

Abstract
Objective
Delayed cerebral ischemia (DCI) is a common, disabling complication of subarachnoid hemorrhage (SAH). Preventing DCI is a key focus of neurocritical care, but interventions carry risk and cannot be applied indiscriminately. While retrospective studies have identified continuous EEG (cEEG) measures associated with DCI, no study has characterized the accuracy of cEEG with sufficient rigor to justify using it to triage patients to interventions or clinical trials. We therefore prospectively assessed the accuracy of cEEG for predicting DCI, following the Standards for Reporting Diagnostic Accuracy Studies.
Methods
We prospectively performed cEEG in non-traumatic, high-grade SAH patients at a single institution. The index test consisted of clinical neurophysiologists prospectively reporting pre-specified EEG alarms: 1) decreasing relative alpha variability, 2) decreasing alpha-delta ratio, 3) worsening focal slowing, or 4) late-appearing epileptiform abnormalities. The diagnostic reference standard was DCI determined by blinded, adjudicated review. Primary outcome measures were sensitivity and specificity of cEEG for subsequent DCI, determined by multi-state survival analysis, adjusted for baseline risk.
Results
103 of 227 consecutive patients were eligible and underwent cEEG monitoring (7.7-day mean duration). EEG alarms occurred in 96.2% of patients with and 19.6% without subsequent DCI (1.9-day median latency [IQR 0.9–4.1]). Among alarm subtypes, late-onset epileptiform abnormalities had the highest predictive value. Pre-specified EEG findings predicted DCI among patients with low (91% sensitivity, 83% specificity) and high (95% sensitivity, 77% specificity) baseline risk.
Interpretation
Continuous EEG accurately predicts DCI following SAH, and may help target therapies to patients at highest risk of secondary brain injury.
Keywords: Subarachnoid hemorrhage, continuous EEG, delayed ischemic neurologic decline, delayed cerebral ischemia, neuromonitoring
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) impacts 600,000 patients worldwide annually, conferring 40% mortality.1 Delayed cerebral ischemia (DCI), a common, disabling complication, usually occurs 4–14 days after onset. Despite tripling the odds of a poor one-year outcome,2 DCI often goes unrecognized, limiting opportunities for intervention. Accurately identifying impending DCI would help target therapies to patients most likely to benefit. However, conventional monitoring methods are intermittent, only modestly accurate,3 and commonly examine only large vessel vasospasm, one of many mechanisms upstream of DCI.4
Electroencephalography (EEG) provides a continuous measure of cerebral function with robust, predictable responses to ischemia.5–10 Several small studies have proposed criteria for predicting DCI based on changes in continuous EEG (cEEG) spectral features,7, 10 including decreasing alpha-to-delta power ratio (ADR)6, 7, 9 or relative alpha power variability (RAV).6, 8 Other findings associated with cerebral ischemia include epileptiform discharges, rhythmic and periodic “ictal-interictal continuum” (IIC) patterns,11–14 and isolated alpha suppression.5 Intracranial electrocorticography suggests that cortical spreading depolarizations may be a phenomenon underlying cEEG changes and DCI itself.15
No study to date has assessed the diagnostic accuracy of cEEG for DCI following the Standards for Reporting of Diagnostic Accuracy Studies (STARD).16 Prior studies employed retrospective, unblinded analysis of cEEG and clinical data, and small cohorts.6–10 Continuous EEG analysis in these studies was performed off-line, outside of real-time clinical practice and employed variable DCI definitions.17 Therefore, the generalizability of previously proposed cEEG criteria for predicting DCI remains uncertain.
We prospectively assessed the sensitivity and specificity of cEEG performed as part of routine medical care for predicting DCI, following STARD criteria. We applied a consensus definition17 of the primary outcome, DCI, using a blinded, adjudicated process, and pre-specified which cEEG findings constitute an alarm for impending DCI. To ensure generalizability, we recruited a large number of consecutively monitored patients.
Methods
Study Design
In this prospective study we assessed the diagnostic accuracy of cEEG performed according to an institutional clinical care guideline18 for consecutive patients with SAH within a single Neurosciences Intensive Care Unit (ICU) over 2.5 years. The clinical guideline recommends cEEG begin within 48 hours of admission and continue for 10 days, among high clinical or radiologic grade patients (Hunt and Hess (HH) grade 4–5 or modified Fisher Scale19 (mFS) grade 3–4). This institutional guideline recommends prophylactic antiseizure medication until ruptured aneurysms are secured.
Inclusion criteria were age ≥18 years; non-traumatic etiology; cEEG lasting ≥72 hours; and, in cases determined to have DCI, cEEG data available within 24 hours of DCI. We excluded patients initially presenting with status epilepticus. Procedures conformed to the STARD criteria (Fig 1 and Supplementary Material Table S1).
Figure 1. Flow diagram of study (STARD diagram).
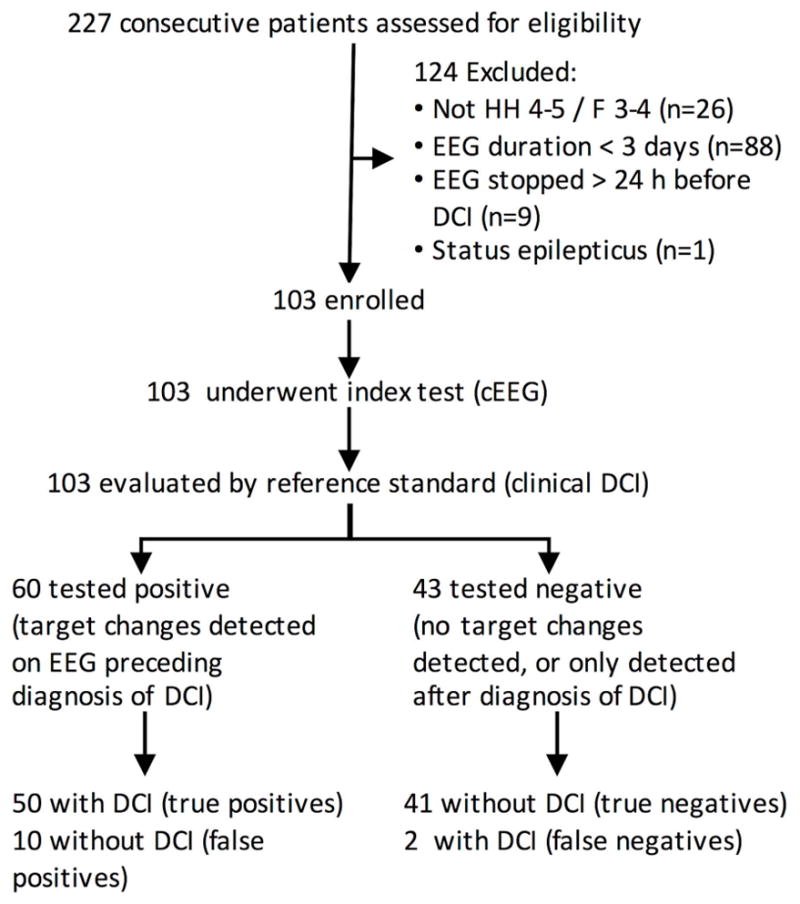
After the index test results, results are sorted based on the reference standard (consensus criteria for the blinded determination of DCI). HH = Hunt and Hess grade. F = Fisher group. cEEG = continuous EEG monitoring. DCI = delayed cerebral ischemia. For the 124 excluded patients, we list only the primary reason for exclusion in the order of exclusion.
