

Abstract
Purpose
Progression of prostate cancer (PC) to the lethal castrate-resistant (CR) stage coincides with loss of responsiveness to androgen deprivation and requires development of novel therapies. We previously provided proof-of-concept that Stat5a/b is a therapeutic target protein for PC. Here we demonstrate that pharmacological targeting of Jak2-dependent Stat5a/b signaling by the Jak2 inhibitor AZD1480 blocks CR growth of PC.
Experimental Design
Efficacy of AZD1480 in disrupting Jak2-Stat5a/b signaling and decreasing PC cell viability was evaluated in PC cells. A unique PC xenograft mouse model (CWR22Pc), which mimics PC clinical progression in patients, was used to assess in vivo responsiveness of primary and CR PC to AZD1480. Patient-derived clinical PCs, grown ex vivo in organ explant cultures, were tested for responsiveness to AZD1480.
Results
AZD1480 robustly inhibited Stat5a/b phosphorylation, dimerization, nuclear translocation, DNA binding and transcriptional activity in PC cells. AZD1480 reduced PC cell viability sustained by Jak2-Stat5a/b signaling through induction of apoptosis, which was rescued by constitutively active Stat5a/b. In mice, pharmacological targeting of Stat5a/b by AZD1480 potently blocked growth of primary androgen-dependent as well as recurrent CR CWR22Pc xenograft tumors, and prolonged survival of tumor-bearing mice vs. vehicle or docetaxel-treated mice. Finally, 9 of 13 clinical PCs responded to AZD1480 by extensive apoptotic epithelial cell loss, concurrent with reduced levels of nuclear Stat5a/b.
Conclusions
We report the first evidence for efficacy of pharmacological targeting of Stat5a/b as a strategy to inhibit CR growth of PC, supporting further clinical development of Stat5a/b inhibitors as therapy for advanced PC.
Keywords: Stat5a/b, Jak2 inhibition, castrate-resistant prostate cancer
Introduction
Prostate cancer (PC) cells initially require androgens and androgen receptor (AR) signaling for sustained growth and survival, a dependency exploited by androgen deprivation as a first-line therapy for advanced PC (1–4). The median duration of response to androgen deprivation of advanced PC is less than three years, after which castrate-resistant prostate cancer (CRPC) emerges (1–4). Additional hormonal manipulations and conventional chemotherapy may be employed but have limited impact on overall survival in CRPC (4) and, therefore, new and more efficient therapies for CRPC are needed. Development of CRPC has been attributed to numerous molecular mechanisms, including: 1) somatic mutations of AR resulting in increased affinity for ligands (5); 2) amplification of the AR gene locus (6); 3) biosynthesis of androgens within PC cells from adrenal steroids and cholesterol (7); 4) expression of constitutively active alternative splice variants of AR which do not require ligand to support PC growth (8–11); and 5) promotion of PC cell growth and survival by protein kinase pathways which act through stimulation of AR (12, 13).
Distinct from these AR-dependent pathways, it is recognized that protein kinase signaling pathways are also capable of stimulating CRPC growth independently of AR signaling. Identification of new therapeutic targets in signaling pathways that bypass AR may provide a novel strategy for treatment of advanced PC. Stat5a/b has been identified and validated as a potential therapeutic target protein in PC (14–22). A rationale for the clinical use of Stat5a/b inhibitors is provided by the finding that Stat5a/b promotes PC growth and tumor progression through both AR-dependent and AR-independent mechanisms (12, 14–17, 20). While Stat5a/b increases transcriptional activity of AR and proliferation of AR-positive PC cells (12), Stat5a/b is also critical for viability of PC cells that are negative for AR (17), suggesting that targeting Stat5a/b may provide a dual strategy to inhibit growth of CRPC by suppressing AR-dependent as well as AR-independent pathways. Activated Stat5a/b and autocrine prolactin (Prl) levels are associated with PCs of high histological grades (21, 23), CRPCs (21, 23) and distant metastases (18, 23), while absent in normal prostate epithelium (14). Stat5a/b is involved in progression to advanced PC, as evidenced by the ability of Stat5a/b to promote metastasis of PC cells as well as enhance hallmarks of the epithelial-to-mesenchymal transition that precedes metastasis (18). Activated Stat5a/b expression in PC was recently identified as a significant predictor of both early disease recurrence and PC-specific death (21, 22), implicating Stat5a/b involvement in growth and progression of metastatic CRPC in patients. Finally, the Stat5a/b gene locus has been shown to undergo amplification in 29% of distant CRPC metastases (24).
Stat5a/b belongs to the Stat family of transcription factors, and is comprised of two highly homologous isoforms, 94-kDa Stat5a and 92-kDa Stat5b (25). Stat5a/b are latent cytoplasmic proteins which become activated by phosphorylation of a conserved tyrosine residue in the carboxy-terminal domain. In PC cells, the predominant tyrosine kinase that activates Stat5a/b is Jak2 (26), which is recruited to the membrane-proximal region of ligand-activated cytokine receptors and is activated through autophosphorylation. Activated Jak2 phosphorylates Stat5a/b on conserved tyrosine residues Y694Stat5a and Y699Stat5b, leading to Stat5a/b dimerization and translocation to the nucleus, where the dimers bind to specific Stat5a/b response elements of target genes for transcriptional regulation (25). Thus, Stat5a/b signaling can be targeted at the level of the transmembrane receptor, further downstream by inhibition of Jak2 kinase or by direct blockade of Stat5a/b activity.
In this study, we investigated the efficacy of pharmacological targeting of the Jak2-Stat5a/b signaling pathway to block primary and CR growth of PC. Jak2-induced phosphorylation and activation of Stat5a/b was inhibited by AZD1480, a potent adenosine triphosphate (ATP)-competitive small-molecule inhibitor of Jak2 kinase (27). We show that AZD1480 effectively suppressed Stat5a/b signaling and decreased viability of not only human PC cell lines and xenograft tumors, but also of clinical PCs ex vivo in organ explant cultures. Most importantly, AZD1480 blocked the growth of primary CWR22Pc tumors, the emergence of CR tumors and the growth of established CRPCs in the CWR22Pc xenograft tumor model.
Materials and Methods
Cell Culture and Reagents
Human prostate cancer cell lines CWR22Rv1, PC-3, DU145, LNCaP (ATCC, Manassas, VA) and CWR22Pc were cultured in RPMI 1640 (Mediatech, Herndon, VA) containing 10% fetal bovine serum (FBS; Quality Biological, Gaithersburg, MD) and penicillin/streptomycin (Mediatech, Inc., 50 IU/ml and 50 μg/ml, respectively). LNCaP and CWR22Pc cells were cultured in the presence of 0.5 and 0.8 nM dihydrotestosterone (DHT; Sigma, St. Louis, MO), respectively. Normal human prostate epithelial cells RC165N (28) were cultured in keratinocyte-serum-free (Gibco, Grand Island, NY) medium supplemented with epidermal growth factor (EGF), bovine pituitary extract (Gibco) and L-glutamine. AZD1480 and bicalutamide were provided by AstraZeneca and docetaxel (20 mg/ml) was purchased from Sanofi-Aventis (Bridgewater, NJ).
Protein Solubilization, Immunoprecipitation and Immunoblotting
CWR22Rv1, CWR22Pc, DU145 and PC-3 cells were solubilized and immunoprecipitations and immunoblottings were performed as described previously (14–18). Antibodies used for immunoprecipitation and immunoblotting are described in Supplementary Materials and Methods.
Detection of Stat5a/b Dimerization by Co-Immunoprecipitation
Generation of Stat5a constructs and the dimerization assay are described in Supplementary Materials and Methods.
Immunofluoresence Staining of Stat5a/b
PC-3 cells were transfected with pStat5a-Flag and pPrlR, serum-starved for 16 h, pretreated with AZD1480 or vehicle for 2 h, stimulated with 10 nM hPrl for 30 min, fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 and incubated with mouse anti-Flag pAb (1:200; Genomics), followed by goat anti-mouse fluorescein IgG (1:200; Vector Laboratories, Burlingame, CA). Immunofluorescence staining was detected using a Zeiss LSM 510 laser-scanning microscope with an Apochromat X63/1.4 oil immersion objective.
Electromobility shift assay (EMSA)
COS-7 cells were transfected with plasmids (3 μg of each) expressing PrlR (pPrlR) and Stat5a (pStat5a) or Stat5b (pStat5b) using FuGENE6, serum-starved for 10 h and pretreated with AZD1480 or vehicle for 2 h, followed by stimulation with 10 nM hPrl for 30 min. Nuclear extracts were prepared and a gel EMSA was performed as described previously (16, 29, 30).
Luciferase Reporter Gene Assay
PC-3 cells (2 × 105) were transiently co-transfected with 0.25 μg of pStat5a or pStat5b, pPrlR (prolactin receptor), 0.5 μg of pStat5a/b-luciferase (β-casein-Luc) and 0.025 μg of pRL-TK (Renilla luciferase). After 24 h, cells were serum-starved for 20 h, pretreated with AZD1480 at indicated concentrations for 1 h and stimulated with 10 nM hPrl for 16 h. The lysates were assayed for firefly and Renilla luciferase activities using the dual-luciferase reporter assay system (Promega) as described previously (12).
Adenoviral Gene Delivery of Dominant-negative (DN) Stat5a/b and DNStat3
Gene delivery and expression of DNStat5a/b and DNStat3 in PC cells was achieved using an adenoviral vector. Generation of adenoviral constructs is described in Supplementary Materials and Methods.
Cell Viability Assay
Cell viability was analyzed by CellTiter 96® AQueous Assay kit (Promega) according to the manufacturer’s protocol.
Cell Cycle Analysis
CWR22Rv1 (data not shown) or CWR22Pc cells were treated with AZD1480 or vehicle for 24 h, 48 h and 72 h. Cells were fixed with 70% ethanol at 4°C overnight and washed with cold PBS twice before staining with propidium iodide (PI) and RNase A (Sigma, USA). PI fluorescence intensity was analyzed by a flow cytometer using FL-2 channel.
Caspase-3 Activation Assay
Caspase-3 activity was determined by a fluorometric immunosorbent enzyme assay (Roche) as described in Supplementary Materials and Methods.
PC Xenograft Tumor Growth Studies
CWR22Rv1 tumor xenografts were grown in male CB-17 SCID mice purchased from Charles River. Mice were maintained under specific pathogen-free conditions and were used in compliance with protocols approved by the Institutional Animal Care and Use Committee of AstraZeneca, which conform to institutional and national regulatory standards on experimental animal usage. Studies were performed using CWR22Rv1 cells implanted subcutaneously (s.c.) or luciferase-tagged CWR22Rv1 cells implanted orthotopically.
