

SUMMARY
UBE2M and UBE2F are two family members of neddylation E2 conjugating enzyme that, together with E3s, activate CRLs (Cullin-RING Ligases) by catalyzing cullin neddylation. However, whether and how two E2s cross-talk with each other are largely unknown. Here, we report that UBE2M is a stress-inducible gene subjected to cis-transactivation by HIF-1α and AP1, and MLN4924, a small molecule inhibitor of E1 NEDD8-activating enzyme (NAE), up-regulates UBE2M via blocking degradation of HIF-1α and AP1. UBE2M is a dual E2 for targeted ubiquitylation and degradation of UBE2F, acting as a neddylation E2 to activate CUL3-Keap1 E3 under physiological condition, but as an ubiquitylation E2 for Parkin-DJ-1 E3 under stressed conditions. UBE2M-induced UBE2F degradation leads to CRL5 inactivation and subsequent NOXA accumulation to suppress the growth of lung cancer cells. Collectively, our study establishes a negative regulatory axis between two neddylation E2s with UBE2M ubiquitylating UBE2F, and two CRLs with CRL3 inactivating CRL5.
Keywords: Cullin 3, DJ-1, Keap1, Neddylation, Parkin, UBE2F/UBE2M, Stress responsiveness, Ubiquitylation
eTOC Blurb
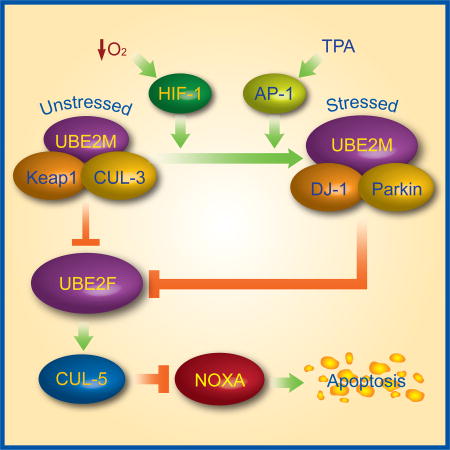
UBE2M and UBE2F are two family members of neddylation conjugating E2 enzymes. Zhou et al. find that UBE2M is a stress inducible protein that acts as a dual E2 for CUL-3 neddylation and Parkin E3 to promote UBE2F degradation, which inactivates CUL-5 to cause substrate NOXA accumulation for apoptosis induction.
INTRODUCTION
Protein neddylation is one type of post-translational modifications that activates or inactivates protein function by conjugation of the ubiquitin-like protein NEDD8 (neural precursor cell expressed, developmentally downregulated 8) to its target proteins (Kamitani et al., 1997). Like ubiquitylation, neddylation is catalyzed by a sequential enzymatic cascade involving one neddylation activating enzyme E1 (NAE, a heterodimer of NAE1/APP-BP1 and NAEβ/UBA3); one of the two neddylation conjugating E2 enzymes (UBE2M, also known as UBC12, and UBE2F); and several neddylation E3 ligases (Zhao et al., 2014). Although numerous proteins have been reported as neddylation substrates, the cullin family proteins, the scaffold component of the cullin-RING ligases (CRLs), have been characterized as physiological substrates of neddylation (Saha and Deshaies, 2008; Sakata et al., 2007). Neddylation of cullins activates the CRL E3s (Duda et al., 2008), the largest family of E3 ubiquitin ligase, which are responsible for ubiquitylation of ~20% cellular proteins for targeted degradation (Soucy et al., 2009), thus precisely regulating many biological processes (Zhao et al., 2014). Abnormal activation of several components of CRLs is frequently found in numerous human cancers, leading to validation of CRL E3s as attractive cancer targets (Zhao and Sun, 2013; Zhou et al., 2018). Indeed, MLN4924, a small molecular inhibitor of NAE, which indirectly inhibits CRLs by blocking cullin neddylation (Soucy et al., 2009), is currently in the Phase I/II clinical trials for anti-cancer applications (Nawrocki et al., 2012; Zhao et al., 2014)
In mammalian cells, neddylation E2s consist of two family members: well-studied UBE2M/UBC12, and less characterized UBE2F. While the core domains and the N-terminal extension of either UBE2M or UBE2F bind to ubiquitin-fold domain of NAE1 subunit and hydrophobic groove of NAEβ subunit of NAE, respectively, the structures of UBE2Fcore and UBE2Mcore do show some unique features (Huang et al., 2009; Huang et al., 2004; Huang et al., 2005). Biochemically, UBE2M pairs with RBX1 to regulate neddylation of CUL1–CUL4, whereas UBE2F is rather specific for RBX2/SAG to mediate CUL5 neddylation (Huang et al., 2009). The transcriptome analysis of siRNA-based knockdown of UBE2M vs. UBE2F revealed largely non-overlapping patterns of gene expression (Huang et al., 2009). Biologically, NIH3T3 cells with knockdown of UBE2M, but not UBE2F, grew much slowly (Huang et al., 2009). Furthermore, while both UBE2M and UBE2F were recruited to DNA-damage sites, depletion of UBE2M, but not UBE2F, significantly sensitized cells to IR (Brown et al., 2015), or to other DNA damaging agents (Cukras et al., 2014). We recently found that UBE2F couples with SAG/RBX2 E3 to facilitate CUL5 neddylation and activation, resulting in NOXA poly-ubiquitylation via K11 linkage for proteasomal degradation and survival of lung cancer cells, thus acting as a cancer survival protein (Zhou et al., 2017). However, whether and how the two neddylation E2s regulate each other are completely unknown.
In the present study, we found that UBE2M is stress-inducible protein, subjected to upregulation by HIF-1 and AP-1, whereas UBE2F is degraded by UBE2M-associated E3 ligases. Specifically, UBE2F is ubiquitylated and degraded by UBE2M-activated CRL3Keap1 E3 under physiological condition, and by Parkin-DJ-1 E3 under stressed conditions. Thus, UBE2M acts as a dual E2 for both neddylation and ubiquitylation to degrade UBE2F, leading to inactivation of CRL5 and accumulation of NOXA, a UBE2F/SAG/CRL5 substrate (Zhou et al., 2017), and growth suppression of lung cancer cells. Together, our study revealed a cross-talk between two neddylation E2s with UBE2M degrading UBE2F, and between two CRLs with CRL3 inactivating CRL5, thus providing an E3 ligase cascade to tightly regulate many cellular processes.
RESULTS
MLN4924 differentially regulates two neddylation E2s, UBE2M and UBE2F
During our recently characterization of pro-apoptotic protein NOXA as a substrate of UBE2F/SAG/CRL5 E3 ligase (Zhou et al., 2017), we unexpectedly and consistently found that MLN4924, a small molecular inhibitor of E1 NEDD8-activating enzyme (Soucy et al., 2009), differentially regulates two neddylation E2s, UBE2M and UBE2F. Specifically, MLN4924 which inhibited neddylation of all cullins, caused a dose- and time-dependent increase of UBE2M, but decrease of UBE2F at protein levels (Figs. 1A&B, S1A&B). MLN4924 also caused a dose-dependent increase of UBE2M mRNA without affecting UBE2F mRNA (Figs. 1C&S1C). This opposite effect on two neddylation E2s were observed in all lung cancer cell lines treated with MLN4924, although to a various extent (Figs. 1D&E). Furthermore, although MLN4924 increased the basal level of UBE2M, it had no effect on its protein half-life, whereas MLN4924 significantly decreased UBE2F basal level and shortened its protein half-life (Figs. 1F–G&S1D–E). Thus, the effect of MLN4924 on UBE2M or UBE2F occurs likely at the transcriptional or post-translational levels, respectively.
Figure 1. MLN4924 differentially regulates UBE2M and UBE2F.

(A and B) A427 cells were treated with MLN4924 at indicated concentrations (24 h) or time points, followed by IB using indicated Abs.
(C) A427 cells were treated with different doses of MLN4924 for 24 h, followed by qRT- PCR. Data are represented as mean ± SEM of three independent experiments. * p < 0.05; *** p < 0.001, based on One-way ANOVA.
(D and E) Various lung cancer cell lines were treated with DMSO or MLN4924 (0.3 µM) for 24 h, followed by IB with indicated Abs (D) or qRT- PCR assay. Data are represented as mean ± SEM of three independent experiments. * p < 0.05; ** p < 0.01, based on the Student’s t test (E).
(F) H358 cells were treated with MLN (0.3 µM) for 24 h and then switched to fresh medium (10% FBS without MLN4924) containing cycloheximide (CHX) and incubated for indicated time periods before being harvested for IB.
(G) Measurement of UBE2M and UBE2F T1/2: the band density was quantified using ImageJ software and plotted.
(H and I) A427 cells were transfected with indicated siRNAs and treated with MLN4924 0.3 µM for 24 h, followed by IB (H) or qRT-PCR assay (I). Data are represented as mean ± SEM of three independent experiments. *** p < 0.001, based on One-way ANOVA.
See also Figure S1.