A pre-specified multi-step process was conducted to determine DCI events including blinded medical record review and adjudication undertaken as part of a prospective quality improvement initiative, as described below. Data archiving and analysis were conducted under an IRB-approved protocol.
Index Test
Continuous EEG was recorded using the International 10–20 system of scalp electrodes (Ives Electrodes; Newburyport, MA). Clinical neurophysiologists reported cEEG findings prospectively in real time, reviewing raw cEEG data (Natus, Inc.; Pleasanton, CA) and displays of quantitative trends (Persyst, Inc.; Scottsdale, AZ) using clinical EEG review software; data were reviewed by both clinical fellows and staff neurophysiologists, according to routine clinical practice. All clinical neurophysiologists received special instruction in interpreting ICU EEG recordings and quantitative EEG trends (described below) used in monitoring SAH patients. Clinical neurophysiologists routinely assessed for medication-induced effects on the cEEG, e.g. anesthetic doses, communicating by telephone and face-to-face about EEG findings.
Neurophysiologists issued twice-daily reports, prospectively documenting the following cEEG findings, pre-specified as electrophysiologic indicators of possible ischemia:5–10, 12, 13, 20, 21 1) focal slowing or dropout of fast activity; 2) decreasing RAV; 3) decreasing ADR; or 4) epileptiform abnormalities, including sporadic epileptiform discharges, lateralized rhythmic delta activity (LRDA), lateralized periodic discharges (LPD), or generalized periodic discharges (GPD). Visual review and reporting of these findings for a single 8–12 hour reporting epoch typically takes our staff 20–30 minutes.
Details regarding the review process and alarm criteria have been previously described by Muñiz et al.18 An EEG “alarm” was defined as description in the published clinical EEG report of clear and persistent worsening or new emergence over the majority of a single 8–12-hour epoch as defined for any of the following EEG features, compared with the previous 8–12-hour epoch, independent of baseline asymmetries, e.g., breach rhythms, or pathological features already present. If more than one alarm occurred, only the first was analyzed. Specific criteria for each feature are:
1. Focal slowing or dropout of fast activity
These focal features on raw EEG were pre-specified as increased power in the delta (<4-Hz) or theta (4–7-Hz) ranges relative to the corresponding contralateral region. An alarm criteria of a “clear and persistent worsening or new emergence” was defined as a qualitative change in the background from the preceding epoch, explicitly described in the official clinical neurophysiology report, persisting the majority of an epoch.
2. Decreasing relative alpha variability (RAV)
Decreasing RAV was pre-specified as a visually apparent decline in the RAV grade over a single 8–12-hour epoch. RAV grades were reported for each 8–12-hour EEG epoch based on the predominant amount of RAV histogram variability best describing the epoch (4, “excellent”; 3, “good”; 2, “fair”; 1, “poor”), as adapted from Vespa et al.8 RAV was displayed with a histogram; bars were given by relative alpha power (8–12-Hz power divided by 1–20-Hz power) computed every 30 seconds. Spectral power was calculated as the squared magnitude of the EEG signal Fourier Transform. RAV was displayed for 6 regions (F3–C3, F4–C4, C3–T3, C4–T4, P3–O1, P4–O2).18 For decreasing RAV, the alarm criteria of a “clear and persistent worsening or new emergence” was defined as worsening by ≥1 grade versus the preceding 8–12-hour epoch (e.g., from 3, “good” to 2, “fair”).
3. Decreasing ADR
Decreasing ADR was pre-specified as a visually apparent decrement in the ADR trend. ADR was calculated using a moving average (2-minute window) of the ratio of 8–13-Hz power (different from the 8–12-Hz used for RAV calculations) divided by 1–4 Hz band power. Power was calculated by the squared magnitude of the EEG signal Fourier transform. Separate ADR trends were computed for each hemisphere and displayed as overlapping lines of different colors to facilitate assessment of hemispheric asymmetry.18 For decreasing ADR, the alarm criteria of a “clear and persistent worsening or new emergence” was defined to be a visually apparent decrease over one or both hemispheres of (1) approximately 10% below baseline persisting at least 6 consecutive hours or (2) at least 50% below baseline for 1 or more hours, compared to the most recent epoch, as adapted from retrospectively derived cutoffs of Claassen et al.9 These criteria were applied in clinical practice based on visual analysis rather than explicit calculations.
4. Increasing Epileptiform abnormalities (EA)
EA were pre-specified as sporadic epileptiform discharges, lateralized rhythmic delta activity (LRDA), lateralized periodic discharges (LPD), or generalized periodic discharges (GPD), defined according to the 2012 Standardized American Clinical Neurophysiology Society (ACNS) ICU EEG Terminology.22 For an increase in EAs, the alarm criteria of a “clear and persistent worsening or new emergence” was defined as appearance of a pattern absent in prior epochs (new emergence), or increase in the ACNS-defined prevalence of the pattern, e.g. from “occasional” LPDs (1–9% of an epoch) to “frequent” (10–49% of an epoch).
EEG review relied on expert visual interpretation of the EEG data as a gold standard. For example, the alarm criteria of a “clear and persistent worsening or new emergence” is qualitative in the case of EEG focal slowing or dropout of fast activity, and is semi-quantitative but still reliant on expert visual pattern recognition for decreasing ADR and epileptiform abnormalities. We pre-specified expert visual review rather than computational analysis for the following reasons: 1) artifacts require verification by raw EEG (e.g., to identify blinking inducing delta waves or breach rhythms), 2) generalization to other clinical practices requires an undistorted clinical workflow practicable at other institutions, and 3) expert visual analysis of EEG in SAH has yet to be matched by computer algorithms.23
We promoted inter-rater reliability in several ways. First, guidelines for reporting EEGs in patients with SAH were developed specifically for this purpose. Educational sessions were held to instruct clinicians on applying the rules before their implementation. Institutional guidelines were posted in the EEG reading area, referred to frequently, and ultimately published in a methodologic manuscript18 available to the clinical neurophysiology community. Additionally, weekly clinical conferences were attended by clinical neurophysiologists to review raw and quantitative EEG data as a group to maintain consistency. Finally, attending physicians reviewed EEG data regularly with clinical trainees to promote consistent application of the EEG guideline’s criteria.
Missing cEEG data (periods of interrupted or uninterpretable EEG) was handled using a carry-forward method; an alarm could not be coded until explicitly recorded in the clinical report.
Primary Outcome Reference Standard
We defined the primary outcome as the first DCI event. In STARD terminology, determination of DCI is the reference standard. DCI was determined based on a recent consensus definition17 as: a) new focal neurological impairment, or b) ≥2-point decrement of the Glasgow Coma Scale (GCS), persisting ≥1 hour, not attributable to other causes including re-rupture, hydrocephalus or elevated intracranial pressure, procedure-related complications, seizures, or systemic or metabolic abnormalities; or c) infarction on follow-up CT or MRI. EEG alarms not followed by DCI were considered false positives; alarms followed by DCI were considered true positives. Late EEG alarms (occurring after DCI) or absent EEG alarms despite a DCI event were considered false negatives.
DCI was determined by a pre-specified blinded consensus review using a multi-step process: (a) independent medical record review (by authors ESR, MBW, SFZ) blinded to cEEG findings, and (b) adjudication in cases of disagreement. Separate daily team interviews were performed to ensure that limitations in medical record documentation did not lead to missed events.