For studies with CWR22Pc tumors (31), castrated male athymic mice (Taconic, Germantown, NY), cared for according to the institutional guidelines of Thomas Jefferson University, were implanted with sustained-release dihydrotestosterone (DHT) pellets (60-day release, 1 pellet/mouse, Innovative Research of America, Sarasota, FL) 3 days before PC cell inoculation. CWR22Pc cells (1.5 × 107) in 0.2 ml of Matrigel (BD Biosciences) were inoculated s.c. into flanks of nude mice (one tumor/mouse). AZD1480 was dissolved in 0.1% Tween 80 (Sigma)/0.5% Methyl Cellulose (HPMC, K4M prep, Dow Chemical), bicalutamide in 0.5% Tween 80/PBS and docetaxel in PBS (1mg/ml).
Details of drug administration and tumor measurements for CWR22Rv1 and CWR22Pc tumor studies are provided in Supplementary Materials and Methods.
Ex Vivo Organ Cultures of Clinical PCs
For organ cultures, PC specimens were obtained from clinical patients (Table 1) with localized or locally advanced prostate cancer undergoing radical prostatectomy and bilateral iliac lymphadenectomy. The use of the de-identified excess tissue specimens for research purposes was approved by the Thomas Jefferson University Institutional Review Board. Details of the ex vivo organ culture conditions and methodology which we have described previously (19, 29, 32–37) are provided in Supplementary Materials and Methods.
Table 1.
Characteristics of the clinical prostate cancers tested ex vivo in explant organ cultures for responsiveness to AZD1480.
| Variables | Responders | Non-responders |
|---|---|---|
| n | 9 | 3 |
| Median (range) | Median (range) | |
| Age at radical prostatectomy | 62 (47-67) | 66 (58-67) |
| AUA score | 6.5 (2-21.5) | 9.5 (5-14) |
| IIEF score | 23 (3-25) | 13.5 (2-25) |
| Gleason score | n (%) | n (%) |
| 4 | 0 (0) | 0 (0) |
| 5 | 0 (0) | 0 (0) |
| 6 | 1 (11.1) | 0 (0) |
| 7 | 7 (77.8) | 2 (66.6) |
| 8 | 0 (0) | 0 (0) |
| 9 | 1 (11.1) | 1 (33.3) |
| 10 | 0 (0) | 0 (0) |
| Unknown | 0 (0) | 0 (0) |
| Stage | n (%) | n (%) |
| T1c | 7 (77.8) | 0 (0) |
| T2a | 1 (11.1) | 0 (0) |
| T2b | 0 (0) | 1 (33.3) |
| T2c | 1 (11.1) | 1 (33.3) |
| T3a | 0 (0) | 1 (33.3) |
| Metastases detected | n (%) | n (%) |
| Yes | 0 (0) | 0 (0) |
| No | 8 (88.9) | 3 (100) |
| Unknown | 1 (11.1) | 0 (0) |
AUA score = American Urological Association symptom score: degree of urinary symptoms – none (0); mild (1-7); moderate (8-19); severe (20-35).
IIEF score = International Index of Erectile Function score: degree of erectile dysfunction – none (22-25); mild (17-21); mild to moderate (12-16); moderate (8-11); severe (5-7).
Immunostaining of Paraffin-embedded Tissue Sections
Immunohistochemistry (IHC) staining of CWR22Pc xenograft tumors and clinical human PCs grown as ex vivo organ cultures was performed as described previously (19, 21, 22, 29, 30, 38). TUNEL assay was performed using the In situ Cell Death Detection Kit (Roche), as described previously (32).
Scoring of Cell Viability, Apoptosis, and Active Stat5a/b/Stat3 Immunostainings
Viable cells, active nuclear Stat5a/b, Stat3 or apoptotic cells with fragmented DNA vs. total number of cells (viable and dead) were counted for 3 views/tumor and 1 view/organ culture explant (20-25 explants per treatment group per patient) and expressed as percentages, as demonstrated previously (32). All percentages within each treatment group (tumor or organ culture explants) were averaged.
Statistics
Statistical analysis of PC xenograft tumors and ex vivo organ cultures of clinical PCs are provided in Supplementary Material and Methods.
Results
AZD1480 disrupts phosphorylation of Stat5a/b in human PC cells
To determine the efficacy of the Jak2 inhibitor AZD1480 in suppressing Stat5a/b-driven growth of PC, we selected the CWR22 cell line/tumor system as our model because it mimics the clinical course of human PC when grown as a xenograft tumor in mice (31). CWR22 tumors were originally derived by Pretlow et al. (39) from a Gleason score 9 PC, and the original tumors could be maintained only by serial regrafting. CWR22Pc is a cell line, established from the original androgen-regulated primary CWR22 tumors (31), the growth of which is strongly up-regulated by androgens (31) and down-regulated by anti-androgens in culture (Suppl. Fig. 1). CWR22Pc cells form tumors in nude mice with 100% tumor take in the presence of androgens (31). Importantly, the tumors regress in response to androgen deprivation but eventually recur as CR tumors (31). CWR22Rv1 is an androgen-independent PC cell line established from one of the original CR CWR22R tumors (40).
Given that Jak1/2 are the primary kinases which phosphorylate both prolactin (Prl)-activated Stat5a/b (19, 23) and IL-6-activated Stat3 (41, 42), we first evaluated the activation status of Stat5a/b vs. Stat3 in exponentially growing CWR22Pc and CWR22Rv1 cells. Stat5a/b was expressed and constitutively activated while Stat3 was not activated in CWR22Rv1 and CWR22Pc cells (Suppl. Fig. 2). In comparison, DU145 cells showed high activation of Stat3 as previously demonstrated (17) (Suppl. Fig. 2). CWR22 tumors and the cell lines derived from them are known to express autocrine Prl (23, 31). AZD1480 inhibited Prl-induced phosphorylation of Stat5a at 15 and 18 nM concentration by approximately 50% (IC50) in serum-starved CWR22Rv1 and CWR22Pc cells, respectively, while the IC50 for Prl-induced Stat5b phosphorylation was 48 nM for CWR22Rv1 and 70 nM for CWR22Pc (Fig. 1A). The efficacy (IC50) of AZD1480 in inhibiting constitutive activation of Stat5a in exponentially growing CWR22Rv1 and CWR22Pc cells was 16 and 7 nM, respectively, while the IC50 for Stat5b phosphorylation was 65 nM for both cell lines (Fig. 1A). In summary, AZD1480 effectively reduced both ligand-induced and constitutive activation of Stat5a/b in a dose-dependent manner in human PC cells.
Figure 1. AZD1480 inhibits Stat5a/b activation and downstream molecular events in prostate cancer (PC) cells. (A) AZD1480 disrupts ligand-induced and constitutive phosphorylation of Stat5a/b in PC cells.
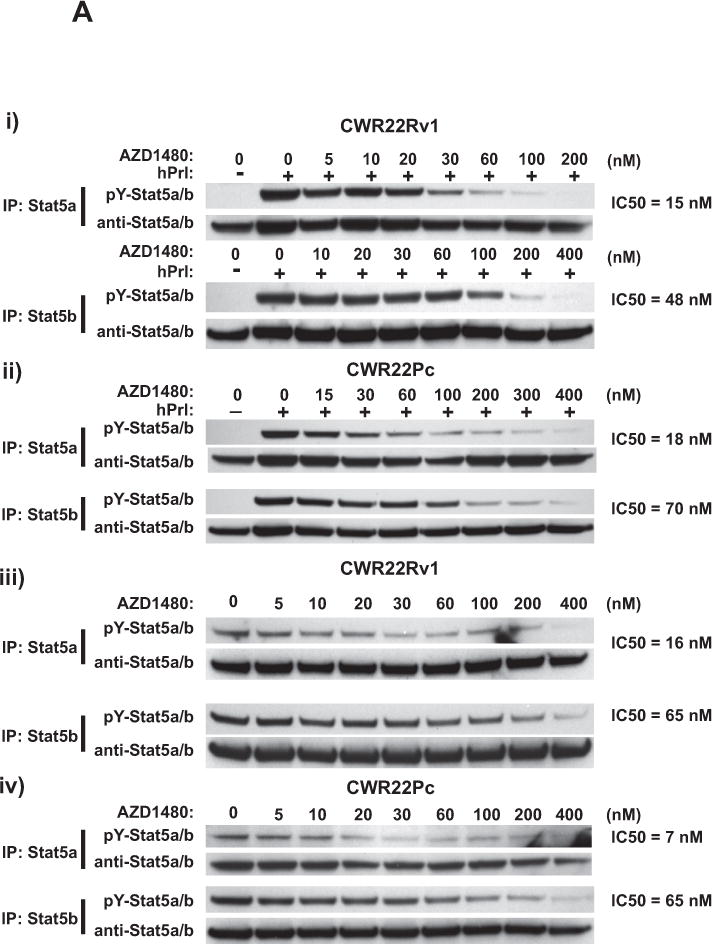
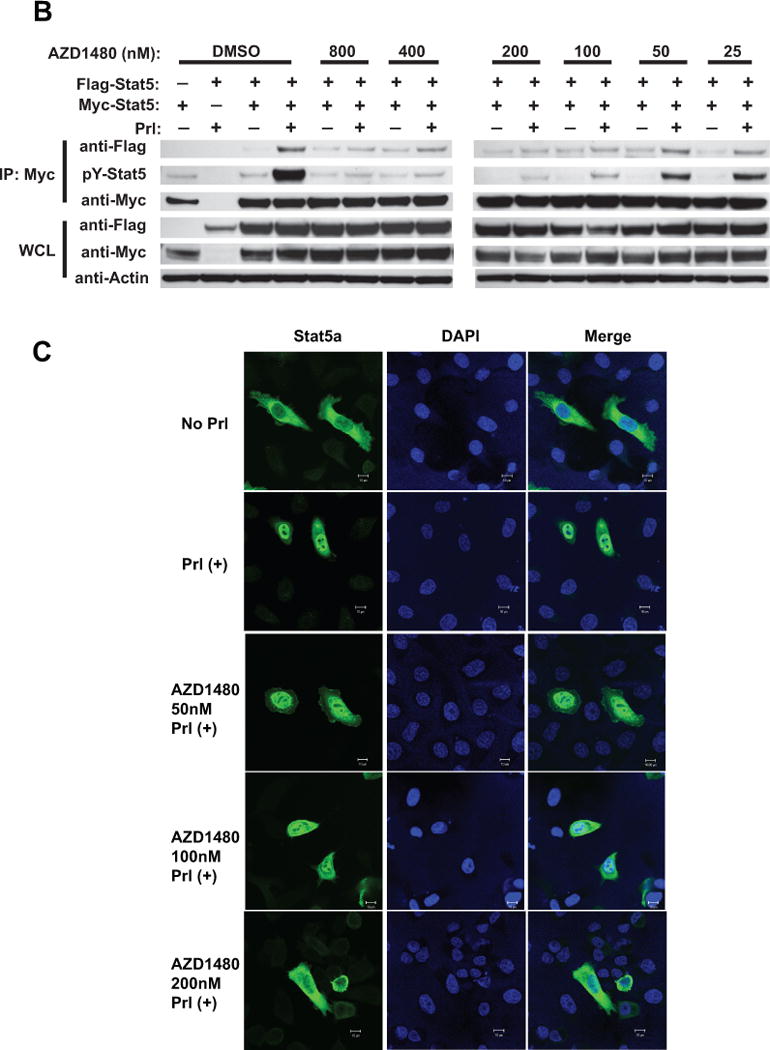
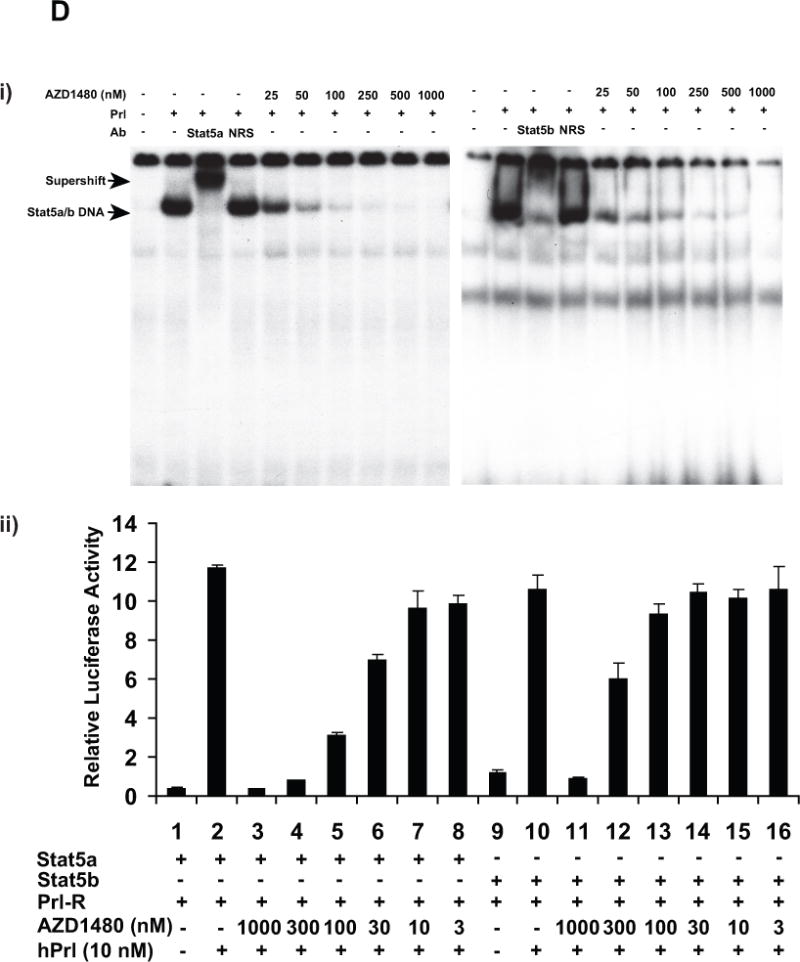
