

Abstract
Terminal iron nitrides (Fe≡N) have been proposed as intermediates of (bio)catalytic nitrogen fixation, yet experimental evidence to support this hypothesis has been lacking. In particular, no prior synthetic examples of terminal Fe≡N species have been derived from N2. Here we show that a nitrogen-fixing Fe—N2 catalyst can be protonated to form a neutral Fe(NNH2) hydrazido(2−) intermediate, which, upon further protonation, heterolytically cleaves the N—N bond to release [FeIV≡N]+ and NH3. These observations provide direct evidence for the viability of a Chatt-type (distal) mechanism for Fe-mediated N2-to-NH3 conversion. The physical oxidation state range of the Fe complexes in this transformation is buffered by covalency with the ligand, a feature of possible relevance to catalyst design in synthetic and natural systems that facilitate multiproton/multielectron redox processes.
The chemistry of high valent iron plays a central role in many challenging chemical transformations.1 For example, the heme oxygenases, as in the cytochrome P450 superfamily, hydroxylate unactivated C—H bonds via oxoferryl (FeIV=O) intermediates derived from the heterolytic reduction of dioxygen (FeII + O2 + 2 H+/2 e−→FeIV=O + H2O; Scheme 1A).1,2 The nitrido ion (N3−) is isolobal to the oxo ion (O2−), and by analogy to Fe-mediated catalytic O2 reduction, we have proposed that Fe-mediated (bio)catalytic nitrogen (N2) reduction to generate ammonia (NH3) might proceed heterolytically at a single Fe site (FeI + N2 + 3 H+/3 e−→FeIV≡N + NH3; Scheme 1B).3 Such a scenario would be similar to that originally proposed by Chatt for molybdenum-mediated N2-to-NH3 conversion4 and thought to be operative in the catalytic cycles of known Mo catalysts for this reaction.5 Whereas terminal Fe≡N complexes have been shown to liberate NH3 via reductive protonation,3a,6 such Fe≡N species have to-date been generated by N atom transfer reactions from azide (N3−) or alternative reagents, not from N2.3a,6,7 Hence, their potential role in synthetic or biological N2-to-NH3 conversion catalysis has remained unclear.
Scheme 1.
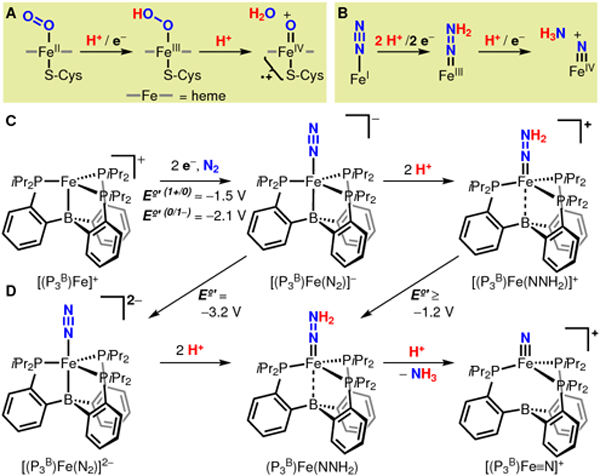
Our recent discovery that the anionic N2 complex [(P3B)Fe(N2)]− (P3b = tris(o-diisopropylphosphinophenyl)borane) catalyzes N2-to-NH3 conversion has positioned us to probe the mechanism(s) by which the key N—N cleavage step occurs in a catalytically functional Fe system.8 Of particular interest has been to distinguish between an early Chatt-type cleavage pathway (a distal mechanism via an Fe≡N intermediate) and a late-stage cleavage pathway (an alternating mechanism via an Fe—N2H4 intermediate).9 While we have considered the possibility that both scenarios might be viable,10 a key feature of the tri(phosphine)borane Fe system is its structurally and electronically flexible Fe—B interaction, which allows access to both reduced trigonal bipyramidal Fe(N2) species as well as, in principle, pseudotetrahedral terminal Fe≡N.11 Here we report the stepwise reduction and protonation of this Fe-based N2 fixation catalyst to yield a terminal Fe(IV) nitride and NH3, derived from N2. This result provides a plausible mechanism for the N—N bond cleavage step under catalytic turnover and highlights a terminally bound FeIV≡N as a viable intermediate of catalytic N2 fixation.
