

Abstract
Recent technological advancements have permitted high-throughput measurement of the human genome, epigenome, metabolome, transcriptome, and proteome at the population level. We hypothesized that subsets of genes identified from omic studies might have closely related biological functions and thus might interact directly at the network level. Therefore, we conducted an integrative analysis of multi-omic datasets of non-small cell lung cancer (NSCLC) to search for association patterns beyond the genome and transcriptome. A large, complex, and robust gene network containing well-known lung cancer-related genes, including EGFR and TERT, was identified from combined gene lists for lung adenocarcinoma. Members of the hypoxia-inducible factor (HIF) gene family were at the center of this network. Subsequent sequencing of network hub genes within a subset of samples from the Transdisciplinary Research in Cancer of the Lung-International Lung Cancer Consortium (TRICL-ILCCO) consortium revealed a SNP (rs12614710) in EPAS1 associated with NSCLC that reached genome-wide significance (OR = 1.50; 95% CI: 1.31–1.72; p = 7.75 × 10−9). Using imputed data, we found that this SNP remained significant in the entire TRICL-ILCCO consortium (p = .03). Additional functional studies are warranted to better understand interrelationships among genetic polymorphisms, DNA methylation status, and EPAS1 expression.
Keywords: Non-small cell lung cancer, Lung adenocarcinoma, Integrated analysis, Network analysis, Hypoxia-inducible factor
-
•
Previous Evidence
-
•
As a major participant of Transdisciplinary Research in Cancer of the Lung-International Lung Cancer Consortium (TRICL-ILCCO), we continued monitoring the progress of genomic and other omics studies of lung cancer through regular communications within the consortium and literature search using the PubMed database. The TRICL-ILCCO is one of the largest international consortium of lung cancer which includes many major ongoing lung cancer case-control and cohort studies with the aim of sharing comparable data. Although GWAS successfully defined many lung cancer-associated genomic loci, the integrative studies of multi-dimensional high throughput “-omics” measurements from tumor tissues and corresponding blood specimens are limited.
-
•
Added value of this study
-
•
We identified a HIFs-EGFR-HDAC4-TERT network associated with lung adenocarcinoma, and subsequent sequencing of network hub genes identified a new locus within EPAS1 that is associated with lung cancer risk. This locus is in hub gene EPAS1, which is a key member of the HIF family involved in every aspect of cancer development and progression.
-
•
Implications of all the available evidence
-
•
We developed a network building approach for the integrative analysis of multi-omic datasets. The integration of multi-dimensional high throughput “-omics” measurements from tumor tissues and corresponding blood specimens, together with new systems strategies for diagnostics, enables the identification of cancer biomarkers that will facilitate pre-symptomatic diagnosis, stratification of disease, assessment of disease progression, evaluation of patient response to therapy, and identification of recurrences.
1. Introduction
Lung cancer is the leading cause of cancer-related mortality worldwide for both men and women [1]. Although advances in cancer prevention, early detection, and treatment have been made in recent decades, the general prognosis for lung cancer remains poor. The high case–fatality ratio of lung cancer has been attributed to advanced stage of disease at diagnosis, poor response to current therapies, and the aggressive biological nature of lung cancer. Non-small cell lung cancer (NSCLC) is the most common type of lung cancer accounting for about 85% of all lung cancers [2,3]. Histologic subtypes of NSCLC include adenocarcinoma, squamous cell carcinoma, and large cell carcinoma [4]. Further, previous studies have demonstrated that heritable factors are significantly important in lung cancer, independent of smoking history or exposure to environmental tobacco smoke [5,6].
More recently, genome-wide association studies (GWAS) have been used to identify multiple independent loci for most diseases, because GWAS can identify common disease susceptibility loci without prior knowledge of locus function or position [7,8]. Several GWAS have identified at least five loci associated with lung cancer risk—on chromosomes 3q28, 5p15, 6p21, 13q13.1 and 15q25—in populations with European ancestry [[9], [10], [11], [12], [13], [14]]. Additional loci at 22q12 and 15q15 have been associated with lung cancer risk [[15], [16], [17]]. GWAS can define lung cancer-associated genomic loci with low to moderate effects, but cannot identify causal mutations given the complicated relationships among disease-associated loci.
Recent technological advances have permitted high-throughput measurement of the human genome, epigenome, metabolome, transcriptome, and proteome at the population level. Each study can offer complementary analyses of a certain biological function, and integrative multi-omics analyses are needed to uncover synergistic interactions [18]. However, because each omic study analyzes a different molecular layer, integrative analyses comparing top-ranked genes from different omic studies might not reveal much overlapping genes.
We hypothesized that there are subsets of genes identified from different omic studies that might have closely related biological functions and thus might directly interact at the network level. Therefore, it is possible to build network(s) with direct interactions among multiple molecular layers, characterized by higher network complexity and larger gene ratios, where network complexity is defined as the ratio of total number of connections between genes to number of genes within a network, and gene ratio is defined as proportion of genes within a network to total number of genes used to build a network. In addition, incorporating biological functionality from different molecular layers, such as RNA, proteome, and metabolome results, can boost the power of genetic mapping.
In this study, we conducted an integrative analysis of GWAS and transcriptomic profiling for NSCLC using network building based on an algorithm that searches for direct interactions from a high-quality, manually curated database of genetic and physical interactions. To evaluate the identified networks, we repeated network building from a large set of randomly generated gene lists for distributions of network complexity and gene ratio. We also used hub genes identified from significant networks for targeted sequencing and further validation in the Transdisciplinary Research in Cancer of the Lung-International Lung Cancer Consortium (TRICO-ILCCO) GWAS meta-analysis.
2. Materials and Methods
2.1. Study Population
This study was based on data derived from 1000 NSCLC cases and 1000 cancer-free controls, frequency-matched by age (±5 years), gender, and smoking status (by packyears) as previously described [19]. All cases were recruited at Massachusetts General Hospital (MGH) from 1992 to 2004, were > 18 years old, and had newly diagnosed, histologically confirmed primary NSCLC. Controls were healthy, non-blood-related family members and friends of patients with cancer or with cardiothoracic conditions undergoing surgery. Histological classification was done by two staff pulmonary pathologists at MGH according to the International Classification of Diseases for Oncology (ICD-O3). For histology analysis, the following codes were used: adenocarcinoma, 8140/3, 8250/3, 8260/3, 8310/3, 8480/3, and 8560/3; large cell carcinoma, 8012/3 and 8031/3; squamous cell carcinoma, 8070/3, 8071/3, 8072/3, and 8074/3; and other non-small cell carcinomas, 8010/3, 8020/3, 8021/3, 8032/3, and 8230/3. The Institutional Review Board of MGH and the Human Subjects Committee of the Harvard School of Public Health approved the study, and all participants signed consent forms.
2.2. GWAS Dataset
DNA was extracted from peripheral white blood cells using standard protocols and was genotyped using the Human610-Quad BeadChip (Illumina, San Diego, CA). Before association tests, we conducted a systematic quality evaluation of raw genotyping data according to a general quality control (QC) procedure described by Anderson et al. [20] Briefly, unqualified samples were excluded if they fit the following QC criteria: (i) overall genotype completion rates <95%; (ii) gender discrepancies; (iii) unexpected duplicates or probable relatives (based on pairwise identity by state value, PI_HAT in PLINK >0.185); or (iv) heterozygosity rates >6 standard deviations from the mean. Unqualified SNPs were excluded if they fit the following QC criteria: (i) overall genotype completion rates <95%; (ii) gender discrepancies; (iii) unexpected duplicates or probable relatives (based on pairwise identity by state value, PI_HAT in PLINK >0.185); (iv) heterozygosity rates >6 standard deviations from the mean; or (v) individuals were non-Caucasians (using the HapMap release 23, including JPT, CEPH, CEU, and YRI populations as reference). Unqualified SNPs were excluded if they fit the following QC criteria: (i) not mapped on autosomes; (ii) call rate < 95% in all GWAS samples; (iii) MAF < 0.01; or (iv) genotype distributions deviated from those expected by Hardy-Weinberg equilibrium (p < 1.0 × 10−6). After quality evaluation, we had a dataset of 984 cases and 970 controls with 543,697 autosomal SNPs for epistasis analysis.