Stat5a and Stat5b were immunoprecipitated (IP) using anti-Stat5a or anti-Stat5b pAbs from (i) CWR22Rv1 and (ii) CWR22Pc cells which had been serum-starved (0% FBS) overnight, pre-treated with AZD1480 at indicated concentrations for 1 h followed by stimulation of the cells with 10 nM human prolactin (hPrl) for 20 min and immunoblotted using anti-phospho-Stat5a/b (anti-pYStat5a/b). Filters were stripped and re-blotted with anti-Stat5a/b mAb. Similarly, Stat5a and Stat5b were IP from exponentially growing (iii) CWR22Rv1 and (iv) CWR22Pc cells and IB for pYStat5a/b and total Stat5a/b. (B) AZD1480 disrupts ligand-induced dimerization of Stat5a/b in PC cells. pCMV-3Flag-Stat5a, pCMV-3Myc-Stat5a and pPrl-receptor (PrlR) plasmids were co-transfected into PC-3 cells. Cells were serum-starved for 16 h, pre-treated with AZD1480 or vehicle (DMSO) at the indicated concentrations for 2 h, followed by stimulation with 10 nM hPrl for 20 minutes. 3Myc-Stat5a was immunoprecipitated with anti-Myc mAb and blotted with anti-Flag mAb or anti-Myc mAb, as indicated. Whole cell lysates were blotted with anti-Myc, anti-Flag mAb or anti-actin pAb to demonstrate the input. (C) AZD1480 inhibits nuclear translocation of ligand-activated Stat5a/b in PC cells. PC-3 cells were transfected with plasmids expressing Stat5a (pStat5a-Flag) and human PrlR (pPrlR). After serum starvation for 16 h, the cells were treated with AZD1480 for 2 h followed by stimulation with 10 nM hPrl for 20 min. Immunostaining of Stat5a/b is demonstrated by indirect immunofluoresence (green), while DAPI staining (blue) shows the nuclei. (D) (i) AZD1480 inhibits binding of Stat5 to DNA, shown by EMSA analysis using the Prl-response element of the beta-casein gene as the probe. COS-7 cells were transiently co-transfected with pStat5a/b and pPrlR, serum-starved for 10 h and pre-treated for 2 h with AZD1480 or vehicle at indicated concentrations followed by stimulation with 10 nM hPrl for 30 min. Nuclear extracts were prepared for EMSA and the specificity of the Stat5-DNA binding complex was demonstrated by supershift with anti-Stat5a pAb vs. normal rabbit serum (NRS). (ii) AZD140 inhibits transcriptional activity of Stat5a/b in PC cells. PC-3 cells were transiently co-transfected with a genomic ß-casein-promoter-luciferase plasmid, pRL-TK (Renilla luciferase), pPrlR, pStat5a or pStat5b, as indicated. Cells were serum-starved for 20 h and pre-treated with AZD1480 at indicated concentrations for 1 h followed by stimulation with hPrl (10 nM) for 16 h. The relative luciferase activities were determined, and the mean values of three independent experiments performed in triplicates +/− SE values are indicated by bars.
AZD1480 decreases dimerization, nuclear translocation, DNA binding and transcriptional activity of Stat5a/b in human PC cells
To investigate if AZD1480 disrupts dimerization of Stat5a/b, we generated Flag-tagged and Myc-tagged Stat5a constructs and co-transfected them into PC-3 cells (negative for endogenous Stat5a/b and AR (43)). The cells were serum-starved and treated with AZD1480 or vehicle for 2 h before induction of Stat5a dimerization by human Prl (hPrl) stimulation for 30 min. The lysates from the cells were immunoprecipitated with anti-Myc antibodies (Abs) and immunoblotted with anti-Flag or anti-Myc Abs (Fig. 1B). Protein input levels were analyzed by immunoblotting of whole cell lysates (WCL) with anti-Flag, anti-Myc and anti-actin Abs. Stimulation of cells by hPrl induced Stat5a/b dimerization, as expected, and this was potently inhibited by AZD1480 treatment (Fig. 1B).
Because AZD1480 inhibited both phosphorylation and dimerization of Stat5a/b, AZD1480 was also expected to disrupt nuclear translocation of Stat5a/b in PC cells. To test this hypothesis, prolactin receptor (PrlR) and Stat5a/b were expressed in PC-3 cells using replication-deficient adenovirus as an expression vector. The cells were serum-starved for 16 h, treated with AZD1480 at indicated concentrations (Fig. 1C) for 2 h and stimulated with hPrl (10 nM) for 20 min. In the absence of hPrl, Stat5a/b was predominantly localized to the cytoplasm of PC-3 cells (Fig. 1C, top panel), with hPrl stimulation inducing nuclear translocation of Stat5a/b (Fig. 1C, second panel from the top). Following pre-treatment of cells with 200 nM AZD1480, there was no ligand-induced nuclear immunostaining of Stat5a/b, indicating that AZD1480 inhibited nuclear translocation of Stat5a/b (Fig. 1C, bottom panel).
To determine if AZD1480 inhibits DNA binding of ligand-induced phosphorylated Stat5, we employed EMSA analysis. The Stat5a/b response element of the β-casein gene promoter was used as a probe and the ability of anti-Stat5a/b Ab to supershift Stat5a/b was verified (Fig. 1D). COS-7 cells transfected with PrlR and Stat5a were starved and pretreated with AZD1480 for 2 h at indicated concentrations followed by hPrl-stimulation (10 nM) for 30 min and EMSA analysis of the nuclear extracts. AZD1480 inhibited binding of Stat5a/b to DNA by more than 50% at a concentration of 25 nM compared to cells treated with vehicle (Fig. 1D). To further assess whether AZD1480 is capable of inhibiting transcriptional activity of Stat5a/b, PC-3 cells transiently transfected with a β-casein-luciferase reporter gene were co-transfected with PrlR, Stat5a or Stat5b. Cells were serum-starved for 16 h and treated with AZD1480 for 1 h, followed by stimulation with hPrl (10 nM) in serum-free medium for 16 h. As demonstrated in Figure 1D, AZD1480 inhibited transcriptional activity of Stat5a and Stat5b with IC50s of 150 and 250 nM, respectively. In summary, the results of the experiments shown in Figure 1 indicate that AZD1480 inhibits transcriptional activity of Stat5a/b through disruption of Stat5a/b phosphorylation, dimerization, nuclear translocation and DNA binding.
AZD1480 suppresses growth of prostate cancer cells through induction of apoptosis
Inhibition of Stat5a/b by various methodological approaches (antisense oligonucleotides, RNA interference, adenoviral expression of a dominant-negative mutant of Stat5a/b) has been shown to inhibit proliferation and induce apoptotic death of human PC cells (14–17, 20). CWR22Rv1, CWR22Pc, PC-3, DU145 and normal prostate epithelial cells (RC165N) (28, 44) cells were cultured for 72 h with increasing concentrations of AZD1480 to determine if Jak1/2 kinase inhibition suppresses cell growth (Fig. 2A). The number of viable cells decreased by 50% (GI50) at concentrations of 482 nM for CWR22Pc cells and 438 nM for CWR22Rv1 cells. The GI50 was significantly higher for PC-3 (1755 nM), DU145 (3517 nM) and normal prostate epithelial cells (2083 nM). Cell death induced by higher than 1000 nM concentration of AZD1480 is considered to be caused by off-target effects of AZD1480 (27, 44). PC-3 cells do not express Stat5a/b (43) or Stat3 (43), and the kinase which activates Stat5a/b in DU145 cells is not Jak2 (18). Furthermore, RC165N normal prostate epithelial cells express low levels of Stat5a/b which are not constitutively activated, and RC165N cells do not undergo cell death in response to Stat5a/b inhibition as previously demonstrated (17), consistent with the lack of responsiveness to AZD1480. To verify that reduction of viable CWR22Rv1 and CWR22Pc cells in response to AZD1480 was caused by inhibition of Stat5a/b rather than Stat3, we inhibited Stat5a/b and Stat3 in parallel by adenoviral (Ad) expression of dominant-negative (DN) Stat5a/b and DNStat3 at multiplicity of infection (MOI) 5 (Suppl. Fig. 3). In line with the results shown in Suppl. Fig. 2 demonstrating lack of Stat3 activation in CWR22Rv1 and CWR22Pc cells, neither cell line responded to AdDNStat3, while AdDNStat5a/b decreased cell number by approximately 40% (Suppl. Fig. 3).