The Fe borane complex [(P3B)Fe(N2)]− , or its oxidized congener [(P3B)Fe]+, catalyze the reduction of N2 to NH3 at low temperature in diethyl ether (Et2O) using various acid/reductant combinations, including HBArF4/KC8 and [Ph2NH2][OTf]/Cp*2Co (HBArF4 = [H(Et2O)2][BArF4]; BArF4 = tetrakis(3,5- bis(trifluoromethyl)phenyl)borate; OTf = trifluoromethane-sulfonate; Cp* = pentamethylcyclopentadienide).8 In a separate synthetic study, it was shown that [(P3B)Fe(N2)]− reacts rapidly with excess HBArF4 at very low temperatures to form the cationic hydrazido(2−) complex [(P3B)Fe(NNH2)]+ (Scheme 1C; potentials shown are measured in THF vs the ferrocenium/ferrocene couple [Fc+/Fc]).12 To probe further steps in the catalytic N2 fixation mechanism, we therefore sought access to the one-electron reduced hydrazido complex (P3B)Fe(NNH2) to evaluate the viability of N—N bond cleavage via subsequent protonation.
Preparations of [(P3B)Fe(NNH2)]+ typically contain several Fe-based impurities,12 so rather than direct reduction of this species, we determined that protonation of the 18 E− dianionic complex [(P3B)Fe(N2)]2− produces (P3B)Fe(NNH2) most cleanly. Reduction of the [Na(12-crown-4)2]+ salt of [(P3B)Fe(N2)]− with KC8 in dimethoxyethane (DME) followed by crystallization enables the isolation of [Na(12-crown-4)2][K(DME)x][(P3B)Fe(N2)] as a black solid, featuring an N—N stretching vibration at 1836 cm−1. For all subsequent studies, [(P3B)Fe(N2)]2− was produced in situ and used immediately. The 57Fe Mössbauer spectrum of [(P3B)Fe(N2)]2− produced from the reduction of 57Fe-enriched [Na(12-crown-4)2][(P3B)Fe(N2)] (Figure 1A) reveals parameters consistent with its diamagnetic ground state (Figure 1E),8b and which are nearly identical with those of the isoelectronic and isostructural silyl complex [(P3Si)Fe(N2)]− (P3Si = tris(o-diisopropylphosphinophenyl)silyl).13
Figure 1.
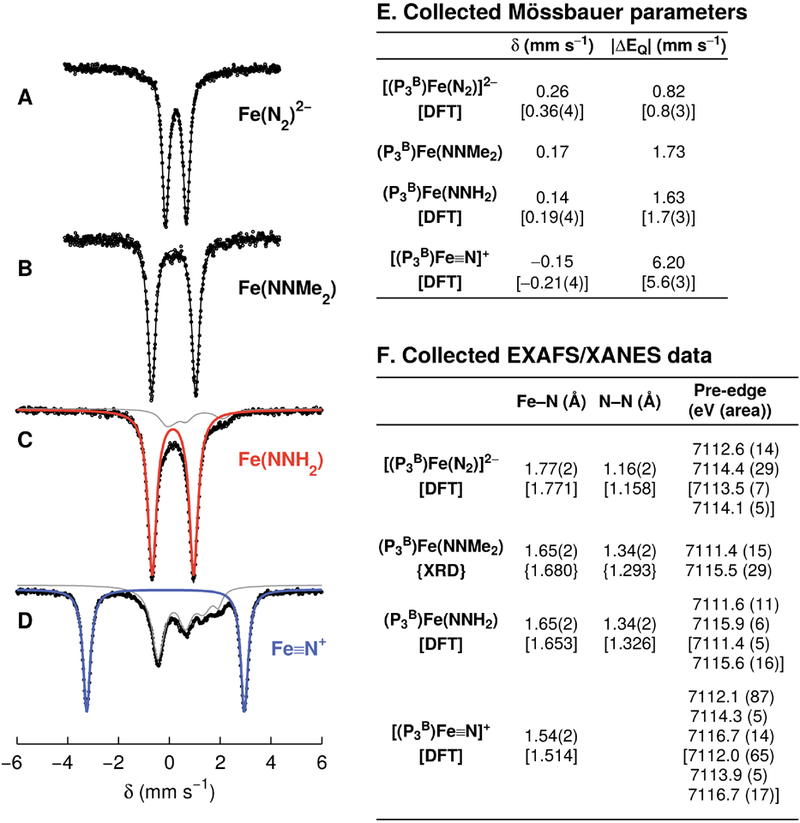
