

Abstract
Zinc (Zn2+) is a pleiotropic modulator of the neuronal and brain activity. The disruption of intraneuronal Zn2+ levels triggers neurotoxic processes and affects neuronal functioning. In this study, we investigated how the pharmacological modulation of brain Zn2+ affects synaptic plasticity and cognition in wild-type mice. To manipulate brain Zn2+ levels, we employed the Zn2+ (and copper) chelator 5-chloro-7-iodo-8-hydroxyquinoline (clioquinol, CQ). CQ was administered for two weeks to 2.5-month-old (m.o.) mice, and effects studied on BDNF-related signaling, metalloproteinase activity as well as learning and memory performances. CQ treatment was found to negatively affect short- and long-term memory performances. The CQ-driven perturbation of brain Zn2+ was found to reduce levels of BDNF, synaptic plasticity-related proteins and dendritic spine density in vivo. Our study highlights the importance of choosing “when”, “where”, and “how much” in the modulation of brain Zn2+ levels. Our findings confirm the importance of targeting Zn2+ as a therapeutic approach against neurodegenerative conditions but, at the same time, underscore the potential drawbacks of reducing brain Zn2+ availability upon the early stages of development.
Introduction
The neurotrophin brain-derived neurotrophic factor (BDNF) critically modulates the plasticity of the Central Nervous System (CNS) with a broad spectrum of activities that spread from neuronal differentiation and survival to synaptogenesis and activity-dependent synaptic plasticity1,2.
BDNF is synthesized as pre-pro-BDNF, further cleaved into pro-BDNF3–5 and into its mature counterpart (mBDNF) in the endoplasmic reticulum (ER) and the trans-Golgi apparatus6. mBDNF is released by an activity-dependent mechanism or first extruded as pro-BDNF in the extracellular space and then processed to mBDNF by extracellular proteases, like plasmin or metalloproteinases (MMPs; and MMP-2 and 9 in particular)7–11. The balance between pro-BDNF and mBDNF has significant biological implications as the immature and mature forms of the protein, by activating distinct class of receptors12,13, exert different actions on synaptic plasticity and neuronal survival14,15. mBDNF promotes neuroprotective activity in in vitro and in vivo preclinical models of neurodegeneration16–18. Preclinical findings are paralleled by evidence in human studies that indicate that high serum levels of mBDNF, as well as high BDNF gene expression, are associated with a reduced risk of dementia in elderly subjects and a milder disease course in dementia patients19–21. mBDNF and its preferred receptor TrkB play a crucial role in the plasticity-related modifications of spines by promoting spine pruning and enlargement. pro-BDNF and its receptor p75NTR exert pro-apoptotic activity and negatively affects the dendritic structure and spine density14,22. The regulation of spine morphology, a phenomenon defined as structural plasticity, is one of the molecular underpinnings of cognition and learning and memory in particular23.
The metalloproteinase, MMP-9, is highly enriched in the hippocampus, the cerebral cortex, and the cerebellum24, brain regions where the enzyme is localized in the extracellular matrix near the dendritic spines of excitatory synapses25. MMP-9 contributes to learning, memory and neuronal plasticity by promoting the spine remodelling and immobilization of ionotropic receptors that occur upon long-term potentiation (LTP) stimulation26,27. In preclinical models of Alzheimer’s disease (AD), the functional overexpression of MMP-9 increases the brain levels of mBDNF and prevents the development of AD-related cognitive deficits28,29. MMP-9 is structurally characterized by a 3-histidine zinc-binding catalytic motif (His-Glu-Xaa-Xaa-His) and requires zinc (Zn2+) to exert its proteolytic activity30.
Zn2+ is largely present in the CNS, stored within synaptic vesicles at several glutamatergic nerve terminals, and synaptically released upon neuronal activity31,32. Zn2+ affects neuronal processes as well as BDNF signaling33. The metal, through MMP activation, promotes the cleavage of pro-BDNF to mBDNF9,10. Zn2+ also modulates BDNF signaling without affecting the MMP activity. This activity is achieved by direct transactivation of TrkB, the preferred and high-affinity BDNF receptor34. In addition, in vivo administration of Zn2+ increases the cortical levels of the BDNF mRNA35, thereby indicating a pro-transcriptional activity of the cation.
In a preclinical transgenic AD model, we have previously shown that long-term dietary supplementation of Zn2+ promotes the activation of MMP-2 and MMP-9, increases the brain levels of mBDNF, and counteracts the development of AD-related pathology and memory deficits36.
In this study, we investigated how the pharmacological modulation of brain Zn2+ affects BDNF signaling, synaptic plasticity, and cognition in young mice. The rationale behind the experimental design and the choice of employing young animals is twofold. First, recent evidence indicate that deficiency in synaptic Zn2+ affects brain development by impairing BDNF signaling10; second, we aimed at evaluating effects related to the perturbation of brain Zn2+ homeostasis in young mice, a setting that, on the contrary of aging- or degeneration driven models37, is devoid of signs of Zn2+ deregulation. To manipulate brain Zn2+ levels, we employed the Zn2+ (and copper) chelator 5-chloro-7-iodo-8-hydroxyquinoline (clioquinol, CQ) as previous studies have shown that the intraperitoneal administration of CQ promotes a rapid reduction in the brain pool of chelatable Zn2+ 38–43. CQ was administered for two weeks to 2.5-month-old (m.o.) mice and effects studied on BDNF-related signaling, MMP activity as well as learning and memory performances.
Results
CQ administration impairs learning, short- and long-term memory performances in young mice
Male C57Bl/6 mice were treated, for 2 weeks, with daily intraperitoneal (i.p.) injections of CQ (30 mg/kg). The rationale for choosing this concentration and route of administration was threefold: (1) this CQ concentration has been shown to be devoid of significant side effects in rodents including changes in body weight (Fig. 1B; 25.8 ± 1.6 g in control vs 24.9 ± 0.91 in CQ treated mice, p = 0.64); (2) the dose matches the one employed in clinical trials; (3) the i.p. route is highly effective in reducing the pool of chelatable Zn2+ in the brain and the hippocampus in particular38,44. In agreement with previous studies38–43, we obtained a significant decrease of hippocampal Zn2+ levels (Supplementary Fig. 1; normalized TSQ fluorescence in control: 1.00 ± 0.08 vs 0.49 ± 0.04 in CQ-treated mice, p < 0.001).
Figure 1.

CQ administration impairs learning and memory performances in mice. Spatial memory performances were evaluated with the MWM test. (A) The pictogram illustrates the study design. (B) The graph illustrates the daily body weight monitoring of vehicle- and CQ-treated mice during the 2-week treatment period (n = 8 per study group). (C) The graph illustrates the learning curve of the vehicle- and CQ-treated mice (n = 9 and n = 10, respectively) as evaluated during the 4-day training session. Compared to vehicle-treated animals, CQ-treated mice showed significantly impaired learning performances in the first 3 trials of the training sessions. Note that the impairment disappeared in the fourth day of training. (D) The bar graph shows CQ-driven decreases in the number of platform crosses (the number of times the mouse crosses the location where the platform used to be) in the 1.5 h (to evaluate STM) and the 24 h (to evaluate LTM) trials. (E) The bar graph shows CQ-driven increases of latency (the time spent to reach the location where the platform used to be) in both the STM and the LTM trials. (F,G) Bar graphs show the absence of drug-related changes in the time spent in the target (the quadrant where the platform used to be) or the opposite (the quadrant opposed to the one where the platform used to be) quadrants, in the STM and LTM trials (F and G, respectively). (H) The bar graph shows no CQ-driven changes in swimming speed in LTM trials in a subset of mice (n = 5 for control and n = 4 for CQ-treated mice). (I) The bar graph shows no CQ-driven changes in distance travelled in LTM trials in a subset of mice (n = 5 for control and n = 4 for CQ-treated mice). Data are presented as mean ± standard error of the mean (SEM). “*” indicates p < 0.05, “**” indicates p < 0.01.