Previous studies have shown that both HIF-1α and c-JUN are substrates of CRL2VHL (Kamura et al., 2000) and CRL1 (SCFFBXW7) (Nateri et al., 2004; Wei et al., 2005), respectively, which are likely accumulated upon MLN4924-mediated deneddylation of cullins and subsequent inactivation of these two CRLs. Here, we confirmed that in lung cancer cell lines, both HIF-1α and c-JUN proteins were accumulated upon MLN4924 treatment in a dose- and time-dependent manners (Figs 1A, B, D&S1A, B). We then determined whether accumulations of HIF-1α and c-JUN are responsible for observed changes in UBE2M and UBE2F via a siRNA knockdown approach. Indeed, MLN4924-mediated increase of UBE2M at both mRNA and protein levels and decrease of UBE2F protein level were remarkably abrogated upon knockdown of either HIF-1α or c-JUN. Double knockdown caused near complete blockage of UBE2M expression at both mRNA and protein levels with restoration of UBE2F level (Figs. 1H&I, S1F&G). These results demonstrated that transcription factors HIF-1α and c-JUN play the essential role in regulation of basal and induced expression of UBE2M and subsequently the level of UBE2F.
UBE2M is a stress inducible protein, transcriptionally activated by HIF-1α or c-JUN/AP1
To define molecular basis of how UBE2M mRNA is subjected to MLN4924 regulation, we searched the promoter and intron-1 sequences of human UBE2M gene with a focus on cis-elements/binding motifs of transcription factors HIF-1α and AP1. Indeed, we found one HIF-1α binding site within the intron-1 and four AP1 binding sites within the promoter of human UBE2M gene (Fig. S2A), suggesting that UBE2M is likely subjected to transcriptional regulation by these two transcript factors.
We followed up this lead and first tested whether endogenous UBE2M and UBE2F are subjected to regulation under two hypoxia conditions triggered by CoCl2, a chemical hypoxia mimetic agent (Piret et al., 2002), or by a hypoxia chamber filled with 1% O2, 5% CO2, and 93% N2, or by TPA, a typical tumor promoter and mitogen stimulator that induces c-JUN (Colburn et al., 1979). Indeed, hypoxia (CoCl2 or hypoxia chamber) (Figs. 2A&B, D&E; Figs. S2B&D), or TPA treatment (Figs. 2C&D; Figs. S2C&E) caused a dose-dependent accumulation of HIF-1α and c-JUN, respectively, with accompanied UBE2M increases in both protein and mRNA levels, and UBE2F decrease at the protein, but not mRNA levels. Importantly, completely different from MLN4924, which inhibits neddylation of all cullins (Figs. 1A&B, S1A&B), hypoxia or TPA increased the neddylation of CUL1–4, whereas decreased the neddylation of CUL5 in time- and dose-dependent manners (Figs. 2A–C and S2B–C). We further determined whether stresses-induced alterations in cullin neddylation affects their ubiquitin E3 ligase activity by measuring the levels of their representative substrates. Indeed, we found a) dose-dependent decrease of p27 (a CRL1 substrate) (Sutterluty et al., 1999), SLBP (a CRL2 substrate, independent of hydroxylation) (Dankert et al., 2017), NRF2 (a CRL3 substrate) (Kobayashi et al., 2004) and CDT1 (a CRL4 substrate) (Hu et al., 2004), reflecting activation of Cullins 1–4, and conversely, b) dose-dependent increase of NOXA (a CRL5 substrate) (Zhou et al., 2017), reflecting inactivation of Cullin 5. Collectively, our results showed that stresses-mediated UBE2M induction (to trigger CUL1–4 neddylation) and UBE2F reduction (to inhibit CUL5 neddylation) indeed has biochemical consequence.
Figure 2. UBE2M is subjected to transactivation by HIF-1α and AP1.

(A–E) H358 cells were treated with different doses of CoCl2 (A) or TPA (C) for 24 h, or incubated in a hypoxia chamber with 1% O2, 5% CO2, and 93% N2, for indicated time period (B), followed by IB with indicated Abs (A–C) or qRT-PCR. Data are represented as mean ± SEM of three independent experiments. * p < 0.05, ** p < 0.01, *** p < 0.001, based on One-way ANOVA (D–E).
(F) Representation of luciferase reporters driven by intron-1 of human UBE2M in the presence or absence of HIF-1α binding site boxed in black.
(G and H) H1299 cells were transiently transfected with indicated luciferase reporter, along with Renilla-luc reporter for normalization of transfection efficiency. Twenty-four hrs post-transfection, cells were treated with CoCl2 (G) or MLN4924 (H) at indicated concentrations for 24 h and then collected for luciferase activity assay. Luciferase activity was presented as fold activation from three independent transfections, each run in triplicate after Renilla-luc normalization.
(I) Representation of a series of luciferase reporters driven by various deletion mutants of human UBE2M promoter with putative AP1 binding site boxed in black.
(J and K) H1299 cells were transiently transfected with indicated luciferase reporters, along with Renilla-luc reporter, and luciferase activity assayed as described above.
(L and M) ChIP analysis: H1299 cells were treated with indicated compounds for 24 h. Cellular DNA was isolated and used directly as PCR templates (input), or subjected to immunoprecipitation in the absence (Ab−) or presence of IgG or Abs against HIF-1α (L) or c-JUN (M). The DNA precipitates were subjected to PCR amplification using primer sets flanking the HIF-1α binding motif (L) or AP1 binding site-2 (M).
See also Figure S2.
We next defined whether CoCl2-induced UBE2M induction is mediated by the cis-HIF-1α binding site identified in the intron of human UBE2M gene (Fig. S2A). To this end, we cloned two luciferase reporters, driven by a partial intron-1 fragment with (UBE2M-WT) or without (UBE2M-MU) HIF-1α binding site (Fig. 2F), and observed a dose-dependent induction of luciferase activity in UBE2M-WT (WT), but not in UBE2M-MU (MU) reporter, upon treatment with two doses of CoCl2 (Fig. 2G) or MLN4924 (Fig. 2H). Thus, this HIF-1α binding site is likely responsible for UBE2M induction at the transcriptional level by CoCl2 or MLN4924.
Similarly, to define the role of four cis-AP1 binding sites found in the promoter of human UBE2M gene (Fig. S2A), in mediating TPA effects, we constructed 4 luciferase reporters, driven by the promoter sequence containing 1) all four binding sites (UBE2M-WT1) or 2) with the first binding site deleted (UBE2M-MU1), or 3) three binding sites (UBE2M-WT2) or 4) with both the first and second binding sites deleted (UBE2M-MU2) (Fig. 2I). Luciferase activity assays showed that upon exposure to TPA or MLN4924, a dose dependent induction of the promoter activity was seen in the first three constructs, whereas a significant loss was observed when UBE2M-MU2 was used (Figs. 2J&K), indicating that the second AP1 binding site is mostly responsible for UBE2M induction by TPA or MLN4924.
We next performed a ChIP experiment to define a causal role of the HIF-1α binding site in the intron-1 of the UBE2M gene by detecting a direct HIF-1α binding. H1299 cells were left untreated or treated with CoCl2 or MLN4924 for 24 h to activate HIF-1α and then subjected to ChIP assays. A 185 bp PCR-amplified fragment, corresponding to the HIF-1α binding site-containing UBE2M intron-1, was detected in the input sample or in the samples immuno-precipitated with HIF-1α antibody upon CoCl2 or MLN4924 treatment, but not in two control samples either without antibody or with IgG for immunoprecipitation (Fig. 2L). Likewise, we performed ChIP assays to measure a direct binding of c-JUN to the first two AP1 binding sites. Consistent with the luciferase reporter finding, c-JUN was only detected in the second but not the first AP1 binding site upon exposure to TPA or MLN4924 (Figs. 2M&S2F). Collectively, we concluded that HIF-1α or c-JUN directly binds to its consensus sequences in the intron-1 or promoter of the human UBE2M gene, respectively, to transactivate its expression, leading to increased levels of UBE2M mRNA and subsequently protein abundance. Thus, UBE2M is stress inducible protein subjected to up-regulation by hypoxia and TPA. It is also noteworthy that in these experimental conditions, elevated UBE2M protein abundance is coupled with a sharp decrease in UBE2F, indicating a cross-talk regulation between these two E2 enzymes.
UBE2M promotes UBE2F ubiquitylation and degradation
Given that MLN4924, hypoxia, and TPA all caused transcriptional up-regulation of UBE2M, whereas simultaneously reduced the protein, but not mRNA levels of UBE2F, we hypothesized that UBE2M might be actively involved in the control of UBE2F stability. To test this hypothesis, we first determined whether UBE2M accumulation is causally related to UBE2F reduction upon exposure to three compounds. To this end, we silenced UBE2M and found UBE2F reduction induced by all three compounds was largely abrogated (Figs. 3A, S3A). Ectopic expression of UBE2M, but not its enzymatic dead mutant (UBE2M-C111S), reduced UBE2F protein levels, which was largely blocked by proteasome inhibitor, MG132, without affecting UBE2F mRNA levels (Figs. S3B&C). Thus, UBE2M appears to promote UBE2F degradation that largely depends on its catalytic activity.
Figure 3. UBE2M negatively regulates UBE2F protein levels.

(A) A427 cells were firstly transfected with si-Cont or si-UBE2M-2 and then treated with indicated compounds. Cells were harvested for IB with indicated Abs.
(B and C) H358 cells were transfected with HA-tagged UBE2M for 48 h, followed by IB with indicated Abs (B), or switched to fresh medium (10% FBS) containing cycloheximide (CHX) and incubated for indicated time periods before being harvested for IB (top panels, C), and the band density was quantified using ImageJ software and plotted (bottom panel, C).