(A–D) Collected Mössbauer data; raw data are shown as circles, and the simulated data as a solid black line with individual subspectra plotted in gray, red, and blue. (A) Spectrum of [(P3B)Fe(N2)]2− prepared in situ from [(P3B)Fe(N2)]−. (B) Spectrum of (P3B)Fe(NNMe2). (C, D) Freeze-quench Mössbauer spectra from the reaction of [(P3B)Fe(N2)]2− with excess TfOH, showing conversion to (P3B)Fe(NNH4) (red subspectrum, ~90%) after mixing for 15 min (C), and subsequent formation of [(P3B)Fe≡N]+ (blue subspectrum, ~60%) after mixing for 120 min (D). Collected Mössbauer and XAS characterization data for these species are given in tables E and F.
An in situ prepared sample of 57Fe-enriched [(P3B)Fe(N2)]2− reacted with an excess of either triflic acid (TfOH) or HBArF4 in supercooled (i.e., between the glass transition at 91 K and the freezing point at 137 K) 2-methyltetrahydrofuran (2-MeTHF), and the products were analyzed by Mössbauer spectroscopy. Dianion [(P3B)Fe(N2)]2− reacts cleanly with both acids to form a new species in ca. 90% yield with δ = 0.14 and |ΔEQ| = 1.63 mm s−1 (Figures 1C and S21A) over the course of ~15 min. These parameters are similar to those of the diamagnetic hydrazido complex [(P3Si)Fe(NNH2)]+ (δ = 0.13 and |ΔEQ| = 1.48 mm s−1),10 and we therefore assign this species as the isoelectronic, isostructural, neutral hydrazido (P3B)Fe(NNH2) in an S = 0 ground state (Scheme 1D). Unlike its silyl analog, (P3B)Fe(NNH2) is very thermally sensitive, decomposing in solution within 15 min upon warming to 195 K. To further cement our assignment, we prepared the isoelectronic but more stable alkylhydrazido(2−) complex (P3B)Fe(NNMe2) as a spectroscopic model. Alkylhydrazido (P3B)Fe(NNMe2) has been structurally characterized, and its Mössbauer spectrum reveals parameters very similar to those observed for (P3B)Fe(NNH2) (Figure 1B, E). We note that although the ground state of (P3B)Fe(NNMe2) has S = 0, this alkylhydrazido possesses a low-energy triplet (S = 1) excited state (Figure S35, Table S14), which is also expected for (P3B)Fe(NNH2) on the basis of computational studies (Table S16).