The effects of CQ or vehicle on hippocampus-dependent spatial learning were assessed, at the end of the second week of treatment, with the Morris Water Maze (MWM) test, a widely employed task that evaluates memory performances in rodents45 (Fig. 1A). Compared to vehicle-treated mice, CQ-treated animals displayed learning impairment. CQ-treated mice showed a statistically-significant increase in the time spent to reach the platform upon the first two training sessions (Fig. 1C; day 1 p < 0.01, day 2 p < 0.001). These learning deficits were transient and disappeared in the final two days of training (Fig. 1C; day 3 p = 0.069, day 4 p = 0.16). The analysis of memory performances revealed that, compared to vehicle-treated animals, CQ-treated mice performed significantly worse in Short- (1.5 h) and Long-Term (24 h) Memory (STM and LTM, respectively) skills. The CQ-treated group showed a decreased number of platform crosses (Fig. 1D; for STM: 4.69 ± 0.69 in control vs 2.15 ± 0.54 in CQ-treated mice, p = 0.008; for LTM: 4.3 ± 0.9 in control vs 1.73 ± 0.38 in CQ-treated mice, p = 0.04) and longer times to reach the platform location (Fig. 1E, escape latency for STM: 8.84 ± 1.5 s in control vs 26.38 ± 5.6 s in CQ-treated mice, p = 0.007; escape latency for LTM: 12.9 ± 5 s in control vs 29.58 ± 5.9 s in CQ-treated mice, p = 0.01). No significant differences, between the two study groups, were observed as far as the time spent in the target or opposite quadrants (Fig. 1F,G, T-target, and T-opposite, respectively). Additional analysis showed no significant differences between the two study groups regarding swimming speed or the total distance travelled upon the LTM probe trial (Fig. 1H,I; p = 0.43 for the swimming speed and p = 0.71 for the distance travelled), thereby indicating that the CQ-mediated effects were not driven by decreased motivational cues or impaired swimming abilities.
CQ reduces BDNF signaling in treated mice
Zn2+ has been shown to promote pro-BDNF maturation in vitro9. Western blot (WB) analysis of brain samples revealed a significant decrease of BDNF levels in CQ-treated mice when compared to vehicle-treated animals (Fig. 2B; see Supplementary Table 1 for the statistics of the WB data). The reduction is region-specific and occurred in the hippocampus, cortex, and striatum but not in the cerebellum. These regional changes were matched by parallel decreased levels of the preferred BDNF receptor, TrkB (Fig. 2C). To assess whether BDNF and TrkB reductions affect BDNF-related signaling, we measured brain levels of the activated -phosphorylated- form of TrkB (pTrkB) and ERK5 (pERK5), a downstream effector of the BDNF-TrkB signaling cascade46. WB analysis showed no significant differences between CQ- and vehicle-treated animals in the pTrkB/TrkB ratio (Fig. 2D) while CQ administration induced a significant decrease in pERK5 levels (Fig. 2F).
Figure 2.
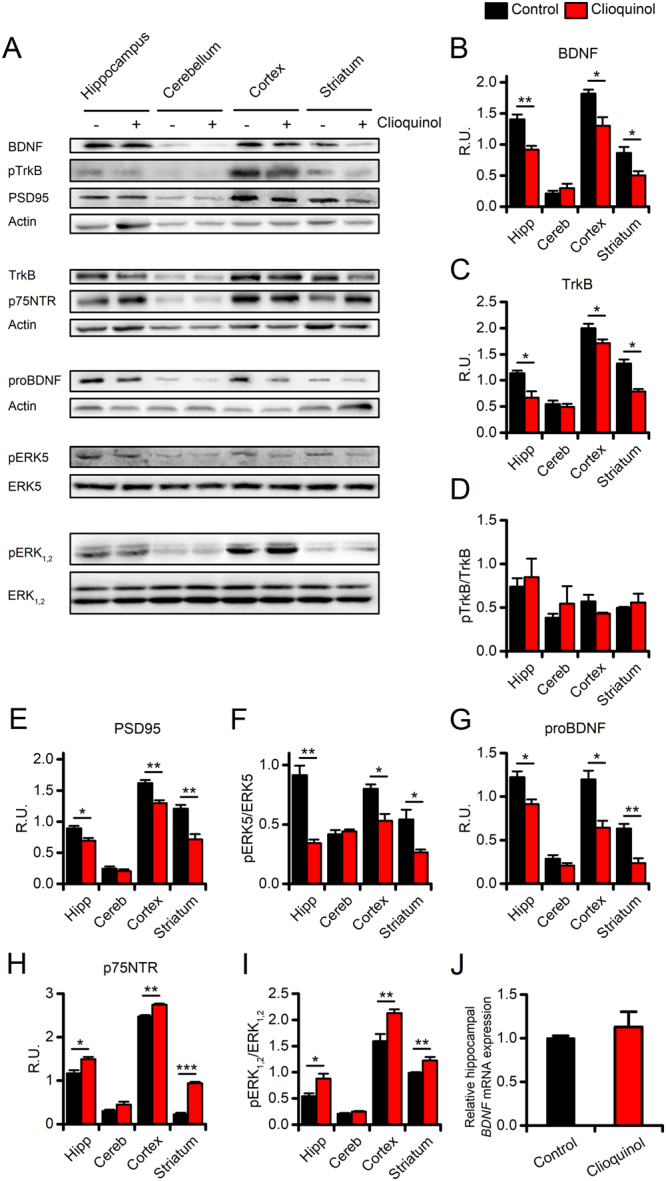
Effects of CQ administration on BDNF signaling. (A) Representative, cropped, images of WB experiments showing vehicle- or CQ-driven effects on BDNF signaling in the hippocampus (Hipp), the cerebellum (Cereb), the cerebral cortex (Cortex), and the striatum of the two study groups. (B,C) Bar graphs show quantification of CQ-driven changes in mBDNF and TrkB expression levels. (D) Bar graphs show no drug-related changes in levels of TrkB phosphorylation (pTrkB/TrkB ratio) in the two study groups. (E) Bar graphs depict CQ-driven changes in PSD95 expression levels in the study groups. (F) Bar graphs depict levels of pERK5 in the study groups. (G,H) Bar graphs depict CQ-driven changes in proBDNF and p75NTR protein levels. (I) Bar graphs depict levels of pERK1,2 in the 2 study groups. All experiments were performed, at least, three times from independent samples. (J) The bar graph depicts mRNA levels of BDNF measured by real-time PCR in the vehicle- and CQ-treated mice (n = 3 per condition). Data are presented as mean ± SEM. “*” indicates p < 0.05, “**” indicates p < 0.01, and “***” indicates p < 0.001.
The neurotrophin BDNF, by activating mechanisms related to functional and structural plasticity, is a potent modulator of LTM47. We, therefore, investigated whether the CQ-driven reduction of BDNF and TrkB translates into changes in PSD95 levels, a marker of the post-synaptic integrity. Matching the CQ-related regional changes in BDNF, we found region-specific decreases of PSD95 in the hippocampus, cortex, striatum, but not in the cerebellum (Fig. 2E).
