the DNMT levels in all the tested cell lines. Although SFN showed complex effects on genome-wide DNA methylation patterns among normal prostate epithelial and prostate cancer cells, the genes of altered methylation status were functionally similar within a single cell line (e.g. cell migration, cell adhesion etc.). In various in vitro and in vivo studies, SFN or PEITC treatments appeared to down-regulate DNMT activity, thereby resulting in promoter demethylation of epigenetically silenced genes, with the concomitant change of gene expressions (reviewed by (64, 65)). Interestingly, DNA demethylation in a promoter region is often found to be associated with local relaxing of histone structure, although the precise mechanism remains to be elucidated. For example, in mouse prostate cancer TRAMP-C1 cells, SFN (1.0 and 2.5 μM) restored the epigenetically suppressed Nrf2 levels by reversing the hypermethylation status of the Nrf2 promoter region via inhibition of DNMT activities, as well as HDACs (66). In mouse epidermal JB6 P+ cells, this change in methylation pattern by SFN is associated with increased Nrf2 level and a phenotype more resistant to 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced neoplastic transformation (67). On the other hand, HDACs are often upregulated in cancers therefore HDAC inhibition is considered as an important strategy in cancer prevention and therapy. Molecular docking experiments have shown the metabolite of SFN and several structural related ITCs directly interact with the HDAC catalytic core to inhibit the enzyme activity (68). In a clinical study, a single dose of 68 g of broccoli sprouts (containing ~105 mg of SFN) inhibited HDAC activity significantly in peripheral blood mononuclear cells (PBMC) 3 and 6 hours post consumption (69). Incubation of BPH-1, LNCaP, and PC3 prostate cancer cells with 15 μM SFN significantly reduces HDAC expression by 30–40%, which is accompanied by a 50–100% increase in the acetylation of histones, as well as G2/M arrest of cell development and induction of apoptosis in a caspase-dependent manner (70).
Epigenetic upregulation of p21 gene expression by PEITC was found to be associated with chromatin remodeling, which compromises dynamic changes in both histone acetylation and methylation (71). To note, PEITC also exhibits the dual functions of CpG demethylation, HDAC inhibition and epigenetic regulation of various genes (72, 73). Last but not least, the anticancer effects of ITCs may be partially attributed to their ability to regulated miRNA. Using an oligonucleotide approach, we identified top altered miRNAs upon 2.5 μM PEITC treatment in prostate cancer cells. Among them, miR-194 was a primary target of PEITC which was able to suppress cell invasion (74).
ITCs have been shown to exert cytoprotection via the activation of Phase II enzymes within the Nrf2 pathway. Rat aortic smooth muscle cells treated with SFN at 0.25–5 μM, resulted in the increase of phase 2 antioxidant enzymes in a concentration-dependent manner. Furthermore, when pre-treated with SFN (0.5, 1, and 5 μM), the cells were protected from oxidative and electrophilic cytotoxicity induced by xanthine oxidase (75). Incubation with SFN, BITC, and PEITC (0–10 μM) protected against oxLDL-induced endothelial damage in a dose-dependent manner through the induction of Nrf2’s target gene HO-1. In addition, the expression of NF-κB, ICAM-1, VCAM-1, and E-selectin were decreased (76). Human peripheral blood mononuclear cells (PBMC) treated with PEITC (1–10 μM) for 24 h, increased the detoxification enzymes GPX1 (3.7-fold increase by 1 μM PEITC treatment) and SOD2 (7.3-fold increase by 10 μM PEITC treatment) (77).
Furthermore, SFN inhibited breast CSCs at concentrations 1–5 μM in vitro which is much lower than the concentration needed to induce apoptosis (78). In an in vivo xenograft model, where 5-week-old female NOD/SCID mice with a xenograft of SUM159 cells received daily injections of 50 mg/kg SFN for 2 weeks, breast CSCs were found to be inhibited mainly due to the down-regulation of Wnt/β-catenin self-renewal pathway (79). SFN is also effective in the treatment of leukemia by enhancing the differentiation of leukemic cells. When human promyelocytic leukemia cells were treated with 0.2–100 μM SFN, SFN induced differentiation in the leukemic cells to granulocytic and macrophagic lineages. This process was mediated mainly through PKC (80).
GATA-3 is a marker for luminal progenitor cell differentiation and can actively promote the differentiation of cancer cells (81, 82). When PyMT transgenic mice were treated with PEITC (8 mmol/kg bw), the progression of tumor size was delayed and there were smaller tumors compared to the control. These findings were accompanied by a low expression of ERα, FOXA1 and GATA-3 (83). PEITC also inhibited CSC growth in vitro (84). PEITC transformed LNCaP floating spheres into prostate cancer stem cells (PCSC) due to the enhancement of H3K4 acetylation, the inhibition of DNMT1 and activation of GSTP1. After androgen deprivation, the PCSCs differentiate into neuroendocrine cells with decreased proliferation, expression of the androgen receptor, and PSA (85)
Extensive studies have shown ITCs are able to inhibit the growth of cancer cells via arresting the cell cycle through the regulation of cell cycle proteins, cyclin-dependent kinase activity, tubulin polymerization and histone acetylation, and the induction of apoptosis (86). PEITC induces extrinsic apoptosis pathway through stimulating death receptors and Fas (87, 88) and the intrinsic apoptosis pathway by regulating BCL2, BID and BAX (89–91). In addition, PEITC can induce G0/G1 arrest via p53 and G2/M cell arrest in a p53 independent manner (90, 92).
PEITC (0 to 10 μM) significantly inhibited human laryngeal carcinoma Hep-2 cell growth and enhanced apoptosis with G2/M cell cycle arrest in a dose- and time-dependent manner while no effect was observed in the growth of normal human bronchial epithelial cells (93). Treating human non-small cell lung cancer L9981 cells with BITC (7.5 and 10 μM) and PEITC (12.5 and 20 μM) resulted in apoptosis through the stimulation of caspase-3 and cell cycle arrest at the G2/M phase via cyclin B1 regulation (94). BITC and PEITC also inhibited the growth of lung cancer L9981 cells with IC50 5.0 and 9.7 μM respectively by suppressing Akt and NF-KB, enhancing ROS production, and reducing GSH (95).
SFN can inhibit the growth of cancer cells by causing cell cycle arrest and apoptosis induction. SFN can stimulate the intrinsic apoptosis pathway by activating BCL-2 and suppressing inhibitors of apoptotic proteins (IAPs) (96). Treating DU145 and PC-3 prostate cancer cells with SFN (10, 20, and 40 μM) enhanced cytochrome c levels by producing more ROS leading to apoptosis (97). Additionally, in human bladder cancer T24 cells, SFN arrested the cell cycle in G0/G1 phase via the p27 pathway (98). It also induced G2/M cell cycle arrest by stimulating p21 pathway and suppressing Cdc2/Cyclin B1 (99, 100).
SFN can inhibit cell growth and induce apoptosis in a dose dependent manner. Incubation of A549 cells treated with 30 μM SFN induced G2/M arrest via p21 pathway (101). Upon treating Caco-2 cells with various concentrations of SFN, 25 μM SFN had the greatest effect on enhancing UGT1A expression via Nrf2 pathway, while 75 μM SFN induced G1/G2 arrest and apoptosis via decreasing bcl-2 level and enhancing bax (102). When treating colorectal cancer (CRC), higher concentrations of SFN (12.5 and 25 μM) produced apoptosis through decreasing caspase-3 and increasing caspase-2, −3, −8, and −9. The low dose SFN generated a mitotic delay (103). SFN inhibited the growth and induced apoptosis in a dose- and time-dependent manner in MDA-MB-231 human breast cancer cells, whereby 30 μM SFN induced apoptosis by increasing caspase-3 and reducing BCL-2. Furthermore, it induced S and G2/M cell-cycle arrest by upregulating p21WAF1 and p27KIP1 expression and down-regulating cyclin A, cyclin B1 and CDC2 levels (104).
In summary, for ITCs, in the context of in vitro cell line dose response, it appears it is dependent on the cell line, biomarker measured, and the chemical structure of the ITC, among others. Nevertheless, there is a dose-dependency of dose response. For instance, in human hepatoma cell line HepG2-C8 expressing the ARE-luciferase reporter, SFN increases ARE activity at concentrations up to 35 μM (105). Beyond 35 μM, ARE activity decreases due to cellular toxicity. This higher dose-dependent cellular toxicity could be blocked by adding exogenous glutathione (GSH). Interestingly, at lower doses of SFN, GSH attenuated ARE activity, however, at higher dose level, GSH enhances ARE activity, due to blockade of caspase 3 activation and apoptosis. These dose-dependency effects of SFN are quite similar to phenolic antioxidants butylated hydroxyanisole (BHA) and its metabolite tert-butylhydroquinone (tBHQ) (106, 107), although SFN in general is more potent by about one order of magnitude. From the above discussion, in our experience, it appears that SFN would activate epigenetic events in low micromolar concentrations, then it would activate Nrf2 signaling in low tenths micromolar and activation of caspases/apoptosis around fifty-one hundred micromolar concentrations. We have also reviewed this dose-dependency effects previously (108).
C. Curcumin
Polyphenols are a group of compounds that have at least one aromatic ring with one or more hydroxyl functional groups attached (109). Natural polyphenols, which are widely present in foods and beverages from plant origin (110), are another category of phytochemicals that have been extensively studied for their health beneficial effects in many diseases, including cancer. It is well accepted that their potent antioxidant and anti-inflammatory activities largely contribute to their anticancer efficacy. In addition, experimental evidence suggests dietary polyphenols are able to modulate molecular targets and signaling pathways regulating detoxification enzymes, cell survival, proliferation, differentiation, migration, and angiogenesis. (111).
While flavonoids and phenolic acids account for over 90% of all the natural polyphenols, curcumin, the bright yellow colored polyphenol rich in rhizomes of Curcuma longa (turmeric) has a distinct chemical structure. Curcumin is considered a highly promising chemopreventive agent since it fulfills several ideal characteristics such as low toxicity, affordability, and easy accessibility. Numerous studies using cell lines and animal models have demonstrated curcumin is effective in inhibiting tumor growth, which warranted clinical trials to test its safety and efficacy. However, phase I/II clinical trials showed poor bioavailability of curcumin in humans. Oral administration at doses up to 8 g resulted in undetectable levels of curcumin in blood (112). It does not seem practically possible to reach the in vitro effective dose of curcumin in humans. Efforts have been made to circumvent the bioavailability challenge by chemical structure modifications (curcumin analogs) and diverse delivery systems (liposome, nanoparticles, and conjugates). However, 17 out of 49 curcumin double-blinded placebo-controlled clinical trial showed efficacy. Another 27 clinical trials of curcumin pointed to the therapeutic benefits (DOI: 10.1038/543040c). Curcumin doses ranged from 180 mg/day to 3,000 mg/day have been used in human. Under the treatment with standard chemotherapy protocols, the bioavailable curcuminoid preparation (180 mg/day) for a period of 8weeks as adjuvant therapy in cancer patients with solid tumors can significantly improve quality of life and suppress systemic inflammation (113). In addition, in curcumin (total 3 g/day) with external-beam radiation therapy of up to 74 Gy patients with prostate cancer group, plasma total antioxidant capacity significantly increased and the activity of superoxide dismutase decreased compared with those at baseline (114). The clinical results still support the use of curcumin as an effective cancer preventive agent, particularly, in several colorectal cancer trials (115, 116). The interactions between curcumin and the host body system are expected to be more complicated. The following sections will focus on the relationship between the exposure and the response of curcumin.
An important molecular switch through which curcumin may mediate its health benefits is the transcription factor nuclear factor 2-related factor (Nrf-2). Curcumin has been shown to induce reactive oxygen species (ROS) scavenging enzymes. ROS is a bi-functional cellular molecule in cancer cells. It can drive DNA mutations in carcinogenesis, and it can trigger mitochondrial apoptosis. In a study where astrocytes were treated with 5–15 μM curcumin expression of NADPH: quinone reductase and glutathioneS-transferase (GST), members of phase II detoxification enzymes, increased significantly. Moreover, HO-1 mRNA and protein expression were elevated after a 6 h incubation with 5–25 μM curcumin. However, higher concentrations of curcumin (50–100 μM) caused a substantial cytotoxic effect with no change in HO-1 protein expression (117). And in renal epithelial cells, curcumin stimulated the expression of Nrf-2 in a dose- and time-dependent manner (118). Conversely, curcumin is able to generate cellular ROS to drive mitochondrial apoptosis to treat malignancies (119). Despite paradoxical roles in regulating cellular ROS, the overall anticancer effect of curcumin has been clearly shown in a number of studies.