(D and E) H358 cells were transfected with two independent siRNAs targeting UBE2M or si-Cont for 48 h, followed by IB with indicated Abs (D), or were transfected with si-UBE2M-2 and then switched to fresh medium (10% FBS) containing cycloheximide (CHX) post-transfection and incubated for indicated time periods before being harvested for IB (top panels, E) and the band density was quantified (bottom panel, E).
(F) A427 and H358 cells were transfected with two independent siRNAs targeting UBE2F or si-Cont for 48 h, followed by IB with indicated Abs.
(G) UBE2F and UBE2M bind to each other physically. H358 cell lysates were immunoprecipitated using UBE2M Ab (left), or UBE2F Ab (right), followed by IB to detect endogenous proteins. The 10% of the extracts was loaded as the input.
(H and I) The 293 cells were co-transfected with indicated plasmids or treated with or without MG132. Ni-NTA affinity-purified fractions (top panel) were analyzed by IB with anti-UBE2F (H) or anti-UBE2M (I); 10% of Ni-NTA affinity-purified fractions or whole cell extracts (bottom panels) were analyzed by HA antibody to detect the exogenous Ub, UBE2F, or UBE2M.
(J) H1299 cells were cotransfected with HA-His-Ub and HA-UBE2F, and then transfected with si-Cont or si-UBE2M-2, followed by treatment with indicated compounds for 24 h. Cell lysates were subjected to Ni-NTA purification, followed by IB with anti-UBE2F Ab (top panel). Whole cell lysates were subjected to IB with indicated Ab (bottom panels).
See also Figure S3.
It has been established that UBE2M pairs with RBX1 to neddylate and activate cullins 1–4, whereas UBE2F couples with RBX2/SAG to neddylate and activate cullin 5 (Huang et al., 2009), which promotes ubiquitylation and degradation of NOXA (Zhou et al., 2017). We therefore determined whether UBE2M, via reducing UBE2F, would inactivate CRL5 E3 ligase activity. While ectopic UBE2M expression increased CUL1 neddylation, as expected, it also reduced CUL5 neddylation to inactivate CRL5, resulting in NOXA accumulation (Figs. 3B, S3D). Consistently, ectopic UBE2M expression substantially shortened protein half-lives of UBE2F and SAG (whose stability is dependent of UBE2F binding) (Zhou et al., 2017), leading to inhibition of CUL5 neddylation and consequent extension of NOXA protein half-life (Figs. 3C, S3E). Conversely, UBE2M knockdown increased the levels of UBE2F, SAG and CUL5 neddylation, and consequently reduced NOXA level without affecting UBE2F mRNA levels (Figs. 3D, S3F–H). Indeed, UBE2M knockdown extended the protein half-lives of UBE2F and SAG, leading to increased CUL5 neddylation and consequent shortening of NOXA protein half-life (Figs. 3E, S3I), indicating that UBE2M negatively regulates UBE2F/SAG/CUL5 complex. On the other hand, UBE2F knockdown had no significant effect on UBE2M (Fig. 3F), excluding a possible regulation of UBE2M by UBE2F.
We further found that two neddylation E2s bound to each other under unstressed physiological conditions (Fig. 3G), and UBE2M-WT, but not its C111S mutant, promoted UBE2F poly-ubiquitylation, which is more evident by MG132 treatment to block degradation (Fig. 3H). On the other hand, neither wild type UBE2F nor its enzymatic dead mutant (UBE2F-C116A) promoted poly-ubiquitylation of UBE2M (Fig. 3I). Finally, we found that three compounds, via inducing UBE2M, could enhance the poly-ubiquitylation of UBE2F, whereas UBE2M knockdown could partially abrogate it, indicating a UBE2M dependent manner (Fig. 3J). Thus, UBE2M shortens protein half-life of UBE2F by promoting its ubiquitylation and degradation.
CUL3, but not other cullins, promotes UBE2F ubiquitylation and degradation
Given that UBE2M is a neddylation E2, it should not directly promote poly-ubiquitylation of UBE2F. It acts, most likely, via activation of one of the downstream CRLs. To this end, we silenced all the cullins individually in either H358 or A427 cells, along with knock-down of UBE2M as a positive control, and found that among all five cullins, only CUL3 knockdown caused the accumulation of UBE2F (Figs. 4A, S4A). Likewise, only overexpression of CUL3, but not other cullins, substantially reduced UBE2F levels (Figs. 4B, S4B). Furthermore, CUL3 knockdown increased UBE2F protein without affecting mRNA levels, while CUL3 overexpression decreased UBE2F protein (but not mRNA), which can be reversed by MG132, (Figs. S4C&D), suggesting that CUL3 promotes UBE2F degradation. Indeed, the protein half-life of UBE2F was extended by CUL3 knockdown, but shortened by ectopically expressed CUL3 (Figs. 4C&D, S4E&F).
Figure 4. CUL3 negatively regulates UBE2F protein levels.

(A and B) H358 cells were transfected with siRNAs oligoes targeting different cullins (A) or either indicated plasmids (B), followed by IB with indicated Abs.
(C and D) A427 cells were transfected with siRNA oligo targeting CUL3 (C) or plasmid expressing CUL3 (D), then switched to fresh medium (10% FBS) containing cycloheximide (CHX) post-transfection and incubated for indicated time periods before being harvested for IB (upper panels, C and D) and the band density was quantified (bottom panels, C and D).
(E and F) H358 cells were transfected with indicated plasmids and cells lysates were then immunoprecipitated using HA antibody (E) or Myc antibody (F), followed by IB with indicated Abs. WCL: Whole cell extracts.
(G–J) The 293 cells were co-transfected with indicated plasmids. HA-His-Ub tagged UBE2F were purified via Ni-NTA affinity, and detected with anti-UBE2F Ab (G & I), or incubated in a reaction mixture containing ATP, ubiquitin, E1, E2 (UBCH5C), E3 (Myc-tagged CUL3) (H) or various Myc-tagged cullins (J) and substrate (purified UBE2F protein), followed by ubiquitylation assay and IB with anti-UBE2F Ab.
See also Figure S4.
We then performed an IP pull-down assay to identify the binding between two neddylation E2s and cullins, respectively, and found that exogenously expressed UBE2M pulled down endogenous CUL1–4, as expected, whereas UBE2F pulled down endogenous CUL5 as well as CUL3 (Fig. 4E). Reciprocally, both ectopically expressed CUL5 or CUL3 bound to endogenous (Fig. 4F) and exogenous UBE2F (Fig. S4G), whereas CUL1–4 bound to endogenous UBE2M (Fig. 4F). To further demonstrate that UBE2F is a bona fide substrate of CUL3, we performed a cell-based (in vivo) ubiquitylation assay and found that CUL3 promoted poly-ubiquitylation of UBE2F, which could be enhanced by MG132 (Fig. 4G). CUL3, along with UBE2M, also promotes poly-ubiquitylation of UBE2F in an in vitro ubiquitylation assay (Fig. 4H). Finally, we demonstrated that among all cullins tested, only CUL3 promoted UBE2F poly-ubiquitylation in both in vivo and in vitro assay systems (Figs. 4I&J), indicating that CUL3 promotes UBE2F poly-ubiquitylation for subsequent proteasomal degradation. Taken together, our results demonstrate that UBE2F is a novel ubiquitin substrate of CRL3.
We further confirmed that neddylation of CUL5, but not CUL1 was decreased upon CUL3 overexpression, but increased upon CUL3 knockdown (Figs. S4H&I). Thus, through UBE2F depletion via promoting its ubiquitylation and degradation, CUL3 inhibits CUL5 activity by blocking its neddylation.
Keap1 is responsible for CUL3-induced UBE2F ubiquitylation and degradation
Previous studies have established that a subset of proteins containing a BTB domain act as substrate-specific adaptors which preferentially bind to CUL3 (Zhao and Sun, 2013), with Keap1 being the first reported mammalian adaptor of the CUL3-based E3 ligase system (Kobayashi et al., 2004). It has been previously reported that Keap1 binds to its substrate via a consensus binding sequence ETGE (Tong et al., 2006). Computer-based search of UBE2F protein indeed found such a motif, which is evolutionarily conserved (Fig. S5A). We then determined whether Keap1 is a substrate-recognizing subunit in CUL3 for targeted ubiquitylation and degradation of UBE2F. Ectopic overexpression of Keap1 decreased UBE2F protein, but not mRNA (Fig. 5A, S5B–D). Furthermore, the protein half-life of UBE2F-WT, but not its ETAA mutant, was significantly shortened by ectopic overexpression of Keap1 (Fig. 5B). Meanwhile, Keap1 knockdown increased UBE2F protein level but not its mRNA level (Figs. 5C, S5E–G), and half-life of endogenous UBE2F was extended by Keap1 knockdown (Figs. 5D, S5H). We also used Keap1−/− MEF cells to demonstrate that UBE2F was accumulated in Keap1−/− cells (Fig. S5I). Finally, we demonstrated that UBE2F reduction by UBE2M overexpression could be partially rescued by single knock-down of Keap1 or CUL3, and completely rescued by double knockdown (Fig. 5E), clearly indicating that Keap1/CUL3 is involved in negative regulation of UBE2F by UBE2M.
Figure 5. Keap1 negatively regulates UBE2F protein levels.