CQ promotes p75NTR signaling in treated mice
Since pro-BDNF-p75NTR activation can modulate neurotoxic activities, we investigated whether the observed CQ-driven impairments of the BDNF-TrkB signaling axis are associated with changes in the pro-BDNF-p75NTR signaling cascade. WB analysis of these pathways showed that, compared to vehicle, CQ treatment promotes a region-specific increase of p75NTR levels along with high levels of phosphorylated ERK1,2 (pERK1,2; Fig. 2H,I). Surprisingly, the CQ-treatment was also found to decrease pro-BDNF levels in the same brain regions (Fig. 2G). The CQ-driven activation of the p75NTR signaling cascade did not produce overt signs of neurotoxicity in vitro and in vivo (Supplementary Fig. 2).
CQ treatment does not affect the hippocampal BDNF mRNA levels
Previous in vivo studies have shown that Zn2+ supplementation increases the mRNA expression of BDNF35. On the contrary, pharmacological manipulations aimed at reducing the brain Zn2+ levels negatively modulate the BDNF mRNA levels48. To address whether CQ-driven Zn2+ chelation interferes with the BDNF expression, we evaluated mRNA levels in the hippocampi of CQ- and vehicle-treated mice. Quantitative Real-Time PCR (qRT-PCR) analysis showed that CQ administration did not affect the hippocampal BDNF gene expression (Fig. 2J; 1.00 ± 0.02 in control vs 1.12 ± 0.17 in CQ treated mice, p = 0.54).
CQ treatment reduces dendritic spine density in vitro and ex vivo
As BDNF potently modulates synaptic plasticity34,49, we first assessed, in an in vitro setting, whether CQ treatment alters the number of dendritic spines and/or their morphology. To that aim, EGFP-transfected primary hippocampal neurons were exposed to CQ (10 µM for 3 days) or vehicle and spines evaluated with confocal microscopy. Confocal imaging analysis showed that, compared to vehicle-treated sister cultures, CQ-treated neurons underwent a significant reduction in dendritic spine density (Fig. 3A,B; spines per µm: 0.51 ± 0.07 in control vs 0.22 ± 0.02 in CQ-treated cultures, p = 0.020). No differences were found, between the two study groups, regarding spine volumes, areas, diameters or lengths (Fig. 3C–F). To assess whether CQ also impairs spine pruning in vivo, we performed Golgi staining on brain slices of treated animals. Compared to vehicle-treated mice, the hippocampal CA1 pyramidal neurons of CQ-treated mice showed a significant decrease (≈−30%) in their dendritic spine density (Fig. 3G,H; spines per µm: 1.41 ± 0.13 in control vs 0.96 ± 0.1 in CQ-treated mice, p = 0.029).
Figure 3.

CQ reduces dendritic spine density in vitro and in ex vivo brain samples. (A) Representative images of primary (left) and secondary (right) proximal dendrites obtained from EGFP-transfected primary hippocampal neurons treated with vehicle (left) or CQ (10 µM for 3 days; right). (B) Bar graph depicts the quantification of dendritic spine density observed with confocal microscopy after the 3-day treatment period (n = 4 for control and n = 3 for CQ-treated neurons). (C–F) Bar graphs depict the quantification of additional parameters related to dendritic spine morphology as spine volumes (C), spine areas (D), spine lengths (E), and spine diameters (F). (G) Representative images of Golgi-stained CA1 pyramidal dendrites obtained from vehicle- and CQ-treated mice. (H) Bar graph depicts the quantification of dendritic spine density as observed at the end of the 2-week treatment period (n = 19–35 neurons from 3 mice per condition). Data are presented as mean ± SEM. “*” indicates p < 0.05.
CQ reduces MMP activity in vitro
MMPs, and MMP-9 in particular, promote BDNF maturation through the proteolytic cleavage of pro-BDNF9. To address whether CQ interferes with MMP activity we performed a gelatin zymographic essay on MMPs isolated from the medium of cultured hippocampal neurons. After gel electrophoresis, the MMP developing buffer was supplemented with CQ (10 µM) or the high-affinity Zn2+ chelator TPEN (100 µM, here employed as a positive control) and results compared with those of control gels. CQ almost completely abolished the MMP-9 and MMP-2 activity (Fig. 4; normalized MMP-9 activity: 1.00 ± 0.09 in control vs 0.08 ± 0.02 for CQ-treated gels, p < 0.001; normalized MMP-2 activity: 1.00 ± 0.17 in control vs 0.02 ± 0.01 for CQ-treated gels, p < 0.001). Gel treatment with TPEN did not induce further MMP reduction, thereby supporting the notion of a maximal inhibitory activity exerted by CQ (Fig. 4A,B).
Figure 4.
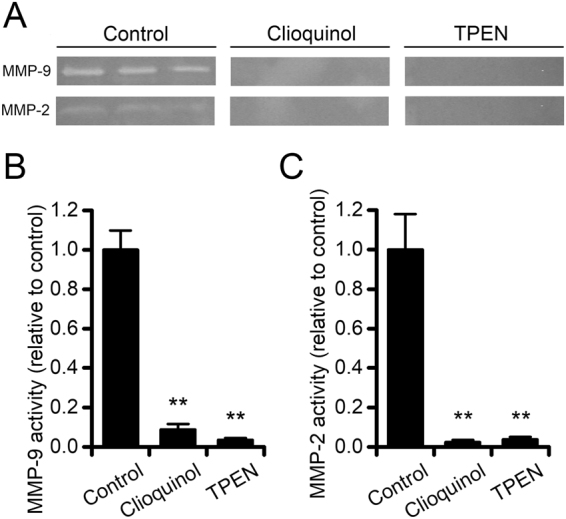
CQ reduces MMPs activity in vitro. (A) Representative images of gelatin zymography showing a CQ-driven decrease of MMP-9 and MMP-2 activity. Note that a CQ concentration as low as 10 µM has the same effect of TPEN (100 µM), here employed as a control. (B,C) Bar graphs show the quantification of images in A for MMP-9 and MMP-2, respectively (n = 6 per condition). Data are presented as mean ± SEM. “**” indicates p < 0.01.
Discussion
In this study, we investigated the cognitive effects of Zn2+ chelation in young mice. Previous studies have shown that intraperitoneal CQ administration promotes a robust reduction of the pool of chelatable Zn2+ in rodents38–43. The main finding of the study is that a 2-week administration of CQ exerts adverse effects on learning and memory performances of treated mice. These results are in line with evidence supporting the critical role played by Zn2+ in the modulation of neuronal functioning32,43,50,51. The study also indicates that the CQ-dependent effects are mediated by the inhibition of the BDNF neurotrophic axis.
CQ administration affects BDNF signaling, synaptic plasticity, and behaviour
Zn2+ chelation profoundly affects mBDNF levels and the related signaling pathway. In CQ-treated animals, we observed a significant reduction in the total content of mBDNF. These changes were associated with a decrease of the total amount of the TrkB receptor. The decreased mBDNF levels that we observed were regionally restricted to the hippocampus, cortex, and striatum (Fig. 2). No effects were detected in the cerebellum, a finding that is in line with the hypothesis that the CQ-driven activity on BDNF signaling is selectively mediated by Zn2+ removal as the BDNF inhibition does not occur in a region, the cerebellum, that is devoid of pools of chelatable Zn2+ 52. These results also parallel the behavioural findings as the CQ-dependent cognitive effects are appreciated on a task, the MWM, that evaluates the hippocampus-related memory performances.
mBDNF exerts its neurotrophic action by binding to the TrkB receptor, and the study shows that CQ promotes a significant decrease in TrkB receptor levels. Unexpectedly, this result was not associated with a parallel reduction in the phosphorylated, activated, form of TrkB as we find that the pTrkB/TrkB ratio is unaffected by the drug treatment. The design of our experimental setting may help to explain the discrepancy. In our model, the molecular effects of CQ administration were assessed at the end of the 2-week treatment period. Within this time frame, it is conceivable that the CQ-mediated decrease in mBDNF is sufficient to induce a significant spine remodelling and/or elimination. This notion is, indirectly, supported by the observed reduction in PSD95 levels and further confirmed by the in vitro and ex vivo decreases of spine density that we observed (Fig. 3). Thus, the CQ-driven reduction of dendritic spines may have led to a decrease in the spine-associated transmembrane proteins including TrkB, thereby producing a net reduction of the receptor levels without affecting the pTrkB/TrkB ratio. The idea of defective TrkB signaling is further corroborated by the observed CQ-driven decrease in pERK5 levels, a kinase that acts downstream of the BDNF-TrkB neurotrophic axis and is known to promote neurogenesis and neuronal survival53,54 but is also implicated in learning- and memory-related processes55,56.