Curcumin is a traditional remedy for inflammatory diseases (120). The anti-inflammatory effects of curcumin have been postulated on the basis of a number of in vitro and in vivo studies(121, 122). Curcumin dose-dependently increased the number of pre-apoptotic and apoptotic cells in phorbol myristate acetate (123) stimulated human neutrophilic granulocytes (124). The application of curcumin significantly inhibited the activity of neutrophilic granulocytes in a rat model of arthritis (an inflammatory arthropathy), which confirmed the antiinflammatory properties of curcumin in vivo. Moreover, a curcumin injection given to mice prior to an intraperitoneal LPS administration led to an inhibition of LPS-induced increased MCP-1 (monocyte chemoattractant protein 1) mRNA levels (125). LPS-induced mRNA and protein levels of MCP-1 and interleukin-8 (IL-8) were reduced by curcumin treatment in human renal epithelial cells HK-2. Furthermore, curcumin prevented LPS-induced NF-kB DNA binding (125).
The cytoprotective effect of curcumin has been well studied. 20 μM curcumin has been reported to protect human proximal tubule HK-2 cells from apoptosis and necrosis induced by Shiga toxin (126). Interestingly, the protective effect of curcumin against stx1 and stx2-induced injury on HK-2 cells is not related to its anti-oxidative properties. Curcumin can attenuate palmitate-induced apoptosis in MIN6 pancreatic β-cells through PI3K/Akt/FoxO1 and mitochondrial survival pathways (127). In this study, 10 μM curcumin improved cell viability and enhanced glucose-induced insulin secretory function. Curcumin treatment neutralizes ROS generated by palmitate induction. The epithelial-to-mesenchymal transition (EMT) of mature tubular epithelial cells in kidney is considered to contribute to the renal accumulation of matrix proteins associated with diabetic nephropathy. Studies suggest 20 μM of curcumin protects renal tubular epithelial cells from high glucose-induced EMT through Nrf2-mediated upregulation of HO-1 (128). Alinejad et al. demonstrated a combination of safranal, thymoquinone and 50 μg/mL of curcumin can block glucose/serum deprivation (GSD)-induced cell death and has the potential to be used for management of cerebral ischemia and neurodegenerative diseases (129). Theracurcumin is a highly bioavailable curcumin analog. It has been found that 10 μM of both theracurcumin and curcumin may have potential protective effects against sodium nitroprusside-induced cytotoxicity by free radical-scavenging and iron-chelating activities (130). Curcumin modulates peroxisome proliferator-activated receptor-γ signaling, which is a key molecule in the etiology of bronchopulmonary dysplasia (BSD). In vivo studies showed curcumin, when given daily at 5 mg/kg bw intraperitoneally, effectively protected against short-term and long-term hyperoxia-induced lung injury. Curcumin prevented hyperoxia-induced increases in cleaved caspase-3 and the phosphorylation of Erk1/2. Molecular effects of curcumin, both structural and cytoprotective, suggest that its actions against hyperoxia-induced lung injury are mediated via Erk1/2 activation and that it is a potential intervention against bronchopulmonary dysplasia (BPD) (131).
Curcumin has been found to alter the differentiation of many different cells. In vitro studies have shown that 0.5 μM curcumin increases the differentiation rate of neurons in neural stem cells via Wnt signaling pathway (132). It’s also reported curcumin can enhance EB directed differentiation of H-9 human embryonic stem cells (hESCs). 10 μM of curcumin significantly increased gene expression of cardiac specific transcription factor NKx2.5, cardiac troponin I, myosin heavy chain, and endothelial nitric oxide synthase during ES cell differentiation through modulation of the nitric oxide-cyclic GMP pathway (133). Myeloid-derived suppressor cells (MDSC) accumulate in the spleen and contribute to tumor growth, angiogenesis, and progression. Curcumin treatment inhibited cell proliferation and colony formation of cancer cells and decreased the secretion of murine IL-6 by MDSCs in a co-culture system. In addition, polarized MDSCs toward a M1-like phenotype with an increased expression of CCR7 and decreased expression of dectin 1 (134). Also, 20 μM curcumin inhibited differentiation of adipocytes and cardiac fibroblasts. Adipocyte differentiation is a key process in determining the number of mature adipocytes in the development of obesity. Curcumin has been reported to have an anti-adipogenic function both in 3T3-L1 murine cells and in human primary preadipocytes (135). The differentiation of cardiac fibroblasts (CFs) into myofibroblasts and the subsequent deposition of the extracellular matrix is associated with myocardial fibrosis following various types of myocardial injury. Treatment with 20 μM curcumin effectively suppressed TGF-β1-induced CF differentiation via Smad-2 and p38 signaling pathways. These findings suggest curcumin may be a potential therapeutic agent for the treatment of cardiac fibrosis (136, 137).
Studies in our laboratory suggest that curcumin increases activity of activator protein (AP-1)-luciferase in a concentration-dependent manner at 1–25 μM in HT-29 cells transfected with an AP-1- luciferase reporter gene. The protein expression of endogenous cyclin D1, a gene that is in downstream of AP-1, increased with 10 μM curcumin treatment (138). Additionally, we found 10 and 50 μM curcumin inhibited LPS-induced NF-κ B-luciferase activity in HT-29 cells stably transfected with a NF-κ B-luciferase construct (126). We found 2.5 and 5 μM curcumin inhibited colony formation of HT-29 cells, whereas, inhibition of colony formation failed in stable knockdown of deleted in lung and esophageal cancer 1 (DLEC1) cells. Furthermore, we observed 5 μM curcumin up-regulated the mRNA expression of DLEC1 and decreased CpG methylation of the DLEC1 promoter in HT-29 cells. We further discovered 5 μM curcumin down-regulated protein expression of DNA methyltransferases and subtypes of histone deacetylases, such as HDAC4, 5, 6 and 8 (139).
Furthermore, treatment with 50 μM curcumin induced apoptosis in colon, leukemia, breast, hepatocellular and ovarian carcinoma cell lines. However, curcumin failed to display cytotoxicity in cell lines established from lung, kidney, cervix, prostate and CNS malignancies. The mechanism of curcumin-mediated apoptosis was determined to be related to the generation of ROS.The addition of N-acetyl cysteine (109), a ROS scavenger, during curcumin treatment resulted in the disappearance of apoptosis. Additionally, curcumin’s failure to exhibit cell death in some cell lines is due to the overexpression of Hsp70 in the cells which protect cells from apoptosis (140). Several studies investigated the relationship of ROS level-effect and apoptosis-induction of curcumin. Different dosage effects of curcumin on cell death types in a human osteoblast cell line were explored. Curcumin at concentrations lower than 25 μM caused apoptosis in human osteoblasts HFOb 1.19 cells, through the activation of JNK and cleavage of caspase-3, PARP and PAK2. However, 50–200 μM curcumin induced necrotic cell death instead of apoptosis in human osteoblasts. In addition, 12.5–25 μM curcumin directly increased oxidative stress demonstrated by the use of the cell permeable dye 2’, 7’-dichlorofluorescin diacetate (DCF-DA), an indicator for intracellular ROS, nevertheless, 50–200 μM curcumin had much less activity. Moreover, NAC or α-tocopherol (ROS scavengers) pretreatment significantly decreased intracellular ROS levels and 12.5–25 μM curcumin-induced apoptosis to necrosis. Pretreatment with antimycin or 2-deoxyglucose reverted apoptosis induced by 12.5–25 μM curcumin to necrosis which could induce ATP (a mediator of apoptosis versus necrotic cell death) depletion (141). Although curcumin caused cell death of HL-60 cells in a concentration- and time-dependent manner, its effects on ROS production differed with fluctuations in concentration. Curcumin at less than 25 μM decreased ROS production, while 50–100 μM enhanced ROS generation. Furthermore, the addition of antioxidant agents, ascorbic acid (ASA), NAC and glutathione (GSH), promoted the antioxidant and anti-cancer activities of curcumin at low concentrations (142). These studies were consistent with reports curcumin at low concentrations (<10M) prevents GSH depletion and higher concentrations decrease GSH levels (143). Proteasome inhibitors have been reported to cause apoptosis in cancer cells (18). Curcumin has been shown to demonstrate biphasic dose-response proteasome activity in human keratinocytes, specifically, 0.3 μM and 1.0 μM curcumin increased proteasome activity by 34% and 46%, respectively. However, curcumin at higher concentrations of 3 and 10 μM decreased proteasome activity by 32% and 46%, respectively (144). It was suggested the biphasic dose-response is through a homeostasis mechanism, in which a low dose of agents stimulates signaling pathways to protect the organism, whereas a high dose displays an inhibitory effect (145) (146). A similar phenomenon has been observed for many natural compounds, such as resveratrol (147) (22), berberine (148), clove and cinnamon essential oils (149).
In a study where dose-dependent differences on DNA-damage and the p53 response of quercetin and curcumin, whose chemical structures are similar, in HT1080 cells (a human cell line with wild-type p53), 8 μM curcumin significantly increased the expression of phosphorylated H2AX, a biomarker of DNA damage, while as much as 20 μM, quercetin could displayed similar activities. Curcumin (4 and 7 μM, respectively) increased the protein expression of p53 and p-p53 (ser15) at lower concentrations than quercetin (30 and 20 μM, respectively). It was suggested even with similar chemical structures, the two natural compounds displayed different effects on DNA-damage response patterns in terms of dose and cell fate (150)
Curcumin is recognized as an epigenetic modulator and plays a major role in the prevention of disease. Studies in our laboratory demonstrated that 10 μM curcumin prevents prostate cancer progression via CpG demethylation in the promoter region of Nrf2 in TRAMP-C1 cells (151). In HT-29 cells, curcumin inhibited anchorage-independent growth by decreasing CpG methylation of the promoter region of the tumor suppressor gene (152). After treatment with curcumin (2.5 and 5.0 μM) for 5 days, protein levels of DNMT1, DNMT3b, HDAC4, HDAC5, HDAC6, and HDAC8 decreased (139). In human prostate LNCaP cells, curcumin treatment decreased the methylation of CpG islands of Neurogl as well as the binding ability of methyl-CpG binding protein 2 (MeCP2). The expression of HDAC1, 4, 5, and 8 increased whereas the expression of HDAC3 and the total HDAC activity decreased upon 2.5μM curcumin treatment. ChIP analysis showed curcumin decreased the enrichment of H3K27Me3 in the Neurogl promoter region (153). In breast cancer cell lines MCF7 and MDA MB 231, DNMT (i.e., DNMT1, DNMT3a, and DNMT3b) transcript levels and the protein levels of DNMT1, HDAC1, and MeCP2 decreased after treatment with 10 μM curcumin (154). In addition to curcumin, the curcumin analogue FN1 and liposomal-formulated curcumin (lipocurc) have been reported to have a protective effect on the development of disease (155, 156). Our previous work demonstrates FN1 is more potent than curcumin in activating the Nrf2-ARE pathway and inducing expression of Nrf2 and its downstream detoxifying enzymes. Not surprisingly, FN1 inhibited colony formation of prostate TRAMP C1 cells by decreasing the expression of Keap1 and CpG hypomethylation of the Nrf2 promoter (155). In a Park 7 (DJ-1)-knockout rat model of Parkinson’s disease, lipocurc was found to improve the motor impairment and prevent neuronal apoptosis by targeting HDACs (156). Approximately 20–40% miRNAs are located close to CpGs and are suppressed by epigenetic mechanisms (157). Epigenetic compounds can induce upregulation of some miRNAs through reducing the percentage of CpG methylation of the promoters of miRNAs. An example is miR-203, a miRNA downregulated in bladder cancer. Restoration of miR-203 expression reduced cell viability, invasiveness and migration, and increased the number of cells in the G0-G1 phase of the cell cycle through Akt2 and Src signaling. Curcumin treatment (10 μmol/L) induced demethylation of miR-203 promoter and subsequent augmentation of miR-203 expression (158).