Other Clinical Testing
We used a composite admission risk score24 for baseline risk stratification in our multistate survival model. We assessed daily transcranial Doppler ultrasound (TCD) velocities for comparison, using institutional peak systolic velocity (PSV) thresholds for mild (>200 cm/s), moderate (>250 cm/s), or severe (>300 cm/s) vasospasm.
Pre-specified Measures of Diagnostic Accuracy
The pre-specified primary statistical measures were the diagnostic sensitivity and specificity of cEEG for predicting DCI. Secondary outcome measures were odds ratios for DCI for two cEEG alarm subtypes (background changes and epileptiform findings); and the latency from cEEG alarm to DCI.
Exploratory Analysis
We additionally explored the association of EEG alarm subtypes DCI neurologic event subtypes.
Post-hoc Analysis for Bias
We examined the baseline characteristics of patients excluded from the study due to lacking sufficient cEEG (≥72 hours) as pre-specified by the inclusion criteria, such that the index test could not adequately be assessed (n=88, Figure 1). We compared both baseline illness severity (age, modified Fisher radiologic grade, Hunt and Hess clinical grade, and composite admission risk score) between these patients and the subjects who were enrolled. In addition to examining baseline characteristics, we examined rates of early withdrawal of life-sustaining therapy (within three days of admission) that would prevent sufficient cEEG from being performed.
Post-hoc Analysis for Confounding
We examined the frequency with which antiseizure medications, anesthetic infusions, and vasopressors were added, increased, or decreased during the baseline period and during the EEG alarm period, to ascertain the impact of management on state transitions. We defined dose or infusion rate increases or decreases as a change of at least 50%, or an increase in medication administration frequency (e.g., from every 12 hours to every 8 hours), compared to the dose or frequency at the beginning of the baseline or EEG alarm state. Infusion changes were required to persist at least 2 hours.
Statistical Analysis
Baseline characteristics in patients with and without DCI were compared utilizing t-tests and Fisher’s exact tests. We used one-tailed tests for radiologic grade, clinical grade, composite admission risk score, and EEG monitoring duration because prior literature19, 24 suggests both symptomatic vasospasm and poor long-term outcome are positively associated with these factors. We used two-tailed tests for age, gender, aneurysm management technique, and time from symptom onset to starting cEEG because no literature consistently associates DCI with these factors.
We measured the univariate time-dependent association between the following findings and subsequent DCI: 1) any documented EEG alarm; 2) EEG alarms due to worsening slowing, ADR or RAV; 3) EEG alarms due to new epileptiform abnormalities; and basic sonographic findings of 4) mild (PSV > 200 cm/sec), 5) moderate (PSV >250 cm/sec), and 6) severe vasospasm (PSV >300 cm/sec). Fisher’s exact test p-values, odds ratio (OR) values, and OR 95% confidence intervals were calculated for baseline characteristics.
A multistate survival analysis model,25 adjusted for baseline composite admission risk score,24 was pre-specified as the statistical measure assessing the time-dependent probability of DCI following SAH onset. In our analysis, patients occupy one of three states at any given time (Fig 2A). Patients begin in the baseline state. Patients with an EEG alarm but without DCI are in the alarm state. Patients who suffer a DCI event are in the DCI state. Transitions are allowed only in one direction. This is known as an “illness-death” model, with “illness” and “death” corresponding to the alarm and DCI states.26, 27 The proportional hazard assumption on state transitions was checked graphically.27 We assumed that the baseline composite admission risk score affects all three transitions, but each transition has a unique hazard rate function. While cEEG monitoring duration varied (Fig 2B), we did not treat data as censored because: (1) all DCI events occurred during or within 24 hours of cEEG monitoring; and (2) DCI is a clinical diagnosis, independent of cEEG. All events were defined with respect to the final state of the patients 20 days after SAH onset (i.e. DCI or no DCI). True positive predictions were defined as cases in which an EEG alarm preceded a diagnosis of DCI, i.e., a baseline→alarm→DCI state transition sequence. Missed events were cases with DCI without a preceding EEG alarm, i.e. a baseline→DCI transition sequence. False alarms were cases with an EEG alarm reported without subsequent DCI, i.e. a baseline→alarm transition sequence. Using this method, sensitivity and specificity were calculated for patients with a low, medium, and high composite admission risk score.
Figure 2. Time-to-event data.
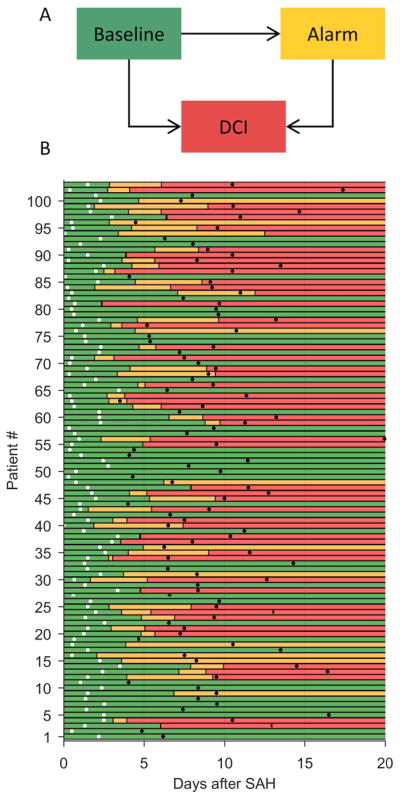
(A) In the multistate survival analysis, patients occupy one of three possible states at any given time. Permissible transitions are indicated by arrows. (B) State occupancy versus time for 103 SAH patients, starting from the time of hemorrhage (t=0), continuing up to 20 days post-hemorrhage. Green bars indicate the baseline state (no EEG alarm or DCI thus far); yellow, the alarm state; and red, the DCI state. White dots indicate when EEG monitoring began, and black dots indicate when EEG monitoring ended.
In addition to sensitivity and specificity, we calculated these measures of test performance: positive predictive value, negative predictive value; false positive rate (1-specificity); positive likelihood ratio; negative likelihood ratio; diagnostic odds ratio; pretest probability of DCI; absolute risk increase (ARI) after a positive test result; absolute risk decrease after a negative test result; and the number needed to monitor to successfully predict one additional DCI event (1/ARI). We estimated these statistics for patients with low, intermediate, and high composite admission risk scores (score = 1, 2.5, 4, respectively). To quantify the precision of our estimates, we calculated Bayesian 95% credible intervals (CI) via 10,000 rounds of bootstrapping, as follows. Bootstrapping was performed by sampling with replacement from the 103 time-to-event entries in our data (one row per case in the cohort), with equal probabilities of drawing each case. Bootstrap samples each had the same number of entries (103) as the original dataset. For each bootstrap sample, we fit a full multistate survival model, and obtained values for parameters in Table 3. In this way, we obtained 10,000 bootstrap values for each of the statistics reported in Table 3. The 10,000 samples can be interpreted as forming an approximate, nonparametric posterior distribution for each parameter. The 95% CI was then computed from each posterior distribution as the narrowest interval, i.e. the interval of highest probability density (including the mode of the distribution) containing 95% of the total probability mass.
We carried out analyses using the mstate package28 in R (The R Foundation) and Statistics Toolbox in MATLAB (MathWorks; Natick, MA).