2.3. Transcriptomic Profiling
FFPE tissues were obtained by surgical biopsy from patients with NSCLC and archived. Histopathologic sections were prepared from tumor and non-affected lung parenchyma tissue by manual microdissection of FFPE blocks. A pathologist who had no knowledge of the study outcome reviewed all tissue sections. Each specimen was evaluated for amount and quality of tumor cells and histologically classified using WHO criteria. Specimens with lower than 70% cancer cellularity were not included for transcriptomic profiling. Sectioned FFPE tissues were sent to Q2 Solutions (formerly Expression Analysis Inc., Morrisville, NC) for RNA extraction, quality assessment, and transcriptomic profiling using whole genome-DASL assay [21]. The Whole-Genome DASL HT assay covered >47,000 annotated transcripts (Illumina, San Diego, CA) [22]. A total of 59 FFPE transcriptomic profiles were obained, including 39 tumor/non-involved tissues from adenocarcinomas, 16 tumor/non-involved tissues from squamous cell carcinomas, and 4 tumor/non-involved tissues from other types of lung carcer. Among them, there were 18 pairs of tumor and matched non-involved tissues of adenocarcinomas and 8 pairs of squamous cell carcinomas, which were used in the transcriptomic analysis.
2.4. External Transcriptomic Data
Two transcriptomic datasets of NSCLC were selected and raw data were downloaded from Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo): GSE10072 and GSE18842. GSE10072 focused on lung adenocarcinoma and included 180 paired adenocarcinoma and non-affected tissue samples [23]. GSE18842 included 91 samples of mainly squamous cell carcinoma [24]. GSE10072 and GSE18842 were generated from fresh snap-frozen samples from surgical resection and profiled on Affymetrix Human Genome U133 array (Affymetrix, Santa Clara, CA).
2.5. Transcriptomic Data Analysis
Although FFPE profiles and external data were generated from different platforms, we used DNA-Chip Analyzer 2006 (dChip, http://www.dchip.org) software, which applied an invariant set of genes for normalization and calculation of expression values across all microarrays, to normalize raw microarray signals. This analysis assumed that a subset of genes had constant expression among all cell subtypes [25] Only paired tumor and non-affected tissue samples were used in the analysis, including 18 FFPE adenocarcinomas, 8 FFPE squamous cell carcinomas, 33 snap-frozen adenocarcinomas (GSE10072), and 32 snap-frozen squamous cell carcinomas (GSE18842).
2.6. Integrated Analysis by Network Building
Gene lists from GWAS and/or transcriptomic profiling were uploaded into MetaCore GeneGo database (https://portal.genego.com, Thomson Reuters, New York, NY) for network building which has >1.7 million molecular interactions, 1600 pathway maps, and 230,000 gene-disease associations [26]. MetaCore is an integrated online software suite for functional analysis of omics data that is based on a high-quality, manually-curated database of molecular interactions, molecular pathways, gene–disease associations, chemical metabolism, and toxicity information. We used direct interaction algorithms on the MetaCore platform to build gene networks consisting only of uploaded genes and their direct interactions, without adding other genes/objects from the GeneGo database. Considering that GWAS and transcriptome were different molecular layers and would not reveal much overlapping genes, we used less stringent criteria to select top-ranked genes from GWAS and transcriptomic profiling without correcting the multiple comparisons.
Significant networks were identified and evaluated by two parameters: network complexity and gene ratio. Network complexity was defined as ratio of total number of connections among genes to number of genes within a network. Gene ratio was defined as proportion of genes within a network to total number of genes used to build a network. These parameters allowed us to distinguish simple networks driven by a few supergenes. In such simple networks, the majority of networked genes only had a single connection to one or several genes, called supergenes. Connections of supergenes usually accounted for the majority of connections within a network, and removing supergenes often dramatically reduced numbers of networked genes or demolished the networks. We also explored different p-value cut offs for the selection of genes from GWAS and transcriptomic profiling. With more stringent cut offs, we could not build a significant network. and with more relaxed cut offs, the network complexities were reduced (data not shown).
2.7. Network Evaluation by Randomly Generated Gene Lists
SNPs of GWAS data and probes of transcriptomic profiling mapped to a total of 24,847 genes. From these genes, we randomly generated 6 sets of gene lists, with each set containing 100 gene lists, for a total of 600 random gene lists. Lists from each set had the same number of genes, but lists from different sets had different numbers of genes—either 50, 100, 200, 300, 400, or 500 genes. Each random gene list was individually uploaded into MetaCore GeneGo database for network building, and network parameters, including network complexity and gene ratio, from the largest networks were recorded. Quantile regression at 95th percentile was performed to estimate the 95% confidence interval.
2.8. Targeted and Whole Exome Sequencing
Targeted and whole exome sequencing was performed at the Center for Inherited Disease Research. Ninety-nine custom regions targeted for a total of 17.26 Mb of custom content was captured and sequenced. BAM files were created by aligning FASTQ files to GRCh37 and joint sample variant calling and variant site filtering was performed. Genotypes for biallelic SNPs were further refined using CalculateGenotypePosteriors and allele frequency information from 1000 genomes phase 3 data as well as the Exome Aggregation Consortium data. Further details were described in Supplementary Methods.
2.9. Statistical Analysis
Clinical characteristics were described as mean ± standard deviation (SD) for continuous variables or n (%) for categorical variables. Student's t-test or Fisher's exact test was used for comparison between groups for continuous or categorical variables, respectively.
We carried out gene-based analysis on GWAS data and targeted sequencing data using SKAT-O [27]. SKAT-O aggregates weighted variance-component score statistics for each SNP/SNV within a set using a kernel function and tests for associations between groups of SNPs/SNVs and a phenotype while adjusting for relevant covariates [28]. For GWAS data, initially all SNPs that passed QC were mapped to human genes within ±20 kb regions based on information curated in the RefSeq database(NCBI build GRCh37.p13). Separate analyses were conducted individually for all SNPs and rare SNVs with MAF <0.01. Models were adjusted for age (years), gender, smoking status, and top significant eigenvectors. SKAT-O analyses were carried out using the SKAT package (R v. 2.13.0).
In meta-analysis of GWAS datasets within TRICL-ILCCO, we combined imputed genotypes from 13,479 lung cancer cases and 43,218 controls undertaken by the previous TRICL-ILCCO GWAS [22,29,30]. We excluded poorly imputed SNPs defined by imputation quality Rsq <0.3 or Info <0.4 for each study and conducted fixed effects meta-analysis with inverse variance weighting and random effects meta-analysis from the DerSimonian-Laird method [31]. We also generated an index of heterogeneity (I2) and p-value of Cochran's Q statistic to assess heterogeneity in meta-analyses. We only considered SNPs with MAF >0.005 and that showed little evidence for effect heterogeneity between studies (Cochran's Q statistic p > .05). All meta-analyses and calculations were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA).