Figure 2. AZD1480 induces apoptotic death of prostate cancer cells.
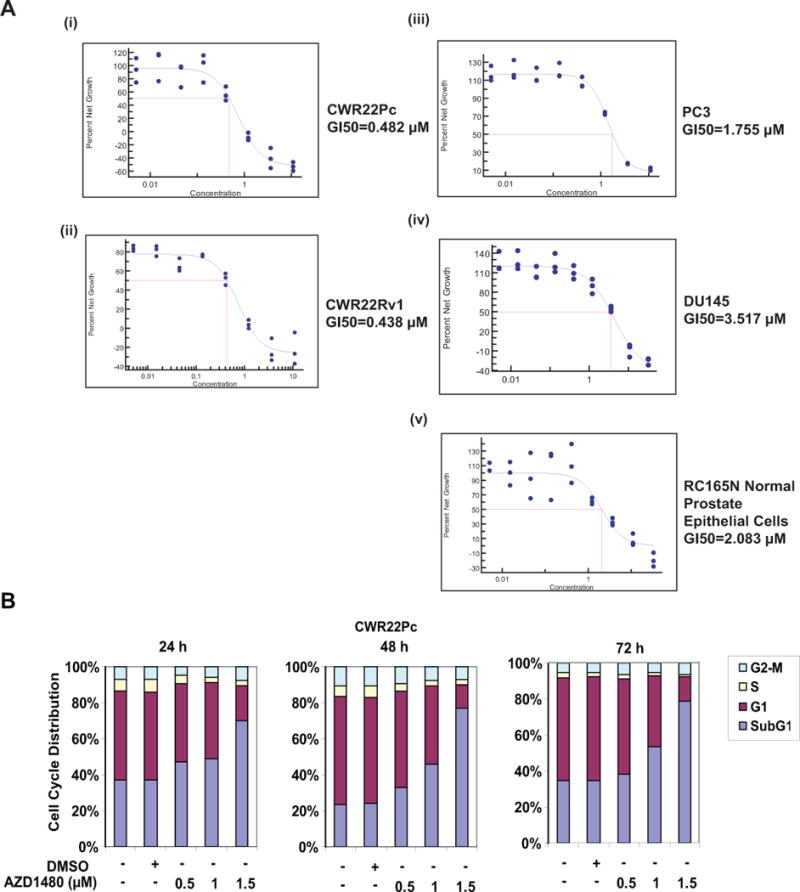
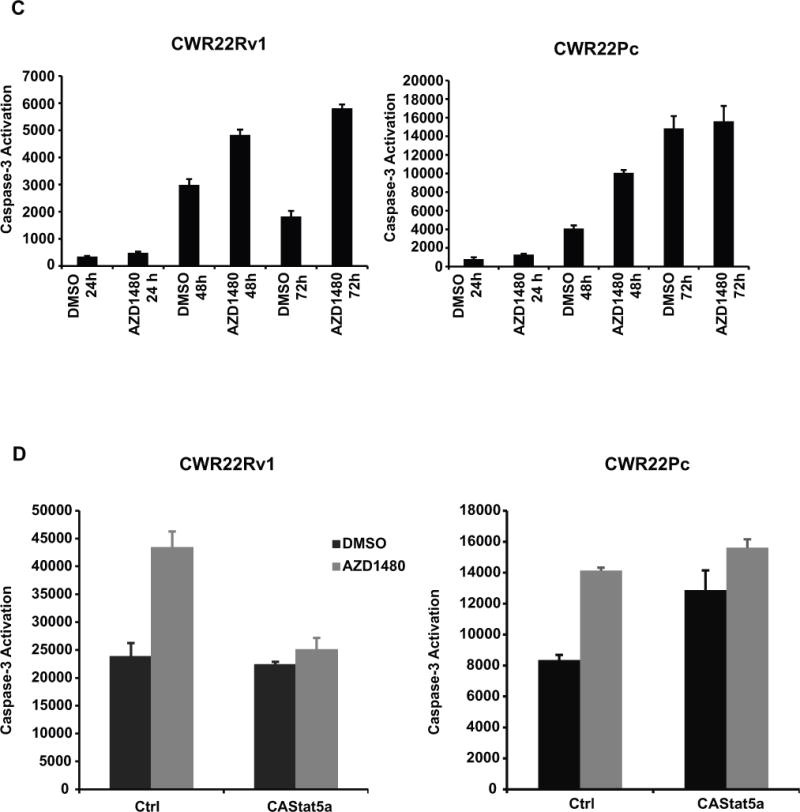
(A) AZD1480 reduces the number of viable CWR22Rv1 and CWR22Pc cells in culture. CWR22Rv1, CWR22Pc, PC3, DU145 and normal prostate epithelial cells (RC165N) were treated with AZD1480 at indicated concentrations for 72 h, and the fraction of viable cells was determined by MTS (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyl-tetrazolium bromide) metabolic activity assay. (B) AZD1480 increases the fraction of dead PC cells in cell cycle analysis. CWR22Pc cells were treated with AZD1480 for 24, 48 or 72 h at indicated concentrations followed by FACS analysis. (C) AZD1480 increases caspase-3 activation in CWR22Rv1 (72 h) and CWR22Pc cells (48 h). CWR22Rv1 cells were treated with AZD1480 (800 nM) or vehicle followed by determination of caspase-3 activation by fluorometric immunosorbent enzyme assay. (D) AZD1480 induction of caspase-3 activation in CWR22Rv1 and CWR22Pc cells is counteracted by expression of constitutively active (CA) Stat5a/b. CWR22Rv1 and CWR22Pc cells were infected with adenovirus expressing constitutively active Stat5a (AdCAStat5a; MOI 2) 6 h prior to the treatment of cells with AZD1480 (800 nM) for 72 h.
To characterize the growth inhibition of CWR22Rv1 (data not shown) and CWR22Pc cells, we conducted cell cycle analysis at 24, 48 and 72 h after treatment of cells with AZD1480 vs. vehicle (Fig. 2B). AZD1480 did not alter the fraction of cells in G1 or G2-M, whereas the fraction of dead cells (sub G1) was increased at all time points in AZD1480-treated CWR22Pc cells. AZD1480 (800 nM) treatment increased caspase-3 activation by 200% in CWR22Rv1 cells at 72 h and by 150% in CWR22Pc cells at 48 h (Fig. 2C). In order to determine if introduction of active Stat5a/b is able to rescue cells from AZD1480-induced apoptosis, constitutively active Stat5a/b (17), which is not dependent on phosphorylation by Jak2 kinase for activation, was overexpressed using an adenoviral (Ad) vector in CWR22Rv1 and CWR22Pc cells 6 h prior to treatment of cells with AZD1480 (800 nM). As shown in Figure 2D, expression of constitutively active Stat5a/b prevented AZD1480-induced caspase-3 activation in both cell lines. Moreover, AZD1480 (800 nM) induced caspase-3 activation to the same extent as genetic knockdown of Jak2 or Stat5 by RNA interference in CWR22Rv1 cells (Suppl. Fig. 4). In summary, the results shown in Figure 2 demonstrate that AZD1480 induces apoptotic cell death in CWR22Rv1 and CWR22Pc cells via inhibition of Stat5a/b.
AZD1480 effectively inhibits both primary and castrate-resistant growth of prostate cancer in vivo
Because targeting Stat5a/b by Jak2 inhibition induced apoptosis of PC cells in culture, we next investigated if AZD1480 affects PC xenograft tumor growth in vivo. Mice inoculated subcutaneously (s.c.) or orthotopically with CWR22Rv1 cells (Fig. 3A) were treated daily with AZD1480 (10 mg/kg, 30 mg/kg) or vehicle. At the end of treatment, growth of s.c. tumors was suppressed by 72% following treatment with 30 mg/kg AZD1480 (p<0.0001) and by 52% following 10 mg/kg AZD1480 (p=0.0002), while orthotopic tumor growth was suppressed by 54% following treatment with 30 mg/kg AZD1480 (p=0.004) and by 6% following 10 mg/kg AZD1480 (p=0.42) (Fig. 3A). Immunoblotting of s.c. xenograft tumor lysates demonstrated significant inhibition of Stat5a/b phosphorylation 2 h following treatment with 30 mg/kg AZD1480 (Fig. 3B, upper panel). Immunohistochemical analysis of tumors harvested after 11 days of drug treatment showed a 70% decrease in nuclear Stat5a/b in tumor tissues relative to the control group; very little expression or modulation of nuclear Stat3 was observed (Fig. 3B, lower panel).
Figure 3. AZD1480 inhibits both primary and castrate-resistant prostate cancer xenograft tumor growth. (A) AZD1480 inhibits growth of s.c. and orthotopic CWR22Rv1 tumors.
