

Abstract
The lissoclimides are unusual succinimide-containing labdane diterpenoids that were reported to be potent cytotoxins. Our short semi-synthesis and analogue-oriented synthesis approaches have provided a series of lissoclimide natural products and analogues that expanded the structure-activity relationships in this family. The semi-synthesis approach yielded significant quantities of chlorolissoclimide that permitted evaluation against the NCI’s 60 cell line panel and allowed us to obtain an X-ray co-crystal structure of the synthetic secondary metabolite with the eukaryotic 80S ribosome. While it shares a binding site with other imide-based natural product translation inhibitors, chlorolissoclimide engages in a particularly interesting and novel face-on halogen-π interaction between the ligand’s alkyl chloride and a guanine residue. Our analogue-oriented synthesis provided many more lissoclimide compounds, which were tested against aggressive human cancer cell lines, and for protein synthesis inhibitory activity. Finally, computational modeling was used to explain the SAR of certain key compounds, setting the stage for structure-guided design of better translation inhibitors.
Table of Contents Summary
Via semi-synthesis and total synthesis, we have made several natural and unnatural lissoclimide cytotoxins. An X-ray co-crystal structure of chlorolissoclimide with the ribosome and evaluation of cytotoxicity and translation inhibition of new compounds in the series improved our understanding of the molecular basis for cytotoxicity.
The ribosome is a large ribonucleoprotein complex with a molecular weight that varies from 2.3 MDa in bacteria to 4.3 MDa in higher eukaryotes.1 Responsible for translating mRNA to polypetides through the complex process of recruiting aminoacyl-tRNAs and catalyzing peptide bond formation, it controls protein expression and thereby sustains all cellular processes. The ribosome has therefore become an important druggable target. Although the core functions of the ribosome are conserved throughout all kingdoms of life, the additional complexity of the eukaryotic ribosome is reflected in differences in terms of structure, function, and regulation.
Natural product inhibitors of eukaryotic protein synthesis have significant therapeutic potential for treating a wide range of human cancers.2–4 As a premier example, in 2012, the FDA approved the first translation inhibitor, the naturally occurring alkaloid homoharringtonine, as Synribo® (1, Fig. 1) for the treatment of chronic myelogenous leukemia (CML).5 Molecules that suppress protein synthesis are also valuable biochemical tools; for example, cycloheximide (CHX; 2) has played an important role in experiments to determine protein half-lives,6 among many other applications.7,8
Figure 1.

Natural product inhibitors of eukaryotic translation are important as potential drugs and biochemical tools. Pictured are Synribo®, a translation inhibitor and approved drug for CML; cycloheximide, a common laboratory reagent for translation inhibition; representative lissoclimides and their cytotoxicity toward P388 murine leukemia cells (IC50); and lactimidomycin, a recently discovered cytotoxin and translation inhibitor. For a complete list of lissoclimide natural products, a few derivatives, and their cytotoxicity toward P388 cells, see ref 14. All data above compiled from refs 13–18.
The high-resolution X-ray structure of the eukaryotic Saccharomyces cerevisiae 80S ribosome, vacant9 or in complex with 16 different translation inhibitors, including CHX,10 has been recently determined, providing a better understanding of molecular mechanisms underlying the action of eukaryotic-specific inhibitors of protein synthesis. While the mechanism of inhibition was known from biochemical experiments for many of these compounds, the advent of detailed structural information from crystallographic studies opens the door to structure-based design of better or more selective inhibitors, as well as the opportunity to rationalize observed structure-activity relationships on the basis of newly understood, critical intermolecular interactions.
The structurally unusual labdane diterpenoids chlorolissoclimide (CL) and dichlorolissoclimide (DCL) (3 and 4, respectively, Fig. 1) are powerful cytotoxins toward murine leukemia and certain human carcinoma cell lines.11–13 Originally isolated from ascidians (sea squirts) by Malochet-Grivois and co-workers in the early 1990s, this small family of secondary metabolites was greatly expanded by the discovery of nearly 20 closely related compounds called the haterumaimides by the groups of Ueda and Schmitz (see representatives 5–12, Fig. 1).14–18 Each of these compounds has been tested against the P388 murine leukemia cell line, with some reaching sub-nM potencies (IC50 values) while some were completely inactive.
CL and DCL were shown by Pelletier and co-workers to be potent inhibitors of eukaryotic translation.19 They each interfere with the elongation step of protein synthesis and prevent tRNA from exiting the ribosome, resulting in polysomal accumulation and eventual cell death. In this same study, these workers noted the structural homology between CL and the well-studied translation inhibitor CHX. Clearly, the glutarimide of CHX and the succinimide of the lissoclimides are structurally similar. CHX and the glutarimide lactimidomycin (LTM, 13, Fig. 3)20–23 have each been shown to stop the translocation process of protein synthesis by inhibiting the entrance of the CCA-end of tRNA to the large ribosomal subunit (LSU) E-site, and the structural studies of Yusupov and co-workers are consistent with this understanding.10 On the other hand, the molecular basis for CL’s interaction with the ribosome remained unknown. We planned a synthesis-driven study of compounds in the lissoclimide family to learn about their cytotoxicity, their translation inhibitory activity, and the structural basis for translation inhibition.
Figure 3.

The X-ray co-crystal structure of CL with the eukaryotic 80S ribosome reveals the molecular basis of translation inhibition. a. Location of CL in a putative in silico model of an actively translating 80S ribosome. b. CL clashes with the CCA-end of the E-site tRNA. The three tRNAs and mRNA (coloured in red) for this model were taken from the PDB entry 4V6F.29 The 60S (light blue) and 40S (light orange) subunits are represented as surfaces. c. CL (yellow) binds to the 60S E-site. CL shares the same binding pocket as cycloheximide (pink) and lactimidomycin (cyan).10 Eukaryotic specific ribosomal protein eL42 is shown in green and 25S rRNA in grey. d. Detail of the interactions occurring between the CL molecule (yellow, represented as sticks) and the neighboring residues. Direct contacts take place with nucleotides G92, C93, U2763, A2802 and G2794 of the 25S rRNA. A hydrogen bond is also observed between CL and the ribosomal protein eL42.