III. Pathways targeted in cancer chemoprevention
As discussed, triterpenoids, ITCs and curcumin exude their chemopreventive properties through a variety of signaling pathways Figure 3. These pathways are explored in greater details in this section.
Figure 3.
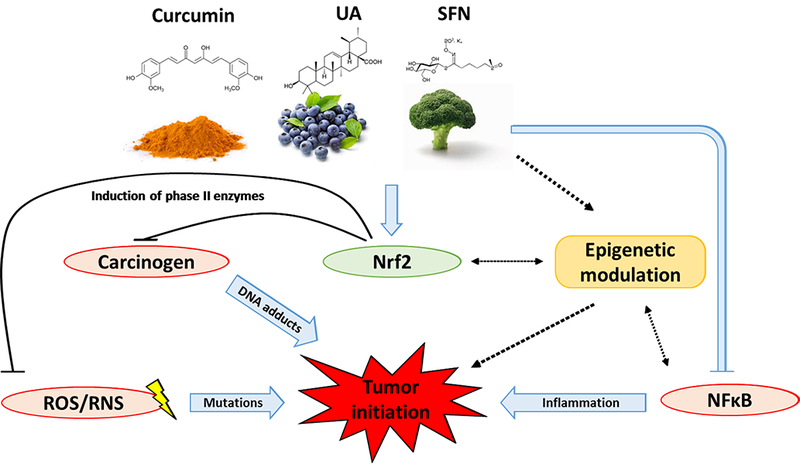
Schematic diagram on mechanisms of phytochemicals (curcumin, UA, SFN) in inhibiting tumor initiation.
A. Nrf2-mediated ARE signaling
The anti-oxidant stress defense system is responsible for the direct inactivation or conjugation of reactive oxygen species (ROS) and reactive nitrogen species (RNS). ROS/RNS into less deleterious molecules. In response to reactive species the cell has implemented an antioxidant defense system encompassing both enzymatic and nonenzymatic mechanisms (159). The enzymatic system includes superoxide dismutases (SODs), catalase, and glutathione peroxidases (GPxs). These enzymes directly inactivate ROS/RNS. In addition to the direct inactivation of ROS/RNS, there are other antioxidant enzymes that facilitate the detoxification of ROS/RNS using reduction/conjugation reactions and the recycling of thiols. The enzymatic soluble products of these reactions are easily excreted. These enzymes include phase II enzymes (e.g. NAD(P)H: quinone oxidoreductase, NQO-1; glutathione S-transferases, GST; UDP-glucuronosyl transferases, UGT; among others). These enzymes play an important role in this protective machinery as detoxifying enzymes that conjugate endogenous polar molecules to the phase I metabolites, thereby facilitating xenobiotics (including carcinogens) elimination and excretion (126). Activation of these cytoprotective enzymes is important for maintaining cellular homeostasis towards environmental challenges. Activation of the genes encoding these enzymes and proteins are regulated in large part by the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), a member of the NF-E2 family of nuclear basic leucine zipper (bZIP) transcription factors. Nuclear factor erythroid 2-related factor 2 (Nrf2), a member of the NF-E2 family of nuclear basic leucine zipper (bZIP) transcription factors, is a master regulator controls the expression of phase II/antioxidant enzymes. Under normal conditions, Nrf2 is bound to Kelch-like erythroid cell-derived protein with CNC homology (82)-associated protein 1 (Keap1). Keap1, an adaptor protein for a Cullin 3 (Cul3)-based ubiquitin E3 ligase, sequesters Nrf2 in the cytosol and ensures its degradation by the proteasome. Upon oxidative stress, Nrf2 is released by Keap1 and translocates to the nucleus where it heterodimerizes with Maf and binds the ARE/EpRE of the antioxidant defense system genes. Unsurprisingly, Nrf2 has been shown to play an essential role in the protection of carcinogenic events and Nrf2 KO mice are susceptible to the initiation, promotion, and progression of cancer (160). As the master regulator of the antioxidant response, unsurprisingly, the induction of Nrf2 has become an attractive target in chemoprevention.
B. Anti-inflammation
Oxidative stress occurs when there is an imbalance between the anti-oxidant defense system and the production of ROS/RNS (161, 162). The general terms ROS and RNS are given to the reactive species generated from the interaction of free radicals such as superoxide anion, hydroxyl radical, and nitric oxide with metals, oxidants, and reductants found in cells. ROS/RNS are important cellular messengers involved in a number of physiological processes: cellular respiration, immune response, ion transport, apoptosis, neuromodulation, and transcription (163). While important secondary messengers, the activities of ROS/RNS can exhibit a double-edged sword. In addition to endogenous production, exogenous production of ROS/RNS can be initiated through UV radiation, environmental pollutants, lipid peroxidation, and inflammatory cytokines (164). The anti-oxidant stress defense system is responsible for the direct inactivation or conjugation of ROS/RNS into less deleterious molecules. An excess of ROS/RNS can cause an imbalance in the system and induce oxidative stress; a hallmark of a number of neurodegenerative diseases and, of most importance in this review, cancer (165, 166). The excess oxidative stress results in the induction of the NFK-B signaling cascade and thus, activation of cytokines and the production of acute inflammation. Nuclear factor kappa B is a transcription factor lays on the molecular node linking inflammation, cell survival, and cancer progression signals (167). is normally sequestrated by its cellular suppressor in the cytosol. Upon activation, IκK phosphorylates IκB consequently leads to the degradation of IκB, accompanied with release and nuclear translocation of A considerable number of target pro-inflammatory genes have been shown to be involved in cancer development, including various cytokines, chemokines, cyclooxygenase-2 (COX-2), and inducible nitric oxide synthase (iNOS) among others. The inability of the cells to eliminate the exogenous culprits results in chronic inflammation. The immune cells involved in the inflammation cascade continuously use ROS/RNS as a means to recruit more immune cells. The excess production can lead to the mutagenesis of oncogenes and tumor suppressing genes and, ultimately, the initiation of cancer.
C. Epigenetic modulation
The term epigenetics is defined as heritable changes in gene expression without changes in the integrity of the DNA sequence (168). Recently, numerous evidences have shown that initiation and progression of carcinogenesis involves aberrant epigenetic alterations. Unlike genetic mutations, changes on the epigenetic level are considered reversible. Given that epigenetics lies on the molecular interface between genetics and environmental factors, there is a growing interest in evaluating the potential of dietary phytochemicals that blocks or reverses the epigenetic abnormity in cancer development. Epigenetic alterations encompass DNA methylation, histone modifications, and microRNA (mRNA) expression changes. Epigenetic alterations such as DNA methylation and histone modifications have been shown to contribute to the development and progression of cancer (169). DNA methylation is the mostly characterized epigenetic event in many cancers (138), which occurs at the 5’ position of the cytosine residues within CG dinucleotides through addition of a methyl group by DNA methyltransferases (DNMTs). CpG dinucleotides tend to be grouped in regions known as CpG islands in the promoters of genes. In normal cells, the majority of CpG islands remain unmethylated leaving an open structure for the transcriptional machinery to bind and induce expression. In cancer cells, certain areas of the promoter region of tumor suppressor genes are hypermethylated leading to the silencing of the tumor suppressor genes (170, 171). In addition, DNA methylation can also serve as a binding site for proteins such as methyl CpG binding domain proteins (MBDs) and methyl CpG binding protein 2 (MeCP2). These proteins can interact with a co-repressor complex to repress transcription. The co-repressor complex includes proteins such as histone deacetylases (HDACs) involved in the modification of histones. Histone modification is tightly associated with DNA methylation. Histone modifications play an important role in chromatin structure. Chromatin is a densely packed macromolecular complex composed of histones, DNA, and nonhistone proteins. Chromatin serves to package a large amount of material into the nucleus of a cell and to influence DNA replication. Histones play an essential role in chromatin structure and post-translational modifications of histones regulate gene expression. Some of these modifications include acetylation, methylation, phosphorylation, ubiquitination, and sumoylation (159). These modifications typically occur at serine, lysine, and arginine resides of N-terminal histone tails. The enzymes responsible for these modifications include histone acetyltransferases (41), histone methyltransferases (HMTs), histone demethylases (HMTs), and HDACs. The influence of these modifications on chromatin structure can either activate or suppress transcription.
The interplay of DNA methylation with histone modifications, transcription factors, transcriptional coactivators, and DNA binding proteins determines the status of gene transcription (126). Epigenetic modifications such as DNA methylation and histone modifications have been shown to be a hallmark of cancers (172–176). The promoter region of human GSTP1 is hypermethylated in approximately 7–100% of prostate cancer specimens (177–179). Aberrant epigenetic modifications have also been associated with the development and progression of skin cancer (49, 180–182). Thus, targeting the reversal of DNA methylation and histone modifications presents a novel strategy for the prevention and treatment of cancer. The FDA has already approved chemotherapeutics targeting DNMTs and HDACs (183). However, their usage has been limited by adverse events. Targeting epigenetic modifications for the prevention or treatment of cancers using dietary phytochemicals has become increasingly more attractive. Dietary phytochemicals may prevent cancer through epigenetic modifications (184–186).
D. Cancer Stem Cells and Apoptosis
Stem cells are characterized by their ability to differentiate into a heterogenous population of specialized cells, their ability to self-renew, and their ability to balance selfrenewal and differentiation based on environmental needs (187). Two of the pathways demonstrated to be involved in stem cell regulation and differentiation include Sonic hedge hog and Notch signaling pathways (188). Similarly, cancer stem cells (CSCs) are able to self-renew and differentiate using common pathways (186). However, CSCs are able to form tumors when implanted into animals (190). For this reason, CSCs are often referred to as tumorigenic cells or tumor initiating cells and are fundamental to the initiation and relapse of many tumor types (189–191) CSCs were first identified in 1997 (192) where CD34+CD38- cells derived from leukemic patients were able to initiate cancer in immunodeficient mice. Currently, cancer stem cells have been identified in a number of cancers including breast and colon (193). Pathways implicated in cancer stem cell renewal include Wnt (194), janus kinase (Jak), bone morphogeneic protein (BMP), and octamer-binding transcription (Oct-4) signaling pathways (193). Natural dietary compounds have been shown to regulate CSCs by increasing their sensitivity to chemotherapeutic agents, enhancing their differentiation, and inhibiting their self-renewal signaling (195, 196).
One of the most important processes involved in regulating the proliferation of cells is apoptosis. The process of apoptosis can be divided into instrinsic and extrinsic pathways. The instrinsic pathway is in large part controlled by Bcl-2 family members, while the extrinsic pathway, is mediated by tumor necrosis factor (TNF) family members. The extrinsic pathway is initiated with the respective ligand binding to death receptors such as TNF-related apoptosis-inducing ligand receptor (TRAILR) and FAS. Oligomerization of the receptors leads to the activation of caspase-8 and caspase-10, which cleave caspase-3 and caspase-7 and ultimately leads to apoptosis (197). The intrinsic pathway is activated when stress stimuli induces BCL-2 homology domain 3 (BH3)-only protein activation which leads to BAX and BAK activity and consequently mitochondrial outer membrane permeabilization (MOMP). This results in the release of cytochrome c which intereacts with apoptotic protease activating factor 1 (APAF1), which activates caspase-9. Caspase-9 then activates caspase-3 and caspase-7, which leads to apoptosis (197). Cancer cells have developed mechanisms by which apoptosis is evaded through the mutation of essential genes involved in regulation of the process. A number of phytochemicals have been shown to induce apoptosis in cancer cells/in vivo models (Table I).
Table I.