Our findings support a role for CQ-driven Zn2+ deprivation in the modulation of synaptic plasticity and dendritic spine pruning. CQ-treated hippocampal neurons show a reduced number of spines when compared with vehicle-treated sister cultures (Fig. 3). Our data are in line with the notion that mBDNF participates in structural plasticity by modulating the spine pruning and morphology57–59. These plasticity-related phenomena represent the biophysical underpinning of the processes controlling the experience-dependent information storage occurring within the brain circuitry. Thus, the CQ-driven reduction of mBDNF levels may serve as one of the molecular mechanisms underlying the behavioural impairments observed in our model.
In the study, we investigated the Zn2+-dependent MMP activity with the aim of providing a mechanistic link between CQ-administration, Zn2+ chelation, and the reduction of mBDNF levels. Among MMPs, MMP-9 plays a permissive role in favouring structural plasticity and promote the dendritic spine expansion60. Experimental evidence indicates that LTP-induced increase in spine size requires MMPs proteolytic activity26 and, conversely, inhibition of MMPs has been shown to impair spatial learning in adult animals61,62. In addition, recent findings have shown that MMP-9 participates, in a Zn2+ dependent manner, to BDNF maturation9,10. In agreement with this notion, our in gel zymography experiments showed that CQ concentrations as low as 10 µM are sufficient to abolish the MMP gelatinolytic activity (Fig. 4). A further mechanistic link relies on the Zn2+-dependent transactivation of TrkB34, an event that is likely occluded by CQ administration. While the mechanism is possible from a theoretical standpoint, it is unlikely to have occurred in our setting as we did not observe significant changes in TrkB phosphorylation and activation. In addition, a recent report has indicated that the receptor transactivation by Zn2+ occurs in in vitro settings and only marginally in vivo49.
This Zn2+-dependent modulation of the MMP activity is of particular relevance when considering the cation role in synaptic plasticity. Zn2+ is particularly enriched within synaptic vesicles of a subset of hippocampal glutamatergic terminals31,32,63. At these synapses, Zn2+ is released upon neuronal activity and plays a crucial role in the modulation of physiological and pathological processes32. Substantiating this critical activity on plasticity and cognition, transgenic mice devoid of the synaptic zinc transporter-3 (ZnT-3 KO mice), and therefore unable to release Zn2+, show behavioural and molecular deficits that closely resemble the ones observed in our CQ-treated mice. Further suggesting a commonality of activities between transgenically- or pharmacologically-driven Zn2+ deprivation, ZnT-3 KO mice, like CQ-treated animals, show alterations in the BDNF-related pathways and plasticity markers64.
In summary, CQ administration, by chelating Zn2+, reduced the MMP activity that, in turn, decreased BDNF maturation and defective structural plasticity, thereby leading to impaired cognitive performances.
CQ administration modulates neurotoxic pathways in a pro-BDNF-independent manner
mBDNF is strongly related to its precursor form pro-BDNF. The decrease of mBDNF that we found was associated with the overexpression of the p75NTR receptor as well as with increased ERK1,2 signaling, a pathway that is activated by the pro-BDNF. Our WB data indicate that the treatment also produced decreased of pro-BDNF levels (Fig. 2G). These unexpected results prompted us to investigate whether CQ administration has affected the BDNF gene expression as previous studies have shown that changes in Zn2+ availability impinge on BDNF expression35,48. qRT-PCR experiments, aiming at investigating the issue, indicated that the drug treatment did not alter BDNF mRNA levels. The additional targets of MMPs offer a possible explanation for these results. In addition to BDNF, MMPs are known to promote the cleavage and maturation of other pro-neurotrophins, including pro-NGF, a process that activates a deleterious p75NTR-related signaling65. An additional, untested, possibility revolves around CQ effects on other intracellular Zn2+-dependent proteases that are involved in the BDNF maturation10,66. Moreover, the reduction of mature neurotrophins, including mBDNF, can lead to a feed-forward increase of p75NTR expression and activation67. Given the fact that TrkB and p75NTR mediate opposite effects13,68, it is therefore conceivable that the mBDNF reduction unleashes a damaging increase in p75NTR levels (Fig. 4H). The hypothesis is unlikely in our setting as the enhanced p75NTR levels did not promote the activation of death pathways. The treatment did not result in increased neuronal toxicity, and the compound was found to be safe and well tolerated (Fig. 1B, and Supplementary Fig. 2). Our p75NTR results, instead fit with previous findings showing that p75NTR-related signaling facilitates the activation of NMDAR-dependent long-term depression22,69, negatively modulates dendritic morphology and spine density in hippocampal pyramidal neurons4,70, and contributes to synaptic and memory deficits associated with neurodegenerative conditions71. Similarly, aberrant activation of ERK1,2 not only results in the activation of death-related pathways but has been recently implicated in memory impairment in a mouse model of autism72.
Conclusions
Our findings support the notion that Zn2+, through the activation of the BDNF signaling cascade, is an important modulator of synaptic plasticity, neuronal functioning and cognitive processes. Impairment in the pro-BDNF/mBDNF signaling cascade may have important clinical implications. Altered neurotrophin biosynthesis and processing has been indicated to be a common feature in several neurological conditions and affect complex human behaviors73–76. Pharmacological tools engineered to restore effective BDNF signaling can, therefore, have a great impact on development- aging- or disease-related physio-pathological settings.
CQ has been employed in preclinical models of neurodegenerative diseases (AD and Huntington’s disease) with encouraging results77–80. These robust preclinical findings were matched by positive effects in clinical trials exploring the use of CQ or other metal-protein-attenuating compounds (MPACs) in AD patients81–85. Our study underscores the importance of choosing “when”, “where”, and “how much” modulation has to be applied when dealing with brain Zn2+ levels. Our study indicates that in young animals, a model conceivably devoid of overt signs of Zn2+ dyshomeostasis, the CQ-driven deregulation of physiological brain levels of the cation (Supplementary Fig. 1) as well as the related impaired BDNF-signaling have strong functional and structural consequences. Conversely, when aging- and/or disease- related Zn2+ deregulation has occurred, the use of pharmacological tools, like CQ, to quench the neurotoxic effects of the metal is highly beneficial80,86,87, thereby supporting the idea that redistributing Zn2+ in these contexts may overrun potentially negative side-effects on synaptic plasticity and neurotrophic signaling37,51. Thus, our findings do not challenge the importance of targeting Zn2+, with CQ or similar MPACs, as a therapeutic approach against neurodegenerative conditions but at the same time highlights the potential drawbacks of reducing brain Zn2+ availability in the early stages of development.