As a first step in our studies, we recently disclosed a short semi-synthesis of CL, haterumaimide Q (hatQ, 8), and two unnatural analogues starting from the commercially available terpenoid sclareolide (14, Fig. 2a).24 This work permitted the preliminary evaluation of their activity against aggressive tumor cell lines (DU145 prostate and A2058 melanoma). In these assays, CL was the most potent of the four compounds, with sub-micromolar IC50 values recorded against each cell line.
Figure 2.

Our two synthesis approaches to the lissoclimides are designed to access a broad range of structurally perturbed analogues for a better understanding of structure-activity relationships. a. Previous short but limited semi-synthesis of lissoclimides from sclareolide; b. An analogue-oriented synthesis plan (* denotes sites of possible variant substitution and configurations)
Access to semi-synthetic CL also enabled the more in-depth investigations described herein, including an X-ray co-crystallography experiment of CL with the eukaryotic ribosome. The resulting high-resolution structure showed the molecular basis for translation inhibition by CL. Still, it was abundantly clear that our semi-synthesis approach would never provide access to numerous natural products in the lissoclimide family and structural analogues that would drive the development of a more comprehensive structure-activity relationship (SAR) model. In light of that limitation, we developed an analogue-oriented synthesis strategy25 to the lissoclimides (Fig. 2b) that was designed to probe specific SAR questions. This approach was predicated on the utility of a π-cyclization to assemble the trans-decalin (15 → 17) that, after dehydration, would bear orthogonal functional groups (A-ring alkene and B-ring ketone in 18) for elaboration to a range of differentially substituted lissoclimide natural products and analogues (16). The results described in this report provide a better understanding of the relationship of chemical structure to in vitro translation inhibition and cytotoxicity. A subsequent computational modeling study permits a rationalization of the SAR and lays the foundation for potential structure-based design of better translation inhibitors.
Results and Discussion
I. The translation inhibition activity of chlorolissoclimide can be attributed to its binding at the LSU ribosomal E-site that features novel attractive interactions
Evaluation of CL against the NCI 60 cell line panel (Supplementary Table 1) revealed relatively consistent and potent cytostatic activity (mean GI50 of 15 nM), but much more selective cytotoxicity (LC50 values above 100 μM for most cell lines, but sub-μM for certain cell lines, including the COLO-205 colon cancer, and the M14 and SK-MEL-28 melanoma cell lines). Not surprisingly, a COMPARE analysis26 yielded high correlations with other known protein synthesis inhibitors, including the quassinoid bruceantin, harringtonine and homoharringtonine, bouvardin, pactamycin, and phyllanthosides, among others. Intererestingly, CL also showed a high correlation with dactinomycin, a known transcription inhibitor27 and romidepsin, an FDA approved cyclic depsipeptide indicated for cutaneous T-cell lymphoma that is a known histone deacetylase inhibitor, and not a translation inhibitor.28
Given the structural relationship among the imide translation inhibitors CHX, LTM, and CL and the similarity in their biochemical activity, it appeared likely that the latter would bind the ribosome with overlap with the CHX and LTM binding site.10 We solved the crystal structure of the S. cerevisiae 80S ribosome in complex with a synthetic sample of the inhibitor CL at a maximal resolution of 3.0 Å (Supplementary Table 2), and found that the binding mode presents novel features. Its binding site is located at the E-site tRNA CCA-end on the large ribosomal subunit (LSU), as it was previously shown for the glutarimide inhibitors CHX and LTM (Figs. 3a and 3b). The CL chemical structure could be fit unambiguously into the difference map (Fobs–Fcalc) generated after the first cycle of refinement, where the model of vacant 80S ribosome (entry 4V88) was fitted into the electron density map as a rigid body (Supplementary Figure 1). The crystal structure was obtained by co-crystallization with a molar ratio CL/80S of 30:1; at this final concentration (33 μM) we did not observe any secondary binding site on the 80S ribosome. A comparison of the structures of the 80S/CL complex and the vacant 80S ribosome did not reveal any conformational changes in the LSU E-site upon binding of the inhibitor.9 Importantly, for the purposes of using this structural information for considerations in anti-tumor agent design, the human and murine 80S ribosomes are nearly structurally identical to the yeast ribosome used in these studies (see Supplementary Figure 2).
Comparison of CL binding with that of CHX and LTM shows a similar network of interactions of the imide-containing moiety with a number of universally conserved nucleotides of the 25S rRNA, namely G92, C93 and U2763 (Fig. 3c). Moreover, the hydroxyl group on the linker between the decalin and the succinimide moieties interacts with the phosphate-oxygen backbone of nucleotide A2802 of the 25S rRNA through another hydrogen bond, completing a tetrad of hydrogen bonds that appears to “anchor” CL into the binding site. We also observed two particularly interesting interactions of CL with the 80S ribosome. The C7-hydroxyl group present on the B-ring forms a hydrogen bond with Pro56 on a stretched loop of the eukaryotic specific ribosomal protein eL42, thus deviating from most other known inhibitors, which exclusively bind rRNA.10 Finally, the chlorine positioned on the decalin ring system interacts with G2794 of the 25S rRNA (and to a lesser extent G2793). The chlorine atom is positioned 3.2 Å away from the center of the 6-membered ring of the purine heterocycle (Fig. 3d), and it appears to form a halogen–π interaction with the guanine residue with a face-on geometry.30
A brief discussion about the apparent face-on halogen–π interaction is warranted. While examples of these attractive forces have been documented in protein–ligand interactions with the aromatic side chains of Phe, Tyr, Trp, and His,30 to the best of our knowledge they have never been reported between halogens and nucleotide bases (according to a search of the Relibase database v3.2.1, Cambridge Crystallographic Data Centre). To estimate the importance of this interaction, we calculated its magnitude in a model system using chloromethane in place of CL. Using the same geometry of the chlorine relative to the guanine ring as obtained from the co-crystal structure shown in Fig. 3, the energetic benefit obtained by calculation was 1.1 kcal/mol when the PBE-D3 method was used; this method takes advantage of a semi-empirical correction for dispersion interactions. The calculated energy benefit for the interaction of the chlorine with both G2794 and G2793 was 1.8 kcal/mol. This favorable dispersion-based interaction appears to afford stabilization to this arrangement of CL in the E-site of the ribosome, and related effects might be leveraged in the design of other nucleic acid ligands.