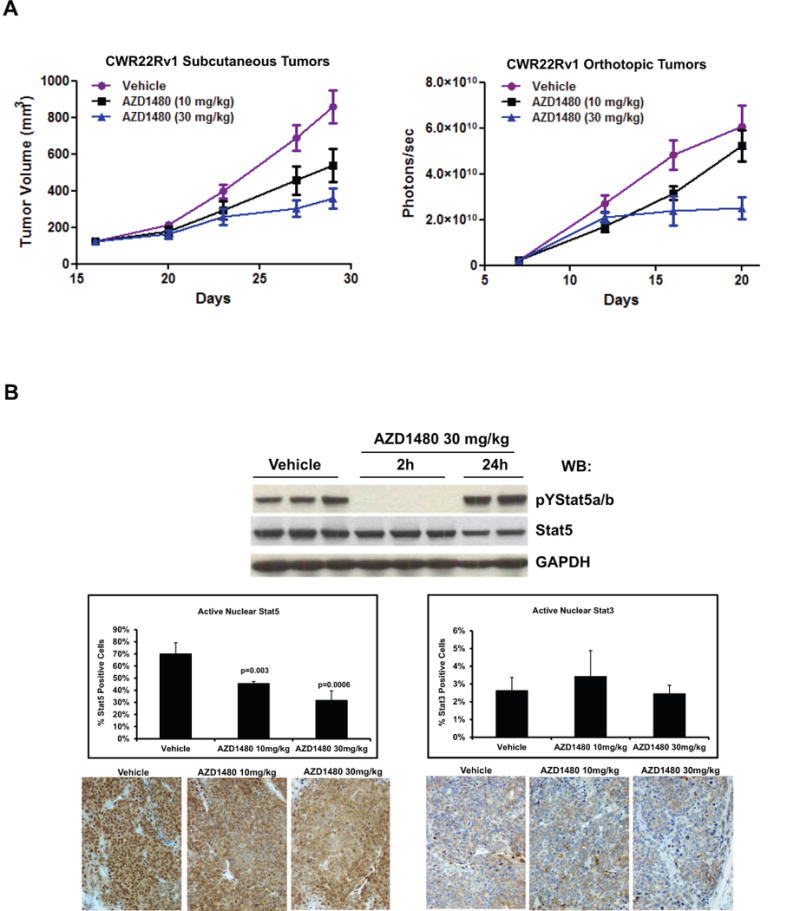
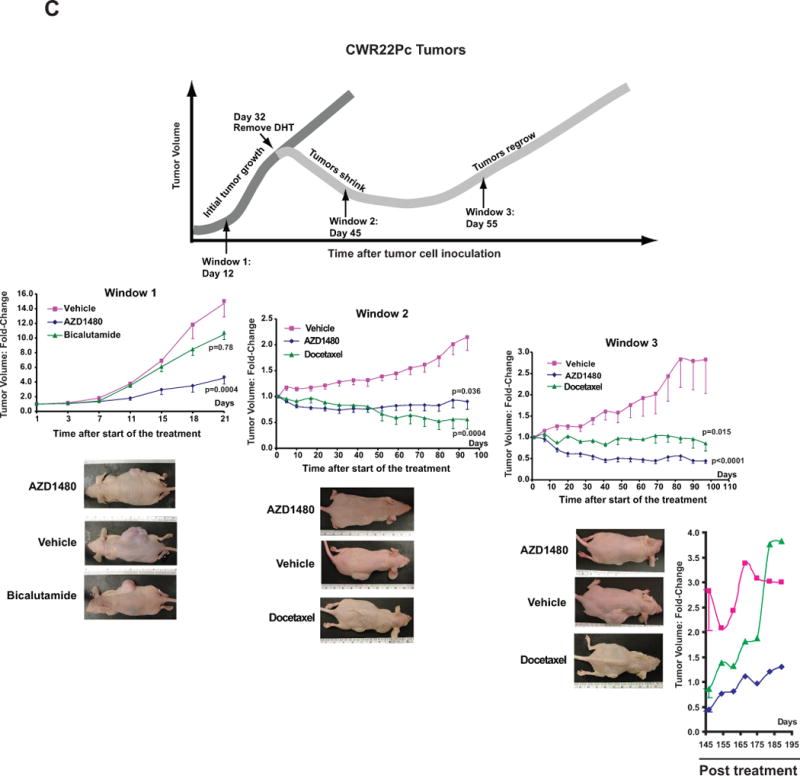
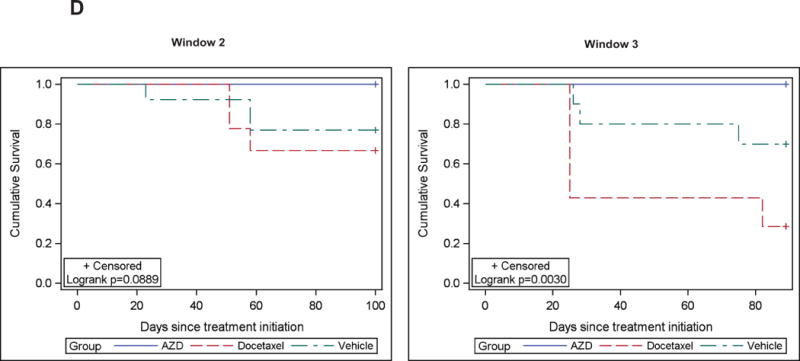
CWR22Rv1 PC cells were inoculated s.c. (left panel) or orthotopically into prostates (right panel) of SCID mice. The mice were treated daily with AZD1480 (10 and 30 mg/kg) or vehicle as control administered by oral gavage. Tumor sizes were measured every 3 days, and the tumor volumes were calculated using the formula 0.5 × (larger diameter) × (smaller diameter) 2. (B) AZD1480 inhibits Stat5a/b activation in CWR22Rv1 tumors. Immunoblotting of tumor (s.c.) cell lysates with anti-pYStat5a/b mAb 2 or 24 h after administration of AZD1480 (30 mg/kg) to the mice (upper panel). Immunohistochemical detection of nuclear Stat5a/b in CWR22Rv1 tumors following 11 days of dosing at the indicated doses of AZD1480 demonstrates a decrease in the percentage of Stat5a/b-positive tumor cells. (C) AZD1480 inhibits both primary androgen-dependent growth of CWR22Pc tumors and castrate-resistant growth of recurrent CWR22Pc tumors after androgen deprivation-induced tumor regression. CWR22Pc cells were inoculated s.c. into the flanks of castrated athymic nude mice supplied with sustained-release DHT-pellets (n=10/treatment group, 1 tumor/mouse, 1.5 × 107 CWR22Pc cells/site). In treatment window (TW) 1 (primary androgen-sensitive PC growth) (left panels), mice were treated daily with vehicle, AZD1480 at 30 mg/kg or bicalutamide at 50 mg/kg for 21 days starting on day 12 by oral gavage. Tumor sizes were measured three times per week. In TWs 2 (middle panels) and 3 (right panels) (castrate-resistant PC growth), the DHT pellets were removed on day 32, and three days after DHT pellet removal, the mice were randomly distributed into three groups. Thirteen days (for TW 2) or 23 days (for TW 3) after DHT removal, treatment of the mice started with AZD1480 at 30 mg/kg or vehicle by oral gavage daily, or docetaxel at 5 mg/kg body weight once a week intravenously. Tumor growth rates were calculated for each treatment group and the fold changes in tumor volume (volume at timepoint/volume at treatment start) for each group are presented. (D) Kaplan-Meier analyses indicate that overall survival of AZD1480-treated mice is longer than docetaxel-treated mice in TWs 2 and 3.
To investigate if AZD1480 is capable of suppressing both primary and CR growth of PC in a more disease-relevant model system, we utilized the CWR22Pc cell line/tumor model (31). CWR22Pc tumors mimic the clinical course of PC in humans, based on formation of androgen-dependent primary PC tumors which regress upon androgen deprivation but recur as castrate-resistant PC (31). CWR22Pc cells were inoculated s.c. into the flanks of nude mice that had been castrated and supplied with androgen pellets to normalize serum androgen levels. To evaluate the efficacy of AZD1480 in suppressing growth of androgen-dependent primary CWR22Pc tumors, mice were treated with AZD1480 (30 mg/kg) (n=10), bicalutamide (50 mg/kg) (n=10) or vehicle (n=10) starting on day 12 for 21 days (Fig. 3C, treatment window (TW) 1). AZD1480 significantly inhibited primary CWR22Pc tumor growth by 73% (p=0.0004) while bicalutamide suppressed tumor growth by only 30% at a non-significant level (p=0.78).
In the next set of experiments, mice carrying CWR22Pc tumors s.c. were androgen-deprived on day 32 by removal of DHT pellets. To evaluate if AZD1480 is able to suppress the emergence of CR CWR22Pc tumors (Fig. 3C, TW 2) or growth of established CR CWR22Pc tumors (TW 3), the mice were treated for 95 days with AZD1480 (30 mg/kg) (n=10), docetaxel (5 mg/kg) (n=10) or vehicle (n=10) starting on day 13 (TW 2) or day 23 (TW 3) after the onset of androgen deprivation. AZD1480 significantly blocked not only the emergence of CR CWR22Pc tumors (Fig. 3C, TW 2; p=0.036) but also significantly blocked growth of established CR CWR22Pc tumors (Fig. 3C, TW 3; p<0.0001). While docetaxel suppressed growth of tumors in TW 2 to a similar degree as AZD1480, docetaxel-treated mice were noticeably more cachectic and lethargic in comparison to AZD1480-treated mice, which maintained healthy body weight and overall activity in both TWs 2 and 3 (Suppl. Fig. 5) and better overall survival (Fig. 3D). Notably, treatment of established CR CWR22Pc tumors in TW 3 with AZD1480 was more effective than docetaxel (Fig. 3C), and AZD1480-treated tumors demonstrated slower regrowth post-treatment than docetaxel-treated tumors (Fig. 3C, post-treatment). Collectively, these data demonstrate that pharmacological inhibition of Jak2 effectively inhibits not only primary androgen-dependent PC growth but also CR growth of CWR22Pc tumors in nude mice.
AZD1480 decreases cell viability and levels of nuclear Stat5a/b in CWR22Pc tumors while not affecting androgen receptor or active Stat3 levels
CWR22Pc tumors harvested at the end of treatment in all TWs were evaluated for nuclear Stat5a/b (Fig. 4A) and Stat3 (Suppl. Fig. 6) by immunohistochemistry, as well as for viable vs. apoptotic cells by analysis of cell morphology (Fig. 4C) and presence of fragmented DNA (Suppl. Fig. 7). AZD1480 decreased nuclear Stat5a/b levels in all TWs significantly (Fig. 4A) while nuclear Stat3 levels were too low in this tumor model to show significant modulation (Suppl. Fig. 6). As expected, bicalutamide or docetaxel had no effect on nuclear Stat5a/b or Stat3 (TWs 2 and 3). Consistent with the inhibition of Stat5a/b nuclear localization by AZD1480, western blots of immunoprecipitated Stat5a/b from tumor lysates demonstrated marked inhibition of Stat5a/b phosphorylation by AZD1480 (Fig. 4B). Importantly, AZD1480 reduced the fraction of viable tumor cells in all TWs (Fig. 4C) and, at the same time, increased the proportion of apoptotic cells (Suppl. Fig. 7). Moreover, AZD1480 down-regulated the Stat5 target gene Bcl-XL expression in CWR22Rv1 cells in culture (Suppl. Fig. 8) and CWR22Pc tumors in vivo (Suppl. Fig. 9). AZD1480 also up-regulated cell surface E-cadherin expression in CWR22Pc tumors (Suppl. Fig. 9) re-cpitulating the effects demonstrated previously using genetic manipulation of Stat5 activity (15, 18). Finally, immunoblotting of tumor lysates with anti-AR Ab demonstrates that AZD1480 did not affect the levels of the full-length AR, the AR splice variants or tissue PSA (Fig. 4D).