3. Results
3.1. Initial Screening of GWAS and Transcriptomic Profiling Data
We produced a genomic dataset of germline polymorphisms (GWAS genotyping data on the Illumina 610 Quad platform) and a transcriptomic dataset of tumor and non-affected tissue (genome-wide expression profiling on Illumina DASL HT platform) from NSCLC samples [29]. The GWAS dataset included 543,697 single nucleotide polymorphisms (SNPs) from 984 NSCLC cases and 970 healthy controls after quality assessment. Transcriptomic profiling was carried out on formalin-fixed paraffin-embedded (FFPE) paired samples of tumor and non-affected tissues from 30 NSCLC cases, including 18 adenocarcinoma, 8 squamous cell carcinoma, and 2 unclassified cases. Eighteen NSCLC cases had both GWAS and transcriptomic data. Patient characteristics are described in Table 1.
Table 1.
Demographic distribution of study populations.
|
GWAS samples |
FFPE samples |
|||
|---|---|---|---|---|
| Case (n = 984) | Control (n = 970) | p | Case (n = 28) | |
| Age (years) | 65.5 ± 10.6 | 59.4 ± 11.6 | <0.001 | 67.1 ± 9.6 |
| ≥65 | 540 (54.9%) | 351 (36.2%) | <0.001 | 20 (71.4%) |
| Sex (female) | 477 (48.5%) | 528 (54.4%) | 0.010 | 12 (42.9%) |
| Smoking pack-years | 49.7 ± 35.7 | 25.0 ± 26.7 | <0.001 | 53.3 ± 34.8 |
| ≥30 | 664 (55.1%) | 326 (33.6%) | <0.001 | 22 (78.6%) |
| Smoking status | <0.001 | |||
| Never | 92 (9.4%) | 161 (16.6%) | 2 (7.1%) | |
| Former | 502 (51.0%) | 555 (57.2%) | 18 (64.3%) | |
| Current | 390 (39.6%) | 254 (26.2%) | 8 (28.6%) | |
| Pathology | ||||
| Adenocarcinomaa | 597 (60.7%) | 18 (64.3%) | ||
| Squamous cell | 216 (22.0%) | 7 (25.0%) | ||
| Small cell | 0 (0.0%) | 0 (0.0%) | ||
| Other | 171 (17.3%) | 3 (10.7%) | ||
Including adenocarcinoma in situ.
Instead of analyzing individual SNP, we conducted gene-based analysis of GWAS data using the optimal unified sequence kernel association test (SKAT-O) method and applied gene lists in the subsequent network analysis [32]. Among 21,981 mapped genes of GWAS data, there were 103 genes with p < .005 (Supplementary Table S1), 232 genes with p < .01, and 1007 genes with p < .05. The top genes associated with risk of developing NSCLC were HYKK (also known as AGPHD1, 15q25, p = 2.30 × 10−6), CLPTM1L (5p15, p = 3.54 × 10−5), CHRNA3 (15q25, p = 6.77 × 10−5), and DNAJC16 (1p36.1, p = 7.12 × 10−5), with 3 genes located within the two previously identified risk loci at 5p15 and 15q25 [29].
We also screened transcriptomic data for differentially expressed genes with >2 fold changes (FC) between tumor and non-affected lung tissues of 18 pairs of FFPE samples (lung adenocarcinoma, including bronchioloalveolar carcinoma) obtained by surgical biopsy. Among 20,818 genes (29,378 probesets total), there were 75 genes with p < .001 (Supplementary Table S2), 252 genes with p < .005, 402 genes with p < .01, and 805 genes with p < .05. Top differentially expressed genes were PTPRB (p = 8.65 × 10−7), SEMA6A (p = 2.03 × 10−6), and PION (p = 4.83 × 10−6).
We compared gene lists from GWAS analysis and transcriptomic profiling and identified 46 common genes with p < .05 in both analyses, which we called core genes (Table 2). Except for SEMA6A [GWAS: p = .004; transcriptome: FC = −4.4 (tumor/non-affected tissue), p = 2.03 × 10−6] and MYLK (GWAS: p = .009; transcriptome: FC = −3.5, p = 3.00 × 10−4), most genes were low-ranked in either GWAS or transcriptomic profiling but high-ranked in the other analysis.
Table 2.
Common genes between GWAS and transcriptomic profiling (p < .05).
| Gene | Chr | Gene ID | Description | Transcriptomea |
GWAS |
||
|---|---|---|---|---|---|---|---|
| Fold change | p | SNP | p | ||||
| ALDH1A1 | 9 | 216 | Aldehyde dehydrogenase 1 family, member A1 | −2.1 | 0.01294 | 29 | 0.02264 |
| ANGPTL2 | 9 | 23,452 | Angiopoietin-like 2 | −2.1 | 0.01364 | 5 | 0.00878 |
| BBS4 | 15 | 585 | Bardet-Biedl syndrome 4 | −2.1 | 0.01240 | 5 | 0.04792 |
| C1orf54 | 1 | 79,630 | Chromosome 1 open reading frame 54 | −2.2 | 0.01047 | 3 | 0.03904 |
| CANX | 5 | 821 | Calnexin | −2.5 | 0.00366 | 1 | 0.04936 |
| CCDC144A | 17 | 9720 | Coiled-coil domain containing 144A | −2.0 | 0.02563 | 1 | 0.04716 |
| CLDN18 | 3 | 51,208 | Claudin 18 | −2.0 | 0.00403 | 7 | 0.03534 |
| CRYZL1 | 21 | 9946 | Crystallin, zeta (quinone reductase)-like 1 | −2.3 | 0.03630 | 3 | 0.03622 |
| CTSS | 1 | 1520 | Cathepsin S | −2.2 | 0.01058 | 2 | 0.00318 |
| DDI2 | 1 | 84,301 | DDI1, DNA-damage inducible 1, homolog 2 (S. cerevisiae) | −2.4 | 0.00707 | 2 | 0.00074 |
| DGKH | 13 | 160,851 | Diacylglycerol kinase, eta | −2.5 | 0.01353 | 71 | 0.04530 |
| DSG2 | 18 | 1829 | Desmoglein 2 | −2.3 | 0.00147 | 12 | 0.01370 |