Materials and Methods
Animal model and treatment paradigm
All the procedures involving the animal handling and their care were approved by the University G. d’Annunzio Institutional Ethics Committee (CEISA protocol no. AD-301). In vivo procedures were performed in compliance with institutional guidelines and in accordance with national and international laws and policies. Mice were bred in the CeSI-MeT animal facility, single-housed, maintained on a 12 h light/dark cycle, and provided with, ad libitum, access to food and water. Behavioral tests were all performed during the early phases of light cycle (8 am–11 am). C57Bl6 male mice were enrolled at 2.5 months of age (m.o.a.) and randomly assigned to a 2-week CQ or vehicle (see below) administration. Animals were treated daily with an intraperitoneal injection of CQ (30 mg/kg body weight) or vehicle. CQ was prepared by first dissolving the drug in DMSO (80 mg/ml) and then diluted in castor oil to a final concentration of 4 mg/ml (with a final 5% DMSO content).
Mice body weight was measured daily as a gross index of the overall safety of the treatment. A cohort of animals was employed for behavioural assessment, a second group which did not perform a behavioural evaluation, was employed for biochemical analysis.
Neuronal cultures
Primary hippocampal neuronal cultures were prepared as previously described56,88. Briefly, hippocampi were obtained from C57Bl6 fetuses at 15 or 16 embryonic days (E15-E16). Alternatively, some experiments were performed on hippocampi obtained from CD1 mice. Hippocampi were dissected on ice and transferred in a 0.25% trypsin/EDTA solution for 10 min at 37 °C, then pelleted by centrifugation, and dissociated with a fire-polished glass pipette. The neuronal suspension was then diluted in Neurobasal medium supplemented with 0.5 mM l-glutamine, 5% fetal bovine serum, 5% horse serum, 1 × B27 and 0.2% penicillin/streptomycin and plated onto pre-treated laminin/poly-dl-lysine coated tissue culture glass coverslips. Three days after plating, non-neuronal cell growth was inhibited by adding 5 μM of cytosine arabinofuranoside, to obtain near-pure hippocampal cultures. 25% of the medium was replaced with fresh Neurobasal two times per week. Experiments were performed on cultures between 12 and 18 days in vitro (12–18 DIV).
Morris Water Maze
The Morris Water Maze (MWM) test was performed as previously described56,89,90. In brief, the MWM test was performed with a circular pool (Panlab; 1.2 m diameter) filled with warm (22 ± 1 °C) water. The pool is placed in a room with several intra- and extra-maze visual cues for mice orientation. Mice were habituated to the testing room and placed on the platform 10 s before the first training session to reduce task-related stress. Mice underwent four trials per day for 4 consecutive days. 1.5 and 24 h after the end of the last training trial mice were tested for spatial memory performances. 1.5 and 24 h probe tests consisted of a 60 s free swim in the pool without the submerged platform. Probe sessions were digitally recorded and analyzed. The following parameters were measured: time to reach the location where the platform used to be (escape latency), number of crosses over the platform location (crosses), time spent in the target (T-target) or opposite (T-opposite) quadrants, average speed (cm/s), and total distance travelled during the LTM session.
Tissue collection and reagents employed
Mice were euthanized after the 2-week treatment and tissue samples collected for further analysis. Brains were halved into two hemispheres, snap frozen by liquid nitrogen, and kept at −80 °C until used. For Western blot (WB) analysis, each hemisphere was dissected into sub-regions (hippocampus, cortex, striatum, and cerebellum), snap frozen in liquid nitrogen, and stored at −80 °C until sampling. For quantitative Real-Time PCR (qRT-PCR) hippocampi obtained from CQ- and vehicle-treated mice were stored in RNAlater solution (ThermoFisher Scientific) until RNA extraction. For Golgi staining hemispheres were rinsed in PBS and immediately transferred to Golgi-Cox solution following manufacturer instructions (see below). All standard chemicals and reagents were, unless otherwise specified, purchased from Sigma-Aldrich.
TSQ staining
TSQ staining was performed as described by Frederickson et al. with slight modifications91. Briefly, 3 h after vehicle or CQ administration, mice were sacrificed, brains extracted, and cryosectioned without fixation. 6 µm thick slices were immediately stained with TSQ (100 µM; Santa Cruz Biotechnology) diluted in borate buffer [BB; containing (in mM): 100 boric acid, 25 sodium tetraborate, 75 NaCl, pH 9.5] for 90 s. Slices were then rinsed in PBS and put on coverslips. Images were acquired with a LSM 800 confocal microscope (Zeiss) by exciting the specimen with a 405 nm LED laser source. DRAQ5 (5 µM) was employed to counterstain the neuronal nuclei.
Protein assay
Protein concentration was assayed by employing the Pierce BCA Protein Assay kit (Thermo scientific). Briefly, the assay is a detergent-compatible formulation based on bicinchoninic acid (BCA) for the colorimetric detection and quantitation of total protein. The method combines the reduction of Cu2+ to Cu+ ions by protein in alkaline medium (the biuret reaction) with the high sensitive and selective colorimetric detection of the cuprous cation by using a reagent containing BCA. The purple-colored reaction product of this assay is formed by the chelation of two molecules of BCA with one cuprous ion. This complex exhibits a strong absorbance at 562 nm.
Western blot analysis
Brain regions were lysated in ice-cold RIPA buffer [pH 7.4 containing (in %): 0.5 sodium deoxycholate, 1 NP-40, 0.1 SDS, 1 protease and phosphatase inhibitor cocktails, and 5 mM EDTA]. Protein lysates (10 µg) were separated by electrophoresis on 9–13% SDS–polyacrylamide gel and blotted onto polyvinyl difluoride (PVDF) membrane. Subsequently, membranes were blocked with 5% non-fat dry milk (Bio-Rad Laboratories) in Tris-buffered saline [TBS; containing (in mM): 20 Tris–HCl, 150 NaCl, pH 7.4] for at least 30 min at room temperature (RT). Membranes were incubated overnight with primary antibodies diluted in TBS containing 0,1% Tween 20 (TBS-T) and 5% non-fat dry milk.
The antibodies and their final concentration used were: anti-pro-BDNF 1:500 (Millipore), rabbit anti-BDNF 1:1000 (Abcam), rabbit anti-TrkB 1:1000 (Abcam), anti-pTrkB (Abcam), rabbit anti-PSD95 1:1000 (Cell Signaling), mouse anti-pERK1,2 1:500 (Santa Cruz), rabbit anti-p75NTR 1:20000 (Abcam), rabbit anti-pERK5 1:1000 (Millipore), and rabbit anti-actin 1:2000 (Sigma). Specie-specific HRP conjugated IgG (1:10000; Vector Laboratories) were employed as secondary antibodies. The obtained chemiluminescent signals were visualized by ECL SuperSignal West Pico PLUS (Thermo scientific) and normalized to actin by using ImageJ software. Values are reported as relative units (R.U.) or phosphoprotein/total protein ratio calculated as: (phosphoprotein/loading control)/(total protein/loading control).
Golgi staining
Hemispheres from vehicle- and CQ-treated mice were stained with an enhanced Golgi-Cox staining system (superGolgi Kit, Bioenno Tech), following manufacturer instructions. Briefly, 150 µm thick brain sections were cut using a vibratome (Leica) and mounted on adhesive microscope slides. Air-dried sections were stained, cleared in xylene, coverslipped in Eukitt mounting medium, and stored in the dark until analyzed. Images from secondary dendrites of CA1 pyramidal neurons were acquired with a DMRD microscope (Leica) equipped with a DFC550 CCD camera (Leica) and a 100× oil-immersion objective (1.3 NA; Leica). A trained experimenter blind to treatment manually counted the spines.