The diverse anti-cancer properties of phytochemicals are driven by dose and model system.
| Cancer/Model Type | Cell Line/Animal Model | Concentration/Dose | Phytochemical | Reference | Process/es Affected |
|---|---|---|---|---|---|
| Bladder | T24 cells | 5–20μM | SFN | Shan, Sun (98) | A1 |
| Breast | SUM159 xeonograph mouse model | 50mg/kg | SFN | Li, Fu (96), (126) | CSCs2 |
| Breast | PyMT transgenic mice | 8mmol/kg | PEITC | Singh and Singh (83) | CSCs2 |
| Breast | MDA-MB-231 | 30μM | SFN | Kanematsu, Uehara (104) | A1 |
| Colon | COLO 25 | 10μM, 50μM | UA | Chun, Kundu (198) | AI3 |
| Colon | DSS-induced acute murine colitis model | 10mg/kg, 20mg/kg | UA | Chun, Lee (20) | AI3 |
| Colon | ApcMin/+ mouse model | 3–13nmol/g | SFN | Hu, Khor (40) | A1, AI3 |
| Colon | (DSS)-induced acute colitis mouse model | 25mg/kg/day | SFN | Wagner, Will (61) | NMAS4 |
| Colorectal | Caco-2 cells | 5μM, 10μM | UA | Ramos, Pereira-Wilson (21) | NMAS4 |
| Colorectal | HT29 cells | 25μM, 50μM | PEITC, SFN | Jeong, Kim (56) | A1, AI3 |
| Colorectal | Caco-2 cells | 25μM, 75μM | SFN | Wang, Chen (102) | NMAS4, A1 (respectively) |
| Colorectal | CRC cells | 12.5μM, 25μM | SFN | Chen, Tang (103) | A1 |
| Gastro-intestinal related | Rat | 40μmol/kg/day | SFN | Munday and Munday (45) | NMAS4 |
| Glioma | A375 cells | 10μM | UA, OA | Bonaccorsi, Altieri (25) | CSCs2 |
| Human study | PBMC | 105mg | SFN | Myzak, Tong (69) | EM5 |
| Laryngeal carcinoma | Hep-2 | 10μM | PEITC | Dai, Wang (93) | A1 |
| Leukemia | HL60, U-937, THP-1 cells | 10μM, 20μM, 30μM | UA | Zhang, He (26) | CSCs2 |
| Leukemia | Promyelocytic leukemic cells | 0.2–100μM | SFN | Fimognari, Lenzi (80) | CSCs2 |
| Liver | Male Wistar rat model | 20mg/kg | UA, OA | Gayathri, Priya (17) | NMAS4 |
| Liver | HepG2 cells and Hepatocytes | 1–10μM | SFN | Bacon, Williamson (46) | NMAS4 |
| Liver | HepG2 cells | 2μM-20μM | SFN | Hong, Freeman (51) | NMAS4 |
| LPS-stimulated Inflammation | RAW 264.7 cells | 25μM, 50μM | SFN | Heiss and Gerhauser (57) | AI3 |
| Melanoma | U87 cells | 15μM | UA, OA | Bonaccorsi, Altieri (25) | CSCs2 |
| Neuronal related | PC12 cells | 20μM, 40μM | UA, OA | Tsai and Yin (22) | CSCs2, NMAS4 |
| Non-small cell lung | A549 Nude Mouse Model | 50mg/kg, 100mg/kg | UA, OA | Cho, Rho (12) | A1 |
| Non-small cell lung | H1299 and A549 cells | 30μM | UA | Wu, Zhao (23) | EM5 |
| Non-small cell lung | L9981 cells | 12.5μM, 20μM | PEITC | Yan, Zhu (94) | A1 |
| Non-small cell lung | L9981 cells | 5μM, 9.7μM | PEITC | Wu, Zhu (95) | AI3 |
| Non-small cell lung | A549 cells | 30μM | SFN | Zuryn, Litwiniec (101) | A1 |
| Ovarian | SKOV3 xenograft athymic BALB/c-nu mouse model | 12.5–50μg/mL | UA | Zhang, Wang (27) | CSCs2 |
| Pancreatic | AsPC-1, MIA, PaCa-2, Panc-28 cells | 5–20μM | UA | Prasad, Yadav (16) | A1, AI3 |
| Pancreatic | Orthotopic Pancreatic Mouse Model | 250mg/kg | UA | Prasad, Yadav (16) | A1, AI3 |
| Prostate | PC3 cells | 80μM | UA | Zhang, Kong (199) | A1, AI3 |
| Prostate | PC3 cells, LNCaP cells | 55μM, 45μM | OA | Kassi, Papoutsi (15) | A1 |
| Prostate | PC3 cells | 20μM & 30μM, 5μM & 7.5μM | SFN, PEITC | Xu, Shen (55) | AI3 |
| Prostate | LNCaP and PC3 cells | 15μM | SFN | Wong, Hsu (63) | EM5 |
| Prostate | TRAMPC1 cells | 1μM, 2.5μM | SFN | Zhang, Su (66) | EM5 |
| Prostate | LNCaP | 0.5–1μM | PEITC | Wang, Beklemisheva (200) | A1, EM5 |
| Prostate | LNCaP, PC3 | 2.5μM | PEITC | Zhang, Shu (74) | EM5 |
| Prostate | DU145 cells, PC3 cells | 10μM, 20μM, 40μM | SFN | Singh, Srivastava (97) | A1 |
| Prostate | BPH-1, LNCaP, PC3 | 15μM | SFN | Myzak, Hardin (70) | A1, EM5 |
| Skin | ICR Mouse Model | 2μmol topical application | UA | Cho, Rho (12) | AI3 |
| Skin | Ca3/7 cells | 5μM, 10μM | UA, OA | Kowalczyk, Walaszek (18) | NMAS4, AI3 |
| Skin | JB6 P+ mouse epidermal cells | 5μM | UA | Kim, Ramirez (201) | EM5 |
| Skin | Nrf2 (+/+) and Nrf2 (−/−) mice | 100nmol topical application | SFN | Saw, Huang (62) | NMAS4, AI3 |
| Skin | JB6 P+ mouse epidermal cells | 2.5μM, 5μM | SFN | Su, Zhang (202) | EM5 |
| T-cell lymphoma | Hut-78 cells | 10–80μM | UA | Yang, Shi ÜU | A1 |
| Thyroid | ARO cells | 20μM | UA, OA | Bonaccorsi, Altieri (25) | CSCs2 |
| Thyroid | MTC-SK cells | 10μM, 20μM | UA | Aguiriano-Moser, Svejda (13) | A1 |
| Immune | Activated T cells, B cells, and macrophages | 5μM | UA | Checker, Sandur(19) | AI3 |
| Anti-oxidative Stress | Nrf2 (+/+) and Nrf2 (−/−) mice | 40mg/kg | PEITC | Hu, Xu (48) | NMAS4 |
| Neuronal | Astrocytes | 5–15μM, 50–100μM | Curcumin | Scapagnini, Colombrita (117) | NMAS4, A1 (respectively) |
| Differentiation | Neuronal stem cells | 0.5μM | Curcumin | Chen, Wang (132) | CSCs2 |
| Differentiation | Embryonic stem cells | 10μM | Curcumin | Mujoo, Nikonoff (133) | CSCs2 |
| Bone | HFOb 1.9 cells | 25μM | Curcumin | Chan, Wu (141) | A1 |
| Leukemia | HL60 cells | 25μM | Curcumin | Chen, Wanming (142) | AI3,NMAS4 |
| Fibrosarcoma | HT1080 cells | 8μM | Curcumin | Sun, Ross (150)) | A1 |
| Colon | HT29 cells | 2.5μM, 5μM | Curcumin | Guo, Shu (139) | EM5 |
| Prostate | LNCaP cells | 2.5μM | Curcumin | Shu, Khor (153)) | EM5 |
| Prostate | TRAMPC1 cells | 10μM | Curcumin | Khor, Huang (151) | EM5 |
| Breast | MCF7 and MDA MB 231 cells | 10μM | Curcumin | Mirza, Sharma (154) | EM5 |
Apoptosis
Cancer Stem Cells
Anti-inflammation
Nrf2-mediated ARE Signaling
Epigenetic Modulation
IV. Perspective
Cancer is one of the leading causes of death in the United States and around the world. Modern diagnostics and treatment regimens have improved patient care, but advanced metastasized cancers remain a challenge to treat. Hence alternative strategies have to be integrated into regimens to reduce the burden of cancer using relatively non-toxic phytochemicals and or pharmaceutical agents such as non-steroidal anti-inflammatory drugs (NSAIDs), selective estrogen receptor modulators (SERMs), aromatase inhibitors, HMG-CoA reductase inhibitors (statins), among others.
The idea of cancer prevention by dietary and nutritional phytochemicals can be further refined to “NutriPrevention” versus “Chemoprevention”. In 2013, the USDA suggested “Myplate” replacing the previous “Food Pyramid”, whereby half of the plate/meal includes fruits and vegetables for healthy living. This could be defined as “NutriPrevention”, where low level phytochemicals would presumably effect and impact the epigenome of “healthy” cellular defense genetic pathways including the Nrf2-regulated anti-oxidative stress/antioxidant pathways and anti-inflammatory pathways discussed above. However, if one were to be exposed to high environmental risk factors such as smoking, “bad/unhealthy diets”, alcohol, environmental pollutants, occupational carcinogens, and/or other environmental factors/insults coupled with inherent genetics/epigenetics “stem cells” that could drive “initiated cells”, then it would logically require higher pharmacological doses of certain dietary phytochemicals and/or nontoxic pharmaceutical agents and this may be classified as “PharmacoPrevention”. During cancer remission, in order to prevent cancer from recurring, or high risk individuals with chronic inflammation diseases such as Inflammatory bowel disease (IBD), then one would use “ChemoPrevention” with relatively higher but non-toxic doses phytochemicals/botanicals alone and/or in combination with relatively nontoxic drugs such as NSAIDs, SERMs, HMG-CoA reductase inhibitors, among others. Analogously, these concepts could be applicable to other chronic diseases which are utilizing similar signaling pathways such as oxidative stress and inflammation.
V. Conclusions
In general, for many phytochemicals, it would appear much higher concentrations are required to elicit biological effects in in vitro cell culture models as compared to in vivo animal models. This phenomenon could be due to a variety of reasons. Most cell lines are tumor cell lines, thus, behave quite differently to their in vivo counterparts. They may possess efflux transporters that can exclude compounds from entering the cells, have very different cellular signaling response pathways as compared to normal cells, lack of active metabolism processes forming potential active metabolites and lack of endocrine-paracrine signaling as compared to in vivo. However, for many epigenetic effects, it appears that lower concentrations of phytochemicals are able to elicit an epigenetic response such as it relates to CpG methylation, DNMTs or HDACs in cell culture models. Further in vitro-in vivo animal and human studies would be warranted to ascertain these observations.
Acknowledgment:
The authors express sincere gratitude to all of the members of Dr. Tony Kong’s laboratory for their helpful discussions. This work was supported in part by institutional funds and by R01-CA200129 from the National Cancer Institute (NCI), R01-AT009152 from the National Center for Complementary and Integrative Health (NCCIH) and R01-AT007065 from NCCIH and the Office of Dietary Supplements (ODS).
Footnotes
The authors declare that there are no conflicts of interest.
References:
- 1.Estimated new cases and deaths from skin (nonmelanoma) cancer in the United States in 2010:National Cancer Institute (NCI); 2010. [Available from: http://www.cancer.gov/cancertopics/types/skin.