(A and B) A427 cells were transfected with indicated plasmids, followed by IB (A) and 293 cells were transfected with indicated plasmids and then switched to fresh medium (10% FBS) containing cycloheximide (CHX) post-transfection and incubated for indicated time periods before being harvested for IB (left panels, B) and the band density was quantified (right panel, B).
(C and D) H358 cells were transfected with indicated siRNA oligoes, followed by IB (C) or subjected to half-life study as described above (D).
(E) H358 cells were transfected with pcDNA3 or HA-UBE2M first, and then silenced with siRNA oligoes targeting control, Keap1, and/or CUL3, followed by IB assay by indicated Abs.
(F) H358 cells were transfected with FLAG-Keap1, followed by IP with FLAG Ab and IB with indicated Abs.
(G) H358 cells were transfected with HA-UBE2F-WT or -ETAA mutant, followed by IP with HA- Ab, and IB with Keap1 Ab.
(H–L) The 293 cells were co-transfected with indicated plasmids. HA-His-Ub tagged UBE2F were purified via Ni-NTA affinity, and detected with anti-UBE2F Ab (H, K & L) or followed by IP with HA-Ab and IB with indicated Abs (J); or incubated in a reaction mixture containing ATP, ubiquitin, E1, E2 (UBCH5C), E3 (FLAG-Keap1) and substrate (purified UBE2F protein), followed by ubiquitylation assay and IB with anti-UBE2F Ab (I). WCL: Whole cell extracts.
See also Figure S5.
We then determined whether Keap1 indeed bound to UBE2F and found that ectopically expressed Keap1 could pull down both endogenous UBE2F and UBE2M, along with CUL3 and RBX1 (Fig. 5F), whereas ectopically expressed CUL3 also pulled down endogenous UBE2F, UBE2M, Keap1, along with RBX1 (Fig. S5J), indicating they form a complex in vivo. We further found that ectopically expressed UBE2F-WT, but not its ETAA mutant, pulled down endogenous Keap1 (Fig. 5G). Using both in vivo (Fig. 5H) and in vitro (Fig. 5I) ubiquitylation assays, we found that Keap1 indeed promoted UBE2F poly-ubiquitylation, which was enhanced by MG132 (Fig. 5H).
The TCGA database search revealed frequent Keap1 mutations in human cancers. We next determined the causal relationship between Keap1-UBE2F binding and Keap1-induced UBE2F ubiquitylation. We tested three Keap1 mutants derived from cancers on the substrate binding surface (Fig. S5K), and found that mutant R554Q, but not G333V and G480W, failed to bind to UBE2F (Fig. 5J), hence failed to promote UBE2F polyubiquitylation (Fig. 5K). Likewise, UBE2F-ETAA mutant, which failed to bind to Keap1 (Fig. 5G), also failed to be ubiquitylated by Keap1 (Fig. 5K). Furthermore, overexpression of Keap1-WT, -G333V, -G480W, but not its binding deficient -R554Q mutant, blocked neddylation of only CUL5 (Fig. S5L), whereas Keap1 knockdown only enhanced CUL5 neddylation (Fig. S5M). Finally, we did a head-to-head comparison of UBE2F ubiquitylation by UBE2M, CUL3 and Keap1, and found that UBE2M is much active than CUL3 and Keap1 (Fig. 5L). Combination of all three did not further enhance UBE2M-induced UBE2F polyubiquitylation (Fig. 5L). Thus, Keap1 promotes UBE2F polyubiquitylation in a manner largely dependent of its UBE2F binding, and UBE2M appears to be a much better promoter of UBE2F polyubiquitylation than CUL3Keap1, suggesting a potential involvement of another E3 besides CUL3Keap1.
Parkin/DJ-1 plays a major role in promoting UBE2F ubiquitylation and degradation, especially under stressed conditions
It is paradoxical that MLN4924, on one hand, inhibits NAE to block the entire neddylation modification and inactivate all CRLs, including CUL3-Keap1. On the other hand, it induces UBE2M expression to promote UBE2F ubiquitylation (Fig. 3J). This effect, therefore, must be independent of the neddylation-dependent CUL3-Keap1 E3, suggesting an involvement of another distinct E3 for UBE2F. Moreover, knockdown of either CUL3 or Keap1 only partially blocked UBE2M-mediated UBE2F ubiquitylation, whereas MLN4924 treatment in CUL3 knockdown cells remains its ability of stimulating UBE2F polyubiquitylation (Fig. S6A). Thus, an UBE2M-associated E3 ligase, independent of neddylation modification, is likely involved in UBE2F polyubiquitylation and degradation.
We then used affinity purification-mass spectrometry method to identify UBE2M binding proteins in H1299 cells with ectopic expression of tagged wild-type UBE2M and UBE2M-C111S mutant. This approach led to the identification of 4 ubiquitin pathway related proteins (PARK7, SUMO2, CUL3, and USP5), which selectively bind to wild-type UBE2M. We focused on PARK7 (also known as DJ-1), which was originally identified as a gene mutated in a recessively inherited form of early-onset Parkinson disease (Bonifati et al., 2003a; Bonifati et al., 2003b), and later found to form a ubiquitin E3 ligase complex with Parkin (PARK2) and PINK1 to promote proteolysis (Xiong et al., 2009). We first confirmed the binding between UBE2M and DJ-1 under overexpressed condition, and found Myc-tagged DJ-1 could pull down HA-UBE2M-WT and vice versa, whereas the binding between DJ-1 with UBE2M mutant is to a much less extent (Fig. S6B&C). Binding between exogenously expressed DJ-1 and Parkin to endogenous UBE2M is also evidential (Fig. 6A). Interestingly, under unstressed physiological condition, endogenous UBE2M could bind to endogenous Parkin and DJ-1 as well as Keap1 and CUL3. Under stressed conditions (with treatment of TPA, CoCl2, or MLN), the UBE2M-DJ-1 and UBE2M-Parkin bindings were significantly enhanced, whereas UBE2M-Keap1 and UBE2M-CUL3 bindings were remarkably decreased (Fig. 6B, left panels; S6D top panels). Stressed conditions, especially CoCl2 or TPA treatments, also induced the levels of DJ-1 and Parkin, but reduced the levels of Keap1 and CUL3 (Fig. 6B, right panels; S6D, bottom panels). Taken together, our data indicate that in addition to forming a complex with CUL3-Keap1, UBE2M also forms a complex with DJ-1-Parkin, which is subjected to stress regulation, with former being reduced and latter enhanced.
Figure 6. DJ-1/Parkin is the major E3 for UBE2F ubiquitylation.

(A) H1299 cells were transfected with indicated plasmids, followed by IP and IB with indicated Abs. WCL: Whole cell extracts.
(B) H1299 cells were treated with indicated compounds for 24 h, followed by IP with UBE2M Ab and IB with indicated Abs. WCL: Whole cell extracts.
(C and D) A427 cells were transfected with indicated siRNAs oligoes, followed by IB with indicated Abs (C), or subjected to half-life study with cycloheximide (CHX) (D). The band density was quantified using ImageJ software and plotted (bottom panel, D).
(E and F) A427 cells were transfected with indicated plasmids, followed by IB with indicated Abs, or subjected to protein half-life study with CHX (F) and the band density was quantified.
(G) H358 cells were transfected with siRNA oligoes targeting control, DJ-1 and/or Parkin, and then treated with DMSO or CoCl2 for 24 h, followed by IB assay.
(H&I) H1299 cells were co-transfected with indicated plasmids. Cells were treated with DMSO vehicle control, or three indicated compounds (I). HA-His-Ub tagged UBE2F were purified via Ni-NTA affinity, and detected with anti-UBE2F Ab
(J) Pure E1, E2 (UBE2M or UBCH5C), E3 (Parkin and/or Pink1), and substrate (UBE2F) were incubated in a reaction mixture containing ATP and ubiquitin for 1 h, followed by IB with UBE2F Ab.
See also Figure S6.
We then individually knocked down DJ-1, Parkin, UBE2M, Keap1 or CUL3, and found that DJ-1 or Parkin knockdown caused the highest accumulation of UBE2F levels (Figs. 6C, S6E) with significant extension of UBE2F protein half-life (Figs. 6D, S6F). Consistently, overexpression of DJ-1 or Parkin caused more reduction of basal levels of UBE2F protein than that of Keap1 or CUL3 (Figs. 6E, S6G), and shortened its half-life significantly (Figs. 6F, S6H). Finally, we demonstrated that UBE2F reduction by CoCl2 exposure (to induce UBE2M) could be partially rescued by single knock-down of Parkin or DJ-1, and completely rescued by double knockdown (Fig. 6G), clearly indicating that UBE2M/DJ-1/Parkin regulate the protein level of UBE2F under stress condition.