Gelatin zymography
Gelatin zymography was performed as previously described36. SDS-PAGE gels (7.5%) were prepared by incorporating 0.1% gelatin during acrylamide/bis-acrylamide polymerization. In all lanes, 10 µl of the supernatant, obtained from cultured hippocampi, was loaded. Gels were subjected to electrophoresis at 125 V constant. After electrophoresis, SDS was removed by washing gels for 30 min with a renaturating solution containing (in mM): 50 Tris-HCl, pH 7.4, and 2.5% Triton X-100. Gels were further washed with large volumes of ddH2O to remove residual Triton X-100. To assess MMPs activity gels were incubated overnight at 37 °C in an activation buffer containing (in mM): 50 Tris-HCI, pH 7.4, 120 NaCl, 5 CaCl2. Accordingly to the experimental setting, activation buffer was supplemented with CQ (10 µM), TPEN (100 µM), or vehicle (DMSO 1%). Gels were then incubated for 30 min with a developing solution containing (in %): 0.1 Coomassie blue, 40 methanol, 10 acetic acid, 50 ddH2O. Staining was stopped when clear gelatinolytic bands appeared. Gels were photographed with a Gel-Doc apparatus (Biorad) and bands quantified by densitometry by using the Fiji distribution of ImageJ (v 1.51)92.
RNA extraction and qRT-PCR
The NucleoSpin Tissue RNA isolation kit (Macherey-Nagel) was employed for total RNA extraction. One µg of RNA was retro-transcribed using the High Capacity RNA-to-cDNA Kit (ThermoFisher Scientific). qRT-PCR was performed in a total volume of 25 μl containing 2 × Maxima SYBR Green/ROX qPCR Master Mix (ThermoFisher Scientific), 1 µL of cDNA and 0.3 µM of each primer on an Abi 7900HT Sequencing Detection System (ThermoFisher Scientific). BDNF relative expression was corrected against GAPDH and HPRT1 that were used as endogenous controls. Amplification conditions were: 2 minutes at 50°, 10 minutes at 95 °C, followed by 40 cycles of 15 seconds at 95 °C and 1 minute at 60 °C. A melting curve was run to assess the specificity of primers employed. Samples were run in triplicate. The genes relative fold changes were calculated by the ΔΔCt method.
The specific primers employed were: BDNF (F 5′-GAGACAAGAACACAGGAGGAAA-3′, R 5′-GACTAGGGAAATGGGCTTAACA-3′), GAPDH (F 5′-AACAGCAACTCCCACTCTTC-3′, R 5′-GTGGTCCAGGGTTTCTTACTC-3′) and HPRT1 (F 5′-GGCCAGACTTTGTTGGATTTG-3′, R 5′-CGCTCATCTTAGGCTTTGTATTTG-3′).
Neuronal viability assay
To exclude a possible neurotoxic effect of the compound, we assessed cell viability in vitro. Primary hippocampal neurons were exposed for up to 3 days to CQ (10 µM) or vehicle (DMSO). CQ toxicity was evaluated by lactate dehydrogenase (LDH) efflux assay as previously described93.
Neuronal viability was also indirectly evaluated in Golgi stained ex vivo brain slices. Briefly, 20× magnification images of hippocampal and cortical areas were acquired and the number of stained neurons per field was counted and averaged.
In vitro analysis of spine morphology
Spine morphology analysis was performed by transfecting primary hippocampal neurons with an EGFP containing construct. Transfection was performed at 7 DIV with Lipofectamine 2000 (ThermoFisher) following manufacturer instructions. Transfected cultures were subsequently treated with CQ or vehicle for 3 days. Primary and secondary proximal dendritic processes of CQ- or vehicle-treated transfected neurons were imaged with a LSM 510 META confocal microscope (Zeiss) by exciting the specimen with a 488 nm Argon laser. z-stack imaging series were acquired and stored for further analysis. Dendrites were considered “proximal” when within 50 μm from the soma. Distances from the neuronal body were measured with the Zeiss proprietary ruler tool. 3-dimensional reconstructions were obtained from stacked images and analyzed with the Fiji 3D Object Counter94 function. Once images were corrected for artifacts (i.e.: fluorescent particles unrelated to the analyzed neuron/region) and background subtracted, the plugin counted the number of the selected 3D particles present in the stack and automatically provided the volume and the surface area of each object. Spine lengths and diameters were, instead, evaluated with a semi-automated approach with the Reconstruct software95.
Statistical analysis
No methods were employed to predetermine sample size. Statistical analysis was performed by two-tailed Student’s t-test. For gelatin zymography ANOVA following Bonferroni post-hoc test was employed. The level of significance was set at p < 0.05. Data were expressed as mean ± SEM.
Data availability
The data of the study are available from the corresponding author upon reasonable request.
Electronic supplementary material
Acknowledgements
The authors are in debt with Angela Maria Vergnano for the helpful discussions and the critical revision of the manuscript. The authors thank Carlo Corona for helping with the gelatin zymography as well as Annalisa Nespoli and Manuela Iezzi for helping with brain cryosectioning. SLS is supported by research grants from the Italian Department of Education (PRIN 2011; 2010M2JARJ_005) and the Italian Department of Health (RF-2013–02358785 and NET-2011-02346784-1).
Author Contributions
S.L.S. and V.F. conceived and designed the experiments. M.B., V.F. and A.G. performed in vivo treatment and sample collection. M.B. performed behavioural testing. V.F. performed in vitro spine analysis. A.G. performed and analyzed gelatin zymography, TSQ staining and LDH assay. A.G. and N.M. performed and analyzed Golgi staining experiment. V.C. and A.C. performed and analyzed WB experiments. M.d.A. and V.G. performed and analyzed qRT-PCR experiment. V.F., A.G., M.P., M.I., A.M., S.D.P. and S.L.S. analyzed and interpreted the data. V.F., A.G. and S.L.S. wrote the manuscript. S.L.S. supervised the study. All authors discussed, revised, and approved the final version of the mansuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Valerio Frazzini and Alberto Granzotto contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-28083-9.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Leal G, Afonso PM, Salazar IL, Duarte CB. Regulation of hippocampal synaptic plasticity by BDNF. Brain Res. 2015;1621:82–101. doi: 10.1016/j.brainres.2014.10.019. [DOI] [PubMed] [Google Scholar]
- 2.Zagrebelsky M, Korte M. Form follows function: BDNF and its involvement in sculpting the? function and structure of synapses. Neuropharmacology. 2014;76:628–638. doi: 10.1016/j.neuropharm.2013.05.029. [DOI] [PubMed] [Google Scholar]
- 3.Pang PT, et al. Cleavage of proBDNF by tPA/Plasmin Is Essential for Long-Term Hippocampal Plasticity. Science (80-.). 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- 4.Yang J, et al. Neuronal release of proBDNF. Nat. Neurosci. 2009;12:113–5. doi: 10.1038/nn.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Je HS, et al. Role of pro-brain-derived neurotrophic factor (proBDNF) to mature BDNF conversion in activity-dependent competition at developing neuromuscular synapses. Proc. Natl. Acad. Sci. USA. 2012;109:15924–9. doi: 10.1073/pnas.1207767109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greenberg ME, Xu B, Lu B, Hempstead BL. New insights in the biology of BDNF synthesis and release: implications in CNS function. J. Neurosci. 2009;29:12764–7. doi: 10.1523/JNEUROSCI.3566-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizoguchi H, et al. Matrix Metalloproteinase-9 Contributes to Kindled Seizure Development in Pentylenetetrazole-Treated Mice by Converting Pro-BDNF to Mature BDNF in the Hippocampus. J. Neurosci. 2011;31:12963–12971. doi: 10.1523/JNEUROSCI.3118-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagappan G, et al. Control of extracellular cleavage of ProBDNF by high frequency neuronal activity. Proc. Natl. Acad. Sci. 2009;106:1267–1272. doi: 10.1073/pnas.0807322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hwang JJ, Park M-H, Choi S-Y, Koh J-Y. Activation of the Trk signaling pathway by extracellular zinc. Role of metalloproteinases. J. Biol. Chem. 2005;280:11995–2001. doi: 10.1074/jbc.M403172200. [DOI] [PubMed] [Google Scholar]