Mixing [(P3B)Fe(N2)]2− with either acid in excess for longer times produces a new species in the Mössbauer spectrum as the major Fe-containing product (≥50% yield with TfOH; Figures 1D, S21B), suggesting a product resulting from the decay of (P3B)Fe(NNH2). This new species (δ = −0.15 and |ΔEQ| = 6.20 mm s−1) has parameters that are diagnostic for Fe(IV) nitrides under C3 symmetry3b,6,7d and is therefore assigned as the S = 0 nitrido cation [(P3B)Fe≡N]+. Negative isomer shifts are observed for Fe species featuring short, covalent interactions, such as those made with terminal N3− and O2− ligands, which drive Fe s-electron density toward the nucleus.14 The observation of quadrupole splittings >5 mm s−1 is limited to C3-symmetric Fe complexes featuring Fe≡L triple bonds, which results in an axial polarization of the electric field gradient due to localization of the Fe 3d electrons to a δ-symmetry e orbital set.3b,6,7d,15 Thus, the simultaneous observation of a negative δ and |ΔEQ| > 6 mm s−1 argues strongly in favor of our assignment of [(P3B)Fe≡N]+, which is also corroborated by XAS studies (vide infra). The absence of magnetic hyperfine splitting in spectra of (P3B)Fe(NNH2) and [(P3B)Fe≡N]+ collected at 5 K in the presence of a 50 mT field is consistent with our assignment of non-Kramers spin states.14 Computational studies reveal a diamagnetic ground state in both cases, and only the S = 0 states accurately reproduce the observed Mössbauer spectroscopic parameters (Figure 1E, Table S16). As with hydrazido (P3B)Fe(NNH2), nitrido [(P3B)Fe≡N]+ is thermally unstable in solution and degrades upon warming to temperatures ≥195 K for longer than 30 min to a mixture of (P3B)Fe(OTf) and unknown species with parameters consistent with high-spin Fe(II).
To gain additional structural characterization of the thermally unstable complexes (P3B)Fe(NNH2) and [(P3B)Fe≡N]+, we turned to Fe K-edge X-ray absorption spectroscopy. The XANES spectrum of (P3B)Fe(NNH2) features two moderate intensity resonances in the pre-edge region separated by 4.3 eV (Figure 2A). The XANES spectrum of (P3B)Fe(NNMe2) displays two resonances of similar intensity separated by 4.1 eV, but red-shifted by ca. 0.3 eV, consistent with replacement of the N-H substituents with more electron-rich N-Me (Figure 2A, Figure 1F). As observed previously for the C3-symmetric Fe(IV) nitrides (PhBPR3)Fe(N) (PhBPR3 = [PhB(CH2PR2)3]−; R = iPr, CH2Cy),16 the pre-edge XANES spectrum of [(P3B)Fe≡N]+ is dominated by an intense resonance at 7112.1 eV integrating to 87 units (Figure 2A). Based on the purity of the XAS sample (Figure S25B), this integration represents a lower limit of the true intensity of the resonance. This resonance is attributable to a transition from the 1s to an a1-symmetry orbital with significant Fe 4p and ligand 2p admixture (vide infra). This imparts dipole-allowed character to the transition and is a hallmark of M-to-N/O multiple bonding.16,17 Furthermore, the pre-edge spectra of (P3B)Fe(NNH2) and [(P3B)Fe≡N]+ predicted by time-dependent density functional theory (TD-DFT) are in very good agreement with those observed experimentally (Figure 2B).
Figure 2.
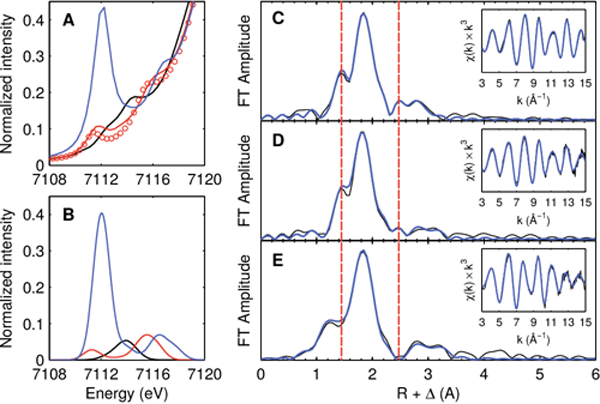
Collected XAS data. (A) Pre-edge regions of the XANES spectra of [(P3B)Fe(N2)]2− (black), (P3B)Fe(NNH2) (red), (P3B)Fe(NNMe2) (red circles), and [(P3B)Fe≡N]+ (blue:). (B) TD-DFT-predicted spectra of [(P3B)Fe(N2)]2− (black), (P3B)Fe(NNH2) (red), and [(P3B)Fe≡N]+ (blue). (C–E) Phase-uncorrected EXAFS data for (C) (P3B)Fe(NNMe2), (D) (P3B)Fe(NNH2), and (E) [(P3B)Fe≡N]+. The data are plotted in black, with simulations in blue.