| EGFR | 7 | 1956 | Epidermal growth factor receptor [erythroblastic leukemia viral (v-erb-b) oncogene homolog, avian) | −2.4 | 0.01896 | 1 | 0.01781 |
| EPAS1 | 2 | 2034 | Endothelial PAS domain protein 1 | −2.6 | 0.04683 | 35 | 0.00262 |
| FOS | 14 | 2353 | V-fos FBJ murine osteosarcoma viral oncogene homolog | −2.9 | 0.02894 | 2 | 0.03475 |
| GPR4 | 19 | 2828 | G protein-coupled receptor 4 | −2.2 | 0.02893 | 5 | 0.01226 |
| GYPC | 2 | 2995 | Glycophorin C (Gerbich blood group) | −2.9 | 0.00198 | 23 | 0.03402 |
| HIF1A | 14 | 3091 | Hypoxia-inducible factor 1, alpha subunit (basic helix-loop-helix transcription factor) | −2.0 | 0.01108 | 8 | 0.00307 |
| HIST1H1A | 6 | 3024 | Histone cluster 1, H1a | 2.1 | 0.00948 | 1 | 0.01148 |
| HSDL1 | 16 | 83,693 | Hydroxysteroid dehydrogenase like 1 | 2.1 | 0.02955 | 1 | 0.01549 |
| IFT81 | 12 | 28,981 | Intraflagellar transport 81 homolog (Chlamydomonas) | −2.2 | 0.04329 | 6 | 0.02961 |
| KIAA1407 | 3 | 57,577 | KIAA1407 | −3.1 | 0.01488 | 6 | 0.00387 |
| LEPR | 1 | 3953 | Leptin receptor | −2.0 | 0.02291 | 1 | 0.03057 |
| LYVE1 | 11 | 10,894 | Lymphatic vessel endothelial hyaluronan receptor 1 | −2.5 | 0.02175 | 2 | 0.02959 |
| MED31 | 17 | 51,003 | Mediator complex subunit 31 | −2.6 | 0.01772 | 1 | 0.02361 |
| MS4A6A | 11 | 64,231 | Membrane-spanning 4-domains, subfamily A, member 6A | −2.0 | 0.04791 | 4 | 0.00202 |
| MYLK | 3 | 4638 | Myosin, light chain kinase | −3.5 | 0.00032 | 1 | 0.00935 |
| NAMPT | 7 | 10,135 | nicotinamide phosphoribosyltransferase | −2.1 | 0.01653 | 5 | 0.00979 |
| NUP50 | 22 | 10,762 | Nucleoporin 50 kDa | −2.6 | 0.00218 | 4 | 0.03813 |
| PAAF1 | 11 | 80,227 | Proteasomal ATPase-associated factor 1 | −2.0 | 0.00515 | 8 | 0.00595 |
| PACRG | 6 | 135,138 | PARK2 co-regulated | −2.0 | 0.00203 | 1 | 0.04082 |
| PARP1 | 1 | 142 | Poly (ADP-ribose) polymerase family, member 1 | 2.2 | 0.01837 | 8 | 0.02273 |
| PDCD2 | 6 | 5134 | Programmed cell death 2 | −2.3 | 0.03616 | 3 | 0.04564 |
| PLEKHB1 | 11 | 58,473 | Pleckstrin homology domain containing, family B (evectins) member 1 | −2.5 | 0.00524 | 5 | 0.04509 |
| PRKAG2 | 7 | 51,422 | Protein kinase, AMP-activated, gamma 2 non-catalytic subunit | −2.3 | 0.02595 | 1 | 0.03415 |
| PRKCQ | 10 | 5588 | Protein kinase C, theta | −2.3 | 0.00363 | 83 | 0.02882 |
| RBMS1 | 2 | 5937 | RNA binding motif, single stranded interacting protein 1 | −2.3 | 0.00746 | 26 | 0.01284 |
| RRM2B | 8 | 50,484 | Ribonucleotide reductase M2 B (TP53 inducible) | −2.0 | 0.01162 | 10 | 0.02509 |
| S1PR5 | 19 | 53,637 | Sphingosine-1-phosphate receptor 5 | −2.0 | 0.00713 | 2 | 0.01977 |
| SEMA6A | 5 | 57,556 | Sema domain, transmembrane domain, and cytoplasmic domain, (semaphorin) 6A | −4.4 | 2.03E-06 | 60 | 0.00432 |
| SMG1 | 16 | 23,049 | PI-3-kinase-related kinase SMG-1 | −2.1 | 0.01381 | 11 | 0.00747 |
| TACC3 | 4 | 10,460 | Transforming, acidic coiled-coil containing protein 3 | 2.2 | 0.03279 | 5 | 0.02719 |
| TCIRG1 | 11 | 10,312 | T-cell, immune regulator 1, ATPase, H+ transporting, lysosomal V0 subunit A3 | −2.7 | 0.02007 | 1 | 0.02523 |
| ZDHHC19 | 3 | 131,540 | Zinc finger, DHHC-type containing 19 | −2.6 | 0.01204 | 8 | 0.04905 |
| ZNF274 | 19 | 10,782 | Zinc finger protein 274 | −3.1 | 0.00116 | 7 | 0.04410 |
| ZRANB2 | 1 | 9406 | Zinc finger, RAN-binding domain containing 2 | −2.3 | 0.00502 | 4 | 0.00498 |
Fold change indicates difference between tumor tissue and non-affected adjacent tissue. P-values are from paired t-tests.
3.2. Networks Built between GWAS and Transcriptomic Profiling Data
No significant network could be built from individual or combined lists of top-ranked genes from GWAS or transcriptomic profiling. Although no significant network could be built from the list of core genes (n = 46), small and simple networks could be built when core genes were combined with either GWAS or transcriptomic profiling data. Further, by combining top-ranked GWAS genes (103 genes with p < .005), top-ranked transcriptomic profiling genes (75 genes with p < .001), and core genes (37 non-overlapped genes out of 46 core genes), we could build a single large complex network (Fig. 1a). The process of integrated analysis is summarized in Supplementary Fig. S1.
Fig. 1.
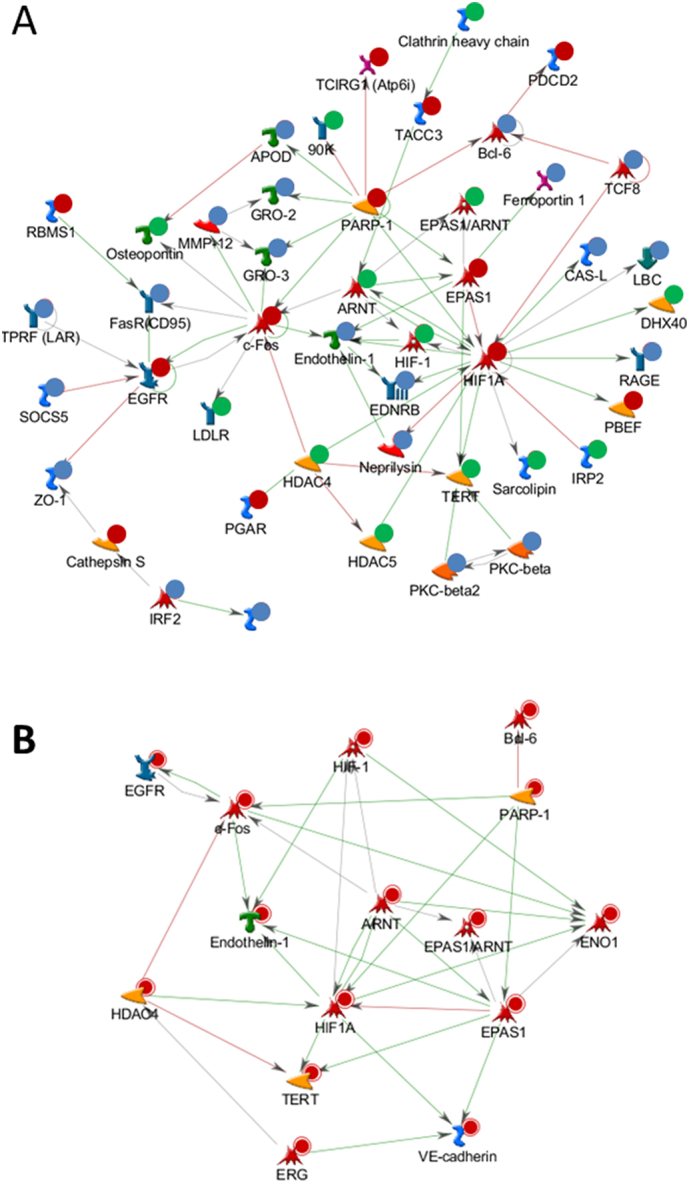
Networks built from GWAS and transcriptomic profiling data. a, Significant network built from combining top-ranked GWAS genes (103 genes with p < .005), top-ranked transcriptomic profiling genes (75 genes with p < .001), and a core list of genes (37 non-overlapping genes out of 46 core genes). Blue indicates genes only identified from transcriptomic profiling; green indicates genes only identified from GWAS; and red indicates genes from the core list. b, Network of 13 hub genes (≥5 connections within the network).