- 10.Yoo MH, Kim T-Y, Yoon YH, Koh J-Y. Autism phenotypes in ZnT3 null mice: Involvement of zinc dyshomeostasis, MMP-9 activation and BDNF upregulation. Sci. Rep. 2016;6:28548. doi: 10.1038/srep28548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orefice LL, Shih C-C, Xu H, Waterhouse EG, Xu B. Control of spine maturation and pruning through proBDNF synthesized and released in dendrites. Mol. Cell. Neurosci. 2016;71:66–79. doi: 10.1016/j.mcn.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr. Opin. Neurobiol. 2001;11:272–80. doi: 10.1016/S0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- 13.Reichardt LF. Neurotrophin-regulated signaling pathways. Philos. Trans. R. Soc. B Biol. Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang J, et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep. 2014;7:796–806. doi: 10.1016/j.celrep.2014.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sasi M, Vignoli B, Canossa M, Blum R. Neurobiology of local and intercellular BDNF signaling. Pflügers Arch. - Eur. J. Physiol. 2017;469:593–610. doi: 10.1007/s00424-017-1964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu D, Pardridge WM. Neuroprotection with noninvasive neurotrophin delivery to the brain. Proc. Natl. Acad. Sci. USA. 1999;96:254–9. doi: 10.1073/pnas.96.1.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beck T, Lindholm D, Castrén E, Wree A. Brain-Derived Neurotrophic Factor Protects against Ischemic Cell Damage in Rat Hippocampus. J. Cereb. Blood Flow Metab. 1994;14:689–692. doi: 10.1038/jcbfm.1994.86. [DOI] [PubMed] [Google Scholar]
- 18.Almeida RD, et al. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 2005;12:1329–1343. doi: 10.1038/sj.cdd.4401662. [DOI] [PubMed] [Google Scholar]
- 19.Weinstein G, et al. Serum brain-derived neurotrophic factor and the risk for dementia: the Framingham Heart Study. JAMA Neurol. 2014;71:55–61. doi: 10.1001/jamaneurol.2013.4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buchman, A. S. et al. Higher brain BDNF gene expression is associated with slower cognitive decline in older adults. Neurology, 10.1212/WNL.0000000000002387 (2016). [DOI] [PMC free article] [PubMed]
- 21.Manna A, et al. Left hippocampus-amygdala complex macro- and microstructural variation is associated with BDNF plasma levels in healthy elderly individuals. Brain Behav. 2015;5:e00334. doi: 10.1002/brb3.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woo NH, et al. Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat. Neurosci. 2005;8:1069–77. doi: 10.1038/nn1510. [DOI] [PubMed] [Google Scholar]
- 23.Lamprecht R, LeDoux J. Structural plasticity and memory. Nat. Rev. Neurosci. 2004;5:45–54. doi: 10.1038/nrn1301. [DOI] [PubMed] [Google Scholar]
- 24.Dzwonek J, Rylski M, Kaczmarek L. Matrix metalloproteinases and their endogenous inhibitors in neuronal physiology of the adult brain. FEBS Lett. 2004;567:129–35. doi: 10.1016/j.febslet.2004.03.070. [DOI] [PubMed] [Google Scholar]
- 25.Wilczynski GM, et al. Important role of matrix metalloproteinase 9 in epileptogenesis. J. Cell Biol. 2008;180:1021–1035. doi: 10.1083/jcb.200708213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szepesi Z, et al. Synaptically Released Matrix Metalloproteinase Activity in Control of Structural Plasticity and the Cell Surface Distribution of GluA1-AMPA Receptors. PLoS One. 2014;9:e98274. doi: 10.1371/journal.pone.0098274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, et al. Extracellular proteolysis by matrix metalloproteinase-9 drives dendritic spine enlargement and long-term potentiation coordinately. Proc. Natl. Acad. Sci. USA. 2008;105:19520–5. doi: 10.1073/pnas.0807248105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fragkouli A, Tsilibary EC, Tzinia AK. Neuroprotective role of MMP-9 overexpression in the brain of Alzheimer’s 5xFAD mice. Neurobiol. Dis. 2014;70:179–89. doi: 10.1016/j.nbd.2014.06.021. [DOI] [PubMed] [Google Scholar]
- 29.Kaminari A, Giannakas N, Tzinia A, Tsilibary EC. Overexpression of matrix metalloproteinase-9 (MMP-9) rescues insulin-mediated impairment in the 5XFAD model of Alzheimer’s disease. Sci. Rep. 2017;7:683. doi: 10.1038/s41598-017-00794-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Apte SS, Parks WC. Metalloproteinases: A parade of functions in matrix biology and an outlook for the future. Matrix Biol. 2015;44–46:1–6. doi: 10.1016/j.matbio.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Paoletti P, Vergnano AM, Barbour B, Casado M. Zinc at glutamatergic synapses. Neuroscience. 2009;158:126–36. doi: 10.1016/j.neuroscience.2008.01.061. [DOI] [PubMed] [Google Scholar]
- 32.Sensi SL, Paoletti P, Bush AI, Sekler I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009;10:780–91. doi: 10.1038/nrn2734. [DOI] [PubMed] [Google Scholar]
- 33.Sensi SL, et al. The neurophysiology and pathology of brain zinc. J. Neurosci. 2011;31:16076–85. doi: 10.1523/JNEUROSCI.3454-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang YZ, Pan E, Xiong ZQ, McNamara JO. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron. 2008;57:546–558. doi: 10.1016/j.neuron.2007.11.026. [DOI] [PubMed] [Google Scholar]
- 35.Nowak G, et al. Zinc treatment induces cortical brain-derived neurotrophic factor gene expression. Eur. J. Pharmacol. 2004;492:57–9. doi: 10.1016/j.ejphar.2004.03.038. [DOI] [PubMed] [Google Scholar]
- 36.Corona C, et al. Dietary zinc supplementation of 3xTg-AD mice increases BDNF levels and prevents cognitive deficits as well as mitochondrial dysfunction. Cell Death Dis. 2010;1:e91. doi: 10.1038/cddis.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCord MC, Aizenman E. The role of intracellular zinc release in aging, oxidative stress, and Alzheimer’s disease. Front. Aging Neurosci. 2014;6:77. doi: 10.3389/fnagi.2014.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nitzan YB, et al. Clioquinol effects on tissue chelatable zinc in mice. J. Mol. Med. 2003;81:637–644. doi: 10.1007/s00109-003-0462-7. [DOI] [PubMed] [Google Scholar]
- 39.Ismail T, Mauerhofer E, Slomianka L. The hippocampal region of rats and mice after a single i.p. dose of clioquinol: Loss of synaptic zinc, cell death and c-Fos induction. Neuroscience. 2008;157:697–707. doi: 10.1016/j.neuroscience.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 40.Choi BY, et al. Zinc chelation reduces traumatic brain injury-induced neurogenesis in the subgranular zone of the hippocampal dentate gyrus. J. Trace Elem. Med. Biol. 2014;28:474–481. doi: 10.1016/j.jtemb.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 41.Wang T, Zheng W, Xu H, Zhou J-M, Wang Z-Y. Clioquinol inhibits zinc-triggered caspase activation in the hippocampal CA1 region of a global ischemic gerbil model. PLoS One. 2010;5:e11888. doi: 10.1371/journal.pone.0011888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suh SW, et al. Decreased Brain Zinc Availability Reduces Hippocampal Neurogenesis in Mice and Rats. J. Cereb. Blood Flow Metab. 2009;29:1579–1588. doi: 10.1038/jcbfm.2009.80. [DOI] [PubMed] [Google Scholar]