With CHX, LTM, and the lissoclimide family, we see a fascinating case of convergent evolution. Very different organisms (Streptomyces for the glutarimide compounds and ascidians of the genus Lissoclinum for the lissoclimides) have developed imide-based inhibitors of eukaryotic translation that function via binding to the ribosomal E-site, despite the rarity of imide functional groups in secondary metabolites.
In short, our synthesis of CL has permitted a broad examination of its activity, revealing that it acts quite generally as a cytostatic but demonstrates some interesting selectivities in its cytotoxic action. Moreover, synthetic CL was used in a crystallographic study showing that it inhibits translation by binding to the LSU E-site of the 80S ribosome with binding characteristics that had not been observed with other E-site binders, including the structurally related glutarimide inhibitors CHX and LTM. Finally, a potentially important and unprecedented interaction between an aliphatic chloride and a guanine residue has been observed.
II. An analogue-oriented synthesis strategy permits greater evaluation of structure-activity relationships and connection of cytotoxicity with translation inhibition
Inspired by the particularly potent activity of naturally occurring C18-oxygenated congeners (see 11 and 12, Fig. 1), and the large changes in reported activity arising from alteration of the substitution on the decalin motif and modifications to the tethered succinimide,14 we aimed to develop a strategy that could produce a range of analogues with changes in all of these key areas. Therefore, we targeted a synthesis design that was at once relatively efficient and readily diversifiable—an analogue-oriented synthesis—that could help us pose questions about the structural determinants of activity in these specific areas of the lissoclimide structure (see 16, Fig. 2b). We report herein the implementation of this design that affords an improved understanding of SAR in the lissoclimide family.
Integral to our design was the incorporation of our recently developed and efficient Evans-aldol-based stereocontrolled succinimide introduction,31–34 first used in the semi-synthesis of CL.24,33 This stereochemically flexible strategy35 would permit us to interrogate the importance of configuration of this part of the molecule via late-stage succinimide incorporation onto aldehyde-bearing decalin intermediates, as performed in our semi-synthesis work. These decalin cores would be produced by a cationic bicyclizations (Fig. 2b). Reduced to practice, this approach in combination with our semi-synthesis work has yielded three natural products and eight key analogues (overall close to 20 lissoclimide-like structures) with changes in all of the key portions of the molecule that we wished to examine.
Synthesis of C18-unoxygenated lissoclimides
For C18 unoxygenated targets, known acylsilane 19 is prepared as previously reported in five steps from geranyl acetate.36,37 Addition of the lithiated sulfone 2038,39 to the acylsilane triggers a Brook rearrangement and subsequent elimination of sulfinate anion,35 leading stereoselectively to E-enoxysilane 21.36,37 Lewis-acid-mediated cationic bicyclization provides trans-decalin 22 after base-mediated equilibration of the C9 substituent to the equatorial orientation. This sequence borrows from the elegant work of Corey and co-workers in their syntheses of scalarenedial36 and α-onocerin37 and perfectly installs the C3 hydroxyl group and the C8 ketone, substituents that can be used orthogonally to decorate the A and B rings, respectively.
With the decalin system built, a carefully orchestrated sequence of manipulations—first of the A ring and then the B ring, and finally succinimide introduction—was deployed. This strategy was based on the likely stability of most of the desired A-ring substitution patterns (protected C3 hydroxyl, C2–C3 alkene, fully reduced, mono- or bis-halide) to B-ring functionalizations via allylsilane chemistry, and the presumed sensitivity of the hydroxyimide, which would be installed last. Therefore, deprotection of the silyl ether and elimination under Mitsunobu conditions provided alkene 24 that can be elaborated in a variety of ways to generate A-ring analogues. For example, hydrogenation delivered an A-ring reduced intermediate that was taken forward by conversion of the C8 ketone to the allylsilane of 25 via enolate triflation and Kumada–Corriu coupling.40 This allylsilane was designed to permit incorporation of varied C7 functionality while simultaneously unveiling the exocyclic alkene via electrophilic SE′ reactions.41 Attempted oxidation of the allylsilane with several oxygen atom transfer reagents led to the unnatural configuration of the C7 hydroxyl group (see below for an analogue incorporating that feature). In a critical sequence, bromination stereoselectively provided a surprisingly robust allylic α-bromide, and aldehyde 26 was obtained after dioxolane cleavage. Application of our Evans-aldol-based succinimide introduction24 delivered 28. Finally, potassium-superoxide-mediated displacement42 completed a total synthesis of hatQ (8).24 In this way, we took advantage of the inherent stereocontrol of allylsilane SE′ functionalization, and carried a remarkably stable allylic bromide through multiple steps as a surrogate for the C7 hydroxyl group. The synthesis of this natural product was achieved via a relatively concise and robust sequence, providing 8 in 1.6% overall yield from acylsilane 19. While not as direct as our semi-synthesis, this independent synthesis of hatQ validates our approach. Furthermore, this strategy by design provides reasonable efficiency in the context of a readily divertible plan that has facilitated access to the collection of A- and B-ring analogues described below, and it therefore satisfies the ideals of an analogue-oriented synthesis.
Synthesis of C18-oxygenated lissoclimides
With the route toward C18 unoxygenated lissoclimides established, this plan was co-opted to incorporate C18 oxygenation, one of the potential sources of the reported potency of hatJ and hatK (Fig. 4b). Intermediate 29 was procured via allylic oxidation and para-methoxybenzyl (PMB) protection of a geraniol derivative, and then by a path parallel to that used in the synthesis of acylsilane 19 (Fig. 4a, see SI for details). Addition of lithiated sulfone 20 to the acylsilane afforded π-cyclization substrate 30. A change in conditions was required for bicyclization in this C18-oxygenated case, and the use of boron trifluoride etherate was found to be optimal,43 yielding decalin 31 after base-mediated equilibration at C9. Under these conditions, silyl group transfer was not observed. Mitsunobu-type elimination and removal of the PMB ether led to alkene 32.
Figure 4.

Our analogue-oriented synthesis approaches to the lissoclimides and congeners are designed to access point changes in the natural product structures at key positions. a. A readily diversifiable synthesis of C18-unoxygenated lissoclimides is exemplified by a second, independent synthesis of hatQ. b. Synthesis of highly functionalized lissoclimide analogues with C18 oxygenation as found in highly cytotoxic natural product hatJ.