We then directly compared the UBE2F poly-ubiquitylation by UBE2M, DJ-1, Parkin, Keap1, or CUL3, individually, under overexpressed condition, and found that UBE2M, DJ-1 or Parkin has similar activity, which is much stronger than Keap1 or CUL3 (Fig. 6H, S6I). We further directly compared UBE2F polyubiquitylation by CUL3 vs. Parkin using endogenous UBE2M (without co-transfection of UBE2M) and found that under unstressed physiological condition, CUL3 is a better E3, whereas under stressed conditions, Parkin plays a major role (Fig. 6I). Finally, we used two in vitro ubiquitylation assays to a) determine whether UBE2M acts as a ubiquitylation E2, and b) compare E2 activity between UBE2M and UBCH5C (a typical ubiquitin E2) for CUL3 vs. Parkin E3 ligase. In the first assay, we used E3 complex of CUL3 or Parkin purified by IP from transfected cells, and incubated them, respectively, with pure E1 and E2 (UBE2M vs. UBCH5C). We found that UBCH5C is a better E2 for CUL3 E3 ligase, whereas UBE2M is better E2 for Parkin E3 ligase in promoting UBE2F polyubiquitylation (Fig. S6J). In second assay, we used all pure proteins to avoid potential interference from CUL3- or Parkin-associated proteins. We found that UBE2M, but not UBCH5C, promoted UBE2F polyubiquitylation by Parkin. Inclusion of PINK1, yet another component of Parkin/DJ-1 E3 complex that is known to activate Parkin E3 (Xiong et al., 2009), further enhanced UBE2F polyubiquitylation by UBE2M without any effect when combined with UBCH5C (Fig. 6J). Thus, UBE2M, but not UBCH5C, acts as an ubiquitin E2 for the Parkin/DJ-1/PINK1 E3 complex. Taken together, our results indicate that UBE2M-DJ-1/Parkin/PINK1 forms an active E2–E3 novel complex and plays a major role in UBE2F poly-ubiquitylation, especially under stressed conditions.
UBE2M or Parkin suppresses lung cancer cell growth by targeting UBE2F
Recently, we showed that UBE2F has oncogenic activity by promoting proliferation and survival of lung cancer cells via activating CRL5 for targeted degradation of pro-apoptotic NOXA (Zhou et al., 2017). Given that UBE2M promotes UBE2F degradation to cause NOXA accumulation, we hypothesized that UBE2M could have tumor suppressive activity. Indeed, overexpression of wild-type UBE2M at the level comparable to endogenous one, caused NOXA accumulation, and inhibited cell proliferation and clonogenic survival, whereas expression of its enzymatic dead C111S mutant reduced NOXA level, and promoted the proliferation and clonogenic survival of lung cancer cells (Figs. 7A–C). The causal role of NOXA in mediating UBE2M effect was demonstrated by simultaneous NOXA knockdown, which largely abrogated UBE2M-induced suppression of proliferation and survival (Figs. 7D&E), and induction of apoptosis (Fig. 7F). Taken together, these results clearly showed that UBE2M has growth suppressive activity, which is largely mediated by causing NOXA accumulation.
Figure 7. UBE2M or Parkin functions as a tumor suppressor against lung cancer.

(A–C) H1299 cells were transfected with indicated plasmids, followed by G418 selection for 2 weeks. The stable clones were pooled and subjected to IB (A), ATPlite assay for proliferation (B) or clonogenic assay for survival (C). Data are represented as mean ± SD (B)/SEM (C) of three independent experiments. ** p < 0.01, *** p < 0.001, based on Two-way ANOVA (B) or One-way ANOVA (C).
(D–F) H358 cells were transfected with indicated plasmids or Lenti-virus targeting NOXA alone or in combination, followed by ATPlite assay for proliferation (D), clonogenic assay for survival (E), and IB with indicated Abs (F). Data are represented as mean ± SEM of three independent experiments. ** p < 0.01, *** p < 0.001, based on Two-way ANOVA (D) or One-way ANOVA (E).
(G and H) H1299 cells (1×106) stably expressing indicated plasmids were inoculated subcutaneously in both flanks of nude mice, with 5 mice in each group. The tumor growth was monitored up to 60 days and growth curve plotted (G). Tumor tissues were weighed and photographed at 60 days (H). Data are represented as mean ± SEM. * p < 0.05, ** p < 0.01, ***, p<0.001, based on Two-way ANOVA (G) or One-way ANOVA (H).
(I–K) A427 cells were transfected with indicated plasmids or Lenti-virus targeting NOXA alone or in combination, followed by IB with indicated Ab (I), ATPlite assay for proliferation (J), or clonogenic assay for survival (K). Data are represented as mean ± SD of three independent experiments. ** p < 0.01, *** p<0.001, based on Two-way ANOVA (J) or One-way ANOVA (K).
(L) Lung cancer tissue microarrays were stained with UBE2F, UBE2M or Parkin, and photographed. Scale bars, 500 µm and 90 µm.
(M) Working model. Under unstressed condition, UBE2M cooperates with CUL3/Keap1 to promote UBE2F ubiquitylation and degradation; upon exposure to hypoxia, or TPA, UBE2M was induced through transcriptional activation by HIF-1 or c-JUN/AP1, and then forms a complex with DJ-1/Parkin to promote UBE2F ubiquitylation and degradation, leading to NOXA accumulation for apoptosis induction.
See also Figure S7.
We next determined growth inhibition activity of UBE2M using an in vivo xenograft tumor model and found that, compared to the vector control, UBE2M-WT inhibits in vivo tumor growth (p<0.05, Fig. 7G), whereas UBE2M-C111S mutant accelerated it (p<0.001, Fig. 7G). The average tumor size and weight at the end of experiment (day 60) was the lowest in UBE2F-WT group and the highest in UBE2F-C111S mutant group, which are statistically different from the vector control and between each other (Fig. 7H). We further confirmed that both UBE2M-WT and UBE2M-C111S mutant were indeed expressed in tumor tissues (Fig. S7A). Immuno-histochemical (IHC) staining of these tumor tissues revealed that compared to that of the vector control, tumors expressing UBE2M-WT had reduced staining in Ki-67 and UBE2F, and increased staining in p21, p27 and NOXA, whereas an opposite was true for tumors expressing UBE2M-C111S mutant (Figs. S7B&C). Taken together, these results from both in vitro cell culture and in vivo xenograft models coherently demonstrated that UBE2M-WT inhibited cell growth and survival, and induced apoptosis, whereas the C111S mutant acted in a dominant negative manner to promote growth and survival, suggesting that UBE2M might be a novel therapeutic target for lung cancer.
Increased lines of evidence have strongly suggested that Parkin acts as a tumor suppressor (Poulogiannis et al., 2010; Xu et al., 2014). In keeping with this notion, we found that like UBE2M, overexpression of Parkin indeed suppressed growth and clonogenic survival of lung cancer cells in a manner dependent of NOXA accumulation (Fig. 7I–K, S7D–F). Finally, we found that in NSCLC tissues the levels of UBE2F is inversely and significantly correlated with UBE2M or Parkin (Fig. 7L & S7G), indicating a potential in vivo association of these three proteins.
DISCUSSION
In this study, we followed up our unexpected observation that MLN4924 differentially regulated two family members of neddylation E2s: positively for UBE2M and negatively for UBE2F (Fig. 1). Mechanistically, MLN4924 induction of UBE2M expression is mediated by c-JUN and HIF-1α, accumulated due to inactivation of CRL1 (SCFFBXW7) and CRL2 (Cul2VHL), respectively (Kamura et al., 2000; Nateri et al., 2004; Wei et al., 2005). UBE2M is directly induced by hypoxia or TPA, which occurs at the transcriptional level, driven by HIF-1α binding to the intron-1, and c-JUN/AP1 binding to the promoter regions of the UBE2M, respectively (Fig. 2). Significantly, UBE2M induction, followed by UBE2F reduction, promotes neddylation of cullins 1–4 (leading to decreased levels of their respective substrates), but inhibits cullin 5 neddylation (thus increasing NOXA level) (Fig. 2), indicating that both neddylation E2 enzymes are NOT present in saturated amounts in cells, and stresses-induced alterations of their levels indeed have biochemical consequence. Together with a previous study reporting that UBE2M is inducible by gemcitabine in a PI3K activity dependent manner (Huang et al., 2011), we concluded that UBE2M is a stress inducible gene which responds to external stimuli. It is interesting to note that the entire process of neddylation modification of cullins is subjected to stimulation by hypoxia and mitogen exposure, since both UBE2M (this study), which promotes neddylation activation of cullins 1–4, and SAG/RBX2, a neddylation E3 which promotes cullin 5 neddylation activation (Huang et al., 2009), are induced by HIF-1α and AP1 via transcriptional activation (Gu et al., 2007; Tan et al., 2008).
Although UBE2M and UBE2F activate different cullins by promoting their neddylation modification (Huang et al., 2009), whether they regulate each other remains totally unknown. Since MLN4924 increased both mRNA and protein levels of UBE2M, but decreased only UBE2F protein level, whereas other UBE2M inducers (hypoxia and TPA) also decreased UBE2F protein level, we tested a working hypothesis that induced UBE2M abundance may promote ubiquitylation and degradation of UBE2F. Indeed, UBE2M knockdown abrogated UBE2F reduction by all three treatments and extended UBE2F protein half-life, whereas UBE2M overexpression directly reduced UBE2F level, shortened its protein half-life by promoting its ubiquitylation (Fig. 3). Thus, we made a novel observation that two neddylation E2s communicate with each other with UBE2M targeting UBE2F for degradation.