Thirteen hub genes (≥5 connections within the network) that significantly contributed to complexity of the network were identified and were related to lung cancer, including FOS, EGFR, HDAC4, and TERT (Fig. 1b). Moreover, the network was centered on important genes belonging to the hypoxia-inducible factor (HIF) family, including hub genes HIF1A, ARNT (also known as HIF1B), and EPAS1 (also known as HIF2A), which are transcription factors that respond to changes in available oxygen in the cellular environment [33]. We therefore named this the HIFs-EGFR-HDAC4-TERT network. Similarly, using top-ranked genes from a GWAS dataset containing only lung adenocarcinomas (597 cases and 970 controls), we built a significant network from a combined list of GWAS genes (90 genes with p < .005), transcriptomic profiling genes (75 genes with p < .001), and core genes.
3.3. Network Evaluation by Random Gene Lists
To examine the possibility that significant networks were formed by random chance of the increased number of genes from combined gene lists, we conducted a series of network analyses using randomly generated lists with different numbers of genes (total list: n = 600) and evaluated the largest network built from each random list. As the number of genes for network building increased, we more frequently observed an increased number of genes and connections within networks, resulting in increased gene ratios. However, the complexity of these networks remained relatively unchanged (data not shown).
Networks were better evaluated in a two-dimensional space of network complexity and gene ratio than any individual parameter we tested. As shown in Fig. 2, the large complex network built from combined gene lists was located far above from a 95% upper-tail conference interval (95% CI) curve, whereas most networks from individual gene lists or combinations of any two lists were located either under or around the 95% CI curve. A similar result was observed for analysis of top-ranked genes of GWAS including only lung adenocarcinomas (data not shown).
Fig. 2.

Distribution of networks built by randomly selected genes. From a total of 24,847 genes, we randomly generated 6 sets of gene lists, with each set containing 100 gene lists. Each set contained 50, 100, 200, 300, 400, or 500 genes, denoted by R50, R100, R200, R300, R400, and R500, respectively. Networks were built from each gene list using MetaCore GeneGo database and were used to calculate a 95% CI curve. GWAS denotes network from GWAS data; FFPE denotes network from transcriptomic profiling data; and GWAS+FFPE+core denotes networks built from combining GWAS data, transcriptomic profiling data, and a core gene list.
We further examined larger gene lists of GWAS and transcriptomic profiling data with a lower p-value cut-off. Networks for individual gene lists were all under the 95% CI curve for both GWAS (gene list: 232 genes, p < .01; network: complexity = 1.36, gene ratio = 0.10) and transcriptomic profiling (gene list: 252 genes, p < .005; network: complexity = 1.30, gene ratio = 0.08) data, and the network for a combination of two lists was just above the 95% CI curve (network: complexity = 1.38, gene ratio = 0.24). Among points outside the 95% CI, the results for the hypoxia network we identified had the highest gene ratio.
3.4. Network Validation by External Transcriptomic Data
An external transcriptomic dataset (GSE10072) was generated on a different platform (Affymetrix Human Genome U133A array) using 33 fresh-frozen pairs of tumor and non-affected tissues from NSCLC adenocarcinomas collected in the Lombardy region of Italy [23]. We identified 85 top-ranked, differentially expressed genes (FC ≥ 2; p < 10−16). Similar to the FFPE dataset, a significant network could only be built from combined lists of top-ranked GWAS genes, top-ranked transcriptomic profiling genes, and a core list of 29 non-overlapping genes (Supplementary Fig. S2).
This network had a complexity and gene ratio comparable with that from the FFPE dataset and also had common hub genes (≥5 connections within the network) shared with the FFPE dataset (Table 3). Moreover, we identified 88 common, differentially expressed genes (FC ≥ 2; p < .05) between FFPE and GSE10072 datasets, with 85 genes (97%) having expression changes in the same direction between tumor and non-affected tissues (Supplementary Table S3). Although only a simple and small network could be built from this common transcriptomic list, a significant network with the same hub genes, including HIF1A, FOS, HDAC4, and EDN1, could be built by direct combination with the GWAS list (Table 3).
Table 3.
Network evaluations.
| Network 1 | Network 2 | Network 3 | Network 4 | |
|---|---|---|---|---|
| Total genes | 215 | 198 | 217 | 191 |
| Genomic list | GWAS | GWAS-AC | GWAS | GWAS |
| Transcriptomic list | FFPE | FFPE | GSE10072 | Common of FFPE & GSE10072 |
| Core list | 37 | 33 | 29 | − |
| Largest network | ||||
| Genes | 46 | 43 | 46 | 40 |
| Connections | 70 | 62 | 69 | 58 |
| Complexity | 1.556 | 1.476 | 1.533 | 1.487 |
| Gene ratio | 0.214 | 0.217 | 0.212 | 0.209 |
| Hub gene of largest network | ||||
| ARNT | + | − | + | − |
| BCL6 | − | + | − | − |
| CDH5 | − | − | + | − |
| EDN1 | + | + | − | + |
| EGFR | + | + | − | − |
| ENO1 | − | − | + | − |
| EPAS1 | + | + | + | − |
| ERG | − | − | + | − |
| FOS | + | − | + | + |
| HDAC4 | + | + | + | + |
| HIF1A | + | + | + | + |
| MMP12 | − | − | − | + |
| PARP1 | + | + | − | − |
| TERT | + | + | − | − |
3.5. Hub Gene Validation by Sequencing and Meta-Analysis of Genotyping Data
As a member of the TRICL-ILCCO consortium, we submitted 13 hub genes as candidates for next-generation targeted and whole exome sequencing [34], which included 1059 NSCLC cases and 900 unrelated controls genetically enriched with young-onset or positive lung cancer family history from four sites (Supplementary Table S4): Harvard School of Public Health, International Agency for Research on Cancer, University of Liverpool, and Mount Sinai Hospital-Princess Margaret Hospital study in Toronto, Canada. Using the SKAT-O method to test combined effects of all common and rare single nucleotide variants (SNVs) within one gene [32], we found that EPAS1 (p = .0009) was significantly associated with NSCLC after adjusting for multiple comparisons by Bonferroni method (p = .05/13 genes = 0.0038). Further, there was no significant aggregation of variants with moderate to high functional impacts (http://useast.ensembl.org/info/genome/variation/predicted_data.html) in either NSCLC cases or controls in the other 12 hub genes (Supplementary Table S5). In EPAS1, sequencing identified 2061 SNVs, including 1617 rare/low-frequency SNVs [minor allele frequency (MAF) ≤ 0.01] and 36 SNVs with moderate–high functional impact. One common SNV (SNP: rs12614710) located within the first intron and identified by sequencing reached genome-wide significance (MAF = 0.45; OR = 1.50; 95% CI: 1.31–1.72; p = 7.75 × 10−9) (Fig. 3).
Fig. 3.

Manhattan plot of EPAS1 in targeted sequencing project.
We further validated SNP rs12614710 in a much larger GWAS dataset using meta-analysis. A fixed-effect model was applied to estimate pooled effects of each SNP using the TRICL-ILCCO GWAS dataset, which included 13,479 lung cancer cases and 43,218 controls (Supplementary Table S6) [34]. Meta-analysis of SNP rs12614710 had a p-value of 0.03 (imputation accuracy: R2 = 0.86).