The A-ring alkene was subjected to dichlorination under several conditions. In contrast to the dichlorination of C18-unoxygenated A-ring alkene 24 (see Fig. 4a), which was high-yielding with Mioskowski’s reagent44 and afforded only the trans-diaxial vicinal dichloride (not shown, see SI), neither dichlorination of homoallylic alcohol 32 nor that of its PMB-protected precursor was straightforward. Our group has significant experience with alkene dichlorination and the remarkable difference that the substituents on proximal alcohols can have on diastereoselection. We have observed one particularly relevant case of selectivity reversal in a homoallylic alcohol superimposed on a cyclohexene ring,45 and others wherein transient installation of electron-deficient acyl groups markedly improved reaction behavior.46,47 In the case of 32, the most interesting outcome came from a one-pot trifluoroacetylation/dichlorination sequence that provided a nearly equimolar mixture of expected diaxial and unexpected diequatorial dichlorides 33 (not separated at this stage). Of course, the formation of the latter contravenes the Fürst–Plattner principle and provides access to the naturally occurring configurations at C2 and C3 as found in DCL and hatA. At this stage, we do not have any explanation for this phenomenon.
Operating on the mixture of dichloride diastereomers, silylation of the hydroxyl group, Wittig olefination, and unmasking of the aldehyde provided aldol substrates 34 and 35 after a facile separation. The aldol-based, stereoselective succinimide introduction described in Fig. 4a was applied to each aldehyde, providing highly functionalized, C18-hydroxylated lissoclimides 36 and 37. The latter has features of both DCL and hatJ, two of the most potent lissoclimides. Of course, similar chemistry as described above can be used to convert the B-ring ketone to the corresponding allylsilane for further functionalization, and the A-ring can be manipulated in several ways other than chlorination. Several examples of such lissoclimide analogues are shown in the description of the SAR, below.
Expanded structure-activity understanding in the lissoclimide family
Compounds obtained via our sclareolide-based semi-synthesis and analogue-oriented synthesis approaches were tested against P388 murine leukemia cell lines (to correlate with the natural products that had previously been tested in this assay14) as well as against the aggressive melanoma (A2058) and prostate cancer (DU145) cell lines (Table 1). Select, key compounds were also tested for eukaryotic translation inhibition. These experiments have provided a greater understanding of SAR among lissoclimide-type compounds.
Table 1.
Cytotoxicity and translation inhibitory activities of synthetic lissoclimide analogues reveal new aspects of structure-activity relationships. Cytotoxicity values shown are measured IC50 values in μM. Translation inhibition assay: cell lysates prepared from Krebs-2 cells containing mRNA for firefly luciferases are treated with compounds at varying doses of drug, and the data are presented as approximate IC50 values. See the Supplementary Information for more details and for a similar assay with renilla luciferases.
 38 |
 39 |
 40 |
 41 |
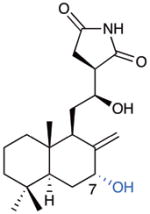 42 |
||
|---|---|---|---|---|---|---|
|
|
||||||
| Cell Line | P388 | 0.43 | 2.4 | >10 | >10 | 3.7 |
| A2058 | 1.91 | >10 | >10 | >10 | >10 | |
| DU145 | 1.91 | >10 | >10 | >10 | 6.8 | |
|
|
||||||
| Translation | ca. 50 μM | |||||
|
|
||||||
 8: hatQ |
 3: CL |
 7: hatN |
 43 |
 44 |
||
|
|
||||||
| Cell Line | P388 | 0.20 (lit = 0.10) | 0.06 (lit = 0.004) | 2.1 (lit = 0.007) | 0.32 | 0.46 |
| A2058 | 2.34 | 0.25 | 3.46 | 0.63 | 0.18 | |
| DU145 | 1.49 | 0.49 | 0.46 | 1.13 | 0.57 | |
|
|
||||||
| Translation | ca. 5 μM | ca. 0.5 μM | 1–5 μM | |||
|
|
||||||
 45 |
 36 |
 37 |
 1: homoharringtonine |
 2: CHX |
||
|
|
||||||
| Cell Line | P388 | 0.14 | 0.70 | 0.33 | 0.05 | 5.6 |
| A2058 | 0.97 | 2.06 | 1.34 | 0.17 | >10 | |
| DU145 | 0.37 | 9.42 | 2.19 | 0.23 | >10 | |
|
|
||||||
| Translation | ca. 1 μM | ca. 1–5 μM* | ca. 0.5 μM | ca. 0.5 μM | ||
|
|
||||||
The value estimated for 36 is based on ca. 37% inhibition at 1 μM; there was insufficient sample to test at higher concentrations.
It is perhaps easiest to unveil SAR components by comparison with the relatively simple lissoclimide analogue 38.24 It shows sub-μM activity against P388 cells, and low μM activity against the invasive melanoma and prostate cell lines. With the previous knowledge that adulteration of the hydroxysuccinimide motif (in the form of N-methylation, O-acetylation, or dehydration) abolishes cytotoxic activity against P388 cells,14 we examined compound 39, with opposite configurations at C12 and C13 and found that this perturbation also negatively impacts activity. The presence of a C3 hydroxy group (as in 40), a vestige of the epoxide-initiated π-cyclization, is completely detrimental; we had wondered whether such an equatorially disposed alcohol might make similar contacts to those potentially made by the C18 hydroxy groups found in hatJ (reported IC50 value against P388 = 600 pM).14 Interestingly, diene 41 was completely inactive, in spite of the relatively small perturbation with respect to 38. The unnatural configuration of C7-hydroxy group in 42 has a negative impact on activity as compared with both the C7-unoxygenated 38 and the naturally configured hatQ (824), which is only slightly more active than 38. In this series of A-ring unchlorinated lissoclimides, the A-ring appears to be best totally unfunctionalized. Further, B-ring C7-oxygenation does not seem to be mandatory for activity; the natural configuration of the C7 hydroxyl group is not detrimental, while the unnaturally configured alcohol depresses activity dramatically. Taken together with SAR known from lissoclimides obtained from nature,14 it appears that there is little flexibility with respect to the hydroxysuccinimide, and oxygenation on the B-ring (C6 or C7) is not critical for activity.