The EXAFS region reveals a short Fe—N bond of 1.65(2) Å in (P3B)Fe(NNH2) (Figure 2D), which compares favorably with that predicted by DFT and observed experimentally for model complex (P3B)Fe(NNMe2) (Figure 2C, Figure 1F). Upon cleavage of the N—N bond in (P3B)Fe(NNH2) to form [(P3B)Fe≡N]+, the Fe—N bond contracts to 1.54(2) Å (Figure 2E, Figure 1F), which is within the range observed in previously characterized C3-symmetric Fe(lV) nitrides (1.51 to 1.55 Å),7d,e,16 and shorter than those observed in C4-symmetric, octahedral Fe(V/Vl) nitrides (1.57 to 1.64 Å).7c,g,h A peak at R + Δ = 2.5 Å in the Fourier transformed EXAFS of (P3B)Fe(NNH2) and (P3B)Fe(NNMe2), due to an Fe—N—N multiple scattering path, is notably absent in the transform of [(P3B)Fe≡N]+ (Figure 2C–E).
Figure 3A shows the calculated frontier orbitals of [(P3B)Fe≡N]+, which has the expected configuration for a C3 symmetric, formally Fe(lV) nitride.3a,7e The low energy of the virtual 1a1 orbital has been explained in terms of (i) an axial distortion which reduces the σ* character of the orbital with respect to the equatorial ligands and (ii) by 3d-4p mixing. In [(P3B)Fe≡N]+, the 1a1 orbital is additionally stabilized by a bonding interaction with the vacant B 2pz orbital (Figure 3B). A Löewdin population analysis of this orbital reveals nearly equal distribution among the Fe, N, and B atoms, with identical Fe 3d (9.5%) and 4p (9.1%) character. The significant amount of predicted ligand 2p character of this orbital (43%) is consistent with the intensity of the first pre-edge transition observed for [(P3B)Fe≡N]+, which is not expected on the basis of the 3d–4p mixing alone.16–18
Figure 3.
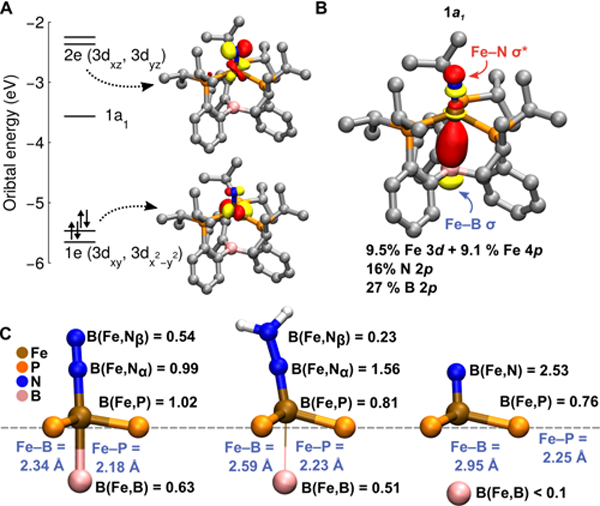
(A) Frontier Kohn—Sham orbitals computed for [(P3B)Fe≡N]+. (B) Löewdin population analysis of the empty 1a1 frontier orbital of [(P3B)Fe≡N]+. (C) Geometric analysis of the bonding in [(P3B)Fe(N2)]2−, (P3B)Fe(NNH2), and [(P3B)Fe≡N]+. The Fe—B distances from DFT models are given, along with the average Fe—P bond length from the EXAFS data. Shown in black are the Mayer bond orders (the average in the case of Fe—P).