- 2.Sporn MB. Perspective: The big C - for Chemoprevention. Nature. 2011;471(7339):S10–1. [DOI] [PubMed] [Google Scholar]
- 3.Anand P, Kunnumakkara AB, Sundaram C, Harikumar KB, Tharakan ST, Lai OS, et al. Cancer is a preventable disease that requires major lifestyle changes. Pharm Res. 2008;25(9):2097–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Advances in Cancer Research. Cameron EFaR, editor. New York, NY: Academic Press, INC; 1980331 p. [Google Scholar]
- 5.Lee JH, Khor TO, Shu L, Su ZY, Fuentes F, Kong AN. Dietary phytochemicals and cancer prevention: Nrf2 signaling, epigenetics, and cell death mechanisms in blocking cancer initiation and progression. Pharmacology & therapeutics. 2013;137(2):153–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010;31(1):90–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nature reviews Cancer. 2007;7(5):357–69. [DOI] [PubMed] [Google Scholar]
- 8.Perl A, Hanczko R, Telarico T, Oaks Z, Landas S. Oxidative stress, inflammation and carcinogenesis are controlled through the pentose phosphate pathway by transaldolase. Trends Mol Med. 2011;17(7):395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shanmugam MK, Dai X, Kumar AP, Tan BK, Sethi G, Bishayee A. Oleanolic acid and its synthetic derivatives for the prevention and therapy of cancer: preclinical and clinical evidence. Cancer letters. 2014;346(2):206–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J. Pharmacology of oleanolic acid and ursolic acid. Journal of ethnopharmacology. 1995;49(2):57–68. [DOI] [PubMed] [Google Scholar]
- 11.Yang L, Shi W, Wang X, Zhou L, Cai Y, Liu H, et al. [Effect of ursolic acid on proliferation of T lymphoma cell lines Hut-78 cells and its mechanism]. Zhonghua Xue Ye Xue Za Zhi. 2015;36(2):153–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho J, Rho O, Junco J, Carbajal S, Siegel D, Slaga TJ, et al. Effect of Combined Treatment with Ursolic Acid and Resveratrol on Skin Tumor Promotion by 12-O-Tetradecanoylphorbol-13-Acetate. Cancer Prev Res (Phila). 2015;8(9):817–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aguiriano-Moser V, Svejda B, Li ZX, Sturm S, Stuppner H, Ingolic E, et al. Ursolic acid from Trailliaedoxa gracilis induces apoptosis in medullary thyroid carcinoma cells. Mol Med Rep. 2015;12(4):5003–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Kong C, Zeng Y, Wang L, Li Z, Wang H, et al. Ursolic acid induces PC-3 cell apoptosis via activation of JNK and inhibition of Akt pathways in vitro. Molecular carcinogenesis. 2010;49(4):374–85. [DOI] [PubMed] [Google Scholar]
- 15.Kassi E, Papoutsi Z, Pratsinis H, Aligiannis N, Manoussakis M, Moutsatsou P. Ursolic acid, a naturally occurring triterpenoid, demonstrates anticancer activity on human prostate cancer cells. J Cancer Res Clin Oncol. 2007;133(7):493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prasad S, Yadav VR, Sung B, Gupta SC, Tyagi AK, Aggarwal BB. Ursolic acid inhibits the growth of human pancreatic cancer and enhances the antitumor potential of gemcitabine in an orthotopic mouse model through suppression of the inflammatory microenvironment. Oncotarget. 2016;7(11):13182–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gayathri R, Priya DK, Gunassekaran GR, Sakthisekaran D. Ursolic acid attenuates oxidative stress-mediated hepatocellular carcinoma induction by diethylnitrosamine in male Wistar rats. Asian Pac J Cancer Prev. 2009;10(5):933–8. [PubMed] [Google Scholar]
- 18.Kowalczyk MC, Walaszek Z, Kowalczyk P, Kinjo T, Hanausek M, Slaga TJ. Differential effects of several phytochemicals and their derivatives on murine keratinocytes in vitro and in vivo: implications for skin cancer prevention. Carcinogenesis. 2009;30(6):1008–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Checker R, Sandur SK, Sharma D, Patwardhan RS, Jayakumar S, Kohli V, et al. Potent antiinflammatory activity of ursolic acid, a triterpenoid antioxidant, is mediated through suppression of NF-kappaB, AP-1 and NF-AT. PLoS One. 2012;7(2):e31318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chun J, Lee C, Hwang SW, Im JP, Kim JS. Ursolic acid inhibits nuclear factor-kappaB signaling in intestinal epithelial cells and macrophages, and attenuates experimental colitis in mice. Life Sci. 2014;110(1):23–34. [DOI] [PubMed] [Google Scholar]
- 21.Ramos AA, Pereira-Wilson C, Collins AR. Protective effects of ursolic acid and luteolin against oxidative DNA damage include enhancement of DNA repair in Caco-2 cells. Mutat Res. 2010;692(1–2):6–11. [DOI] [PubMed] [Google Scholar]
- 22.Tsai SJ, Yin MC. Antioxidative and anti-inflammatory protection of oleanolic acid and ursolic acid in PC12 cells. J Food Sci. 2008;73(7):H174–8. [DOI] [PubMed] [Google Scholar]
- 23.Wu J, Zhao S, Tang Q, Zheng F, Chen Y, Yang L, et al. Activation of SAPK/JNK mediated the inhibition and reciprocal interaction of DNA methyltransferase 1 and EZH2 by ursolic acid in human lung cancer cells. J Exp Clin Cancer Res. 2015;34:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen IH, Lu MC, Du YC, Yen MH, Wu CC, Chen YH, et al. Cytotoxic triterpenoids from the stems of Microtropis japonica. Journal of natural products. 2009;72(7):1231–6. [DOI] [PubMed] [Google Scholar]
- 25.Bonaccorsi I, Altieri F, Sciamanna I, Oricchio E, Grillo C, Contartese G, et al. Endogenous reverse transcriptase as a mediator of ursolic acid’s anti-proliferative and differentiating effects in human cancer cell lines. Cancer Lett. 2008;263(1):130–9. [DOI] [PubMed] [Google Scholar]
- 26.Zhang T, He YM, Wang JS, Shen J, Xing YY, Xi T. Ursolic acid induces HL60 monocytic differentiation and upregulates C/EBPbeta expression by ERK pathway activation. Anticancer Drugs. 2011;22(2):158–65. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Wang W, Qian L, Zhang Q, Lai D, Qi C. Ursolic acid inhibits the proliferation of human ovarian cancer stem-like cells through epithelial-mesenchymal transition. Oncology reports. 2015;34(5):2375–84. [DOI] [PubMed] [Google Scholar]
- 28.Ramirez-Rodriguez AM, Gonzalez-Ortiz M, Martinez-Abundis E, Acuna Ortega N. Effect of Ursolic Acid on Metabolic Syndrome, Insulin Sensitivity, and Inflammation. Journal of medicinal food. 2017;20(9):882–6. [DOI] [PubMed] [Google Scholar]
- 29.Cho YH, Lee SY, Kim CM, Kim ND, Choe S, Lee CH, et al. Effect of Loquat Leaf Extract on MuscleStrength, Muscle Mass, and Muscle Function in Healthy Adults: A Randomized, Double-Blinded, and Placebo-Controlled Trial. Evidence-based complementary and alternative medicine : eCAM. 2016;2016:4301621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang L, Zirpoli GR, Guru K, Moysich KB, Zhang Y, Ambrosone CB, et al. Intake of cruciferous vegetables modifies bladder cancer survival. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2010;19(7):1806–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palmer S. Diet, nutrition, and cancer. Progress in food & nutrition science. 1985;9(3–4):283–341. [PubMed] [Google Scholar]
- 32.Fahey JW, Zalcmann AT, Talalay P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry. 2001;56(1):5–51. [DOI] [PubMed] [Google Scholar]
- 33.Kliebenstein DJ, Kroymann J, Mitchell-Olds T. The glucosinolate-myrosinase system in an ecological and evolutionary context. Current opinion in plant biology. 2005;8(3):264–71. [DOI] [PubMed] [Google Scholar]
- 34.Gupta P, Kim B, Kim SH, Srivastava SK. Molecular targets of isothiocyanates in cancer: recent advances. Molecular nutrition & food research. 2014;58(8):1685–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keum YS, Jeong WS, Kong AN. Chemopreventive functions of isothiocyanates. Drug news & perspectives. 2005;18(7):445–51. [DOI] [PubMed] [Google Scholar]
- 36.Hayes JD, Kelleher MO, Eggleston IM. The cancer chemopreventive actions of phytochemicals derived from glucosinolates. European journal of nutrition. 2008;47 Suppl 2:73–88. [DOI] [PubMed] [Google Scholar]
- 37.Ji Y, Kuo Y, Morris ME. Pharmacokinetics of dietary phenethyl isothiocyanate in rats. Pharmaceutical research. 2005;22(10):1658–66. [DOI] [PubMed] [Google Scholar]
- 38.Mi L, Wang X, Govind S, Hood BL, Veenstra TD, Conrads TP, et al. The role of protein binding in induction of apoptosis by phenethyl isothiocyanate and sulforaphane in human non-small lung cancer cells. Cancer Res. 2007;67(13):6409–16. [DOI] [PubMed] [Google Scholar]
- 39.Hu R, Hebbar V, Kim BR, Chen C, Winnik B, Buckley B, et al. In vivo pharmacokinetics and regulation of gene expression profiles by isothiocyanate sulforaphane in the rat. The Journal of pharmacology and experimental therapeutics. 2004;310(1):263–71. [DOI] [PubMed] [Google Scholar]
- 40.Hu R, Khor TO, Shen G, Jeong WS, Hebbar V, Chen C, et al. Cancer chemoprevention of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis. 2006;27(10):2038–46. [DOI] [PubMed] [Google Scholar]
- 41.Yuan JM, Stepanov I, Murphy SE, Wang R, Allen S, Jensen J, et al. Clinical Trial of 2-Phenethyl Isothiocyanate as an Inhibitor of Metabolic Activation of a Tobacco-Specific Lung Carcinogen in Cigarette Smokers. Cancer prevention research. 2016;9(5):396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cipolla BG, Mandron E, Lefort JM, Coadou Y, Della Negra E, Corbel L, et al. Effect of Sulforaphane in Men with Biochemical Recurrence after Radical Prostatectomy. Cancer prevention research. 2015;8(8):712–9. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Z, Atwell LL, Farris PE, Ho E, Shannon J. Associations between cruciferous vegetable intake and selected biomarkers among women scheduled for breast biopsies. Public health nutrition. 2016;19(7):1288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheung KL, Kong AN. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. The AAPS journal. 2010;12(1):87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Munday R, Munday CM. Induction of phase II detoxification enzymes in rats by plant-derived isothiocyanates: comparison of allyl isothiocyanate with sulforaphane and related compounds. J Agric Food Chem. 2004;52(7):1867–71. [DOI] [PubMed] [Google Scholar]
- 46.Bacon JR, Williamson G, Garner RC, Lappin G, Langouet S, Bao Y. Sulforaphane and quercetin modulate PhIP-DNA adduct formation in human HepG2 cells and hepatocytes. Carcinogenesis. 2003;24(12):1903–11. [DOI] [PubMed] [Google Scholar]
- 47.Dingley KH, Ubick EA, Chiarappa-Zucca ML, Nowell S, Abel S, Ebeler SE, et al. Effect of dietary constituents with chemopreventive potential on adduct formation of a low dose of the heterocyclic amines PhIP and IQ and phase II hepatic enzymes. Nutrition and cancer. 2003;46(2):212–21. [DOI] [PubMed] [Google Scholar]
- 48.Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, et al. Identification of Nrf2-regulated genes induced by chemopreventive isothiocyanate PEITC by oligonucleotide microarray. Life sciences. 2006;79(20):1944–55. [DOI] [PubMed] [Google Scholar]