Results
Of 227 consecutive patients screened (April 2013 to November 2015), 103 met criteria (75.7% women; mean age 57.7 years) (Table 1) and were enrolled. Fifty-two [50.5%] developed DCI (Fig 1). Patients with DCI had higher baseline mFS, HH, and composite admission risk scores (Table 1). Subjects with and without DCI did not differ statistically in age, gender, or proportion undergoing endovascular aneurysm management. Patients who were excluded due to insufficient EEG (Supplementary Table S2; n=88) did not differ in illness severity (age, modified Fisher radiologic grade, Hunt and Hess clinical grade, or the composite admission risk score). Patients with early withdrawal of life-sustaining therapy (WLST) were unlikely to undergo the index test; 21 (23.9%) of the 88 patients not enrolled had early WLST, while none of the 103 subjects enrolled had early WLST (p<0.0001).
Table 1.
Patient Characteristics
| DCI (n=52) | No DCI (n=51) | p-value | |
|---|---|---|---|
| Age, mean ± SD | 59.6 ±14.6 | 55.8 ±12.7 | 0.16 |
| Female (%) | 82.7% | 68.6% | 0.11 |
| Modified Fisher Scale, mean ± SD | 3.3 ± 0.5 | 3.1 ± 0.3 | 0.03 |
| Hunt and Hess grade > 3 (%) | 50.0% | 21.6% | <0.01 |
| Composite admission risk score ± SD | 2.4 ± 0.82 | 2.0 ± 0.8 | 0.01 |
| Aneurysm location | 0.42 | ||
| Anterior cerebral artery (%) | 28.9% | 30.8% | |
| Middle cerebral artery (%) | 19.2% | 7.7% | |
| Internal carotid artery (%) | 19.2% | 13.5% | |
| Posterior communicating artery (%) | 17.3% | 19.23% | |
| Vertebrobasilar and posterior cerebral artery (%) | 11.5% | 17.3% | |
| Other (%) | 3.9% | 9.6% | |
| Endovascular aneurysm management (%) | 48.1% | 58.8% | 0.32 |
| EEG monitoring start time, days after SAH onset ± SD | 1.5 ± 0.9 | 1.5 ± 1.3 | 0.99 |
| EEG monitoring duration, days, mean ± SD | 9.1 ± 3.3 | 6.3 ± 2.4 | <0.01 |
| Time of DCI, days after SAH onset, mean ± SD | 6.6 ± 2.4 | --- | --- |
DCI = delayed cerebral ischemia; EEG = electroencephalography; SAH = subarachnoid hemorrhage.
Most DCI events were preceded by EEG alarms (Fig 2). EEG alarms were documented in 60 patients. Of these, 50 preceded DCI, and 10 were false positives. 43 patients had no EEG alarms; 41 were true negatives, and two developed DCI without a preceding EEG alarm. From these numbers, unadjusted sample statistics for cEEG as a predictor for DCI were: sensitivity, 96.2%; specificity, 80.4%; positive predictive value, 83.3%; and negative predictive value, 95.4%.
We examined the predictive value of pre-specified EEG alarm subtypes (Table 2). Background deterioration (new slowing, decreasing ADR, or decreasing RAV) strongly predicted DCI (63.5% vs. 17.7%; OR 8.11 [3.25–20.2]; p<0.01). EEG alarms due to new or worsening epileptiform abnormalities showed a stronger association (63.5% vs. 7.84%, OR 20.4 [6.36–65.5]; p<0.01). A post-hoc analysis of background deterioration showed that RAV had a particularly low false positive rate (2%) and the highest odds ratio within the subtypes of EEG background deterioration. Deterioration of the raw EEG alone was reported in only 15.4% of patients with subsequent DCI, while the overall rate of background abnormalities rose to 63.5% when including all three indicators of background deterioration. Background deterioration and epileptiform alarm subtypes were also synergistic: detection of any EEG alarm conferred substantially higher risk (96.2% vs. 19.6%; OR 102.5 [21.3–494]; p <0.01).
Table 2.
Univariate association between time-dependent predictors and subsequent DCI
| DCI (n=52) | No DCI (n=51) | OR [95% CI] | p-value | |
|---|---|---|---|---|
| Any EEG alarm documented | 96.2% | 19.6% | 102.5 [21.3, 494] | <0.01 |
| Worsening slowing, ADR or RAV | 63.5% | 17.7% | 8.11 [3.25, 20.2] | <0.01 |
| Worsening focal slowing | 15.4% | 7.8% | 2.14 [0.60, 7.60] | 0.19 |
| Worsening ADR | 32.7% | 9.8% | 4.47 [1.50, 13.3] | <0.01 |
| Worsening RAV | 42.3% | 2.0% | 36.7 [4.70, 286.11] | <0.01 |
| New epileptiform abnormality | 63.5% | 7.84% | 20.4 [6.36, 65.5] | <0.01 |
| Sonographic vasospasm | ||||
| Maximum PSV > 200 cm/sec | 75.0% | 45.1% | 3.65 [1.58, 8.42] | <0.01 |
| Maximum PSV > 250 cm/sec | 57.7% | 33.3% | 2.73 [1.2, 6.1] | 0.01 |
| Maximum PSV > 300 cm/sec | 30.8% | 19.6% | 1.80 [0.73, 4.52] | 0.14 |
ARD = alpha-to-delta ratio; CI = confidence interval; DCI = delayed cerebral ischemia; EEG = electroencephalography; PSV = transcranial Doppler ultrasound peak systolic velocity; RAV = relative alpha variability.
For comparison, we examined the association of TCD vasospasm with DCI (Table 2). Mild vasospasm (PSV >200 cm/s) was significantly more common in DCI cases (75.0% vs. 45.1%); however, compared with EEG, the association was substantially weaker (OR 3.65 [1.58–8.42], p<0.01). Moderate vasospasm (PSV >250 cm/s) predicted DCI more weakly (57.7% vs. 33.3%, OR=2.73 [1.2–6.1], p=0.01). Severe vasospasm (PSV >300 cm/s) was not a significant predictor (30.8% vs. 19.6%, OR = 1.80 [0.73–4.52], p=0.14).
To investigate diagnostic accuracy accounting for time-dependence, monitoring duration, and baseline risk, we fit a multistate survival model to the time-to-event data, using the composite admission risk score as a baseline covariate. Table 3 reports the resulting diagnostic accuracy, estimated from the proportion of patients predicted to be in each state 20 days after SAH. The plots in Figure 3 show how these values arise from the evolution of state probabilities (Figure 3A–C). Transitions to the EEG alarm state begin around day 2; transitions to DCI begin around day 3. State transitions cease by day 15. In high risk patients (3A,D), the high prevalence of DCI (79% [61–92]) and high positive predictive value (PPV) of EEG alarms (94% [79–100]) arise from a high rate of transitions to the alarm state, and the fact that nearly all patients with alarms subsequently transition to DCI. Conversely, for low-risk patients (3C,F) the low DCI prevalence (37% [25–50]) and lower PPV (76% [58–90]) arise from a lower rate of transitions into the alarm state, and the fact that some patients of low admission risk with EEG alarms never develop DCI. Similar considerations explain the higher negative predictive value (NPV) of EEG alarms for patients with low (94% [87–99]) compared to those with high baseline risk (81% [47–97]).
Table 3.