Based on these collective data, we propose the sequence of reactions shown in Scheme 1D. Rapid protonation of [(P3B)Fe(N2)]2− at low temperature results in formation of hydrazido (P3B)Fe(NNH2) (via presumed diazenido complex [(P3B)Fe(N2H)]−). In a kinetically slow step, (P3B)Fe(NNH2) is protonated to form an unobserved transient (or transition state) hydrazidium cation [(P3B)Fe(NNH3)]+, which decays via heterolytic rupture of the N—N bond to yield NH3 and [(P3B)Fe≡N]+. In a larger-scale experiment, protonation of [(P3B)Fe(N2)]2− with TfOH in supercooled 2-MeTHF produced NH3 in 36.0(5)% isolated yield, comparable to the observed yield of [(P3B)Fe≡N]+ under identical conditions (~50% Figure S24A). Under catalytic conditions (i.e., with a reductant present), we propose that [(P3B)Fe≡N]+ can be reduced by 3 H+/3 e− to form a second equivalent of NH3 and [(P3B)Fe]+ (or [(P3B)Fe(NH3)]+),3a,6 from which [(P3B)Fe(N2)]− can be regenerated in turn upon reduction.19 Indeed, sequential reaction of [(P3B)Fe(N2)]2− with TfOH and Cp*2Co in supercooled 2-MeTHF doubles the isolated yield of NH3 to 73(17)%.
We have shown that two reductants, KC8 and Cp*2Co, can drive catalytic N2 fixation in this system, yet only KC8 is sufficiently reducing to access dianion [(P3B)Fe(N2)]2− under catalytic turnover. The most reduced state of the catalyst accessible with Cp*2Co is the anion [(P3B)Fe(N2)]−,8c and this state appears to be catalytically relevant under all conditions canvassed,8b including with TfOH and Cp*2Co as the acid/reductant combination (Table S11). An alternative pathway to form [(P3B)Fe≡N]+ under these milder conditions is shown in Scheme 1C, D via the known cationic hydrazido [(P3B)Fe(NNH2)]+.12 We estimate the reduction potential of this species to be ≥ −1.2 V based on the alkyl congener (Figure S32), and thus [(P3B)Fe(N2)]− (or its oxidized congener (P3B)Fe(N2)) is a sufficiently strong reductant to produce (P3B)Fe(NNH2) from [(P3B)Fe(NNH2)]+. The viability of this pathway is demonstrated by a low temperature protonation experiment of [(P3B)Fe(N2)]− with excess TfOH (i.e., no exogenous reductant), which produced appreciable quantities of [(P3B)Fe≡N]+ (~20%) and NH3 (34(3)%) along with competitive oxidation to (P3B)Fe(OTf). The same reaction sequence is thermodynamically accessible under turnover conditions using Cp*2Co as the terminal source of reducing equivalents. Other routes to [(P3B)Fe≡N]+ via metallocene-mediated proton-coupled electron transfer reactions are also conceivable, as are late-stage N-N cleavage pathways.8b,c
In the transformation from [(P3B)Fe(N2)]2− to [(P3B)Fe≡N]+, the Fe center spans six formal oxidation states, from d10 Fe(−II) to d4 Fe(IV). However, these formal assignments do not account for Z-type Fe-to-B σ-backbonding,20 in addition to π-backbonding with the phosphines. Indeed, the presence of preedge transitions in the XANES spectrum of [(P3B)Fe(N2)]2−, which is reproduced by TD-DFT, requires a physical oxidation state dn with n < 10 (Figure 2A, B). The physical oxidation state range of Fe is buffered by these soft electron-accepting interactions, which allow e− to be stored in covalent Fe—B/P backbonding interactions