- 49.Millington GW. Epigenetics and dermatological disease. Pharmacogenomics. 2008;9(12):1835–50. [DOI] [PubMed] [Google Scholar]
- 50.Xu C, Yuan X, Pan Z, Shen G, Kim JH, Yu S, et al. Mechanism of action of isothiocyanates: the induction of ARE-regulated genes is associated with activation of ERK and JNK and the phosphorylation and nuclear translocation of Nrf2. Molecular cancer therapeutics. 2006;5(8):1918–26. [DOI] [PubMed] [Google Scholar]
- 51.Hong F, Freeman ML, Liebler DC. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem Res Toxicol. 2005;18(12):1917–26. [DOI] [PubMed] [Google Scholar]
- 52.Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98(6):3410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khor TO, Huang MT, Prawan A, Liu Y, Hao X, Yu S, et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer prevention research. 2008;1(3):187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu C, Huang MT, Shen G, Yuan X, Lin W, Khor TO, et al. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res. 2006;66(16):8293–6. [DOI] [PubMed] [Google Scholar]
- 55.Xu C, Shen G, Chen C, Gelinas C, Kong AN. Suppression of NF-kappaB and NF-kappaB-regulated gene expression by sulforaphane and PEITC through IkappaBalpha, IKK pathway in human prostate cancer PC-3 cells. Oncogene. 2005;24(28):4486–95. [DOI] [PubMed] [Google Scholar]
- 56.Jeong WS, Kim IW, Hu R, Kong AN. Modulatory properties of various natural chemopreventive agents on the activation of NF-kappaB signaling pathway. Pharm Res. 2004;21(4):661–70. [DOI] [PubMed] [Google Scholar]
- 57.Heiss E, Gerhauser C. Time-dependent modulation of thioredoxin reductase activity might contribute to sulforaphane-mediated inhibition of NF-kappaB binding to DNA. Antioxid Redox Signal. 2005;7(11–12):1601–11. [DOI] [PubMed] [Google Scholar]
- 58.Heiss E, Herhaus C, Klimo K, Bartsch H, Gerhauser C. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. The Journal of biological chemistry. 2001;276(34):32008–15. [DOI] [PubMed] [Google Scholar]
- 59.Liao BC, Hsieh CW, Lin YC, Wung BS. The glutaredoxin/glutathione system modulates NF-kappaB activity by glutathionylation of p65 in cinnamaldehyde-treated endothelial cells. Toxicological sciences : an official journal of the Society of Toxicology. 2010;116(1):151–63. [DOI] [PubMed] [Google Scholar]
- 60.Bellezza I, Tucci A, Galli F, Grottelli S, Mierla AL, Pilolli F, et al. Inhibition of NF-kappaB nuclear translocation via HO-1 activation underlies alpha-tocopheryl succinate toxicity. The Journal of nutritional biochemistry. 2012;23(12):1583–91. [DOI] [PubMed] [Google Scholar]
- 61.Wagner AE, Will O, Sturm C, Lipinski S, Rosenstiel P, Rimbach G. DSS-induced acute colitis in C57BL/6 mice is mitigated by sulforaphane pre-treatment. J Nutr Biochem. 2013;24(12):2085–91. [DOI] [PubMed] [Google Scholar]
- 62.Saw CL, Huang MT, Liu Y, Khor TO, Conney AH, Kong AN. Impact of Nrf2 on UVB-induced skin inflammation/photoprotection and photoprotective effect of sulforaphane. Molecular carcinogenesis. 2011;50(6):479–86. [DOI] [PubMed] [Google Scholar]
- 63.Wong CP, Hsu A, Buchanan A, Palomera-Sanchez Z, Beaver LM, Houseman EA, et al. Effects of sulforaphane and 3,3’-diindolylmethane on genome-wide promoter methylation in normal prostate epithelial cells and prostate cancer cells. PLoS One. 2014;9(1):e86787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang LG, Chiao JW. Prostate cancer chemopreventive activity of phenethyl isothiocyanate through epigenetic regulation (review). International journal of oncology. 2010;37(3):533–9. [DOI] [PubMed] [Google Scholar]
- 65.Fuentes F, Paredes-Gonzalez X, Kong AT. Dietary Glucosinolates Sulforaphane, Phenethyl Isothiocyanate, Indole-3-Carbinol/3,3’-Diindolylmethane: Anti-Oxidative Stress/Inflammation, Nrf2, Epigenetics/Epigenomics and Cancer Chemopreventive Efficacy. Current pharmacology reports. 2015;1(3):179–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang C, Su ZY, Khor TO, Shu L, Kong AN. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochem Pharmacol. 2013;85(9):1398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Su ZY, Zhang C, Lee JH, Shu L, Wu TY, Khor TO, et al. Requirement and epigenetics reprogramming of Nrf2 in suppression of tumor promoter TPA-induced mouse skin cell transformation by sulforaphane. Cancer prevention research. 2014;7(3):319–29. [DOI] [PubMed] [Google Scholar]
- 68.Ho E, Clarke JD, Dashwood RH. Dietary sulforaphane, a histone deacetylase inhibitor for cancer prevention. The Journal of nutrition. 2009;139(12):2393–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med (Maywood). 2007;232(2):227–34. [PMC free article] [PubMed] [Google Scholar]
- 70.Myzak MC, Hardin K, Wang R, Dashwood RH, Ho E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis. 2006;27(4):811–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang LG, Liu XM, Fang Y, Dai W, Chiao FB, Puccio GM, et al. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int J Oncol. 2008;33(2):375–80. [PubMed] [Google Scholar]
- 72.Liu K, Cang S, Ma Y, Chiao JW. Synergistic effect of paclitaxel and epigenetic agent phenethyl isothiocyanate on growth inhibition, cell cycle arrest and apoptosis in breast cancer cells. Cancer cell international. 2013;13(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cang S, Ma Y, Chiao JW, Liu D. Phenethyl isothiocyanate and paclitaxel synergistically enhanced apoptosis and alpha-tubulin hyperacetylation in breast cancer cells. Experimental hematology & oncology. 2014;3(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang C, Shu L, Kim H, Khor TO, Wu R, Li W, et al. Phenethyl isothiocyanate (PEITC) suppresses prostate cancer cell invasion epigenetically through regulating microRNA-194. Mol Nutr Food Res. 2016;60(6):1427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhu H, Jia Z, Strobl JS, Ehrich M, Misra HP, Li Y. Potent induction of total cellular and mitochondrial antioxidants and phase 2 enzymes by cruciferous sulforaphane in rat aortic smooth muscle cells: cytoprotection against oxidative and electrophilic stress. Cardiovascular toxicology. 2008;8(3):115–25. [DOI] [PubMed] [Google Scholar]
- 76.Huang CS, Lin AH, Liu CT, Tsai CW, Chang IS, Chen HW, et al. Isothiocyanates protect against oxidized LDL-induced endothelial dysfunction by upregulating Nrf2-dependent antioxidation and suppressing NFkappaB activation. Molecular nutrition & food research. 2013;57(11):1918–30. [DOI] [PubMed] [Google Scholar]
- 77.Hofmann T, Kuhnert A, Schubert A, Gill C, Rowland IR, Pool-Zobel BL, et al. Modulation of detoxification enzymes by watercress: in vitro and in vivo investigations in human peripheral blood cells. European journal of nutrition. 2009;48(8):483–91. [DOI] [PubMed] [Google Scholar]
- 78.Ullah MF. Sulforaphane (SFN): An Isothiocyanate in a Cancer Chemoprevention Paradigm. Medicines. 2015;2(3):141–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li Y, Zhang T, Korkaya H, Liu S, Lee HF, Newman B, et al. Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells. Clin Cancer Res. 2010;16(9):2580–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fimognari C, Lenzi M, Cantelli-Forti G, Hrelia P. Induction of differentiation in human promyelocytic cells by the isothiocyanate sulforaphane. In Vivo. 2008;22(3):317–20. [PubMed] [Google Scholar]
- 81.Asselin-Labat ML, Sutherland KD, Barker H, Thomas R, Shackleton M, Forrest NC, et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol. 2007;9(2):201–9. [DOI] [PubMed] [Google Scholar]
- 82.Kouros-Mehr H, Bechis SK, Slorach EM, Littlepage LE, Egeblad M, Ewald AJ, et al. GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell. 2008;13(2):141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Singh SV, Singh K. Cancer chemoprevention with dietary isothiocyanates mature for clinical translational research. Carcinogenesis. 2012;33(10):1833–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McCune K, Mehta R, Thorat MA, Badve S, Nakshatri H. Loss of ERalpha and FOXA1 expression in a progression model of luminal type breast cancer: insights from PyMT transgenic mouse model. Oncol Rep. 2010;24(5):1233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dvorankova B, Smetana K Jr., Chovanec M, Lacina L, Stork J, Plzakova Z, et al. Transient expression of keratin 19 is induced in originally negative interfollicular epidermal cells by adhesion of suspended cells. Int J Mol Med. 2005;16(4):525–31. [PubMed] [Google Scholar]
- 86.Gamet-Payrastre L. Signaling pathways and intracellular targets of sulforaphane mediating cell cycle arrest and apoptosis. Curr Cancer Drug Targets. 2006;6(2):135–45. [DOI] [PubMed] [Google Scholar]
- 87.Huong LD, Shin JA, Choi ES, Cho NP, Kim HM, Leem DH, et al. beta-Phenethyl isothiocyanate induces death receptor 5 to induce apoptosis in human oral cancer cells via p38. Oral Dis. 2012;18(5):513–9. [DOI] [PubMed] [Google Scholar]
- 88.Huong le D, Shim JH, Choi KH, Shin JA, Choi ES, Kim HS, et al. Effect of beta-phenylethyl isothiocyanate from cruciferous vegetables on growth inhibition and apoptosis of cervical cancer cells through the induction of death receptors 4 and 5. J Agric Food Chem. 2011;59(15):8124–31. [DOI] [PubMed] [Google Scholar]
- 89.Gupta P, Adkins C, Lockman P, Srivastava SK. Metastasis of Breast Tumor Cells to Brain Is Suppressed by Phenethyl Isothiocyanate in a Novel Metastasis Model. PLoS One. 2013;8(6):e67278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen PY, Lin KC, Lin JP, Tang NY, Yang JS, Lu KW, et al. Phenethyl Isothiocyanate (PEITC) Inhibits the Growth of Human Oral Squamous Carcinoma HSC-3 Cells through G(0)/G(1) Phase Arrest and Mitochondria-Mediated Apoptotic Cell Death. Evid Based Complement Alternat Med. 2012;2012:718320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tang NY, Huang YT, Yu CS, Ko YC, Wu SH, Ji BC, et al. Phenethyl isothiocyanate (PEITC) promotes G2/M phase arrest via p53 expression and induces apoptosis through caspase- and mitochondria-dependent signaling pathways in human prostate cancer DU 145 cells. Anticancer Res. 2011;31(5):1691–702. [PubMed] [Google Scholar]
- 92.Jakubikova J, Cervi D, Ooi M, Kim K, Nahar S, Klippel S, et al. Anti-tumor activity and signaling events triggered by the isothiocyanates, sulforaphane and phenethyl isothiocyanate, in multiple myeloma. Haematologica. 2011;96(8):1170–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dai MY, Wang Y, Chen C, Li F, Xiao BK, Chen SM, et al. Phenethyl isothiocyanate induces apoptosis and inhibits cell proliferation and invasion in Hep-2 laryngeal cancer cells. Oncol Rep. 2016;35(5):2657–64. [DOI] [PubMed] [Google Scholar]
- 94.Yan H, Zhu Y, Liu B, Wu H, Li Y, Wu X, et al. Mitogen-activated protein kinase mediates the apoptosis of highly metastatic human non-small cell lung cancer cells induced by isothiocyanates. Br J Nutr. 2011;106(12):1779–91. [DOI] [PubMed] [Google Scholar]