4. Discussion
In this study, we conducted an integrative analysis of multi-omic datasets of NSCLC to assess associations beyond the genome and transcriptome. A large, complex, and robust gene network containing well-known lung cancer-related genes, including EGFR and TERT, was identified for lung adenocarcinoma from the combined gene lists. However, the framework of this network was built by key members of the HIF gene family. Subsequent sequencing of network hub genes within a subset of consortium samples revealed a SNP (rs12614710) in EPAS1 associated with NSCLC that reached genome-wide significance based on whole exome sequencing data. Although this SNP was not covered in any GWAS dataset, we used imputed data to find that this SNP is borderline significant in the entire TRICL-ILCCO GWAS dataset. This discrepancy could be due to differential associations among genetically enriched individuals as those in the whole exome sequencing project.
HIFs are a family of proteins that sense and respond to oxygen deficiency by acting as heterodimeric transcription factors that regulate expression of multiple genes involved in the adaptation and progression of cancer. Hypoxia is a typical cancer microenvironment, particularly in rapidly growing tumors, and activation of HIFs is the first step of tumor cells' adaptive responses to hypoxic surroundings [33]. HIFs are involved in every aspect of cancer development and progression, including cell proliferation, apoptosis, metabolism, immune responses, genomic instability, vascularization, invasion, and metastasis.
HIFs consist of two subunits: an oxygen-sensitive α subunit, including HIF-1α (HIF1A), HIF-2α (EPAS1 or HIF2A), and HIF-3α (HIF3A) isoforms; and a ubiquitously expressed β subunit (HIF1B or ARNT). Hypoxic conditions result in HIF-α stabilization, nuclear translocation, and dimerization with HIF-1β to form the HIF transcription factor, which can bind to hypoxia-response elements (A/GCGTG consensus motif) in numerous target gene promoter regions [35].
HIFs are attractive therapeutic targets in cancer [33]. HIF-1α and HIF-2α are the predominant regulators of hypoxic responses at both cellular and organismal levels. Although they share highly conserved structural features, each isoform mediates a unique set of target genes and even oppositely influences some critical factors, such as c-Myc, p53, and nitric oxide [36]. Expression of HIF-2α has been identified in human lung cells, including type II pneumocytes and pulmonary endothelial cells, in response to hypoxia, as well as in epithelium and mesenchymal structures that give rise to the vascular endothelium [37]. Additional studies report that HIF-2α plays a vital role in malignant behavior. In murine models of lung cancer, increased tumor size, invasion, and angiogenesis correlate with high levels of HIF-2α expression cooperating with RAS [38]. Further, high levels of HIF-2α in NSCLC tumor tissue are associated with significantly poor patient prognosis [39,40]. However, in our transcriptomic data from FFPE samples, tumor tissue had low EPAS1 expression compared with non-affected tissue. A similar low EPAS1 expression profile was also observed in fresh-frozen samples.
The most significant SNP (rs12614710, p = 7.75 × 10−9) of EPAS1 identified from sequencing was located in the first intron, and several adjacent SNPs within this intron had p-values of 10−5–10−7. Previous studies have reported associations of EPAS1 polymorphisms with development of renal cell carcinoma (rs11894252, p = 1.8 × 10−8; rs9679290, p = 5.75 × 10−8; rs4953346, p = 4.09 × 10−14) and prostate cancer [[40], [41], [42]]. In a small study of 346 NSCLC patients and 247 controls from a Japanese population, SNP rs4953354 was associated with increased risk of lung adenocarcinoma (OR = 1.80; 95% CI, 1.16–2.79; p = .008) [42]. In our TRICL-ILCCO GWAS dataset of 13,479 cases and 43,218 controls [34], all of these SNPs were significantly associated with NSCLC (rs11894252, p = .043; rs9679290, p = .0011; rs4953346, p = .0015; rs4953354, p = .025). All previously reported SNPs are located in the first intron, except for rs4953354, which is located in the third intron. In addition, some SNPs are not correlated [40], including rs12614710 from our sequencing project.
Moreover, bioinformatic analyses using Genome Browser (http://genome.ucsc.edu) suggest that most of these first intron SNPs are located in histone mark H3K27Ac, which is defined by a ChIP-seq assay related to enhanced gene transcription [43]. Further, analysis of ChIP-seq datasets from ENCODE identified binding sites and binding activities for C/EBP-β, AP-1, and MYC families of transcription factors in many cancer cell types within the first intron of EPAS1. Further, the A allele of rs13419896 is associated with enhanced EPAS1 expression and poor prognosis of 76 NSCLC patients [44]. It is likely that genetic polymorphism of EPAS1 may lead to varied gene expression through either changes in binding sites and binding activities for certain transcription factors or modification of histone epigenetic regulation. In a study of chronic obstructive pulmonary disease, hypermethylation of EPAS1 is correlated with decreased EPAS1 expression and is significantly associated with disease severity [45].
Although GWAS has provided useful insights into the genetic architecture of complex diseases, there is weak evidence for how GWAS findings improve understanding of molecular pathways involved in disease, thus bringing post-GWAS challenges to the characterization of molecular data. Therefore, it is important to assess how diverse omic datasets at different biological levels can be integrated to exploit the full potential of information to identify causal genes and networks, regulatory genes and networks, and predictive markers for complex traits. Using direct interaction algorithms for network building, we successfully conducted an integrated study of multi-omic data for exploration beyond GWAS. This approach implemented a stringent criterion of only searching for direct gene–gene interactions within a manually curated database (MetaCore, https://portal.genego.com), while using less strict p-value cut-offs to select gene lists from different omic datasets. Thus, we could explore less significant genes, which often do not reach genome-wide significance, in omic datasets. The underlying hypothesis is that, because genes are highly networked and coordinated and do not act alone, polymorphisms of several genes in one biological process might not reach genome-wide significance.
Initially, we could not find any meaningful networks (with relatively higher complexities and gene ratios) from gene lists selected from single omic datasets. This result is reasonable in that a list of top genes with small p-values is more likely to contain discrete genes, especially for transcriptomic data, as differentially large expressions were often downstream target genes in a transcription pathway and might not have direct interactions. Only combined gene lists from both GWAS and transcriptomic datasets plus a core list of common genes could build a large, complex HIFs-EGFR-HDAC4-TERT network. However, with even less stringent p-value cut-offs, we built a large network from genes selected from a single omic dataset. All such networks were simple, with the majority of networked genes only having a single connection to one or several hub genes, called supergenes. Connections of supergenes usually accounted for the majority of connections within a network, and removing supergenes often dramatically reduced numbers of networked genes or demolished the networks.
To distinguish from supergene networks, we evaluated the HIFs-EGFR-HDAC4-TERT network by investigating networks built from 600 randomly selected gene lists of different sizes. We found that a gene network had to be evaluated by two factors: size and complexity. Network size was measured by gene ratio of number of networked genes to number of total genes used to build a network. Network complexity was measured by the ratio of total number of network connections to total number of networked genes. A supergene network always was large in size but low in complexity. The HIFs-EGFR-HDAC4-TERT network had a moderate size but high complexity without supergenes. From the distribution of network properties of 600 randomly selected gene lists plotted for network size against network complexity, the HIFs-EGFR-HDAC4-TERT network was above the 95% CI curve, suggesting that this network was not randomly built.