until transferred to the N—N unit upon protonation. The three sequential H+ transfers to the distal N atom of [(P3B)Fe(N2)]2− to form [(P3B)Fe≡N]+ and NH3 result in a lengthening of the Fe—B distance by 0.61 Å (distances from DFT) and a lengthening of the average Fe—P distance by 0.07 Å (distances from EXAFS), reflecting loss of Fe—B/P covalency (Figure 3C). Owing to the highly flexible Fe—borane interaction, which increases from 2.34 Å ([(P3B)Fe(N2)]2−) to 2.59 Å ((P3B)Fe(NNH2)) to 2.95 Å ([(P3B)Fe≡N]+), the valency of Fe is largely conserved, as it distorts out of the P3 plane to form π bonds with the NNH22− and N3− ions. This is reflected in the nearly constant sum of the bond orders about Fe for [(P3B)Fe(N2)]2− (5.2), (P3B)Fe(NNH2) (4.7), and [(P3B)Fe≡N]+ (4.8), a notion supported by the fact that the change in isomer shift (Δδ) is only 0.38 mm s−1 (0.06 mm s−1/e−). For comparison, Δδ is nearly 1 mm s−1 for a series of isostructural Fe complexes ranging over only five formal oxidation states (ca. 0.2 mm s−1/e−).7c
The demanding multielectron process, [(P3B)FeI]+ + N2 + 3H+/3 e− → [(P3B)FeIV≡N]+ + NH3 (Scheme 1C, D), is thus facilitated by load-sharing of the reducing equivalents between Fe and the ligand. In this way, the redox behavior of the (P3B)Fe unit crudely models that of a metallocluster, in which the potential range of multielectron processes is compressed by delocalization of electrons/holes among many metals.21
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Latimer, E. Nelson, C. Krest, C. Miller, and K. Mittra for assistance with synchrotron measurements; This work was supported by the Resnick Sustainability Institute at Caltech (Graduate Fellowship, N.B.T.), as well as the NIH (GM 070757). Use of the Stanford Synchrotron Radiation Light-source, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02–76SF00515.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b09364.
Crystallographic data for (P3B)Fe(NNMe2) (CIF)
Computational models (PDF)
Experimental procedures, characterization data (PDF)
The authors declare no competing financial interest.
Additional notes in the Supporting Information.
REFERENCES
- (1).Hohenberger J; Ray K; Meyer K Nat. Commun 2012, 3, 720. [DOI] [PubMed] [Google Scholar]
- (2).(a) Poulos TL Chem. Rev 2014, 114, 3919. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Que L Acc. Chem. Res 2007, 40, 493. [DOI] [PubMed] [Google Scholar]
- (3).(a) Betley TA; Peters JC J. Am. Chem. Soc 2004, 126, 6252. [DOI] [PubMed] [Google Scholar]; (b) Hendrich MP; Gunderson W; Behan RK; Green MT; Mehn MP; Betley TA; Lu CC; Peters JC Proc. Natl. Acad. Sci. U. S. A 2006, 103, 17107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chatt J; Dilworth JR; Richards RL Chem. Rev 1978, 78, 589. [Google Scholar]
- (5).(a) Yandulov DV; Schrock RR J. Am. Chem. Soc 2002, 124, 6252. [DOI] [PubMed] [Google Scholar]; (b) Yandulov DV; Schrock RR Science 2003, 301, 76. [DOI] [PubMed] [Google Scholar]; (c) Tanaka H; Arashiba K; Kuriyama S; Sasada A; Nakajima K; Yoshizawa K; Nishibayashi Y Nat. Commun 2014, 5, DOI: 10.1038/ncomms4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Scepaniak JJ; Vogel CA; Khusniyarov MM; Heinemann FW; Meyer K; Smith JM Science 2011, 331, 1049. [DOI] [PubMed] [Google Scholar]