Performance measures for DCI prediction following SAH using cEEG monitoring, stratified by composite admission risk score
| Low Risk [risk score = 1] | Medium Risk [risk score = 2.5] | High Risk [risk score = 4] | |
|---|---|---|---|
| Sensitivity (%) | 91 [81–98] | 94 [88–99] | 95 [87–99] |
| Specificity (%) | 83 [71–93] | 80 [69–90] | 77 [36–99] |
| LR+ | 5.37 [3.1–13.2] | 4.7 [3.0–9.8] | 4.2 [1.5–66.3] |
| LR− | 0.1 [0.02–0.24] | 0.08 [0.02–0.15] | 0.06 [0.01–0.22] |
| PTP (%) | 37 [25–50] | 58 [50–66] | 79 [61–92] |
| ARI (%) | 39 [26–51] | 29 [21–36] | 15 [4–28] |
| PPV (%) | 76 [58–90] | 87 [79–94] | 94 [79–100] |
| ARR (%) | 32 [20–43] | 48 [39–57] | 60 [30–77] |
| NPV (%) | 94 [87–99] | 90 [81–98] | 81 [47–97] |
| DOR | 52 [16–448] | 57 [24–321] | 69 [8 – 4204] |
| NNM | 2.6 [2.0–3.8] | 3.5 [2.8–4.8] | 6.7 [3.6–25.3] |
Statistical measures given in the table (parameter estimate [Bayesian 95% credible interval]) are determined using multistate survival analysis. Low, medium and high baseline risk levels correspond to composite admission risk scores of 1, 2.5, and 4. ARI = absolute risk increase; ARR = absolute risk reduction; LR = likelihood ratio; DOR = diagnostic odds ratio; NNM = number needed to monitor (1/ARI); NPV = negative predictive value; PPV = positive predictive value; PTP = pre-test probability.
Figure 3. Delayed cerebral ischemia (DCI) probability as a function of postbleed time for patients with low (A,D), medium (B,E) and high (C,F) baseline risk.

(A–C) Multi-state survival model used to quantify the time-dependent risk of transitioning 1) from the baseline state, without DCI and without a cEEG warning (green), to the alarm state, in which there has been a cEEG warning but no DCI (yellow); 2) from the alarm state to DCI following a cEEG warning (pink), thus becoming a true positive case; or 3) from the baseline state to DCI without a preceding cEEG warning (red), thus becoming a false negative case. From the stacked probability plots one can calculate that cEEG has a positive predictive value ranging from 76% in patients with low baseline risk to 94% in patients with high baseline risk. (D–F) Examples of individualized risk curves, derived from the multistate survival model. The probability of subsequent DCI decreases each day following SAH, starting from a point that depends on the composite admission risk score (green curve). Patients for whom the clinical neurophysiologist detects a cEEG alarm are at increased risk for DCI, and move to the yellow probability curve which reflects a higher probability of DCI. Subfigure (d) illustrates a case with no alarm and no DCI in a low risk patient (true positive); (e), a case in a medium-risk patient with an alarm on day 6 but ultimately no DCI (false positive); and (f) a case in a high-risk patient with an alarm on day 6, followed by a DCI on day 8 (true positive). SAH = subarachnoid hemorrhage. DCI = delayed cerebral ischemia. cEEG = continuous EEG monitoring.
The model also assigns a time-dependent risk of DCI, individualized by baseline admission risk and the specific time a cEEG alarm (if any) is reported (Fig 3D–F). Of the low-risk subgroup (composite admission risk score = 1), 0% had an HH score of 4–5 at admission, whereas 86% of the high-risk subgroup (composite admission risk score = 4) had an HH score of 4–5 at admission.
The number needed to monitor to predict one additional case of DCI (NNM) ranged from 2.6 among patients with low admission risk, to 6.7 among those at high risk (Table 3). The lower NNM in low-risk, non-comatose patients reflects the larger change from pre- to post-test probability when an EEG alarm occurs (ARI: 39%) compared with a high-risk patient (ARI: 15%).
The latency from an EEG alarm to DCI in true positive cases ranged from 30 minutes to 9.1 days (median 1.9 days [IQR 0.9–4.1]; Fig 4) and exceeded 12-hours in 82% of cases (n=41).
Figure 4. Latency from EEG alarm to diagnosis of DCI.
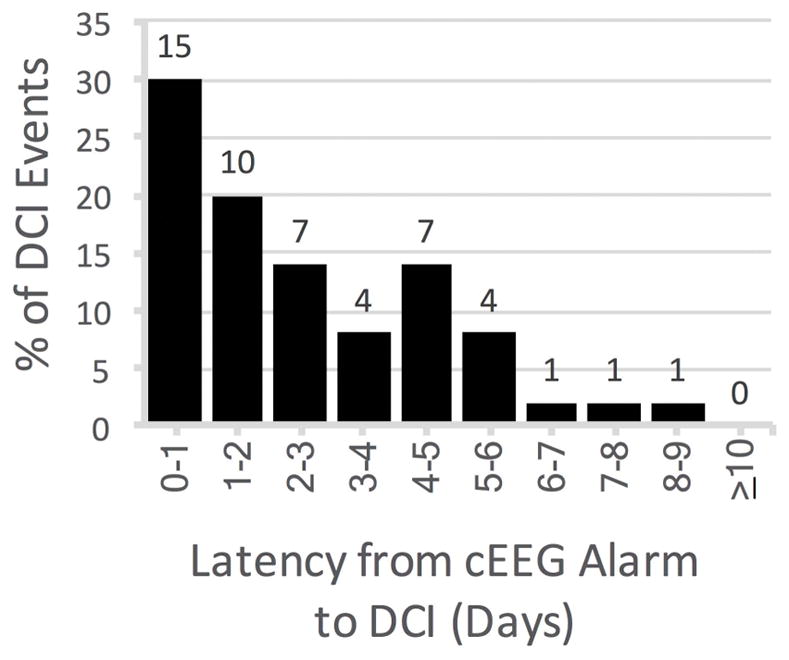
The histogram shows the proportion of the 50 true positive cases (y-axis) occurring within each time latency interval (x-axis) from EEG alarm to DCI. The 2 false negative DCI cases had no EEG alarm and are not shown. The median duration from EEG alarm to DCI was 1.9 days (minimum 30 min, maximum 9.1 days). 41 [82%] of 50 true positive cases had a lead time longer than 12-hours. cEEG = continuous EEG monitoring. DCI = delayed cerebral ischemia.
Clinical presentations for the 52 DCI events varied. Presenting signs occurring in isolation were new or worsening focal weakness [38%; n=20], decline in level of consciousness (LOC) [33%; n=17], or aphasia [8%; n=4]. DCI was silent in four cases, discovered incidentally upon neuroimaging. DCI presented in 7 cases (13%) with combinations of signs, including focal weakness plus either decreased LOC [n=4]; infarction on imaging [n=1]; aphasia [n=1]; or decreased LOC accompanied by a new radiologic infarct [n=1].
Figure 5 categorizes EEG findings preceding DCI among the 50 true positive cases by EEG alarm subtypes and DCI subtypes. While more than one EEG feature of worsening was often present, focal EEG abnormalities were evident for most true-positive DCI predictions, including 18 of 27 [67%] with focal weakness, 17 of 22 [77%] with decline in LOC, all 6 [100%] with radiologic infarction, and 4 [80%] of 5 with aphasia. Among true positive alarms from epileptiform abnormalities (n=33), most [82%; n=27] were in patients without prior epileptiform activity; the rest were due to increasing frequency of epileptiform discharges or periodic patterns (e.g., from 1-Hz to 2-Hz) [n=4; 12%], or increasing prevalence (e.g., from intermittent to continuous) [n=3; 9%]. While status epilepticus prior to enrollment was an exclusion criteria for the study, 1 patient developed status epilepticus during the study period, which was nonconvulsive. In this patient, the last seizure recorded was 5 days before an EEG alarm, and the EEG alarm in this patient was due to background deterioration rather than epileptiform activity.