Next, different combinations of “natural substituents” were assessed. Comparison of hatQ (8) to CL (324) showed the significant impact of a C2 chloride; CL emerged as the most active compound that we made in this work (ca. 60 nM, compare 4.4 nM reported previously).11 Unexpectedly, we note that inclusion of a C2 chloride on the C7-unoxygenated scaffold, giving 43,24 provides little increase in activity (compare 8). A small sample of natural DCL (4) was obtained and tested (IC50 of ca. 400 nM;11 compare 2.4 nM reported previously, data not shown in Figure 5). Acetylation of the C7 hydroxy group as in hatN (7) was largely detrimental to activity while excision of C7 oxygenation detracted moderately (compare pair 38/3, wherein this change had little consequence). HatN is poorly active against P388, with a large discrepancy from the previously reported cytotoxicity of 7 nM, but shows sub-μM activity against DU145. Having been unable to generate the C2/C3 diequatorial (natural) dichloride in the C18-unoxygenated series, we compared the diaxial diastereomer of DCL (44) and found activity similar to A-ring unchlorinated hatQ. Without the C7 hydroxy group, diaxial dichloride 45 maintains sub-μM activity across the three cell lines. Overall, a C2 equatorial chloride appears to lead to increased potency and with its presence, the C7 hydroxyl adds further potency; most surprisingly, diaxial dichlorides are well tolerated.
Figure 5.

Crystallographically derived binding arrangement of CL in the 80S ribosome and predicted binding poses of three analogue structures to rationalize aspects of the experimentally determined SAR. a. Co-crystal structure of CL with the 80S ribosome. b. Docking-based structure of analogue 39 bound to the 80S ribosome. c. Docking-based structure of analogue 45 bound to the 80S ribosome. d. Docking-based structure of analogue 37 bound to the 80S ribosome.
Having ruled out critically important roles for the C7 hydroxy group, and the C2 and C3 chloride groups, we hoped that C18 oxidation was a uniquely important contributor to the potent reported cytotoxicity (sub-nM activity reported against P388) of hatJ and hatK (11 and 12, respectively). However, the relatively modest cytotoxicity of 36 and 37, especially the latter, was very surprising. Diequatorial dichloride 37 is effectively C3-chloro-hatJ, which we anticipated would be incredibly potent; unfortunately and surprisingly, it proved less active than CL.
Finally, we tested several of the more intriguing compounds for protein synthesis inhibition in the firefly luciferase and renilla luciferase translation assays (see Fig. 5 for IC50 data for the former; titration data for both assays are found in the SI).19,48 Although most of the lissoclimides and analogues were at best poorly active, CL proved to be a potent inhibitor, and 45 and 36 also showed reasonable activity. We note that our value for translation inhibition obtained with synthetic CL matches that obtained previously with natural CL (IC50 values for CL ~ 0.7 μM, DCL ~ 1.25 μM).19 This rough correlation of translation inhibition to cytotoxicity supports the implication of this activity as the main driver of cytotoxicity. That there is an imperfect correlation is not so surprising; the differing physical properties of the compounds could lead to different outcomes in the biochemical and the cellular assays. Further, as with any bioactive compound, the possibility exists for multiple mechanisms of action.
We also compared our compounds to homoharringtonine (1), the alkaloid that is a marketed drug for CML, and the translation inhibitor CHX (2). With respect to the former, we find that CL is about equivalently cytotoxic to homoharringtonine in the three cell lines examined, and also matches it in translation inhibitory activity. CHX was significantly less cytotoxic than many of our synthetic lissoclimides, but its translation inhibitory activity was equivalent to CL and homoharringtonine. It is worthy of note that, in our hands, the cytotoxicity against P388 cells of hatQ differed by a factor of two, CL by an order of magnitude, and both DCL and hatN by over two orders of magnitude compared with the data obtained using natural lissoclimides previously disclosed by the Malochet-Grivois and Ueda groups.11,14,16
This first round of implementation of our analogue-oriented synthesis study has shown that both C18 oxygenation and C7 hydroxylation are not critical for potency. However, A-ring chlorination patterns have variable effects, and the configurations of the hydroxysuccinimide are important. With respect to cytotoxicity, we have learned that CL is at least a local maximum, and obvious perturbations of its structure either fail to improve or abolish its activity. Furthermore, it has permitted a rough correlation of cytotoxicity with protein synthesis inhibitory activity, which further supports that translation is the dominant target of these compounds. CL and analogue 45 demonstrate potent cytotoxicity and translation inhibition, and we wished to learn more about the determinants of activity in these cases.
Computational modeling sheds light on important interactions
The co-crystal structure of CL (3) with the eukaryotic 80S ribosome provides a starting point to rationalize our observed SAR with computational tools. Furthermore, because CL was the most potent of all of our compounds, it was a logical starting point to explain how structural deviations might account for decreased activity relative to this benchmark. It is clear from the co-crystal structure (Figs. 3 and 5) that each functional group in CL plays a role in binding; contacts are made between the rRNA and the hydroxysuccinimide motif (four points of contact) and the C2 chlorine. The latter appears to make halogen–π interactions with G2794 and G2793, enabling the hydrophobic A-ring of CL to adopt a stable conformation in a groove between the two guanine residues. Further, the C7 hydroxy group makes a contact to a proline amide oxygen on the eL42 protein component of the ribosome.
Our computational approach involved several different sets of docking calculations, as well as a hybrid between docking and shape overlays, to explain the observed trends in cytotoxicity (see the SI for details).49–51 Prior to the availability of the co-crystal structure shown above in Fig. 3, we used these methods and the co-crystal structure of CHX/80S10 for guidance to obtain a remarkably similar binding pose of CL (not shown) to the one obtained crystallographically. This result provided confidence in our computational methods. Starting from our co-crystal structure of 3, we exchanged substituents to evaluate the fit of our analogues in the binding pocket (Fig 5). New functionality present in many analogues, including 39, 45, and 37, required conformational changes in the ligand or translation of the ligand for reasonable binding.
Analogue 39, epimeric at C12/13, retains modest activity despite changes to the C12-hydroxy/C13-succinimide stereochemistry (2.4 μM, P388). Our predicted binding pose (Fig. 5) suggests that the C12-OH forms a hydrogen bond with the same C2 cytosine carbonyl (C2765) as CHX (see also Fig. 3c).