How does UBE2M, a neddylation E2, not an E3, promote UBE2F ubiquitylation? Although UBE2M was recently reported to serve as a substrate for auto-neddylation (Coleman et al., 2017), its only well-known biochemical function is to activate cullins 1–4, acting as an E2 (Huang et al., 2009). Among CULs1–5, CUL3 is the only cullin that negatively regulates UBE2F levels by shortening its protein half-life via promoting ubiquitylation (Fig. 4). CUL3-induced substrate ubiquitylation is mediated through a substrate recognizing and BTB-domain containing subunit (Furukawa et al., 2003). We identified an evolutionally conserved Keap1 consensus binding motif on UBE2F and confirmed that Keap1 indeed promotes UBE2F ubiquitylation and shortens its protein half-life in a binding domain dependent manner (Fig. 5). Thus, UBE2F is a novel substrate of UBE2M-CRL3Keap1 E3 ubiquitylation ligase. Targeted ubiquitylation and degradation of UBE2F by CRL3 established yet another inter-regulation axis among CRL family members, namely CRL3 inactivation of CRL5, in analogues to CRL5 inactivation of CRL1 via SAG-mediated ubiquitylation of β-TrCP1 (Kuang et al., 2016).
The observation that MLN4924, on one hand, inactivates all CRLs, and on the other hand, induces UBE2M expression to target UBE2F for degradation, is paradoxical, but strongly suggests that UBE2M may serve as an ubiquitylation E2 for a CRL3 independent E3. Following this lead, we identified DJ-1 as a UBE2M binding protein via affinity purification-mass spectrometry. DJ-1 (also known as PARK7) and Parkin are the causative genes of familial Parkinson’s disease (Bonifati et al., 2003b; Kim and Alcalay, 2017). Together with PINK1, DJ-1 and Parkin form a triple E3 complex to promote proteolysis (Xiong et al., 2009). We confirmed an endogenous binding between UBE2M and DJ-1 or Parkin, which is induced under stressed conditions with treatment of MLN4924, CoCl2 or TPA. Significantly, the same treatments inhibited UBE2M-Keap1 or UBE2M-CUL3 binding (Fig. 6 & S6). Furthermore, it appears that for targeted UBE2F ubiquitylation and degradation, UBE2M mainly serves as a neddylation E2 to activate CRL3Keap1 under unstressed physiological condition, but as an ubiquitylation E2 to complex with DJ-1/Parkin E3 under stress conditions (Fig. 6 & S6). The switch between two E3s for UBE2F ubiquitylation is likely due to the combination of stress-mediated induction of UBE2M and DJ-1/Parkin, and reduction of Keap1/CUL3 (Fig. 6 & S6). Previous studies have also shown that various stresses could up-regulate DJ-1/Parkin (Mitsumoto and Nakagawa, 2001; Won et al., 2015; Xiao et al., 2017), but impair Keap1 pathway (Jaramillo and Zhang, 2013).
Our finding that UBE2M is a stress-inducible protein may also explain our previous report of an elevated UBE2M level detected in lung cancer tissues (Li et al., 2014), which is likely due to the cellular response to environmental stimuli, such as hypoxia, occurring frequently in cancer tissues. However, the biological function of UBE2M in lung cancer cells remains elusive. Here, we reported that UBE2M actually has tumor suppressive function in both cell culture and in vivo xenograft models by inducing apoptosis via NOXA accumulation as a result of targeted degradation of UBE2F and subsequent inactivation of CRL5, an E3 responsible for NOXA degradation (Zhou et al., 2017), while catalytic dead UBE2M mutant acts in a dominant negative manner.
Increasing lines of evidence have strongly suggested that PARK2 (the Parkin encoding gene) is a tumor suppressor (Xu et al., 2014), which is altered in over a third of all human cancers (Gupta et al., 2017). Here, we showed that similar to UBE2M, Parkin overexpression suppressed proliferation and clonogenic survival in a manner largely dependent of NOXA in lung cancer cell models (Fig. 7). Thus, tumor suppressor function of Parkin is through not only blockage of PI3K/AKT activation to inhibit survival, as recently reported (Gupta et al., 2017), but also inactivation of NOXA degradation to induce apoptosis. Finally, we found that the levels of UBE2M and Parkin are positively correlated to each other, but negatively associated with UBE2F levels in human lung cancer tissues (Fig. 7 & S7), suggesting their relevance in lung tumorigenesis.
In summary, our study supports the following model: Under basal unstressed physiological conditions, UBE2M acts as a neddylation E2 to activate CRL3Keap1 to keep UBE2F level in check. Upon stress stimuli, such as hypoxia or mitogen stimuli, UBE2M is induced and serves as an ubiquitin E2 to complex with DJ-1/Parkin, to promote ubiquitylation and degradation of UBE2F, leading to CRL5 inactivation and, subsequent NOXA accumulation and apoptosis induction (Fig. 7M). Our study provides experimental evidence that one neddylation E2 (UBE2M) regulates the other (UBE2F) via two E3s, leading to one CRL E3 (CRL3) inactivating the other (CRL5), thus establishing a cross-talk at both E2 and E3 levels.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
- EXPERIMENTAL MODEL AND SUBJECT DETAILS
-
◦Cell culture
-
◦Xenograft Tumor Growth
-
◦Patient Tumor Tissue Samples and Immunohistochemical Staining
-
◦
- METHOD DETAILS
-
◦Site-directed Mutagenesis
-
◦ATPlite cell proliferation assay
-
◦Clonogenic survival assay
-
◦Antibodies and Immunoblotting
-
◦Cloning of intron-1 and promoter, luciferase reporter construction and luciferaseactivity assay
-
◦Chromatin immunoprecipitation assay
-
◦siRNA, and transfection
-
◦Immunoprecipitation
-
◦The in vivo and in vitro ubiquitylation assay
-
◦
QUANTIFICATION AND STATISTICAL ANALYSIS
DATA AND SOFTWARE AVAILABILITY
STAR★METHODS
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to, and will be fulfilled by Yi Sun (sunyi@umich.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
A427, H358, SK-MES-1, H2170and H1299 lung cancer cell lines and HEK293 cell line were all purchased from American Type Culture Collection. A427, H2170 and H358 were grown in RPMI-1640 with 10% FBS; H1299 and HEK293 were cultured in DMEM with 10% FBS; SK-MES-1 cells were cultured in EMEM with 10% FBS. All cell lines were tested to be free of mycoplasma contamination.
Xenograft Tumor Growth
For assaying tumor growth in the xenograft model, Four- to six-week-old BALB/c athymic nude mice (nu/nu, female) were used with each experimental group consisting of five mice. All animal experiments were carried out according to a protocol approved by the University of Michigan, Committee for Use and Care of Animals. 1×106 H1299 stable cell lines (Vector; HA-UBE2M-WT; HA-UBE2M-C111S) were mixed 1:1 with matrigel (BD biosciences) in a total volume of 0.2 ml and were injected subcutaneously into both flanks of mice The growth of tumours was measured twice a week and average tumour volume (TV) was calculated according to the equation: TV=(L×W2)/2.
Patient Tumor Tissue Samples and Immunohistochemical Staining
The human lung cancer tissue arrays were purchased from Guge biotechnology co. LTD, Wuhan, China. The immunohistochemical staining was assessed and scored as described previously (Xu et al., 2017; Zhou et al., 2012). Briefly, expression of UBE2M, UBE2F, or Parkin was evaluated by combined assessing of staining intensity and extent. The intensity of staining was graded on the following scale: 0, no staining; 1, weak; 2, moderate; 3, strong staining. The area of staining was evaluated as follows: 0, no staining of cells in any microscopic fields; 1+, <30% of tissue stained positively; 2+, between 30% and 60% of tissues stained positively; 3+, >60% of tissues stained positively. The minimum score when summed (extension + intensity) was 0, and the maximum was 6.
METHOD DETAILS
Site-directed Mutagenesis
Site directed mutagenesis to generate various Keap1 mutants were performed using the QuikChange XL Site-Directed Mutagenesis Kit (Agilent) according to the manufacturer’s instructions.
ATPlite cell proliferation assay
Cells were seeded into 96-well plates with 3,000 cells per well in triplicate and grew for 24, 48, 72, 96, and 120 h, followed by ATPlite cell proliferation assay (Perkin-Elmer), according to the manufacturer's instructions.
Clonogenic survival assay
Cells were seeded into 60-mm dishes at 800 cells (H358) or 600 cells (A427 and H1299) per dish in triplicate, followed by incubation at 37°C for 14 d. The colonies were fixed with 10% acidic acid in methanol, stained with 0.05% methylene blue, and counted.
Antibodies and Immunoblotting
For direct IB analysis, cells were lysed in a Triton X-100 or RIPA buffer with phosphatase inhibitors. The antibodies used were as follows: SAG (Jia et al., 2010), RBX1 (Jia et al., 2009), UBE2F (Abcam), UBE2M (mouse for IB from Santa Cruz, and rabbit for IHC from Abcam), UBCH5C (Cell Signalling), HIF-1α (Santa Cruz), c-JUN (Santa Cruz), HA (Roche), FLAG (Sigma), CUL1 (Santa Cruz), CUL2 (BD Bioscience), CUL3 (Cell Signal), CUL4A (Cell Signalling), CUL4B (Proteintech), CUL5 (Santa Cruz), Myc-tag (Santa Cruz), Keap1 (Cell Signalling), DJ-1 (Cell Signalling), Parkin (Cell Signalling), His (Cell Signalling), NOXA (Millipore), and β-actin (Sigma).