We further validated the HIFs-EGFR-HDAC4-TERT network generated from multi-omic datasets by using different transcriptomic datasets. The GSE10072 dataset was from a study of gene expression signatures of cigarette smoking and its role in lung adenocarcinoma development and survival, and it contained 33 fresh-frozen pairs of tumor and non-affected tissues from NSCLC adenocarcinomas [23]. Combined with our GWAS dataset, which contained a majority of NSCLC adenocarcinomas, and a core gene list, we also built a network with network size and complexity comparable to the HIFs-EGFR-HDAC4-TERT network and several shared hub genes. Different combinations of the GWAS dataset, transcriptomic dataset, and core gene list provided similar results.
We also analyzed a transcriptomic dataset of squamous cell carcinoma, including our 8 pairs of tumor and non-affected tissues from FFPE samples and 32 pairs of fresh-frozen samples from GSE18842 [24], and found that no complex networks except several supergene networks could be built (data not shown). This might be because a majority of our GWAS samples were lung adenocarcinomas (60.7%), and squamous cell carcinomas only accounted for ~22% of samples. Meanwhile, no identified network from squamous cell GWAS transcriptomic datasets suggests that our integrated approach of multi-omic data was sensitive to tumor histology.
Sequencing of hub genes identified a new locus in EPAS1 that reached genome-wide significance and was validated in the largest lung cancer consortium, providing additional evidence that the HIFs-EGFR-HDAC4-TERT network is associated with NSCLC adenocarcinoma. Further, a recent study reported that EPAS1 could specifically bind to tyrosine kinase inhibitor (TKI)-resistant T790 M EGFR in NSCLC cell lines and enhance amplification of MET [46]. These findings suggest that EPAS1 is a key factor in EGFR-MET crosstalk in conferring TKI resistance in NSCLC cases and provide in vitro support of the HIFs-EGFR-HDAC4-TERT network.
At present, FFPE samples, which represent the greatest stock of archived disease entities, are limited mainly to investigations of a small number of genes using quantitative real-time PCR or global micro-RNA profiling, which is much more stable than mRNA [47,48]. The main reason for this restriction is that RNA is often altered and degraded within FFPE samples from the impact of collection and storage [49,50]. In our study, from the top 808 differentially expressed genes from FFPE samples, we identified 88 common, differentially expressed genes between FFPE and the GSE10072 dataset, with 85 genes (97%) having expression changes in the same direction between tumor and non-affected tissues. This common gene list could be used as a transcriptomic list to build a network containing HIFs without incorporating a core gene list. These results demonstrate that FFPE samples could generate a transcriptomic profile for integrated analysis, as we found similar networks with shared hub genes as compared to fresh-frozen samples.
During the analyses, we evaluated different network results by exploring different p-value cut offs for the selection of genes from different omics datasets. We didn't find any significant networks as all networks were under the 95% CI curve in random gene list evaluation. We noticed there was a limitation that we hadn't control this level of multiple comparisons. Currently, we didn't have a proper analytic method to adjust the multiple comparisons. Therefore, we used external data to evaluate the network results.
Through integrated analysis and subsequent sequencing of the identified network, we identified a new locus associated with lung cancer risk. This locus is in hub gene EPAS1, which is a key member of the HIF family involved in every aspect of cancer development and progression. Because this locus has potential functions related to epigenetic regulation, the observation of low EPAS1 expression in tumor compared to non-affected tissues warrants additional functional studies to further illustrate interrelationships among genetic polymorphisms, DNA methylation status, and EPAS1 expression.
Acknowledgments
Acknowledgements
The authors thank The Cancer Genome Atlas for contributing clinical, DNA methylation, and RNA sequencing data as well as all subjects who participated in Harvard LCS and external replication studies. The authors also thank Dr. Lucian R. Chirieac, for reviewing FFPE samples.
Funding Sources
This work was supported by the National Institutes of Health (NIH CA092824, CA090578, CA074386, and CA209414). Funding source for KFD, EWP, KNH, BAM (affiliation 7) is NIH contract HHSN268201200008I. The Toronto MSH-PMH study was supported by The Canadian Cancer Society Research Institute (020214), Ontario Institute of Cancer and Cancer Care Ontario Chair Award to R.J.H. and G.L. and the Alan Brown Chair and Lusi Wong Programs at the Princess Margaret Hospital Foundation. The data harmonization of the epidemiological variables across the studies is supported by National Institute of Health (U19-CA148127) and Lunenfeld-Tanenbaum Research Institute, Sinai Health System.
Conflict of Interest
The authors declare no potential conflicts of interest.
Author Contributions
Conceived and designed the project: ZW, YW, DCC.
Drafting of the manuscript: ZW, RZ.
Project coordination: DCC, CIA, JDM, LS.
Network analysis: ZW, YW.
Statistical analysis: ZW, RZ, XL.
Sequencing, genomic annotation of variants, and genotyping: SMG, GL, PB, JKF, XZ, JL, BS, VJ, CB, MK, GS, DZ, CCL, KFD, EKP, BAM, KNH, XX, CP, RJH, CIA.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2018.05.024.
Appendix A. Supplementary data
Supplementary material
References
- 1.ACS Cancer Facts & Figures 2009 . 2009. Estimated new cancer cases and deaths by sex U. [Google Scholar]
- 2.Mountain C.F., Lukeman J.M., Hammar S.P. Lung cancer classification: the relationship of disease extent and cell type to survival in a clinical trials population. J Surg Oncol. 1987;35(3):147–156. doi: 10.1002/jso.2930350302. [DOI] [PubMed] [Google Scholar]
- 3.AJCC Cancer Staging . sixth ed. Springer-Verlag; New York: 2002. Handbook. [Google Scholar]
- 4.Ginsberg R., Vokes E., Raben A. Non-small cell lung cancer: Diagnosis and staging. In: Devita V.J., Hellman S., Rosenberg S., editors. Cancer: principles and practice of oncology. 5th ed. Lippincott-Raven; Philadelphia: 1997. pp. 868–876. [Google Scholar]
- 5.Matakidou A., Eisen T., Houlston R.S. Systematic review of the relationship between family history and lung cancer risk. Br J Cancer. 2005;93(7):825–833. doi: 10.1038/sj.bjc.6602769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hung R.J., Christiani D.C., Risch A. International lung Cancer consortium: pooled analysis of sequence variants in DNA repair and cell cycle pathways. Cancer Epidemiol Biomarkers Prev. 2008;17(11):3081–3089. doi: 10.1158/1055-9965.EPI-08-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altshuler D., Daly M.J., Lander E.S. Genetic mapping in human disease. Science. 2008;322(5903):881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craddock N., Hurles M.E., Cardin N. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464(7289):713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKay J.D., Hung R.J., Gaborieau V. Lung cancer susceptibility locus at 5p15.33. Nat Genet. 2008;40(12):1404–1406. doi: 10.1038/ng.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y., Broderick P., Webb E. Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat Genet. 2008;40(12):1407–1409. doi: 10.1038/ng.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hung R.J., McKay J.D., Gaborieau V. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452(7187):633–637. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- 12.Amos C.I., Wu X., Broderick P. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40(5):616–622. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Landi M.T., Chatterjee N., Yu K. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am J Hum Genet. 2009;85(5):679–691. doi: 10.1016/j.ajhg.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Broderick P., Wang Y., Vijayakrishnan J. Deciphering the impact of common genetic variation on lung cancer risk: a genome-wide association study. Cancer Res. 2009;69(16):6633–6641. doi: 10.1158/0008-5472.CAN-09-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Truong T., Sauter W., McKay J.D. International lung Cancer consortium: coordinated association study of 10 potential lung cancer susceptibility variants. Carcinogenesis. 2010;31(4):625–633. doi: 10.1093/carcin/bgq001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rafnar T., Sulem P., Besenbacher S. Genome-wide significant association between a sequence variant at 15q15.2 and lung cancer risk. Cancer Res. 2011;71(4):1356–1361. doi: 10.1158/0008-5472.CAN-10-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cybulski C., Masojc B., Oszutowska D. Constitutional CHEK2 mutations are associated with a decreased risk of lung and laryngeal cancers. Carcinogenesis. 2008;29(4):762–765. doi: 10.1093/carcin/bgn044. [DOI] [PubMed] [Google Scholar]