We were surprised to find that analogue 45, bearing C2/C3 diaxial chlorides, is the second most potent compound we have tested despite the unnatural stereochemistry of the chloride-bearing centers as compared with DCL (4, Fig. 1). As shown in Fig. 6, the B-ring of 45 adopts a twist-boat conformation where the then pseudo-equatorial chlorides are poised for two halogen-π interactions on facing guanines (G2793/4). We have performed ab initio calculations on the C2/C3 diepi dichloride-bearing trans-decalin substructure (see SI for details). The calculated energy difference between the chair (axial chlorides) and twist-boat conformation that reorients the chlorides in a pseudo-equatorial fashion is less than 1.5 kcal/mol, presumably owing to alleviation of multiple 1,3-diaxial interactions. It is plausible that halogen-π interactions with both chlorines could compensate for the small energy requirement to induce this conformational change.
We expected 37 to be much more potent than 45 because it has features of both DCL (4) and hatJ (11)—two natural products with a greater reported activity than 3. Surprisingly, 37 was less potent than 45. When a C3 chlorine substituent is placed on our co-crystal structure of 3, this chloride clashes with guanine N3 (G2793). In our hybrid binding pose (37, Fig. 6) the B-ring is therefore pushed away from the guanine, disrupting the C2 halogen–π interaction. Additionally we found that the C18 hydroxyl group does not make any strong hydrogen bonding interactions, although a contact is made with the proximal guanine moiety. In combination with the modest activity of 37 and 38, these studies suggest that C18 oxygenation has little effect on potency. We remain hopeful that activity would increase with removal of the C3 chlorine of 37 and are currently working on a novel route towards hatJ (11), the most potent naturally occurring lissoclimide.
Conclusions
Via a combination of semi-synthesis and analogue-oriented synthesis, supported by computational modeling, we have expanded our understanding of SAR in the lissoclimide family of translation-inhibiting cytostatic agents. We have also uncovered the structural basis for inhibition of protein synthesis with a co-crystallographic study of synthetic CL bound to the ribosome. The fascinating binding mode includes a novel face-on chlorine-π interaction of the ligand with a guanine residue.
We designed a robust analogue-oriented synthesis route to efficiently access a diverse range of lissoclimide analogues. A synthesis of hatQ validated this route and extrapolation to the production of analogues bearing key functional groups permitted us to glean information on the pharmacophoric elements of the family. Strategic features of this reliable synthesis include (1) a polyene cyclization; (2) the incorporation of an A-ring alkene and a B-ring allylsilane for robust diversification; and (3) a dependable aldol-based, late-stage introduction of the critical hydroxysuccinimide motif. Other interesting aspects of the chemistry include the retention of an allylic bromide through multiple operations only to displace it with KO2 in the final step, and a context-dependent cyclohexene chlorination to give both diaxial and diequatorial chlorides. We evaluated the cytotoxicity of three synthetic lissoclimide natural products and 10 analogues against P388 murine leukemia and two aggressive human cancer cell lines. Many of our compounds showed promising activity against the latter, more important cell lines. We also determined translation inhibitory activity for selected compounds. CL was the most potent cytotoxin tested (IC50 = 59 nM against P388) and exhibited greater translation inhibitory activity than the marketed CML drug Synribo®. Finally, the most likely binding poses of key analogues were predicted computationally to help rationalize the observed activities.
Altogether, the combination of a robust but divergent synthesis, the important structural information from the crystallographic analysis, and our computational studies has permitted a greater understanding of the translation inhibition and cell-killing ability of the lissoclimides.
Supplementary Material
Acknowledgments
Preliminary studies in the Vanderwal laboratory were supported by grants from the National Institutes of Health and the UC Cancer Research Coordinating Committee (GM-086483 and UCCRCC-55179, respectively, to C.D.V.). We acknowledge the Developmental Therapeutics Program of the National Cancer Institute for the evaluations of CL in the NCI-60 panel. The work in the Yusupov laboratory was supported by the French National Research Agency ANR-15-CE11-0021-01 (to G.Y), and by European Research Council advanced grant 294312 (to S.P., M.M., M.Y.). M.Y. thanks the Russian Government Program of Competitive Growth of Kazan Federal University. The Yusupov group is grateful to the staff of PROXIMA 1 beamline at the synchrotron SOLEIL (France) and, in particular, to Leonard Chavas and Pierre Legrand for providing rapid access and assisting with data collection. D.L.M. and C.Z. appreciate support from the NIH (GM-108889), and C.Z. was supported by a Brazilian Science Without Borders fellowship administered by Capes/LASPAU. The results from the Horne laboratory reported in this publication derived from work performed in the Drug Discovery and Structural Biology Core of City of Hope Comprehensive Cancer Center supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The work of Dr. Vamsee Voora from the laboratory of Prof. Filipp Furche (UCI Chemistry) was supported by the NSF (CHE-1464828).
Footnotes
Data availability statement
All X-ray crystallographic coordinates have been deposited with the CCD. All other relevant data not directly provided in the paper of the Supplementary Information are available from the authors upon request.
Author contributions
C.D.V. and M.Y. designed the research with assistance from Z.A.K., G.Y., D.L.M., J.P., and D.E.H. All synthetic chemistry was performed by Z.A.K., A.R.S., and S.E.M. M.M. purified and crystallized the yeast 80S ribosome. S.P. and M.M. performed data collection at the synchrotron source. S.P. carried out data processing, structure determination and interpretation of the CL/80S structure, with inputs from M.M, G.Y. and M.Y. Computational studies were carried out by C.Z. and V.V., cytotoxicity experiments were performed by S.N., and translation inhibition data were obtained by R.M. C.D.V. wrote the manuscript with contributions from all authors; all of the authors helped with refining the manuscript and approved the final version.