Figure 5. EEG changes reported before a clinical diagnosis of DCI.
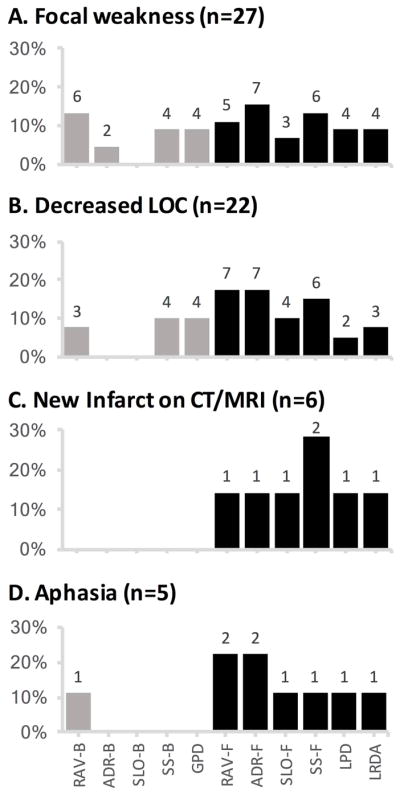
EEG alarms for the 50 true positive predictions of DCI are divided by clinical presentation with (A) focal motor deficits (n=27), (B) decreased level of consciousness (n=22), (C) new infarct on CT or MRI (n=6), or (D) aphasia (n=5). Black bars indicate the percentage of cases with focal EEG abnormalities, including decreases in relative alpha variability (RAV-F) or alpha-delta ratio (ADR-F), new or increased focal slowing (SLO-F), focal sporadic epileptiform spikes or sharp waves (SS-F), and new or worsening lateralized periodic discharges (LPD) or lateralized rhythmic delta activity (LRDA). Gray bars indicate the percentage of cases with cEEG worsening in a bilaterally symmetric distribution, including symmetric decreases in relative alpha variability (RAV-B) or alpha-delta ratio (ADR-B), new or increased generalized slowing (SLO-B), bilaterally synchronous (usually frontally predominant) sporadic epileptiform spikes or sharp waves (SS-B), and new or worsening generalized periodic discharges (GPD). Numbers above the bars indicate the number of DCI subtype events preceded by a given EEG finding. Note that alarms may include multiple EEG abnormalities, thus the numbers above the black and gray bars do not add up to the total number of DCI events with a given presenting sign. Moreover, some DCI events presented with more than one clinical finding, thus the total number of presenting signs is larger than 50.
The two undetected (false negative) DCI events included one patient developing left hemiparesis that was transient but long enough to meet the DCI consensus definition, and one with re-emergence of leg weakness that had been present days prior.
There was no association to suggest that state transitions (baseline to alarm, or alarm to DCI) were due to adding, increasing or decreasing antiseizure medications, anesthetic infusions, or vasopressor infusions (Supplementary Material Table S3). To the contrary, the only apparent association that reached statistical significance was that patients who transitioned from baseline to the alarm state were less likely to have had new antiseizure medications started compared with patients who did not have EEG deterioration (6.67% vs. 23.3%; p=0.02). These results suggest that changes in medications do not cause the baseline to alarm or alarm to DCI state transitions.
Discussion
This prospective study, performed according to STARD criteria, demonstrates that continuous EEG monitoring accurately predicts DCI following SAH. Routine reporting of visually identified cEEG features (new or worsening ADR trends, RAV trends, focal slowing, epileptiform discharges, or rhythmic and periodic patterns) showed excellent sensitivity and high specificity across baseline risk levels. The number of patients needed to monitor to predict one additional case of DCI prior to clinical symptoms was between 3 and 7.
Continuous EEG predicted DCI over 24 hours before clinical symptoms in the majority of cases, prospectively validating findings from previous retrospective studies8,29 and demonstrating the opportunity for cEEG to aid early detection of and rescue from impending secondary brain injury. We speculate that early cEEG findings which predict DCI represent a pre-symptomatic early phase of subclinical ischemia ultimately becoming persistent or severe enough to manifest. One mechanism by which this may occur is through cortical spreading depolarizations, which may manifest in intracranial EEG as transient electrographic suppressions associated with local transient brain tissue hypoxia, ultimately culminating in infarction when spreading depolarizations are repeated over time, cluster, or occur in the setting of vasospasm or hypotension.15,30
An important secondary finding is that EEG predicts DCI with greater sensitivity and specificity than TCD criteria employing absolute velocities, which identify DCI with poor sensitivity,31 late detection,10 and high false positive rates.9 TCD interrogates vascular supply alone, whereas the “supply-demand mismatch model”32 of DCI pathogenesis posits that when increased metabolic demand due to depolarizing events (e.g. spreading depolarization or epileptiform activity) exceeds the blood supply due to ischemia or vascular dysfunction (e.g. large- and small-vessel vasospasm, microthrombosis, inflammation, or inverse neurovascular coupling), a mismatch in neuronal metabolism occurs. This mismatch might cause epileptiform activity to progress towards and limit recovery from spreading depolarizations,30 triggering toxic intracellular changes, additional depolarizing events, and ultimately neuronal death, infarction, and disability.
While using the entire battery of pre-specified EEG features produced the best predictions, late-appearing epileptiform abnormalities were especially strong predictors of DCI. This new finding, together with experience that seizures occur later following SAH than after other neurocritical care illnesses,33 raises the possibility that some DCI cases may reflect seizures detectable by invasive but not scalp electrodes.34 Another consideration is that secondary ischemic brain injury may both cause and be caused by epileptiform activity either via spreading depolarizations or direct metabolic strain.11–14, 35 Biophysical models of ischemia suggest epileptiform discharges may arise from selective ischemic vulnerability of inhibitory neurons, yielding disinhibition of glutaminergic pyramidal cells and a feed-forward cycle of excitation.36 The current study raises the hypothesis that preventing DCI might be possible in some cases by pharmacologically suppressing epileptiform EEG abnormalities or by augmenting cerebral perfusion and oxygen delivery.
EEG changes predicted DCI despite pathology that was often severe at admission (early infarcts, intracerebral hematoma etc.), which can disturb EEG recordings such that detecting further deterioration might seem difficult (i.e., a “floor” effect). Nevertheless, our findings suggest that both low- and high-risk patients do usually first exhibit EEG deterioration prior to developing DCI.