- 95.Wu X, Zhu Y, Yan H, Liu B, Li Y, Zhou Q, et al. Isothiocyanates induce oxidative stress and suppress the metastasis potential of human non-small cell lung cancer cells. BMC Cancer. 2010;10:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li SH, Fu J, Watkins DN, Srivastava RK, Shankar S. Sulforaphane regulates self-renewal of pancreatic cancer stem cells through the modulation of Sonic hedgehog-GLI pathway. Mol Cell Biochem. 2013;373(1–2):217–27. [DOI] [PubMed] [Google Scholar]
- 97.Singh SV, Srivastava SK, Choi S, Lew KL, Antosiewicz J, Xiao D, et al. Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J Biol Chem. 2005;280(20):19911–24. [DOI] [PubMed] [Google Scholar]
- 98.Shan Y, Sun C, Zhao X, Wu K, Cassidy A, Bao Y. Effect of sulforaphane on cell growth, G(0)/G(1) phase cell progression and apoptosis in human bladder cancer T24 cells. Int J Oncol. 2006;29(4):883–8. [PubMed] [Google Scholar]
- 99.Gamet-Payrastre L, Li P, Lumeau S, Cassar G, Dupont MA, Chevolleau S, et al. Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res. 2000;60(5):1426–33. [PubMed] [Google Scholar]
- 100.Suppipat K, Park CS, Shen Y, Zhu X, Lacorazza HD. Sulforaphane induces cell cycle arrest and apoptosis in acute lymphoblastic leukemia cells. PLoS One. 2012;7(12):e51251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zuryn A, Litwiniec A, Safiejko-Mroczka B, Klimaszewska-Wisniewska A, Gagat M, Krajewski A, et al. The effect of sulforaphane on the cell cycle, apoptosis and expression of cyclin D1 and p21 in the A549 non-small cell lung cancer cell line. Int J Oncol. 2016. [DOI] [PubMed] [Google Scholar]
- 102.Wang M, Chen S, Wang S, Sun D, Chen J, Li Y, et al. Effects of phytochemicals sulforaphane on uridine diphosphate-glucuronosyltransferase expression as well as cell-cycle arrest and apoptosis in human colon cancer Caco-2 cells. Chin J Physiol. 2012;55(2):134–44. [DOI] [PubMed] [Google Scholar]
- 103.Chen MJ, Tang WY, Hsu CW, Tsai YT, Wu JF, Lin CW, et al. Apoptosis Induction in Primary Human Colorectal Cancer Cell Lines and Retarded Tumor Growth in SCID Mice by Sulforaphane. Evid Based Complement Alternat Med. 2012;2012:415231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kanematsu S, Uehara N, Miki H, Yoshizawa K, Kawanaka A, Yuri T, et al. Autophagy inhibition enhances sulforaphane-induced apoptosis in human breast cancer cells. Anticancer Res. 2010;30(9):3381–90. [PubMed] [Google Scholar]
- 105.Kim BR, Hu R, Keum YS, Hebbar V, Shen G, Nair SS, et al. Effects of glutathione on antioxidant response element-mediated gene expression and apoptosis elicited by sulforaphane. Cancer Res. 2003;63(21):7520–5. [PubMed] [Google Scholar]
- 106.Yu R, Tan TH, Kong AN. Butylated hydroxyanisole and its metabolite tert-butylhydroquinone differentially regulate mitogen-activated protein kinases. The role of oxidative stress in the activation of mitogen-activated protein kinases by phenolic antioxidants. The Journal of biological chemistry. 1997;272(46):28962–70. [DOI] [PubMed] [Google Scholar]
- 107.Yu R, Lei W, Mandlekar S, Weber MJ, Der CJ, Wu J, et al. Role of a mitogen-activated protein kinase pathway in the induction of phase II detoxifying enzymes by chemicals. The Journal of biological chemistry. 1999;274(39):27545–52. [DOI] [PubMed] [Google Scholar]
- 108.Kong AN, Yu R, Lei W, Mandlekar S, Tan TH, Ucker DS. Differential activation of MAPK and ICE/Ced-3 protease in chemical-induced apoptosis. The role of oxidative stress in the regulation of mitogen-activated protein kinases (MAPKs) leading to gene expression and survival or activation of caspases leading to apoptosis. Restorative neurology and neuroscience. 1998;12(2–3):63–70. [PubMed] [Google Scholar]
- 109.Manach C, Scalbert A, Morand C, Remesy C, Jimenez L. Polyphenols: food sources and bioavailability. The American journal of clinical nutrition. 2004;79(5):727–47. [DOI] [PubMed] [Google Scholar]
- 110.Neveu V, Perez-Jimenez J, Vos F, Crespy V, du Chaffaut L, Mennen L, et al. Phenol-Explorer: an online comprehensive database on polyphenol contents in foods. Database : the journal of biological databases and curation. 2010;2010:bap024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhou Y, Zheng J, Li Y, Xu DP, Li S, Chen YM, et al. Natural Polyphenols for Prevention and Treatment of Cancer. Nutrients. 2016;8(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cheng AL, Hsu CH, Lin JK, Hsu MM, Ho YF, Shen TS, et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer research. 2001;21(4B):2895–900. [PubMed] [Google Scholar]
- 113.Panahi Y, Saadat A, Beiraghdar F, Sahebkar A. Adjuvant therapy with bioavailability-boosted curcuminoids suppresses systemic inflammation and improves quality of life in patients with solid tumors: a randomized double-blind placebo-controlled trial. Phytotherapy research : PTR. 2014;28(10):1461–7. [DOI] [PubMed] [Google Scholar]
- 114.Hejazi J, Rastmanesh R, Taleban FA, Molana SH, Hejazi E, Ehtejab G, et al. Effect of Curcumin Supplementation During Radiotherapy on Oxidative Status of Patients with Prostate Cancer: A Double Blinded, Randomized, Placebo-Controlled Study. Nutrition and cancer. 2016;68(1):77–85. [DOI] [PubMed] [Google Scholar]
- 115.Cruz-Correa M, Shoskes DA, Sanchez P, Zhao R, Hylind LM, Wexner SD, et al. Combination treatment with curcumin and quercetin of adenomas in familial adenomatous polyposis. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2006;4(8):1035–8. [DOI] [PubMed] [Google Scholar]
- 116.Carroll RE, Benya RV, Turgeon DK, Vareed S, Neuman M, Rodriguez L, et al. Phase IIa clinical trial of curcumin for the prevention of colorectal neoplasia. Cancer prevention research. 2011;4(3):354–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Scapagnini G, Colombrita C, Amadio M, D’Agata V, Arcelli E, Sapienza M, et al. Curcumin activates defensive genes and protects neurons against oxidative stress. Antioxid Redox Signal. 2006;8(3–4):395–403. [DOI] [PubMed] [Google Scholar]
- 118.Shishodia S, Chaturvedi MM, Aggarwal BB. Role of curcumin in cancer therapy. Current problems in cancer. 2007;31(4):243–305. [DOI] [PubMed] [Google Scholar]
- 119.Woo JH, Kim YH, Choi YJ, Kim DG, Lee KS, Bae JH, et al. Molecular mechanisms of curcumin-induced cytotoxicity: induction of apoptosis through generation of reactive oxygen species, down-regulation of Bcl-XL and IAP, the release of cytochrome c and inhibition of Akt. Carcinogenesis. 2003;24(7):1199–208. [DOI] [PubMed] [Google Scholar]
- 120.Rajasekaran SA. Therapeutic potential of curcumin in gastrointestinal diseases. World journal of gastrointestinal pathophysiology. 2011;2(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Basnet P, Skalko-Basnet N. Curcumin: an anti-inflammatory molecule from a curry spice on the path to cancer treatment. Molecules. 2011;16(6):4567–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Strimpakos AS, Sharma RA. Curcumin: preventive and therapeutic properties in laboratory studies and clinical trials. Antioxidants & redox signaling. 2008;10(3):511–45. [DOI] [PubMed] [Google Scholar]
- 123.Brinkmann J, Stolpmann K, Trappe S, Otter T, Genkinger D, Bock U, et al. Metabolically competent human skin models: activation and genotoxicity of benzo[a]pyrene. Toxicol Sci. 2013;131(2):351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jancinova V, Perecko T, Nosal R, Mihalova D, Bauerova K, Drabikova K. Pharmacological regulation of neutrophil activity and apoptosis: Contribution to new strategy for modulation of inflammatory processes. Interdisciplinary toxicology. 2011;4(1):11–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhong F, Chen H, Han L, Jin Y, Wang W. Curcumin attenuates lipopolysaccharide-induced renal inflammation. Biological & pharmaceutical bulletin. 2011;34(2):226–32. [DOI] [PubMed] [Google Scholar]
- 126.http://www.skincancer.org/publications/sun-and-skin-news/summer-2010-27-2/nonmelanoma-skin-cancer-incidence SCF. Nonmelanoma Skin Cancer Incidence Increases Dramatically. Sun & Skin News Summer 2010.
- 127.Hao F, Kang J, Cao Y, Fan S, Yang H, An Y, et al. Curcumin attenuates palmitate-induced apoptosis in MIN6 pancreatic β-cells through PI3K/Akt/FoxO1 and mitochondrial survival pathways. Apoptosis. 2015;20(11):1420–32. [DOI] [PubMed] [Google Scholar]
- 128.Zhang X, Liang D, Guo L, Liang W, Jiang Y, Li H, et al. Curcumin protects renal tubular epithelial cells from high glucose-induced epithelial-to-mesenchymal transition through Nrf2-mediated upregulation of heme oxygenase-1. Mol Med Rep. 2015;12(1):1347–55. [DOI] [PubMed] [Google Scholar]
- 129.Alinejad B, Ghorbani A, Sadeghnia HR. Effects of combinations of curcumin, linalool, rutin, safranal, and thymoquinone on glucose/serum deprivation-induced cell death. Avicenna journal of phytomedicine. 2013;3(4):321–8. [PMC free article] [PubMed] [Google Scholar]
- 130.Nazari QA, Kume T, Izuo N, Takada-Takatori Y, Imaizumi A, Hashimoto T, et al. Neuroprotective effects of curcumin and highly bioavailable curcumin on oxidative stress induced by sodium nitroprusside in rat striatal cell culture. Biol Pharm Bull. 2013;36(8):1356–62. [DOI] [PubMed] [Google Scholar]
- 131.Sakurai R, Villarreal P, Husain S, Liu J, Sakurai T, Tou E, et al. Curcumin protects the developing lung against long-term hyperoxic injury. Am J Physiol Lung Cell Mol Physiol. 2013;305(4):L301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen F, Wang H, Xiang X, Yuan J, Chu W, Xue X, et al. Curcumin increased the differentiation rate of neurons in neural stem cells via wnt signaling in vitro study. J Surg Res. 2014;192(2):298–304. [DOI] [PubMed] [Google Scholar]
- 133.Mujoo K, Nikonoff LE, Sharin VG, Bryan NS, Kots AY, Murad F. Curcumin induces differentiation of embryonic stem cells through possible modulation of nitric oxide-cyclic GMP pathway. Protein Cell. 2012;3(7):535–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tu SP, Jin H, Shi JD, Zhu LM, Suo Y, Lu G, et al. Curcumin induces the differentiation of myeloid-derived suppressor cells and inhibits their interaction with cancer cells and related tumor growth.Cancer prevention research. 2012;5(2):205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kim CY, Le TT, Chen C, Cheng JX, Kim KH. Curcumin inhibits adipocyte differentiation through modulation of mitotic clonal expansion. J Nutr Biochem. 2011;22(10):910–20. [DOI] [PubMed] [Google Scholar]
- 136.Liu H, Liu A, Shi C, Li B. Curcumin suppresses transforming growth factor-beta1-induced cardiac fibroblast differentiation via inhibition of Smad-2 and p38 MAPK signaling pathways. Exp Ther Med. 2016;11(3):998–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ma J, Ma SY, Ding CH. Curcumin reduces cardiac fibrosis by inhibiting myofibroblast differentiation and decreasing transforming growth factor beta1 and matrix metalloproteinase 9 / tissue inhibitor of metalloproteinase 1. Chin J Integr Med. 2016. [DOI] [PubMed] [Google Scholar]
- 138.http://www.healthguideinfo.com/skin-cancer/p90830/ HC. Metastasis of Squamous Cell Carcinoma.