Figure 4. AZD1480 decreases levels of nuclear Stat5a/b, resulting in loss of CWR22Pc xenograft tumor cell viability. (A) AZD1480 decreases levels of nuclear Stat5a/b in CWR22Pc tumors.
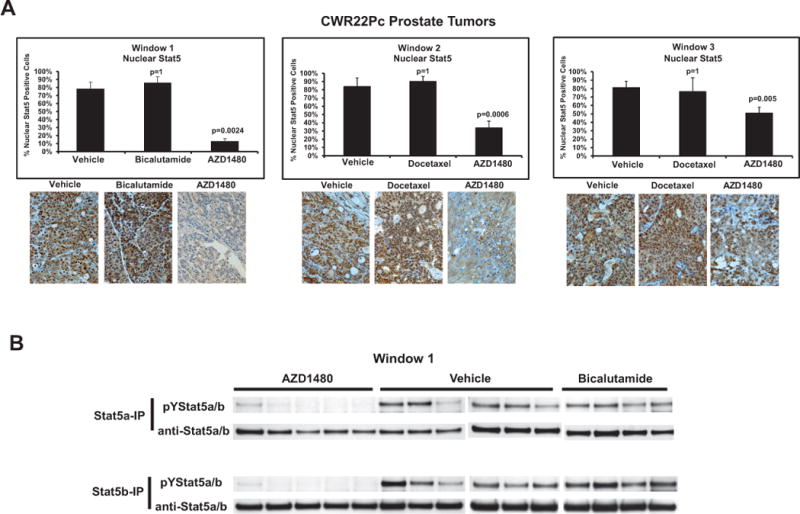
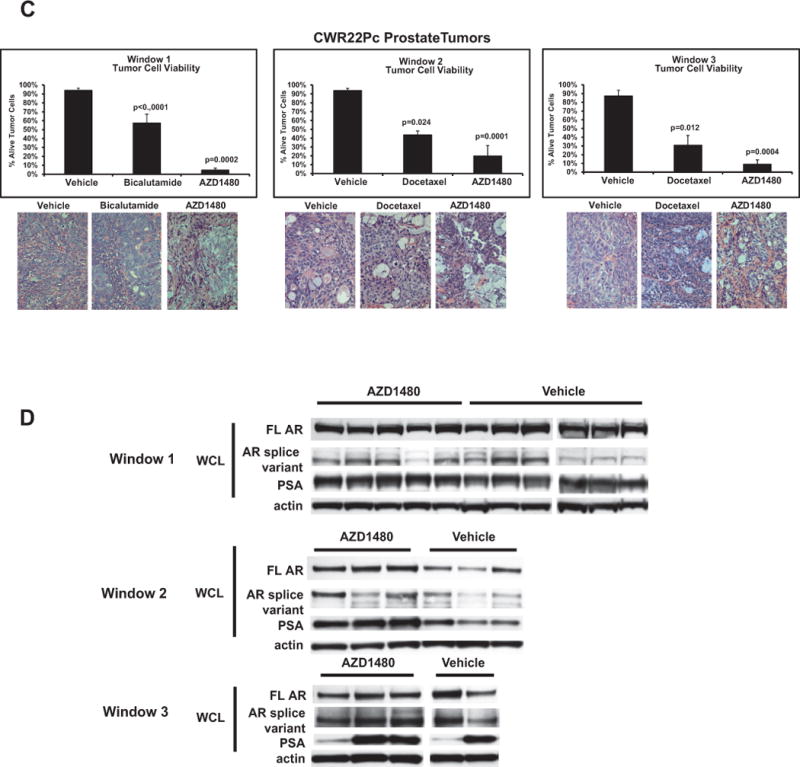
(A) AZD1480 decreased the levels of nuclear Stat5a/b in CWR22Pc xenograft tumors in all treatment windows (TWs). Nuclear Stat5a/b levels were detected by immunohistochemistry in paraffin-embedded tissue sections of the tumors using biotin-streptavidin-amplified peroxidase-antiperoxidase immunodetection. (B) Immunoprecipitation of Stat5a/b from tumor lysates, normalized by protein content and followed by immunoblotting with anti-pYStat5a/b mAb. The filters were stripped and re-blotted with anti-Stat5a/b mAb to demonstrate equal loading. (C) AZD1480 decreases cell viability in CWR22Pc xenograft tumors in all TWs. Hematoxylin-eosin staining of the CWR22Pc tumor sections demonstrated a loss of viable tumor cells and accumulation of dead cells in AZD1480-treated CWR22Rv1 xenograft tumors vs. controls in TWs 1, 2 and 3. (D) Full-length androgen receptor (AR) and AR splice variant protein levels are unaffected by AZD1480 treatment. CWR22Pc tumors in TWs 1, 2 and 3 were evaluated for protein levels of full-length AR, AR splice variants and PSA by immunoblotting of tumor cell lysates normalized for equal protein content with anti-AR mAb, anti-PSA pAb and anti-actin pAb.
AZD1480 inhibits Stat5a/b activation and epithelial cell viability of clinical PCs ex vivo in organ explant cultures
To investigate the efficacy of AZD1480 in inhibiting Stat5a/b activation and inducing cell death in clinical PCs, we exploited a previously described ex vivo organ explant culture system of PCs obtained from radical prostatectomies (19, 29, 32–37). We have extensively characterized this experimental model system previously for hormone/autocrine/paracrine regulation of normal and malignant prostate tissue at transcriptional and post-transcriptional levels (19, 29, 32–37). The advantages of explant organ cultures over cell lines are that all tissue components of PC are present in this ex vivo system and therefore the interactions of epithelium and stroma are maintained, which is critical for tissue-specific functions and for predicting the behavior of an individual tumor and its therapeutic response.
To determine responsiveness of clinical PCs to AZD1480 in ex vivo organ explant cultures, 12 PCs (Table 1) were cultured for 7 days in the presence of AZD1480 or vehicle at the indicated concentrations (Fig. 5). At the end of each culture period, viable and apoptotic carcinoma cells were assessed, as previously described (32), and Stat5a/b and Stat3 activation were analyzed by immunohistochemistry (14, 30). Nine of 12 PCs responded to AZD1480 by extensive loss of viable epithelium starting at a concentration of 25 μM (Fig. 5A, left panel; p<0.0001). At the same time, 3 of the 12 PCs did not respond to AZD1480 by reduced epithelial cell viability (Fig. 5A, right panel). It is important to note that a 25 μM concentration of hormones or pharmacological agents in organ explant culture of PC is equivalent to a concentration approximately 100 times lower (250 nM) in cell culture, as has been established previously (19, 29, 32–37). This is because diffusion of hormones or small-molecule pharmacological agents occurs much less efficiently in the tissue compartments of PC explants compared to single-layer cell culture, where cells are fully immersed in culture medium. Stat5a/b immunostaining of the explants showed that nuclear Stat5a/b levels were significantly decreased (p<0.0001) in both AZD1480 treatment groups for the 9 PCs demonstrating responsiveness to AZD1480 by reduced epithelial cell viability (Fig. 5B, left panel). In contrast, nuclear Stat5a/b levels remained constant in the non-responsive PCs (Fig. 5B, right panel). Importantly, AZD1480 induced apoptotic death of epithelial cells in cancer acini of the 9 AZD1480-responsive PCs compared to explants cultured in the presence of vehicle (Fig. 5C) (25 μM p=0.011, 50 μM p=0.023). This was evidenced by accumulation of dead cells in acinar lumens and an increase in apoptotic cells demonstrated by in situ end labeling of fragmented DNA. Collectively, these data suggest that a majority of clinical PCs from patients are responsive to pharmacological Jak1/2-inhibition, demonstrating decreased Stat5a/b activation and cell viability with increased apoptosis following AZD1480 treatment.
Figure 5. AZD1480 induces cancer cell death in clinical prostate cancers ex vivo in organ explant cultures.
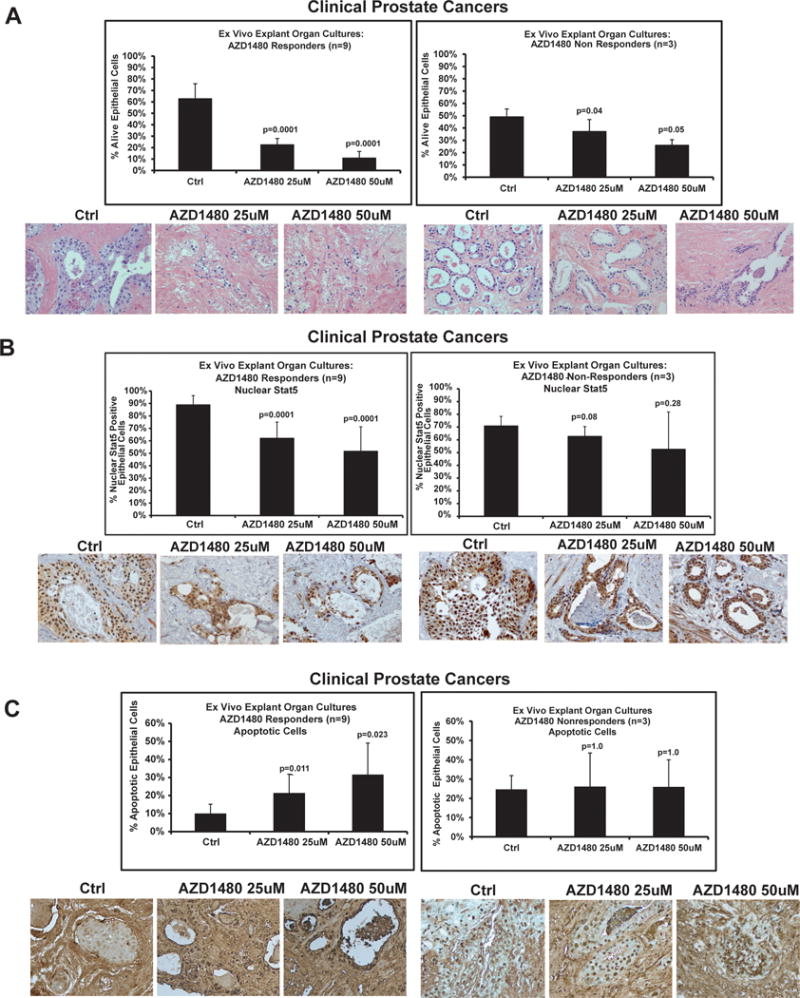
To test the responsiveness of clinical PCs to AZD1480, 12 localized PCs (Table 1) were cultured for 7 days ex vivo in explant organ cultures in the presence of AZD1480 at indicated concentrations vs. vehicle. (A) Nine individual PCs responded to AZD1480 by excessive loss of viable acinar epithelium (responders, left panel), while in three PCs the epithelial viability remained intact and unaffected in AZD1480-treated explants (non-responders, right panel). Viability of epithelial cells in the PC explants at the end of the cultures was scored by counting the fraction of viable cancer cells. A representative histology of an individual PC that responded to AZD1480 by extensive loss of acinar epithelium is depicted. (B) Levels of nuclear active Stat5a/b were determined by immunohistochemistry at the end of the organ explant cultures. Nuclear Stat5a/b indices were determined by counting Stat5a/b-positive cells per 500 epithelial cells in the cultured explants in each treatment group (20-25 explants). A representative immunostaining of nuclear Stat5a/b in a PC that was responsive to AZD1480 is depicted. Biotin-streptavidin-amplified peroxidase-antiperoxidase immunodetection showed intensive positive immunostaining for nuclear Stat5a/b in explants cultured in the presence of vehicle, while AZD1480 treatment reduced the levels of nuclear Stat5a/b expression. (C) AZD1480 increased the fraction of apoptotic cells with fragmented DNA in the AZD1480 responder group (left panel). Apoptotic cells were detected by In Situ End Labeling of fragmented DNA.