References and Footnotes
- 1.Melnikov S, Ben-Shem A, Garreau de Loubresse N, Jenner L, Yusupova G, Yusupov M. One core, two shells: bacterial and eukaryotic ribosomes. Nat Struct Mol Bio. 2012;19:560–567. doi: 10.1038/nsmb.2313. [DOI] [PubMed] [Google Scholar]
- 2.Mamane Y, Petroulakis E, Rong L, Yoshida K, Ler LW, Sonenberg N. eIF4E–from translation to transformation. Oncogene. 2004;23:3172–3179. doi: 10.1038/sj.onc.1207549. [DOI] [PubMed] [Google Scholar]
- 3.Heys SD, Park KGM, McNurlan MA, Calder AG, Buchan V, Blessing K, Eremin O, Garlick PJ. Measurement of tumour protein synthesis in vivo in human colorectal and breast cancer and its variability in separate biopsies from the same tumour. Clin Sci. 1991;80:587–593. doi: 10.1042/cs0800587. [DOI] [PubMed] [Google Scholar]
- 4.Malina A, Mills JR, Pelletier J. Emerging therapeutics targeting mRNA translation. Cold Spring Harb Perspect Biol. 2014 doi: 10.1101/cshperspect.a012377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gandhi V, Plunkett W, Cortes JE. Omacetaxine: a protein translation inhibitor for treatment of chronic myelogenous leukemia. Clin Cancer Res. 2014;20:1735–1740. doi: 10.1158/1078-0432.CCR-13-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belle A, Tanay A, Bitincka L, Shamir R, O’Shea EK. Quantification of protein half-lives in the budding yeast proteome. Proc Natl Acad Sci USA. 2006;103:13004–13009. doi: 10.1073/pnas.0605420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stern-Ginossar N, Weisburd B, Michalski A, Le VTK, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, Mann M, Ingolia NT, Weissman JS. Decoding human cytomegalovirus. Science. 2012;338:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science. 2011;334:1524–1529. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- 10.Garreau de Loubresse N, Prokhorova I, Holtkamp W, Rodnina MV, Yusupova G, Yusupov M. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014;513:517–522. doi: 10.1038/nature13737. [DOI] [PubMed] [Google Scholar]
- 11.Malochet-Grivois C, Cotelle P, Biard JF, Hénichart JP, Debitus C, Roussakis C, Verbist JF. Dichlorolissoclimide, a new cytotoxic labdane derivative from Lissoclinum voeltzkowi Michaelson (Urochordata) Tetrahedron Lett. 1991;32:6701–6702. [Google Scholar]
- 12.Toupet L, Biard JF, Verbist JF. Dichlorolissoclimide from Lissoclinum voeltzkowi Michaelson (Urochordata): crystal structure and absolute stereochemistry. J Nat Prod. 1996;59:1203–1204. [Google Scholar]
- 13.Malochet-Grivois C, Roussakis C, Robillard N, Biard JF, Riou D, Debitus C, Verbist JF. Effects in vitro of two marine substances, chlorolissoclimide and dichlorolissoclimide, on a non-small-cell bronchopulmonary carcinoma cell line (MSCLC-N6) Anti-Cancer Drug Design. 1992;7:493–502. [PubMed] [Google Scholar]
- 14.Uddin J, Ueda K, Siwu ERO, Kita M, Uemura D. Cytotoxic labdane alkaloids from an ascidian Lissoclinum sp.: isolation, structure elucidation, and structure-activity relationship. Bioorg Med Chem. 2006;14:6954–6961. doi: 10.1016/j.bmc.2006.06.043. [DOI] [PubMed] [Google Scholar]
- 15.Fu X, Palomar AJ, Hong EP, Schmitz FJ, Valeriote FA. Cytotoxic lissoclimide-type diterpenes from the molluscs Pleurobranchus albiguttatus and Pleurobranchus forskalii. J Nat Prod. 2004;67:1415–1418. doi: 10.1021/np0499620. [DOI] [PubMed] [Google Scholar]
- 16.Uddin MJ, Kokubo S, Ueda K, Suenaga K, Uemura D. Haterumaimides J and K, potent cytotoxic diterpene alkaloids from the ascidian Lissoclinum species. Chem Lett. 2002;10:1028–1029. doi: 10.1021/np010066n. [DOI] [PubMed] [Google Scholar]
- 17.Uddin MJ, Kokubo S, Ueda K, Suenaga K, Uemura D. Haterumaimides F–I, four new cytotoxic diterpene alkaloids from an ascidian Lissoclinum species. J Nat Prod. 2001;64:1169–1173. doi: 10.1021/np010066n. [DOI] [PubMed] [Google Scholar]
- 18.Uddin MJ, Kokubo S, Suenaga K, Ueda K, Uemura D. Haterumaimides A–E, five new dichlorolissoclimide-type diterpenoids from an ascidian Lissoclinum species. Heterocycles. 2001;54:1039–1047. [Google Scholar]
- 19.Robert F, Gao HQ, Donia M, Merrick WC, Hamann MT, Pelletier J. Chlorolissoclimides: new inhibitors of eukaryotic protein synthesis. RNA. 2006;12:717–724. doi: 10.1261/rna.2346806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Kiu JO. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Bio. 2010;6:209–217. doi: 10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sugawara K, Nishiyama Y, Toda S, Komiyama N, Hatori M, Moriyama T, Sawada Y, Kamei H, Konishi M, Oki T. Lactimidomycin, a new glutarimide group antibiotic. Production, isolation, structure and biological activity. J Antibiot. 1992;45:1433–1441. doi: 10.7164/antibiotics.45.1433. [DOI] [PubMed] [Google Scholar]
- 22.Ju J, Lim SK, Jiang H, Seo JW, Shen B. Iso-migrastatin congeners from Streptomyces platensis and generation of a glutarimide polyketide library featuring the dorrigocin, lactimidomycin, migrastatin, and NK30424 scaffolds. J Am Chem Soc. 2005;127:11930–11931. doi: 10.1021/ja053118u. [DOI] [PubMed] [Google Scholar]
- 23.Micoine K, Persich P, Llaveria J, Lam MH, Maderna A, Loganzo F, Fürstner A. Total syntheses and biological reassessment of lactidomycin, isomigrastatin and congener glutarimide antibiotics. Chem Eur J. 2013;19:7370–7383. doi: 10.1002/chem.201300393. [DOI] [PubMed] [Google Scholar]
- 24.Quinn RK, Könst ZA, Michalak SE, Schmidt Y, Szklarski AR, Flores AR, Nam S, Horne DA, Vanderwal CD, Alexanian EJ. Site-selective aliphatic C–H chlorination using N-chloroamides enables a synthesis of chlorolissoclimide. J Am Chem Soc. 2016;138:696–702. doi: 10.1021/jacs.5b12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seiple IB, Zhang Z, Jakubec P, Langlois-Mercier A, Wright PM, Hog DT, Yabu K, Allu SR, Fukuzaki T, Carlsen PN, Kitamura Y, Zhou X, Condakes ML, Szczypiński FT, Green WD, Myers AG. A platform for the discovery of new macrolide antibiotics. Nature. 2016;533:338–345. doi: 10.1038/nature17967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. [accessed 24 March 2017]; https://dtp.cancer.gov/databases_tools/docs/compare/compare.htm.