Several limitations apply to our study. First, we were only able to evaluate the diagnostic performance of the index test among patients undergoing sufficient cEEG. However, the composite admission risk score and its sub-scores were not systematically associated with the probability of undergoing the index test itself (Supplementary Table S2). Moreover, we have shown through stratified analysis that EEG deterioration predicted DCI independent of the composite admission risk score. These findings suggest that our results are unlikely to be biased by excluding patients who did not undergo sufficient EEG monitoring. While patients not undergoing the index test had a high rate of undergoing early withdrawal of life-sustaining therapy, this did not occur in those undergoing the index test, possibly due to the requirement for continuous EEG lasting beyond this 3-day period. Second, interventions in response to EEG changes were not standardized. Nevertheless, we found no evidence that state transitions (baseline to alarm, or alarm to DCI) were caused by changes in antiseizure, anesthetic, or vasopressor medications. Third, this was a single center study although our findings are concordant with prior retrospective studies.8, 9 To ensure consistent application across centers in the future, a curriculum and certification test may help ensure uniform implementation, inter-rater agreement, and replication. Fourth, we did not perform routine neuroimaging; imaging was performed at clinicians’ discretion. This limitation is partly balanced by the observation that DCI does not entirely overlap with imaging findings; some patients have clinical worsening without radiologic infarction, potentially due to successful interventions,3 while others have silent radiologic infarction.37 Fifth, we did not routinely perform invasive EEG, and could not detect seizures lacking manifestations in scalp EEG34, 35 or cortical spreading depolarizations,38 which may underlie DCI events. Sixth, the latency from EEG alarm to DCI exceeded 2 days in 50% of cases. Our data do not discriminate between a biological explanation for such prolonged latencies such as an early pre-symptomatic phase of DCI versus a more mundane explanation, delayed bedside recognition of DCI due to coma. Seventh, it is possible that information from cEEG reports increased the vigilance of the neurocritical care team documenting neurologic examinations. However, DCI was not determined by the clinical team; rather DCI was determined by blinded investigators employing strict criteria for focal neurologic changes, persistent GCS decline, and radiologic infarcts. Additionally, most DCI events occurred more than 36 hours after an EEG alarm, suggesting that EEG alarms did not influence exam findings. Similarly, EEG more accurately predicted DCI events than TCD velocities, which were also available to treating clinicians. Sharing of information in the opposite direction through awareness of clinical changes by the clinical neurophysiology team is not a possible source of bias; EEG alarms were required to precede clinical and radiologic DCI events to be considered true positive predictions. Eighth, treating physicians may have prevented DCI in some cases by modifying treatment following EEG alarms. However, our institutional guideline recommended treatment for DCI, not for cEEG alarms in isolation. Additionally, if clinicians did treat cEEG alarms, then the excellent performance statistics we found for cEEG are conservative: interventions following alarms that prevented DCI would have produced false alarms. Finally, while prior literature suggests that DCI triples the odds of poor long-term outcomes,2 we did not independently investigate the impact of DCI detected by cEEG on long-term outcome; rather, our study was limited to assessing the diagnostic accuracy of cEEG for predicting DCI.
Delayed diagnosis of DCI remains a major challenge in SAH management. Evidence supports the concept of hemodynamic or endovascular rescue therapy,3 but clinical efficacy depends critically on early detection. Our data suggest that reliance on serial neurologic exams and vascular studies, the current standard of care, could lead to delays and missed opportunities for medical intervention, while failing to measure additional pathophysiologic mechanisms that result in neuronal injury. Continuous EEG monitoring might enable more timely identification of impending DCI before irreversible injury ensues, focusing clinical interventions and clinical trials on patients at greatest risk of secondary brain injury and poor long-term outcomes.
Supplementary Material
Acknowledgments
We would like to acknowledge funding support from the Andrew David Heitman Neuroendovascular Research Foundation; NIH 1K23NS090900-01; and the Phyllis & Jerome Lyle Rappaport Foundation as well as nurses, technologists, clinical fellows, and faculty who contributed to the study.
Footnotes
Author Contributions:
Study concept and design: ESR, SFZ, MMS, CA, AJC, NG, BBF, EJG, MBW
Data acquisition and analysis: ESR, SFZ, SB1, SB2, EJB, KLO, AVS, TLM, MBW
Drafting the manuscript and figures: ESR, SB1, SFZ, KLO, SB2, AVS, EJB, MMS, EJG, BPF, NG, TML, JR, DBH, CA, SSC, AJC, ABP, MBW
Potential Conflicts of Interest:
The authors have no conflicts of interest to report.
References
- 1.Feigin VL, Rinkel GJ, Algra A, Vermeulen M, van Gijn J. Calcium antagonists for aneurysmal subarachnoid haemorrhage. The Cochrane Database of Systematic Reviews. 2000;(2):CD000277. doi: 10.1002/14651858.CD000277. [DOI] [PubMed] [Google Scholar]
- 2.Dorhout Mees SM, Kerr RS, Rinkel GJ, Algra A, Molyneux AJ. Occurrence and impact of delayed cerebral ischemia after coiling and after clipping in the International Subarachnoid Aneurysm Trial (ISAT) Journal of Neurology. 2012 Apr;259(4):679–83. doi: 10.1007/s00415-011-6243-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diringer MN, Bleck TP, Claude Hemphill J, 3rd, et al. Critical care management of patients following aneurysmal subarachnoid hemorrhage: Recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocritical Care. 2011 Sep;15(2):211–40. doi: 10.1007/s12028-011-9605-9. [DOI] [PubMed] [Google Scholar]
- 4.Macdonald RL, Higashida RT, Keller E, et al. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: a randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2) Lancet Neurol. 2011 Jul;10(7):618–25. doi: 10.1016/S1474-4422(11)70108-9. [DOI] [PubMed] [Google Scholar]
- 5.Nuwer MR, Jordan SE, Ahn SS. Evaluation of stroke using EEG frequency analysis and topographic mapping. Neurology. 1987:37. doi: 10.1212/wnl.37.7.1153. [DOI] [PubMed] [Google Scholar]
- 6.Rots ML, van Putten MJ, Hoedemaekers CW, Horn J. Continuous EEG Monitoring for Early Detection of Delayed Cerebral Ischemia in Subarachnoid Hemorrhage: A Pilot Study. Neurocritical Care. 2015 Oct 2; doi: 10.1007/s12028-015-0205-y. [DOI] [PubMed] [Google Scholar]
- 7.Labar DR, Fisch BJ, Pedley TA, Fink ME, Solomon RA. Quantitative EEG monitoring for patients with subarachnoid hemorrhage. Electroencephalogr Clin Neurophysiol. 1991 May;78(5):325–32. doi: 10.1016/0013-4694(91)90094-k. [DOI] [PubMed] [Google Scholar]
- 8.Vespa PM, Nuwer MR, Juhasz C, et al. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr Clin Neurophysiol. 1997 Dec;103(6):607–15. doi: 10.1016/s0013-4694(97)00071-0. [DOI] [PubMed] [Google Scholar]