Cloning of intron-1 and promoter, luciferase reporter construction and luciferase activity assay
For the UBE2M intron-1 cloning, the primers used are UBE2M-H01 (5’-GGGGTACCGCGTGGGGGGGGTCGGAGGTC-3’) and UBE2M-H02 (5’-CCC AAGCTTCTGGGTTTGGGTAGCAGAGAG-3’) with HIF-1-binding site 996 bp); The primers UBE2M-H03 (5’-GGGGTACCGGGGGGGTCGGAGGTCAAGTC-3’) and UBE2M-H02 were used to generate a 991 bp DNA fragment with HIF-1-binding site deleted. For the UBE2M promoter cloning, the primer set of UBE2M-AP1-01 (5’-CGGGGTACCGTTAAGTCAATCTGTCTTAGAAAT-3’) and UBE2M-AP1-02 (5’-CCGCTCGAGGTAAACTAACAAAACATGTAT-3’) were used to generate a 2073 bp fragment containing all 4 AP1 binding sites (WT1); the primer set of UBE2M-AP1-03 (5’-CGGGGTACCATCTGTCTTAGAAATGGGAAAGTT-3’) and UBE2M-AP1-02 were used to generate a 2064 bp DNA fragment with AP1-1-binding site deleted (MU1); the primer set of UBE2M-AP1-04 (5’-CGGGGTACCT CCAAGTCAGCGTTAATGA TTCATT-3’) and UBE2M-AP1-02 were used to generate a 1438 bp fragment including 3 AP1 binding sites (WT2); the primer set of UBE2M-AP1-05 (5’-CGGGGTACCGCGTTAATGATTCATTCCCATGATAC-3’) and UBE2M-AP1-02 were used to generate a 1429 bp DNA fragment with AP1-2-binding site deleted (MU2). The above fragments were subcloned into pGL3-basic luciferase reporter and luciferase activity measured in the absence or presence of various stress-inducers, as previously described (Gu et al., 2007).
Chromatin immunoprecipitation assay
The assay was performed as described (Gu et al., 2007). The primers used for UBE2M-intron-1 HIF-1binding site are UBE2M-CHIP01 (5’-TGGGAGTCAGGA AGGGGTAAT-3’) and UBE2M-CHIP02 (5’-AGTCCCTAGTAGCAGACACC-3’), which generated a 185 bp fragment; the primers used for UBE2M-AP1-1 binding site are UBE2M-CHIP03 (5’-TGCCTGATCTGATTGAGTCCC-3’) and UBE2M-ChIP04 (5’-TTGGAGCTTCTAAGTTGCAAT-3’) to generate a 180 bp fragment; the primers used for UBE2M-AP1-2 binding site are UBE2M-CHIP05 (5’-TGATGAAATCCCA TGGGATTT-3’) and UBE2M-ChIP06 (5’-GTAGAAAGCTTTGGCCGCTCA-3’) to generate a 240 bp fragment. The IP antibodies used were HIF-1α, and c-JUN, respectively, along with normal IgG as negative control. One-tenth original DNA lysates was used as input controls.
siRNA, and transfection
UBE2M/RNAi-1 is a pool of 2 target-specific 19–25 nt siRNAs from Santa Cruz and the sequence for UBE2M/RNAi-2 is 5’-GGGCTTCTACAAGAGTGGG AAGT-3’ (Meyer-Schaller et al., 2009). The siRNAs targeting CUL5 (Tan et al., 2011) and CUL1 (Tan et al., 2011) were used previously in our lab. The sequence for CUL2 is 5’-GCCCUUACGUCAGUUGUAAAUUACA-3’(Cukras et al., 2014), for CUL4A is 5’-GAAGCUGGUCAUCAAGAAC-3’ (Yu et al., 2015) and CUL4B is 5’-AAGCCUAAAUUACCAGAAA-3’ (Hu et al., 2004). The siRNA targeting Keap1, HIF-1α, c-JUN and CUL3 are all pools purchased from Santa Cruz. The sequence for the scrambled control siRNA is 5’-AUUGUAUGCGAUCGCAGACUU-3’ (Zhou et al., 2014). Transfection of siRNAs was carried out using Lipofectamine 2000 (Invitrogen).
Immunoprecipitation
The immunoprecipitation assay was performed as previously described (Zhou et al., 2014). Briefly, the pre-cleared cell lysates were incubated with indicated antibodies for 3 h, followed by incubation with protein A&G beads (Santa Cruz) at 4°C overnight with rotation. The beads were washed three times with lysis buffer, and the immunoprecipitation complexes were subjected to SDS-PAGE.
The in vivo and in vitro ubiquitylation assay
The 293 or H1299 cells were co-transfected with various plasmids. The in vivo assay was performed as previously described using Ni-beads pull-down (Zhou et al., 2017; Zhou et al., 2014). For the in vitro ubiquitylation assays, two E3 sources were used: one was generated by transfection of E3 encoding plasmids, followed by IP pull-down; the other was pure E3 components purchased from commercial sources with Parkin from Millipore and PINK1 from Boston Biochem. Reaction mixture, containing reaction buffer, different combinations of E1, E2, E3 and substrates, was incubated for 60 min under constant vortexing and then resuspended in 2× SDS-PAGE sample buffer for SDS-PAGE and detected by Western blotting, as described (Xu et al., 2017).
QUANTIFICATION AND STATISTICAL ANALYSIS
The Student’s t test was used for the comparison of parameters between two groups. One-way ANOVA was used to compare the parameters among groups greater than two, and Two-way ANOVA was used to compare the parameters of the growth curves. The correlation of the expression of two given proteins was assessed by Pearson correlation analysis. Statistical Program for Social Sciences software 20.0 (SPSS, Chicago, IL) was used. All statistical tests were two-sided.
DATA AND SOFTWARE AVAILABILITY
Raw data have been deposited to Mendeley Data and are available at http://dx.doi.org/10.17632/g3xxjdhwkt.1
Supplementary Material
HIGHLIGHTS.
Neddylation inhibitor MLN4924 increases UBE2M levels, but decreases UBE2F levels
UBE2M is stress-inducible by hypoxia and mitogen stimulation via HIF-1α and AP1
UBE2M acts as a neddylation E2 to activates CUL3/Keap1 E3 for UBE2F degradation
UBE2M acts as an ubiquitin E2 for Parkin/DJ-1 E3 to degrade UBE2F upon stress stimuli
Acknowledgments
We thank Takeda Pharmaceuticals, Inc. for providing us MLN4924. This work is supported in part by the National Key R&D Program of China (2016YFA0501800) (YS), by the NCI grants CA156744 and CA171277 (Y. Sun), and by the Chinese NSFC Grants 81572718 and 81630076 (Y. Sun), and 31528015 (W. Wei).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Conception and design: WZ, WW, YS
Experiment execution: WZ, JX.
Data acquisition: WZ, JX, HL, HL, MT.
Data analysis and interpretation: WZ, JX, YS
Manuscript writing: WZ, YS
Study supervision: YS
DECLARATION OF INTERESTS
The authors declare no conflict of interest.