- 27.Hollstein U. Actinomycin. Chemistry and mechanism of action. Chem Rev. 1974;74:625–652. [Google Scholar]
- 28.Nakajima H, Kim YB, Terano H, Yoshida M, Horinouchi S. FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exper Cell Res. 1998;241:126–133. doi: 10.1006/excr.1998.4027. [DOI] [PubMed] [Google Scholar]
- 29.Jenner LB, Demeshkina N, Yusupova G, Yusupov M. Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nat Struct Mol Biol. 2010;17:555–560. doi: 10.1038/nsmb.1790. [DOI] [PubMed] [Google Scholar]
- 30.Imai YN, Inoue Y, Nakanishi I, Kitaura K. Cl–π interactions in protein–ligand complexes. Protein Sci. 2008;17:1129–1137. doi: 10.1110/ps.033910.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evans DA, Bartroli J, Shih TL. Enantioselective aldol condensations. 2 Erythro-selective chiral aldol condensations via boron enolates. J Am Chem Soc. 1981;103:2127–2129. [Google Scholar]
- 32.Hajra S, Giri AK, Karmakar A, Khatua S. Asymmetric aldol reactions under normal and inverse addition modes of the reagents. Chem Commun. 2007:2408–2410. doi: 10.1039/b618699h. [DOI] [PubMed] [Google Scholar]
- 33.Nguyen TM, Vu NQ, Youte JJ, Lau J, Cheong A, Ho YS, Tan BSW, Yogonathan K, Butler MS, Chai CLL. A fast and straightforward route towards the synthesis of the lissoclimide class of anti-tumour agents. Tetrahedron. 2010;66:9270–9276. [Google Scholar]
- 34.González MA, Romero D, Zapata B, Betancur-Galvis L. First synthesis of lissoclimide-type alkaloids. Lett Org Chem. 2009;6:289–292. [Google Scholar]
- 35.Walker MA, Heathcock CH. Extending the scope of the Evans asymmetric aldol reaction: preparation of anti and “non-Evans” syn aldols. J Org Chem. 1991;56:5747–5750. [Google Scholar]
- 36.Corey EJ, Luo G, Lin LS. A simple enantioselective synthesis of the biologically active tetracyclic marine sesterterpene Scalarenedial. J Am Chem Soc. 1997;119:9927–9928. [Google Scholar]
- 37.Mi Y, Schreiber JV, Corey EJ. Total synthesis of (+)-α-onocerin in four steps via four-component coupling and tetracyclization steps. J Am Chem Soc. 2002;124:11290–11291. doi: 10.1021/ja027373f. [DOI] [PubMed] [Google Scholar]
- 38.Craig D, Pennington MW, Warner P. Template-directed intramolecular C-glycosidation. Total synthesis of 2,3-dideoxy-D-manno-2-octopyranosonic acid. Tetrahedron Lett. 1995;36:5815–5818. [Google Scholar]
- 39.Bonete P, Nájera C. Lithiated 3-tosylpropanal and 4-tosyl-2-butanone dimethyl acetals as β-acylvinyl anion equivalents for the synthesis of unsaturated 1,4-dicarbonyl compounds and α,β-butenolides. Tetrahedron. 1995;51:2763–2776. [Google Scholar]
- 40.Takahashi K, Takeda K, Honda T. Efficient formal synthesis of (±)-axamide-1 and (±)-axisonitrile-1 via an intramolecular Hosomi–Sakurai reaction. Tetrahedron Lett. 2010;51:3542–3544. [Google Scholar]
- 41.Dowling MS, Vanderwal CD. Ring-closing metathesis of allylsilanes as a flexible strategy toward cyclic terpenes. Short syntheses of teucladiol, isoteucladiol, poitediol and dactylol, and an attempted synthesis of caryophyllene. J Org Chem. 2010;75:6908–6922. doi: 10.1021/jo101439h. [DOI] [PubMed] [Google Scholar]
- 42.San Filippo J, Jr, Chern C-I, Valentine JS. The reaction of superoxide with alkyl halides and tosylates. J Org Chem. 1975;40:1678–1680. [Google Scholar]
- 43.Tanis SP, Deaton MV, Dixon LA, McMills MC, Raggon JW, Collins MA. Furan-terminated N-acyliminium ion initiated cyclizations in alkaloid synthesis. J Org Chem. 1998;63:6914–6928. doi: 10.1021/jo980718l. [DOI] [PubMed] [Google Scholar]
- 44.Schlama T, Gabriel K, Gouverneur V, Mioskowski C. Tetraethylammonium trichloride: a versatile reagent for chlorinations and oxidations. Angew Chem Int Ed. 1997;36:2342–2344. [Google Scholar]
- 45.Tartakoff SS, Vanderwal CD. A synthesis of the ABC tricyclic core of the clionastatins serves to corroborate their proposed structures. Org Lett. 2014;16:1458–1461. doi: 10.1021/ol500265v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vogel CV, Pietraskiewicz H, Sabry OM, Gerwick WH, Valeriote FA, Vanderwal CD. Enantioselective, divergent syntheses of several polyhalogenated Plocamium monoterpenes and evaluation of their selectivity for solid tumors. Angew Chem Int Ed. 2014;53:12205–12209. doi: 10.1002/anie.201407726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shibuya GM, Kanady JS, Vanderwal CD. Stereoselective dichlorination of allylic alcohol derivatives to access key stereochemical arrays of the chlorosulfolipids. J Am Chem Soc. 2008;130:12514–12518. doi: 10.1021/ja804167v. [DOI] [PubMed] [Google Scholar]
- 48.Novac O, Guenier AS, Pelletier J. Inhibitors of protein synthesis identified by a high throughput multiplexed translation screen. Nucleic Acids Res. 2004;32:902–915. doi: 10.1093/nar/gkh235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.ROCS 3.2.1.4. OpenEye Scientific Software; Sante Fe, NM: http://www.eyesopen.com. [Google Scholar]
- 50.OEdocking v3.0.1: McGann M. FRED pose prediction and virtual screening accuracy. J Chem Inf Model. 2011;51:578–596. doi: 10.1021/ci100436p.
- 51.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comp Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.