- (7).(a) Wagner WD; Nakamoto K J. Am. Chem. Soc 1988, 110, 4044. [Google Scholar]; (b) Meyer K; Bill E; Mienert B; Weyhermüller T; Wieghardt K J.Am. Chem. Soc 1999, 121, 4859. [Google Scholar]; (c) Berry JF; Bill E; Bothe E; George SD; Mienert B; Neese F; Wieghardt K Science 2006, 312, 1937. [DOI] [PubMed] [Google Scholar]; (d) Vogel C; Heinemann FW; Sutter J; Anthon C; Meyer K Angew. Chem. Int. Ed 2008,47, 2681. [DOI] [PubMed] [Google Scholar]; (e) Scepaniak JJ; Fulton MD; Bontchev RP; Duesler EN; Kirk ML; Smith JM J. Am. Chem. Soc 2008, 130, 10515. [DOI] [PubMed] [Google Scholar]; (f) Rodriguez MM; Bill E; Brennessel WW; Holland PL Science 2011, 334, 780. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Aliaga-Alcalde N; DeBeer George S; Mienert B; Bill E; Wieghardt K; Neese F Angew. Chem., Int. Ed 2005, 44, 2908. [DOI] [PubMed] [Google Scholar]; (h) Sabenya G; Lazaro L; Gamba I; Martin-Diaconescu V; Andris E; Weyhermüller T; Neese F; Roithova J; Bill E; Lloret-Fillol J; Costas M J. Am. Chem. Soc 2017, 139, 9168. [DOI] [PubMed] [Google Scholar]
- (8).(a) Anderson JS; Rittle J; Peters JC Nature 2013, 501, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Del Castillo TJ; Thompson NB; Peters JC J. Am. Chem. Soc 2016, 138, 5341. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chalkley MJ; Del Castillo TJ; Matson BD; Roddy JP; Peters JC ACS Cent. Sci 2017, 3, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Seefeldt LC; Hoffman BM; Dean DR Annu. Rev. Biochem 2009, 78, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rittle J; Peters JC J. Am. Chem. Soc 2016, 138, 4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Moret M-E; Peters JC Angew. Chem., Int. Ed 2011, 50, 2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Anderson JS; Cutsail GE III; Rittle J; Connor BA; Gunderson WA; Zhang L; Hoffman BM; Peters JC J. Am. Chem. Soc 2015, 137,7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lee YH; Mankad NP; Peters JC Nat. Chem 2010, 2, 558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Gütlich P; Bill E; Trautwein AX Mössbauer Spectroscopy and Transition Metal Chemistry; Springer Berlin Heidelberg: New York, 2011. [Google Scholar]
- (15).Rudd PA; Liu S; Planas N; Bill E; Gagliardi L; Lu CC Angew. Chem. Int. Ed 2013, 52, 4449. [DOI] [PubMed] [Google Scholar]
- (16).Rohde J-U; Betley TA; Jackson TA; Saouma CT; Peters JC; Que L Jr. Inorg. Chem 2007, 46, 5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Weng T-C; Hsieh W-Y; Uffelman ES; Gordon-Wylie SW; Collins TJ; Pecoraro VL; Penner-Hahn JE J. Am. Chem. Soc 2004, 126, 8070. [DOI] [PubMed] [Google Scholar]
- (18).Westre TE; Kennepohl P; DeWitt JG; Hedman B; Hodgson KO; Solomon EI J. Am. Chem. Soc 1997, 119, 6297. [Google Scholar]
- (19).Anderson JS; Moret ME; Peters JC J. Am. Chem. Soc 2013, 135, 534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bouhadir G; Bourissou D Chem. Soc. Rev 2016, 45, 1065. [DOI] [PubMed] [Google Scholar]
- (21).Hernández Sánchez R; Zheng S-L; Betley TA J. Am. Chem. Soc 2015, 137, 11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.