Discussion
Therapeutic options for advanced PC patients are limited. Current therapeutic strategies for metastatic PC are directed against AR signaling or utilize non-targeted cytotoxic regimens (1, 4). In this work, we show that PC growth in pre-clinical models can be effectively suppressed by targeting Stat5a/b signaling using pharmacological inhibition of Jak2. The pharmacological Jak2 inhibitor AZD1480 disrupted phosphorylation, dimerization, nuclear translocation, DNA binding and transcriptional activity of Stat5a/b in PC cells, and induced apoptotic death of Jak2-Stat5a/b-driven PC cells in culture. Most importantly, AZD1480 inhibited not only primary androgen-dependent PC growth, but also CR growth of recurrent PC which emerged after androgen deprivation-induced regression of the original tumors in mice. Finally, we demonstrate that pharmacological targeting of Stat5a/b in clinical PCs ex vivo in organ explant cultures induced extensive epithelial cancer cell death.
Targeting Stat5a/b by AZD1480 inhibited growth of Jak2-Stat5a/b-driven PC cells in culture. We have shown previously that Stat5a/b is critical for the viability of human PC cells in vitro and for human PC tumor growth in nude mice using multiple alternative methods to genetically disrupt Stat5a/b action in PC cells (adenoviral delivery of DNStat5a/b, antisense oligonucleotides, or Stat5a/b siRNA) (14–17, 20). These observations have been validated and extended to the TRAMP mouse PC model (45) and C4-2 model (20) by others. In this work, we show for the first time that pharmacological inhibition of Jak2-Stat5a/b signaling inhibited PC cell viability. AZD1480 induced cell death through the mechanism of apoptosis and viability was rescued by overexpression of constitutively active Stat5a/b, which are results consistent with the type of cell death previously shown to be induced by genetic disruption of Stat5a/b activity in PC cells (14, 15).
Pharmacological inhibition of Stat5a/b by AZD1480 effectively suppressed both primary androgen-dependent PC growth as well as CRPC growth in nude mice after androgen deprivation-induced regression of the original tumors. Stat5a/b has been demonstrated previously to be involved in progression of PC to CR disseminated disease. Notably, high nuclear Stat5a/b expression in clinical PCs was identified as a significant predictor of both early recurrence and PC-specific death (21, 22) which both typically result from development of CR metastatic disease. Moreover, Stat5a/b has been shown to promote development of disseminated PC in in vitro and in vivo models (18). In this work, we demonstrated that inhibition of Stat5a/b signaling suppressed CRPC growth in vivo. AR transcriptional activity is believed to recover in CRPC despite pharmacological AR blockade and depletion of circulating and local androgens. Reactivation of AR in CRPC has been attributed to various molecular mechanisms, yet none completely account for progression to CRPC. Recently, we introduced the novel concept of a biologically significant, reciprocal physical interaction between Stat5a/b and AR, with each protein enhancing the other’s nuclear localization and transcriptional activity in AR-positive PC cells (12). Consistent with our findings, a recent report showed that knockdown of Stat5a/b by antisense oligonucleotides reduced stability of the AR protein in PC cells (20). However, in recurrent CR CWR22Pc tumors AZD1480 did not suppress transcriptional activity of AR, as evidenced by unaltered expression of PSA in the AZD1480-treated tumors vs. control group. Furthermore, AZD1480 did not down-regulate expression of full-length AR or AR splice variants, which have been suggested to mediate CR growth of PC (8–11). Collectively, these results suggest that AZD1480-induced suppression of primary and CR CWR22Pc tumor growth was not mediated through Jak2-Stat5a/b regulation of the AR. In line with this finding, we have previously shown that Stat5a/b inhibition induced rapid death of DU145 PC cells which do not express AR (17). Finally, AZD1480 is a potent inhibitor of not only Jak2-induced Stat5a/b activation, but also Stat3 activation in PC cells. However, since we intended to focus our study on determining the importance of targeting Stat5a/b in CRPC growth by AZD1480, we selected the CWR22Pc cell line/tumor system as our experimental model because of low Stat3 activation levels in CWR22Pc cells and tumors. The results of the present study suggest that AZD1480 inhibited both primary and CR growth of CWR22Pc tumors independently of Stat3. Future work will focus on identifying Stat5-regulated, AR/Stat3-independent molecular mechanisms governing regulation of PC cell viability.
AZD1480 induced apoptotic cell death and loss of viable epithelial cells in the majority of clinical PCs ex vivo in organ explant cultures. In addition to the Prl-Jak2-Stat5 pathway, Stat5a/b in PC is potentially activated by multiple kinases other than Jak2 such as Src (17) and potentially by the EGF-receptor family (46–49). Analysis of Stat5a/b activation status in PC samples prior to organ culture testing revealed that some of the AZD1480 non-responsive PCs had high levels of active Stat5a/b. This may be a result of kinases other than Jak2 activating Stat5a/b in those given non-responsive PCs. This suggests that direct inhibition of Stat5 by small-molecule Stat5 inhibitors may target larger fraction of PCs with active Stat5. In future studies, it will also be important to determine if expression of components of the Prl-Jak2-Stat5a/b autocrine loop, such as Prl and Jak2, will predict responsiveness of clinical PCs to pharmacological Jak2 inhibition. Clinical data available for Jak2 inhibitors in the treatment of myelofibrosis suggest that the adverse effects on hematopoiesis are manageable (50).
In summary, the findings presented in this work demonstrate, for the first time, responsiveness of primary and CRPC to pharmacological targeting of the Jak2-Stat5a/b signaling pathway. These findings are important and of high translational value since they pave the way for clinical efficacy studies assessing the pharmacological targeting of Stat5a/b in PC.
Supplementary Material
Translational Relevance.
A paucity of therapeutic options exist for patients with advanced prostate cancer (PC), which ultimately fails androgen deprivation and progresses to lethal castrate-resistant PC (CRPC). Inhibition of Stat5a/b, a validated therapeutic target protein in PC, represents a novel strategy to bypass androgen receptor (AR) signaling and eliminate CRPC growth. In the present study, we demonstrate proof-of-concept that pharmacological targeting of Stat5a/b signaling by AZD1480, a small-molecule inhibitor of upstream Jak2 kinase, blocks growth of not only androgen-dependent primary PC but also CRPC. Our results provide mechanistic evidence that AZD1480 robustly inhibits the molecular events leading to Stat5a/b activation and transcriptional regulation, decreasing PC cell viability by induction of apoptosis. Using human PC cell lines, xenograft tumors and ex vivo clinical PCs, we show that AZD1480 specifically disrupts Jak2-Stat5a/b signaling required for PC cell survival. These findings provide a strong rationale for further development of Stat5a/b inhibitors as therapy for CRPC.
Acknowledgments
This work was supported by grants from NCI (2RO1CA11358-06), AstraZeneca and Pennsylvania Department of Health to MTN. Shared Resources of KCC are partially supported by NIH Grant CA56036-08 (Cancer Center Support Grant, to KCC). We thank Geraldine Bebermitz, Shenghua Wen, Denis Hughes and Corinne Remer at AstraZeneca for their contributions.
Footnotes
Conflict of Interest: Dennis Huszar and Michael Zinda are employees of AstraZeneca Pharmaceuticals.