- 18.Civelek M., Lusis A.J. Systems genetics approaches to understand complex traits. Nat Rev Genet. 2014;15(1):34–48. doi: 10.1038/nrg3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asomaning K., Miller D.P., Liu G. Second hand smoke, age of exposure and lung cancer risk. Lung Cancer. 2008;61(1):13–20. doi: 10.1016/j.lungcan.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson C.A., Pettersson F.H., Clarke G.M., Cardon L.R., Morris A.P., Zondervan K.T. Data quality control in genetic case-control association studies. Nat Protoc. 2010;5(9):1564–1573. doi: 10.1038/nprot.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abramovitz M., Ordanic-Kodani M., Wang Y. Optimization of RNA extraction from FFPE tissues for expression profiling in the DASL assay. Biotechniques. 2008;44(3):417–423. doi: 10.2144/000112703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y., Wei Y., Gaborieau V. Deciphering associations for lung cancer risk through imputation and analysis of 12,316 cases and 16,831 controls. Eur J Hum Genet. 2015;23(12):1723–1728. doi: 10.1038/ejhg.2015.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landi M.T., Dracheva T., Rotunno M. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One. 2008;3(2) doi: 10.1371/journal.pone.0001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanchez-Palencia A., Gomez-Morales M., Gomez-Capilla J.A. Gene expression profiling reveals novel biomarkers in nonsmall cell lung cancer. Int J Cancer. 2011;129(2):355–364. doi: 10.1002/ijc.25704. [DOI] [PubMed] [Google Scholar]
- 25.Sorlie T., Perou C.M., Tibshirani R. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reinholz M.M., Eckel-Passow J.E., Anderson S.K. Expression profiling of formalin-fixed paraffin-embedded primary breast tumors using cancer-specific and whole genome gene panels on the DASL(R) platform. BMC Med Genomics. 2010;3:60. doi: 10.1186/1755-8794-3-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee S., Emond M.J., Bamshad M.J. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012;91(2):224–237. doi: 10.1016/j.ajhg.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kachuri L., Amos C.I., McKay J.D. Fine mapping of chromosome 5p15.33 based on a targeted deep sequencing and high density genotyping identifies novel lung cancer susceptibility loci. Carcinogenesis. 2016;37(1):96–105. doi: 10.1093/carcin/bgv165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Timofeeva M.N., Hung R.J., Rafnar T. Influence of common genetic variation on lung cancer risk: meta-analysis of 14 900 cases and 29 485 controls. Hum Mol Genet. 2012;21(22):4980–4995. doi: 10.1093/hmg/dds334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y., McKay J.D., Rafnar T. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat Genet. 2014;46(7):736–741. doi: 10.1038/ng.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viechtbauer W. Conducting meta-analyses in R with the metafor package. J Stat Softw. 2010;36(1):1–48. [Google Scholar]
- 32.Ionita-Laza I., Lee S., Makarov V., Buxbaum J.D., Lin X. Sequence kernel association tests for the combined effect of rare and common variants. Am J Hum Genet. 2013;92(6):841–853. doi: 10.1016/j.ajhg.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wigerup C., Pahlman S., Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther. 2016;164:152–169. doi: 10.1016/j.pharmthera.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 34.Brenner D.R., Amos C.I., Brhane Y. Identification of lung cancer histology-specific variants applying Bayesian framework variant prioritization approaches within the TRICL and ILCCO consortia. Carcinogenesis. 2015;36(11):1314–1326. doi: 10.1093/carcin/bgv128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keith B., Johnson R.S., Simon M.C. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12(1):9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takeda N., O'Dea E.L., Doedens A. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24(5):491–501. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiesener M.S., Jurgensen J.S., Rosenberger C. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003;17(2):271–273. doi: 10.1096/fj.02-0445fje. [DOI] [PubMed] [Google Scholar]
- 38.Kim W.Y., Perera S., Zhou B. HIF2alpha cooperates with RAS to promote lung tumorigenesis in mice. J Clin Invest. 2009;119(8):2160–2170. doi: 10.1172/JCI38443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giatromanolaki A., Koukourakis M.I., Sivridis E. Relation of hypoxia inducible factor 1 alpha and 2 alpha in operable non-small cell lung cancer to angiogenic/molecular profile of tumours and survival. Br J Cancer. 2001;85(6):881–890. doi: 10.1054/bjoc.2001.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han S.S., Yeager M., Moore L.E. The chromosome 2p21 region harbors a complex genetic architecture for association with risk for renal cell carcinoma. Hum Mol Genet. 2012;21(5):1190–1200. doi: 10.1093/hmg/ddr551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Purdue M.P., Johansson M., Zelenika D. Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3. Nat Genet. 2011;43(1):60–65. doi: 10.1038/ng.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ciampa J., Yeager M., Amundadottir L. Large-scale exploration of gene-gene interactions in prostate cancer using a multistage genome-wide association study. Cancer Res. 2011;71(9):3287–3295. doi: 10.1158/0008-5472.CAN-10-2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ernst J., Kheradpour P., Mikkelsen T.S. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Putra A.C., Eguchi H., Lee K.L. The a allele at rs13419896 of EPAS1 is associated with enhanced expression and poor prognosis for non-small cell lung Cancer. PLoS One. 2015;10(8) doi: 10.1371/journal.pone.0134496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoo S., Takikawa S., Geraghty P. Integrative analysis of DNA methylation and gene expression data identifies EPAS1 as a key regulator of COPD. PLoS Genet. 2015;11(1) doi: 10.1371/journal.pgen.1004898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhen Q., Liu J.F., Liu J.B. Endothelial PAS domain-containing protein 1 confers TKI-resistance by mediating EGFR and MET pathways in non-small cell lung cancer cells. Cancer Biol Ther. 2015;16(4):549–557. doi: 10.1080/15384047.2015.1016689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma X.J., Wang Z., Ryan P.D. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell. 2004;5(6):607–616. doi: 10.1016/j.ccr.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 48.Munding J.B., Adai A.T., Maghnouj A. Global microRNA expression profiling of microdissected tissues identifies miR-135b as a novel biomarker for pancreatic ductal adenocarcinoma. Int J Cancer. 2012;131(2):E86–E95. doi: 10.1002/ijc.26466. [DOI] [PubMed] [Google Scholar]
- 49.Freidin M.B., Bhudia N., Lim E., Nicholson A.G., Cookson W.O., Moffatt M.F. Impact of collection and storage of lung tumor tissue on whole genome expression profiling. J Mol Diagn. 2012;14(2):140–148. doi: 10.1016/j.jmoldx.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frank M., Doring C., Metzler D., Eckerle S., Hansmann M.L. Global gene expression profiling of formalin-fixed paraffin-embedded tumor samples: a comparison to snap-frozen material using oligonucleotide microarrays. Virchows Arch. 2007;450(6):699–711. doi: 10.1007/s00428-007-0412-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material