References
- Bonifati V, Rizzu P, Squitieri F, Krieger E, Vanacore N, van Swieten JC, Brice A, van Duijn CM, Oostra B, Meco G, et al. DJ-1(PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2003a;24:159–160. doi: 10.1007/s10072-003-0108-0. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003b;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Brown JS, Lukashchuk N, Sczaniecka-Clift M, Britton S, le Sage C, Calsou P, Beli P, Galanty Y, Jackson SP. Neddylation promotes ubiquitylation and release of Ku from DNA-damage sites. Cell reports. 2015;11:704–714. doi: 10.1016/j.celrep.2015.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colburn NH, Former BF, Nelson KA, Yuspa SH. Tumour promoter induces anchorage independence irreversibly. Nature. 1979;281:589–591. doi: 10.1038/281589a0. [DOI] [PubMed] [Google Scholar]
- Coleman KE, Bekes M, Chapman JR, Crist SB, Jones MJ, Ueberheide BM, Huang TT. SENP8 limits aberrant neddylation of NEDD8 pathway components to promote cullin-RING ubiquitin ligase function. Elife. 2017;6 doi: 10.7554/eLife.24325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukras S, Morffy N, Ohn T, Kee Y. Inactivating UBE2M impacts the DNA damage response and genome integrity involving multiple cullin ligases. PLoS One. 2014;9:e101844. doi: 10.1371/journal.pone.0101844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankert JF, Pagan JK, Starostina NG, Kipreos ET, Pagano M. FEM1 proteins are ancient regulators of SLBP degradation. Cell Cycle. 2017;16:556–564. doi: 10.1080/15384101.2017.1284715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134:995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa M, He YJ, Borchers C, Xiong Y. Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nature cell biology. 2003;5:1001–1007. doi: 10.1038/ncb1056. [DOI] [PubMed] [Google Scholar]
- Gu Q, Tan M, Sun Y. SAG/ROC2/Rbx2 is a novel activator protein-1 target that promotes c-Jun degradation and inhibits 12-O-tetradecanoylphorbol-13-acetate-induced neoplastic transformation. Cancer research. 2007;67:3616–3625. doi: 10.1158/0008-5472.CAN-06-4020. [DOI] [PubMed] [Google Scholar]
- Gupta A, Anjomani-Virmouni S, Koundouros N, Dimitriadi M, Choo-Wing R, Valle A, Zheng Y, Chiu YH, Agnihotri S, Zadeh G, et al. PARK2 Depletion Connects Energy and Oxidative Stress to PI3K/Akt Activation via PTEN S-Nitrosylation. Mol Cell. 2017;65:999–1013. e1017. doi: 10.1016/j.molcel.2017.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, McCall CM, Ohta T, Xiong Y. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nature cell biology. 2004;6:1003–1009. doi: 10.1038/ncb1172. [DOI] [PubMed] [Google Scholar]
- Huang AM, Kao YT, Toh S, Lin PY, Chou CH, Hu HT, Lu CY, Liou JY, Chao SY, Hour TC, et al. UBE2M-mediated p27(Kip1) degradation in gemcitabine cytotoxicity. Biochem Pharmacol. 2011;82:35–42. doi: 10.1016/j.bcp.2011.03.026. [DOI] [PubMed] [Google Scholar]
- Huang DT, Ayrault O, Hunt HW, Taherbhoy AM, Duda DM, Scott DC, Borg LA, Neale G, Murray PJ, Roussel MF, et al. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Molecular cell. 2009;33:483–495. doi: 10.1016/j.molcel.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DT, Miller DW, Mathew R, Cassell R, Holton JM, Roussel MF, Schulman BA. A unique E1-E2 interaction required for optimal conjugation of the ubiquitin-like protein NEDD8. Nature structural & molecular biology. 2004;11:927–935. doi: 10.1038/nsmb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DT, Paydar A, Zhuang M, Waddell MB, Holton JM, Schulman BA. Structural basis for recruitment of Ubc12 by an E2 binding domain in NEDD8's E1. Molecular cell. 2005;17:341–350. doi: 10.1016/j.molcel.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes & development. 2013;27:2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Soengas MS, Sun Y. ROC1/RBX1 E3 ubiquitin ligase silencing suppresses tumor cell growth via sequential induction of G2-M arrest, apoptosis, and senescence. Cancer research. 2009;69:4974–4982. doi: 10.1158/0008-5472.CAN-08-4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Yang J, Hao X, Zheng M, He H, Xiong X, Xu L, Sun Y. Validation of SAG/RBX2/ROC2 E3 ubiquitin ligase as an anticancer and radiosensitizing target. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:814–824. doi: 10.1158/1078-0432.CCR-09-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani T, Kito K, Nguyen HP, Yeh ET. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. The Journal of biological chemistry. 1997;272:28557–28562. doi: 10.1074/jbc.272.45.28557. [DOI] [PubMed] [Google Scholar]
- Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci U S A. 2000;97:10430–10435. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CY, Alcalay RN. Genetic Forms of Parkinson's Disease. Semin Neurol. 2017;37:135–146. doi: 10.1055/s-0037-1601567. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang P, Tan M, Zhou W, Zhang Q, Sun Y. SAG/RBX2 E3 ligase complexes with UBCH10 and UBE2S E2s to ubiquitylate beta-TrCP1 via K11-linkage for degradation. Sci Rep. 2016;6:37441. doi: 10.1038/srep37441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Wang M, Yu G, Chen P, Li H, Wei D, Zhu J, Xie L, Jia H, Shi J, et al. Overactivated neddylation pathway as a therapeutic target in lung cancer. Journal of the National Cancer Institute. 2014;106:dju083. doi: 10.1093/jnci/dju083. [DOI] [PubMed] [Google Scholar]
- Meyer-Schaller N, Chou YC, Sumara I, Martin DD, Kurz T, Katheder N, Hofmann K, Berthiaume LG, Sicheri F, Peter M. The human Dcn1-like protein DCNL3 promotes Cul3 neddylation at membranes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12365–12370. doi: 10.1073/pnas.0812528106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsumoto A, Nakagawa Y. DJ-1 is an indicator for endogenous reactive oxygen species elicited by endotoxin. Free radical research. 2001;35:885–893. doi: 10.1080/10715760100301381. [DOI] [PubMed] [Google Scholar]
- Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004;303:1374–1378. doi: 10.1126/science.1092880. [DOI] [PubMed] [Google Scholar]
- Nawrocki ST, Griffin P, Kelly KR, Carew JS. MLN4924 : a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin Investig Drugs. 2012;21:1563–1573. doi: 10.1517/13543784.2012.707192. [DOI] [PubMed] [Google Scholar]
- Piret JP, Mottet D, Raes M, Michiels C. CoCl2, a chemical inducer of hypoxia-inducible factor-1, and hypoxia reduce apoptotic cell death in hepatoma cell line HepG2. Ann N Y Acad Sci. 2002;973:443–447. doi: 10.1111/j.1749-6632.2002.tb04680.x. [DOI] [PubMed] [Google Scholar]
- Poulogiannis G, McIntyre RE, Dimitriadi M, Apps JR, Wilson CH, Ichimura K, Luo F, Cantley LC, Wyllie AH, Adams DJ, et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc Natl Acad Sci U S A. 2010;107:15145–15150. doi: 10.1073/pnas.1009941107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Molecular cell. 2008;32:21–31. doi: 10.1016/j.molcel.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata E, Yamaguchi Y, Miyauchi Y, Iwai K, Chiba T, Saeki Y, Matsuda N, Tanaka K, Kato K. Direct interactions between NEDD8 and ubiquitin E2 conjugating enzymes upregulate cullin-based E3 ligase activity. Nature structural & molecular biology. 2007;14:167–168. doi: 10.1038/nsmb1191. [DOI] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Muller U, Krek W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nature Cell Biol. 1999;1:207–214. doi: 10.1038/12027. [DOI] [PubMed] [Google Scholar]
- Tan M, Gu Q, He H, Pamarthy D, Semenza GL, Sun Y. SAG/ROC2/RBX2 is a HIF-1 target gene that promotes HIF-1alpha ubiquitination and degradation. Oncogene. 2008;27:1404–1411. doi: 10.1038/sj.onc.1210780. [DOI] [PubMed] [Google Scholar]
- Tan M, Zhao Y, Kim SJ, Liu M, Jia L, Saunders TL, Zhu Y, Sun Y. SAG/RBX2/ROC2 E3 ubiquitin ligase is essential for vascular and neural development by targeting NF1 for degradation. Developmental cell. 2011;21:1062–1076. doi: 10.1016/j.devcel.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Molecular and cellular biology. 2006;26:2887–2900. doi: 10.1128/MCB.26.8.2887-2900.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Won KJ, Lee KP, Yu S, Lee D, Lee DY, Lee HM, Kim J, Jung SH, Baek S, Kim B. Ketoconazole induces apoptosis in rat cardiomyocytes through reactive oxygen species-mediated parkin overexpression. Archives of toxicology. 2015;89:1871–1880. doi: 10.1007/s00204-015-1502-0. [DOI] [PubMed] [Google Scholar]
- Xiao B, Goh JY, Xiao L, Xian H, Lim KL, Liou YC. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. The Journal of biological chemistry. 2017 doi: 10.1074/jbc.M117.787739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H, Wang D, Chen L, Choo YS, Ma H, Tang C, Xia K, Jiang W, Ronai Z, Zhuang X, et al. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. The Journal of clinical investigation. 2009;119:650–660. doi: 10.1172/JCI37617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhou W, Yang F, Chen G, Li H, Zhao Y, Liu P, Li H, Tan M, Xiong X, et al. The beta-TrCP-FBXW2-SKP2 axis regulates lung cancer cell growth with FBXW2 acting as a tumour suppressor. Nature communications. 2017;8:14002. doi: 10.1038/ncomms14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Lin DC, Yin D, Koeffler HP. An emerging role of PARK2 in cancer. J Mol Med (Berl) 2014;92:31–42. doi: 10.1007/s00109-013-1107-0. [DOI] [PubMed] [Google Scholar]
- Yu C, Ji SY, Sha QQ, Sun QY, Fan HY. CRL4-DCAF1 ubiquitin E3 ligase directs protein phosphatase 2A degradation to control oocyte meiotic maturation. Nature communications. 2015;6:8017. doi: 10.1038/ncomms9017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Morgan MA, Sun Y. Targeting neddylation pathways to inactivate Cullin-RING ligases for anti-cancer therapy. Antioxid Redox Signal. 2014;21:2383–2400. doi: 10.1089/ars.2013.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Sun Y. Cullin-RING Ligases as Attractive Anti-cancer Targets. Curr Pharm Des. 2013;19:3215–3225. doi: 10.2174/13816128113199990300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Zhang W, Sun Y, Jia L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal. 2018;44:92–102. doi: 10.1016/j.cellsig.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Xu J, Li H, Xu M, Chen ZJ, Wei W, Pan ZQ, Sun Y. Neddylation E2 UBE2F promotes the survival of lung cancer cells by activating CRL5 to degrade NOXA via the K11 linkage. Clin Cancer Res. 2017;23:1104–1116. doi: 10.1158/1078-0432.CCR-16-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Xu J, Zhao Y, Sun Y. SAG/RBX2 is a novel substrate of NEDD4-1 E3 ubiquitin ligase and mediates NEDD4-1 induced chemosensitization. Oncotarget. 2014;5:6746–6755. doi: 10.18632/oncotarget.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou WH, Tang F, Xu J, Wu X, Yang SB, Feng ZY, Ding YG, Wan XB, Guan Z, Li HG, et al. Low expression of Beclin 1, associated with high Bcl-xL, predicts a malignant phenotype and poor prognosis of gastric cancer. Autophagy. 2012;8:389–400. doi: 10.4161/auto.18641. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.