References
- 1.Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol. 2009;10:981–91. doi: 10.1016/S1470-2045(09)70229-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loblaw DA, Virgo KS, Nam R, Somerfield MR, Ben-Josef E, Mendelson DS, et al. Initial hormonal management of androgen-sensitive metastatic, recurrent, or progressive prostate cancer: 2006 update of an American Society of Clinical Oncology practice guideline. J Clin Oncol. 2007;25:1596–605. doi: 10.1200/JCO.2006.10.1949. [DOI] [PubMed] [Google Scholar]
- 3.Pestell RG, Nevalainen MT. Prostate Cancer: Signaling Networks, Genetics and New Treatment Strategies. Totowa: Humana Press; 2008. [Google Scholar]
- 4.Yap TA, Zivi A, Omlin A, de Bono JS. The changing therapeutic landscape of castration-resistant prostate cancer. Nat Rev Clin Oncol. 2011;8:597–610. doi: 10.1038/nrclinonc.2011.117. [DOI] [PubMed] [Google Scholar]
- 5.Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, et al. Mutation of the androgen-receptor gene in metastatic androgen- independent prostate cancer. N Engl J Med. 1995;332:1393–8. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 6.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Kein鋘en R, Palmberg C, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nature Gen. 1995;9:401–6. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 7.Penning TM, Byrns MC. Steroid hormone transforming aldo-keto reductases and cancer. Ann N Y Acad Sci. 2009;1155:33–42. doi: 10.1111/j.1749-6632.2009.03700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–77. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–13. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–30. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tan SH, Dagvadorj A, Shen F, Gu L, Liao Z, Abdulghani J, et al. Transcription factor Stat5 synergizes with androgen receptor in prostate cancer cells. Cancer Res. 2008;68:236–48. doi: 10.1158/0008-5472.CAN-07-2972. [DOI] [PubMed] [Google Scholar]
- 13.Zhu ML, Kyprianou N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr Relat Cancer. 2008;15:841–9. doi: 10.1677/ERC-08-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahonen TJ, Xie J, LeBaron MJ, Zhu J, Nurmi M, Alanen K, et al. Inhibition of transcription factor Stat5 induces cell death of human prostate cancer cells. J Biol Chem. 2003;278:27287–92. doi: 10.1074/jbc.M304307200. [DOI] [PubMed] [Google Scholar]
- 15.Dagvadorj A, Kirken RA, Leiby B, Karras J, Nevalainen MT. Transcription factor signal transducer and activator of transcription 5 promotes growth of human prostate cancer cells in vivo. Clin Cancer Res. 2008;14:1317–24. doi: 10.1158/1078-0432.CCR-07-2024. [DOI] [PubMed] [Google Scholar]
- 16.Dagvadorj A, Tan SH, Liao Z, Xie J, Nurmi M, Alanen K, et al. N-Terminal Truncation of Stat5a/b Circumvents PIAS3-Mediated Transcriptional Inhibition of Stat5 in Prostate Cancer Cells. Int J Biochem Cell Biol. 2011;42:2037–46. doi: 10.1016/j.biocel.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, Lisanti MP, et al. Transcription factor Stat3 stimulates metastatic behavior of human prostate cancer cells in vivo, whereas Stat5b has a preferential role in the promotion of prostate cancer cell viability and tumor growth. Am J Pathol. 2010;176:1959–72. doi: 10.2353/ajpath.2010.090653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu L, Vogiatzi P, Puhr M, Dagvadorj A, Lutz J, Ryder A, et al. Stat5 promotes metastatic behavior of human prostate cancer cells in vitro and in vivo. Endocr Relat Cancer. 2010;17:481–93. doi: 10.1677/ERC-09-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Ahonen TJ, Alanen K, Xie J, LeBaron MJ, Pretlow TG, et al. Activation of signal transducer and activator of transcription 5 in human prostate cancer is associated with high histological grade. Cancer Res. 2004;64:4774–82. doi: 10.1158/0008-5472.CAN-03-3499. [DOI] [PubMed] [Google Scholar]
- 20.Thomas C, Zoubeidi A, Kuruma H, Fazli L, Lamoureux F, Beraldi E, et al. Transcription factor Stat5 knockdown enhances androgen receptor degradation and delays castration-resistant prostate cancer progression in vivo. Mol Cancer Ther. 2010;10:347–59. doi: 10.1158/1535-7163.MCT-10-0850. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Zhang Y, Glass A, Zellweger T, Gehan E, Bubendorf L, et al. Activation of signal transducer and activator of transcription-5 in prostate cancer predicts early recurrence. Clin Cancer Res. 2005;11:5863–8. doi: 10.1158/1078-0432.CCR-05-0562. [DOI] [PubMed] [Google Scholar]
- 22.Mirtti T, Leiby BE, Abdulghani J, Aaltonen E, Pavela M, Mamtani A, et al. Nuclear Stat5a/b predicts early recurrence and prostate cancer-specific death in patients treated by radical prostatectomy. Hum Pathol. 2013;44:310–9. doi: 10.1016/j.humpath.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dagvadorj A, Collins S, Jomain JB, Abdulghani J, Karras J, Zellweger T, et al. Autocrine prolactin promotes prostate cancer cell growth via Janus kinase-2-signal transducer and activator of transcription-5a/b signaling pathway. Endocrinology. 2007;148:3089–101. doi: 10.1210/en.2006-1761. [DOI] [PubMed] [Google Scholar]
- 24.Haddad BR, Gu L, Mirtti T, Dagvadorj A, Bajaj R, Vogiatzi P, et al. Stat5a/b gene locus undergoes amplification during human prostate cancer progression. Am J Pathol. 2013;182:2264–2275. doi: 10.1016/j.ajpath.2013.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 26.Rui H, Djeu JY, Evans GA, Kelly PA, Farrar WL. Prolactin receptor triggering. Evidence for rapid tyrosine kinase activation. J Biol Chem. 1992;267:24076–81. [PubMed] [Google Scholar]
- 27.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–97. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu Y, Li H, Miki J, Kim KH, Furusato B, Sesterhenn IA, et al. Phenotypic characterization of telomerase-immortalized primary non-malignant and malignant tumor-derived human prostate epithelial cell lines. Exp Cell Res. 2006;312:831–43. doi: 10.1016/j.yexcr.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 29.Ahonen TJ, Harkonen PL, Rui H, Nevalainen MT. PRL signal transduction in the epithelial compartment of rat prostate maintained as long-term organ cultures in vitro. Endocrinology. 2002;143:228–38. doi: 10.1210/endo.143.1.8576. [DOI] [PubMed] [Google Scholar]
- 30.Nevalainen MT, Xie J, Bubendorf L, Wagner KU, Rui H. Basal activation of transcription factor signal transducer and activator of transcription (Stat5) in nonpregnant mouse and human breast epithelium. Mol Endocrinol. 2002;16:1108–24. doi: 10.1210/mend.16.5.0839. [DOI] [PubMed] [Google Scholar]
- 31.Dagvadorj A, Tan SH, Liao Z, Cavalli LR, Haddad BR, Nevalainen MT. Androgen-regulated and highly tumorigenic human prostate cancer cell line established from a transplantable primary CWR22 tumor. Clin Cancer Res. 2008;14:6062–72. doi: 10.1158/1078-0432.CCR-08-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahonen TJ, Harkonen PL, Laine J, Rui H, Martikainen PM, Nevalainen MT. Prolactin is a survival factor for androgen-deprived rat dorsal and lateral prostate epithelium in organ culture. Endocrinology. 1999;140:5412–21. doi: 10.1210/endo.140.11.7090. [DOI] [PubMed] [Google Scholar]
- 33.Nevalainen MT, Harkonen PL, Valve EM, Ping W, Nurmi M, Martikainen PM. Hormone regulation of human prostate in organ culture. Cancer Res. 1993;53:5199–207. [PubMed] [Google Scholar]
- 34.Nevalainen MT, Valve EM, Ahonen T, Yagi A, Paranko J, Harkonen PL. Androgen-dependent expression of prolactin in rat prostate epithelium in vivo and in organ culture. Faseb J. 1997;11:1297–307. doi: 10.1096/fasebj.11.14.9409549. [DOI] [PubMed] [Google Scholar]
- 35.Nevalainen MT, Valve EM, Ingleton PM, Harkonen PL. Expression and hormone regulation of prolactin receptors in rat dorsal and lateral prostate. Endocrinology. 1996;137:3078–88. doi: 10.1210/endo.137.7.8770934. [DOI] [PubMed] [Google Scholar]
- 36.Nevalainen MT, Valve EM, Ingleton PM, Nurmi M, Martikainen PM, Harkonen PL. Prolactin and prolactin receptors are expressed and functioning in human prostate. J Clin Invest. 1997;99:618–27. doi: 10.1172/JCI119204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nevalainen MT, Valve EM, Makela SI, Blauer M, Tuohimaa PJ, Harkonen PL. Estrogen and prolactin regulation of rat dorsal and lateral prostate in organ culture. Endocrinology. 1991;129:612–22. doi: 10.1210/endo-129-2-612. [DOI] [PubMed] [Google Scholar]
- 38.Nevalainen MT, Xie J, Torhorst J, Bubendorf L, Haas P, Kononen J, et al. Signal transducer and activator of transcription-5 activation and breast cancer prognosis. J Clin Oncol. 2004;22:2053–60. doi: 10.1200/JCO.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 39.Pretlow TG, Wolman SR, Micale MA, Pelley RJ, Kursh ED, Resnick MI, et al. Xenografts of primary human prostatic carcinoma. J Natl Cancer Inst. 1993;85:394–8. doi: 10.1093/jnci/85.5.394. [DOI] [PubMed] [Google Scholar]
- 40.Sramkoski R, Pretlow Tn, Giaconia J, Pretlow T, Schwartz S, Sy M, et al. A new human prostate carcinoma cell line, 22Rv1. In Vitro Cell Dev Biol Anim. 1999;35:403–9. doi: 10.1007/s11626-999-0115-4. [DOI] [PubMed] [Google Scholar]
- 41.Culig Z, Steiner H, Bartsch G, Hobisch A. Interleukin-6 regulation of prostate cancer cell growth. J Cell Biochem. 2005;95:497–505. doi: 10.1002/jcb.20477. [DOI] [PubMed] [Google Scholar]
- 42.Lou W, Ni Z, Dyer K, Tweardy DJ, Gao AC. Interleukin-6 induces prostate cancer cell growth accompanied by activation of stat3 signaling pathway. Prostate. 2000;42:239–42. doi: 10.1002/(sici)1097-0045(20000215)42:3<239::aid-pros10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 43.Clark J, Edwards S, Feber A, Flohr P, John M, Giddings I, et al. Genome-wide screening for complete genetic loss in prostate cancer by comparative hybridization onto cDNA microarrays. Oncogene. 2003;22:1247–52. doi: 10.1038/sj.onc.1206247. [DOI] [PubMed] [Google Scholar]
- 44.Ioannidis S, Lamb ML, Wang T, Almeida L, Block MH, Davies AM, et al. Discovery of 5-Chloro-N(2)-[(1S)-1-(5-fluoropyrimidin-2-yl)ethyl]-N(4)-(5-methyl-1H-pyr azol-3-yl)pyrimidine-2,4-diamine (AZD1480) as a Novel Inhibitor of the Jak/Stat Pathway. J Med Chem. 2011;54:262–76. doi: 10.1021/jm1011319. [DOI] [PubMed] [Google Scholar]
- 45.Kazansky AV, Spencer DM, Greenberg NM. Activation of signal transducer and activator of transcription 5 is required for progression of autochthonous prostate cancer: evidence from the transgenic adenocarcinoma of the mouse prostate system. Cancer Res. 2003;63:8757–62. [PubMed] [Google Scholar]
- 46.Arcasoy MO, Jiang X, Haroon ZA. Expression of erythropoietin receptor splice variants in human cancer. Biochem Biophys Res Commun. 2003;307:999–1007. doi: 10.1016/s0006-291x(03)01303-2. [DOI] [PubMed] [Google Scholar]
- 47.Bidosee M, Karry R, Weiss-Messer E, Barkey RJ. Regulation of growth hormone receptors in human prostate cancer cell lines. Mol Cell Endocrinol. 2009;309:82–92. doi: 10.1016/j.mce.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 48.Feldman L, Wang Y, Rhim JS, Bhattacharya N, Loda M, Sytkowski AJ. Erythropoietin stimulates growth and STAT5 phosphorylation in human prostate epithelial and prostate cancer cells. Prostate. 2006;66:135–45. doi: 10.1002/pros.20310. [DOI] [PubMed] [Google Scholar]
- 49.Wang Z, Luque RM, Kineman RD, Ray VH, Christov KT, Lantvit DD, et al. Disruption of growth hormone signaling retards prostate carcinogenesis in the Probasin/TAg rat. Endocrinology. 2008;149:1366–76. doi: 10.1210/en.2007-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.