- 9.Claassen J, Hirsch LJ, Kreiter KT, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol. 2004 Dec;115(12):2699–710. doi: 10.1016/j.clinph.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 10.Gollwitzer S, Groemer T, Rampp S, et al. Early prediction of delayed cerebral ischemia in subarachnoid hemorrhage based on quantitative EEG: A prospective study in adults. Clin Neurophysiol. 2015 Aug;126(8):1514–23. doi: 10.1016/j.clinph.2014.10.215. [DOI] [PubMed] [Google Scholar]
- 11.Echlin FA, Arnett V, Zoll J. Paroxysmal high voltage discharges from isolated and partially isolated human and animal cerebral cortex. Electroencephalogr Clin Neurophysiol. 1952 May;4(2):147–64. doi: 10.1016/0013-4694(52)90004-7. [DOI] [PubMed] [Google Scholar]
- 12.Chatrian GE, Shaw CM, Luttrell CN. Focal Electroencephalographic Seizure Discharges in Acute Cerebral Infarction. Electrographic, Clinical, and Pathological Observations. Neurology. 1965 Feb;15:123–31. doi: 10.1212/wnl.15.2.123. [DOI] [PubMed] [Google Scholar]
- 13.Hartings JA, Williams AJ, Tortella FC. Occurrence of nonconvulsive seizures, periodic epileptiform discharges, and intermittent rhythmic delta activity in rat focal ischemia. Exp Neurol. 2003 Feb;179(2):139–49. doi: 10.1016/s0014-4886(02)00013-4. [DOI] [PubMed] [Google Scholar]
- 14.Dreier JP, Major S, Pannek HW, et al. Spreading convulsions, spreading depolarization and epileptogenesis in human cerebral cortex. Brain. 2012 Jan;135(Pt 1):259–75. doi: 10.1093/brain/awr303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dreier JP, Fabricius M, Ayata C, et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. Journal of Cerebral Blood Flow and Metabolism. 2016 Jun 17; doi: 10.1177/0271678X16654496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bossuyt PM, Reitsma JB, Bruns DE, et al. STARD 2015: an updated list of essential items for reporting diagnostic accuracy studies. BMJ. 2015;351:h5527. doi: 10.1136/bmj.h5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke. 2010 Oct;41(10):2391–5. doi: 10.1161/STROKEAHA.110.589275. [DOI] [PubMed] [Google Scholar]
- 18.Muniz CF, Shenoy AV, O’Connor KL, et al. Clinical Development and Implementation of an Institutional Guideline for Prospective EEG Monitoring and Reporting of Delayed Cerebral Ischemia. Journal of Clinical Neurophysiology. 2016 Jun;33(3):217–26. doi: 10.1097/WNP.0000000000000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery. 2006 Jul;59(1):21–7. doi: 10.1227/01.neu.0000243277.86222.6c. discussion -7. [DOI] [PubMed] [Google Scholar]
- 20.Echlin FA, Arnett V, Zoll J. Paroxysmal high-voltage and rhythmic low-voltage discharges from isolated and partially isolated human cortex. Archives of Neurology and Psychiatry. 1952 May;67(5):692–3. [PubMed] [Google Scholar]
- 21.Rando T, Ricci D, Mercuri E, et al. Periodic lateralized epileptiform discharges (PLEDs) as early indicator of stroke in full-term newborns. Neuropediatrics. 2000 Aug;31(4):202–5. doi: 10.1055/s-2000-7454. [DOI] [PubMed] [Google Scholar]
- 22.Hirsch LJ, LaRoche SM, Gaspard N, et al. American Clinical Neurophysiology Society’s Standardized Critical Care EEG Terminology: 2012 version. Journal of Clinical Neurophysiology. 2013 Feb;30(1):1–27. doi: 10.1097/WNP.0b013e3182784729. [DOI] [PubMed] [Google Scholar]
- 23.Wickering E, Gaspard N, Zafar S, et al. Automation of Classical QEEG Trending Methods for Early Detection of Delayed Cerebral Ischemia: More Work to Do. Journal of Clinical Neurophysiology. 2016 Jun;33(3):227–34. doi: 10.1097/WNP.0000000000000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogilvy CS, Carter BS. A proposed comprehensive grading system to predict outcome for surgical management of intracranial aneurysms. Neurosurgery. 1998 May;42(5):959–68. doi: 10.1097/00006123-199805000-00001. discussion 68–70. [DOI] [PubMed] [Google Scholar]
- 25.Putter H, Fiocco M, Geskus RB. Tutorial in biostatistics: competing risks and multi-state models. Statistics in Medicine. 2007 May 20;26(11):2389–430. doi: 10.1002/sim.2712. [DOI] [PubMed] [Google Scholar]
- 26.Joly P, Commenges D, Helmer C, Letenneur L. A penalized likelihood approach for an illness-death model with interval-censored data: application to age-specific incidence of dementia. Biostatistics. 2002 Sep;3(3):433–43. doi: 10.1093/biostatistics/3.3.433. [DOI] [PubMed] [Google Scholar]
- 27.Leffondre K, Touraine C, Helmer C, Joly P. Interval-censored time-to-event and competing risk with death: is the illness-death model more accurate than the Cox model? International Journal of Epidemiology. 2013 Aug;42(4):1177–86. doi: 10.1093/ije/dyt126. [DOI] [PubMed] [Google Scholar]
- 28.de Wreede LC, Fiocco M, Putter H. The mstate package for estimation and prediction in non- and semi-parametric multi-state and competing risks models. Computer Methods and Programs in Biomedicine. 2010 Sep;99(3):261–74. doi: 10.1016/j.cmpb.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Kondziella D, Friberg CK, Wellwood I, Reiffurth C, Fabricius M, Dreier JP. Continuous EEG monitoring in aneurysmal subarachnoid hemorrhage: a systematic review. Neurocritical Care. 2015 Jun;22(3):450–61. doi: 10.1007/s12028-014-0068-7. [DOI] [PubMed] [Google Scholar]
- 30.Oka F, Hoffmann U, Lee JH, et al. Requisite ischemia for spreading depolarization occurrence after subarachnoid hemorrhage in rodents. Journal of Cerebral Blood Flow and Metabolism. 2016 Jul 18; doi: 10.1177/0271678X16659303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suarez JI, Qureshi AI, Yahia AB, et al. Symptomatic vasospasm diagnosis after subarachnoid hemorrhage: evaluation of transcranial Doppler ultrasound and cerebral angiography as related to compromised vascular distribution. Crit Care Med. 2002 Jun;30(6):1348–55. doi: 10.1097/00003246-200206000-00035. [DOI] [PubMed] [Google Scholar]
- 32.Foreman B. The Pathophysiology of Delayed Cerebral Ischemia. Journal of Clinical Neurophysiology. 2016 Jun;33(3):174–82. doi: 10.1097/WNP.0000000000000273. [DOI] [PubMed] [Google Scholar]
- 33.O’Connor KL, Westover MB, Phillips MT, et al. High risk for seizures following subarachnoid hemorrhage regardless of referral bias. Neurocritical Care. 2014 Dec;21(3):476–82. doi: 10.1007/s12028-014-9974-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Claassen J, Perotte A, Albers D, et al. Nonconvulsive seizures after subarachnoid hemorrhage: Multimodal detection and outcomes. Ann Neurol. 2013 Jul;74(1):53–64. doi: 10.1002/ana.23859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Struck AF, Westover MB, Hall LT, Deck GM, Cole AJ, Rosenthal ES. Metabolic Correlates of the Ictal-Interictal Continuum: FDG-PET During Continuous EEG. Neurocritical Care. 2016 Jun;24(3):324–31. doi: 10.1007/s12028-016-0245-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Putten MJ, Hofmeijer J. Generalized periodic discharges: Pathophysiology and clinical considerations. Epilepsy & Behavior. 2015 Aug;49:228–33. doi: 10.1016/j.yebeh.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt JM, Wartenberg KE, Fernandez A, et al. Frequency and clinical impact of asymptomatic cerebral infarction due to vasospasm after subarachnoid hemorrhage. J Neurosurg. 2008 Dec;109(6):1052–9. doi: 10.3171/JNS.2008.109.12.1052. [DOI] [PubMed] [Google Scholar]
- 38.Woitzik J, Dreier JP, Hecht N, et al. Delayed cerebral ischemia and spreading depolarization in absence of angiographic vasospasm after subarachnoid hemorrhage. Journal of Cerebral Blood Flow and Metabolism. 2012 Feb;32(2):203–12. doi: 10.1038/jcbfm.2011.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.