- 139.Guo Y, Shu L, Zhang C, Su ZY, Kong AN. Curcumin inhibits anchorage-independent growth of HT29 human colon cancer cells by targeting epigenetic restoration of the tumor suppressor gene DLEC1. Biochem Pharmacol. 2015;94(2):69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Khar A, Ali AM, Pardhasaradhi BV, Varalakshmi CH, Anjum R, Kumari AL. Induction of stress response renders human tumor cell lines resistant to curcumin-mediated apoptosis: role of reactive oxygen intermediates. Cell stress & chaperones. 2001;6(4):368–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chan WH, Wu HY, Chang WH. Dosage effects of curcumin on cell death types in a human osteoblast cell line. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association. 2006;44(8):1362–71. [DOI] [PubMed] [Google Scholar]
- 142.Chen J, Wanming D, Zhang D, Liu Q, Kang J. Water-soluble antioxidants improve the antioxidant and anticancer activity of low concentrations of curcumin in human leukemia cells. Pharmazie. 2005;60(1):57–61. [PubMed] [Google Scholar]
- 143.Banerjee A, Kunwar A, Mishra B, Priyadarsini KI. Concentration dependent antioxidant/prooxidant activity of curcumin studies from AAPH induced hemolysis of RBCs. Chemico-biological interactions. 2008;174(2):134–9. [DOI] [PubMed] [Google Scholar]
- 144.Ali RE, Rattan SI. Curcumin’s biphasic hormetic response on proteasome activity and heat-shock protein synthesis in human keratinocytes. Annals of the New York Academy of Sciences. 2006;1067:394–9. [DOI] [PubMed] [Google Scholar]
- 145.Calabrese EJ, Baldwin LA. Hormesis: the dose-response revolution. Annual review of pharmacology and toxicology. 2003;43:175–97. [DOI] [PubMed] [Google Scholar]
- 146.Calabrese EJ. Hormesis: why it is important to toxicology and toxicologists. Environmental toxicology and chemistry / SETAC. 2008;27(7):1451–74. [DOI] [PubMed] [Google Scholar]
- 147.Borriello A, Bencivenga D, Caldarelli I, Tramontano A, Borgia A, Pirozzi AV, et al. Resveratrol and cancer treatment: is hormesis a yet unsolved matter? Current pharmaceutical design. 2013;19(30):5384–93. [DOI] [PubMed] [Google Scholar]
- 148.Bao J, Huang B, Zou L, Chen S, Zhang C, Zhang Y, et al. Hormetic Effect of Berberine Attenuates the Anticancer Activity of Chemotherapeutic Agents. PloS one. 2015;10(9):e0139298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Haddi K, Oliveira EE, Faroni LR, Guedes DC, Miranda NN. Sublethal Exposure to Clove and Cinnamon Essential Oils Induces Hormetic-Like Responses and Disturbs Behavioral and Respiratory Responses in Sitophilus zeamais (Coleoptera: Curculionidae). Journal of economic entomology. 2015;108(6):2815–22. [DOI] [PubMed] [Google Scholar]
- 150.Sun B, Ross SM, Trask OJ, Carmichael PL, Dent M, White A, et al. Assessing dose-dependent differences in DNA-damage, p53 response and genotoxicity for quercetin and curcumin. Toxicol In Vitro. 2013;27(6):1877–87. [DOI] [PubMed] [Google Scholar]
- 151.Khor TO, Huang Y, Wu TY, Shu L, Lee J, Kong AN. Pharmacodynamics of curcumin as DNA hypomethylation agent in restoring the expression of Nrf2 via promoter CpGs demethylation. Biochem Pharmacol. 2011;82(9):1073–8. [DOI] [PubMed] [Google Scholar]
- 152.Hager B, Bickenbach JR, Fleckman P. Long-term culture of murine epidermal keratinocytes. J Invest Dermatol. 1999;112(6):971–6. [DOI] [PubMed] [Google Scholar]
- 153.Shu L, Khor TO, Lee JH, Boyanapalli SS, Huang Y, Wu TY, et al. Epigenetic CpG demethylation of the promoter and reactivation of the expression of Neurog1 by curcumin in prostate LNCaP cells. Aaps J. 2011;13(4):606–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mirza S, Sharma G, Parshad R, Gupta SD, Pandya P, Ralhan R. Expression of DNA methyltransferases in breast cancer patients and to analyze the effect of natural compounds on DNA methyltransferases and associated proteins. J Breast Cancer. 2013;16(1):23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li W, Pung D, Su ZY, Guo Y, Zhang C, Yang AY, et al. Epigenetics Reactivation of Nrf2 in Prostate TRAMP C1 Cells by Curcumin Analogue FN1. Chemical research in toxicology. 2016;29(4):694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chiu S, Terpstra KJ, Bureau Y, Hou J, Raheb H, Cernvosky Z, et al. Liposomal-formulated curcumin [Lipocurc] targeting HDAC (histone deacetylase) prevents apoptosis and improves motor deficits in Park 7 (DJ-1)-knockout rat model of Parkinson’s disease: implications for epigenetics-based nanotechnology-driven drug platform. Journal of complementary & integrative medicine. 2013;10. [DOI] [PubMed] [Google Scholar]
- 157.Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101(9):2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Saini S, Arora S, Majid S, Shahryari V, Chen Y, Deng G, et al. Curcumin modulates microRNA-203-mediated regulation of the Src-Akt axis in bladder cancer. Cancer prevention research. 2011;4(10):1698–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Su ZY, Shu L, Khor TO, Lee JH, Fuentes F, Kong AN. A perspective on dietary phytochemicals and cancer chemoprevention: oxidative stress, nrf2, and epigenomics. Top Curr Chem. 2013;329:133–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chen C, Kong AN. Dietary cancer-chemopreventive compounds: from signaling and gene expression to pharmacological effects. Trends in pharmacological sciences. 2005;26(6):318–26. [DOI] [PubMed] [Google Scholar]
- 161.Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78(2):547–81. [DOI] [PubMed] [Google Scholar]
- 162.Halliwell BGJ. Free radicals in biology and medicine. Oxford: Oxford University Press; 1999. [Google Scholar]
- 163.Lander HM. An essential role for free radicals and derived species in signal transduction. FASEB J. 1997;11(2):118–24. [PubMed] [Google Scholar]
- 164.Yu BP. Cellular defenses against damage from reactive oxygen species. Physiol Rev. 1994;74(1):139–62. [DOI] [PubMed] [Google Scholar]
- 165.Emerit J, Edeas M, Bricaire F. Neurodegenerative diseases and oxidative stress. Biomed Pharmacother. 2004;58(1):39–46. [DOI] [PubMed] [Google Scholar]
- 166.Cataldi A. Cell responses to oxidative stressors. Curr Pharm Des. 2010;16(12):1387–95. [DOI] [PubMed] [Google Scholar]
- 167.Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009;45(4–5):324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286(5439):481–6. [DOI] [PubMed] [Google Scholar]
- 169.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–59. [DOI] [PubMed] [Google Scholar]
- 170.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genetics. 2005;37(4):391–400. [DOI] [PubMed] [Google Scholar]
- 171.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3(6):415–28. [DOI] [PubMed] [Google Scholar]
- 172.Nakayama M, Gonzalgo ML, Yegnasubramanian S, Lin X, De Marzo AM, Nelson WG. GSTP1 CpG island hypermethylation as a molecular biomarker for prostate cancer. J Cell Biochem. 2004;91(3):540–52. [DOI] [PubMed] [Google Scholar]
- 173.Nelson WG, Yegnasubramanian S, Agoston AT, Bastian PJ, Lee BH, Nakayama M, et al. Abnormal DNA methylation, epigenetics, and prostate cancer. Front Biosci. 2007;12:4254–66. [DOI] [PubMed] [Google Scholar]
- 174.Nelson WG, De Marzo AM, Yegnasubramanian S. Epigenetic alterations in human prostate cancers. Endocrinology. 2009;150(9):3991–4002. Epub 2009 Jun 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Jeronimo C, Henrique R. Epigenetic biomarkers in urological tumors: A systematic review. Cancer letters. 2014;342(2):264–74. [DOI] [PubMed] [Google Scholar]
- 176.Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev. 2012;22(1):50–5. [DOI] [PubMed] [Google Scholar]
- 177.Lee WH, Morton RA, Epstein JI, Brooks JD, Campbell PA, Bova GS, et al. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc Natl Acad Sci U S A. 1994;91(24):11733–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lee WH, Isaacs WB, Bova GS, Nelson WG. CG island methylation changes near the GSTP1 gene in prostatic carcinoma cells detected using the polymerase chain reaction: a new prostate cancer biomarker. Cancer Epidemiol Biomarkers Prev. 1997;6(6):443–50. [PubMed] [Google Scholar]
- 179.Santourlidis S, Florl A, Ackermann R, Wirtz HC, Schulz WA. High frequency of alterations in DNA methylation in adenocarcinoma of the prostate. Prostate. 1999;39(3):166–74. [DOI] [PubMed] [Google Scholar]
- 180.van Doorn R, Gruis NA, Willemze R, van der Velden PA, Tensen CP. Aberrant DNA methylation in cutaneous malignancies. Semin Oncol. 2005;32(5):479–87. [DOI] [PubMed] [Google Scholar]
- 181.Bachman AN, Curtin GM, Doolittle DJ, Goodman JI. Altered methylation in gene-specific and GC-rich regions of DNA is progressive and nonrandom during promotion of skin tumorigenesis. Toxicol Sci. 2006;91(2):406–18. [DOI] [PubMed] [Google Scholar]
- 182.Schinke C, Mo Y, Yu Y, Amiri K, Sosman J, Greally J, et al. Aberrant DNA methylation in malignant melanoma. Melanoma Res. 2010;20(4):253–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502(7472):480–8. [DOI] [PubMed] [Google Scholar]
- 184.Davis CD, Uthus EO. DNA methylation, cancer susceptibility, and nutrient interactions. Exp Biol Med (Maywood). 2004;229(10):988–95. [DOI] [PubMed] [Google Scholar]
- 185.Fang M, Chen D, Yang CS. Dietary polyphenols may affect DNA methylation. J Nutr. 2007;137(1 Suppl): 223S–8S. [DOI] [PubMed] [Google Scholar]
- 186.Yang CS, Fang M, Lambert JD, Yan P, Huang TH. Reversal of hypermethylation and reactivation of genes by dietary polyphenolic compounds. Nutr Rev. 2008;66 Suppl 1:S18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rajasekhar VK. Cancer stem cells. Hoboken, New Jersey: John Wiley & Sons; 2014. xxxv, 508 pages, 32 pages of plates p. [Google Scholar]
- 188.Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411(6835):349–54. [DOI] [PubMed] [Google Scholar]
- 189.Katoh M. Networking of WNT, FGF, Notch, BMP, and Hedgehog signaling pathways during carcinogenesis. Stem Cell Rev. 2007;3(1):30–8. [DOI] [PubMed] [Google Scholar]
- 190.Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009;7(6):e1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005;7(3):86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–7. [DOI] [PubMed] [Google Scholar]
- 193.Dawood S, Austin L, Cristofanilli M. Cancer stem cells: implications for cancer therapy. Oncology (Williston Park). 2014;28(12):1101–7, 10. [PubMed] [Google Scholar]
- 194.Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int J Oncol. 2017;51(5):1357–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov. 2009;8(10):806–23. [DOI] [PubMed] [Google Scholar]
- 196.Kawasaki BT, Hurt EM, Mistree T, Farrar WL. Targeting cancer stem cells with phytochemicals. Mol Interv. 2008;8(4):174–84. [DOI] [PubMed] [Google Scholar]
- 197.Ichim G, Tait SW. A fate worse than death: apoptosis as an oncogenic process. Nature reviews Cancer. 2016;16(8):539–48. [DOI] [PubMed] [Google Scholar]
- 198.Chun KS, Kundu J, Kundu JK, Surh YJ. Targeting Nrf2-Keap1 signaling for chemoprevention of skin carcinogenesis with bioactive phytochemicals. Toxicology letters. 2014;229(1):73–84. [DOI] [PubMed] [Google Scholar]
- 199.Zhang Y, Kong C, Zeng Y, Wang L, Li Z, Wang H, et al. Ursolic acid induces PC-3 cell apoptosis via activation of JNK and inhibition of Akt pathways in vitro. Molecular carcinogenesis.49(4):374–85. [DOI] [PubMed] [Google Scholar]
- 200.Wang LG, Beklemisheva A, Liu XM, Ferrari AC, Feng J, Chiao JW. Dual action on promoter demethylation and chromatin by an isothiocyanate restored GSTP1 silenced in prostate cancer. Molecular carcinogenesis. 2007;46(1):24–31. [DOI] [PubMed] [Google Scholar]
- 201.Kim H, Ramirez CN, Su ZY, Kong AN. Epigenetic modifications of triterpenoid ursolic acid in activating Nrf2 and blocking cellular transformation of mouse epidermal cells. J Nutr Biochem. 2016;33:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Su ZY, Zhang C, Lee JH, Shu L, Wu TY, Khor TO, et al. Requirement and Epigenetics Reprogramming of Nrf2 in Suppression of Tumor Promoter TPA-Induced Mouse Skin Cell Transformation by Sulforaphane. Cancer prevention research. 2014. [DOI] [PubMed] [Google Scholar]