- 43.Takeda A, et al. Impairment of recognition memory and hippocampal long-term potentiation after acute exposure to clioquinol. Neuroscience. 2010;171:443–450. doi: 10.1016/j.neuroscience.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 44.Mao X, Schimmer AD. The toxicology of Clioquinol. Toxicology Letters. 2008;182:1–6. doi: 10.1016/j.toxlet.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 45.Morris RGM, Garrud P, Rawlins JNP, O’Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- 46.Su C, Sun F, Cunningham RL, Rybalchenko N, Singh M. ERK5/KLF4 signaling as a common mediator of the neuroprotective effects of both nerve growth factor and hydrogen peroxide preconditioning. Age (Dordr). 2014;36:9685. doi: 10.1007/s11357-014-9685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bekinschtein P, et al. BDNF is essential to promote persistence of long-term memory storage. Proc. Natl. Acad. Sci. USA. 2008;105:2711–6. doi: 10.1073/pnas.0711863105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Y, et al. High Dose Zinc Supplementation Induces Hippocampal Zinc Deficiency and Memory Impairment with Inhibition of BDNF Signaling. PLoS One. 2013;8:e55384. doi: 10.1371/journal.pone.0055384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Helgager J, Huang YZ, Mcnamara JO. Brain-derived neurotrophic factor but not vesicular zinc promotes TrkB activation within mossy fibers of mouse hippocampus in vivo. J. Comp. Neurol. 2014;522:3885–3899. doi: 10.1002/cne.23647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hagmeyer S, Haderspeck JC, Grabrucker AM. Behavioral impairments in animal models for zinc deficiency. Front. Behav. Neurosci. 2014;8:443. doi: 10.3389/fnbeh.2014.00443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corona C, Pensalfini A, Frazzini V, Sensi SL. New therapeutic targets in Alzheimer’s disease: brain deregulation of calcium and zinc. Cell Death Dis. 2011;2:e176. doi: 10.1038/cddis.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wall M. A role for zinc in cerebellar synaptic transmission? The Cerebellum. 2005;4:224–229. doi: 10.1080/14734220500242084. [DOI] [PubMed] [Google Scholar]
- 53.Obara, Y. & Nakahata, N. The signaling pathway leading to ERK5 activation via G-proteins and ERK5-dependent neurotrophic effects. Mol. Pharmacol. (2009). [DOI] [PubMed]
- 54.Shalizi, A. et al. Characterization of a Neurotrophin Signaling Mechanism that Mediates Neuron Survival in a Temporally Specific Pattern. J. Neurosci. 23 (2003). [DOI] [PMC free article] [PubMed]
- 55.Wang W, et al. Genetic activation of ERK5 MAP kinase enhances adult neurogenesis and extends hippocampus-dependent long-term memory. J. Neurosci. 2014;34:2130–47. doi: 10.1523/JNEUROSCI.3324-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bomba M, et al. Exenatide exerts cognitive effects by modulating the BDNF-TrkB neurotrophic axis in adult mice. Neurobiol. Aging. 2018;64:33–43. doi: 10.1016/j.neurobiolaging.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 57.Cohen-Cory S, Kidane AH, Shirkey NJ, Marshak S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev. Neurobiol. 2010;70:271–88. doi: 10.1002/dneu.20774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.An JJ, et al. Distinct Role of Long 3′ UTR BDNF mRNA in Spine Morphology and Synaptic Plasticity in Hippocampal Neurons. Cell. 2008;134:175–187. doi: 10.1016/j.cell.2008.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harward SC, et al. Autocrine BDNF–TrkB signaling within a single dendritic spine. Nature. 2016;538:99–103. doi: 10.1038/nature19766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X-b, et al. Extracellular proteolysis by matrix metalloproteinase-9 drives dendritic spine enlargement and long-term potentiation coordinately. Proc. Natl. Acad. Sci. 2008;105:19520–19525. doi: 10.1073/pnas.0807248105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meighan SE, et al. Effects of extracellular matrix-degrading proteases matrix metalloproteinases 3 and 9 on spatial learning and synaptic plasticity. J. Neurochem. 2006;96:1227–1241. doi: 10.1111/j.1471-4159.2005.03565.x. [DOI] [PubMed] [Google Scholar]
- 62.Stawarski M, Stefaniuk M, Wlodarczyk J. Matrix metalloproteinase-9 involvement in the structural plasticity of dendritic spines. Front. Neuroanat. 2014;8:68. doi: 10.3389/fnana.2014.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vergnano AM, et al. Zinc dynamics and action at excitatory synapses. Neuron. 2014;82:1101–1114. doi: 10.1016/j.neuron.2014.04.034. [DOI] [PubMed] [Google Scholar]
- 64.Adlard PA, Parncutt JM, Finkelstein DI, Bush AI. Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 2010;30:1631–6. doi: 10.1523/JNEUROSCI.5255-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Le AP, Friedman WJ. Matrix metalloproteinase-7 regulates cleavage of pro-nerve growth factor and is neuroprotective following kainic acid-induced seizures. J. Neurosci. 2012;32:703–12. doi: 10.1523/JNEUROSCI.4128-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Podsiadlo P, Komiyama T, Fuller RS, Blum O. Furin Inhibition by Compounds of Copper and Zinc. J. Biol. Chem. 2004;279:36219–36227. doi: 10.1074/jbc.M400338200. [DOI] [PubMed] [Google Scholar]
- 67.Bamji SX, et al. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J. Cell Biol. 1998;140:911–23. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV. Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J. Neurosci. 1998;18:3273–81. doi: 10.1523/JNEUROSCI.18-09-03273.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rosch H, Schweigreiter R, Bonhoeffer T, Barde Y-A, Korte M. The neurotrophin receptor p75NTR modulates long-term depression and regulates the expression of AMPA receptor subunits in the hippocampus. Proc. Natl. Acad. Sci. 2005;102:7362–7367. doi: 10.1073/pnas.0502460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matsumoto T, et al. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat. Neurosci. 2008;11:131–133. doi: 10.1038/nn2038. [DOI] [PubMed] [Google Scholar]
- 71.Brito V, et al. Neurotrophin receptor p75NTR mediates Huntington’s disease–associated synaptic and memory dysfunction. J. Clin. Invest. 2014;124:4411–4428. doi: 10.1172/JCI74809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seese RR, Maske AR, Lynch G, Gall CM. Long-term memory deficits are associated with elevated synaptic ERK1/2 activation and reversed by mGluR5 antagonism in an animal model of autism. Neuropsychopharmacology. 2014;39:1664–73. doi: 10.1038/npp.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carlino D, De Vanna M, Tongiorgi E. Is Altered BDNF Biosynthesis a General Feature in Patients with Cognitive Dysfunctions? Neurosci. 2013;19:345–353. doi: 10.1177/1073858412469444. [DOI] [PubMed] [Google Scholar]
- 74.Hempstead BL. Brain-Derived Neurotrophic Factor: Three Ligands, Many Actions. Trans. Am. Clin. Climatol. Assoc. 2015;126:9–19. [PMC free article] [PubMed] [Google Scholar]
- 75.Garcia KLP, et al. Altered balance of proteolytic isoforms of pro-brain-derived neurotrophic factor in autism. J. Neuropathol. Exp. Neurol. 2012;71:289–97. doi: 10.1097/NEN.0b013e31824b27e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mizui T, Ishikawa Y, Kumanogoh H, Kojima M. Neurobiological actions by three distinct subtypes of brain-derived neurotrophic factor: Multi-ligand model of growth factor signaling. Pharmacol. Res. 2016;105:93–8. doi: 10.1016/j.phrs.2015.12.019. [DOI] [PubMed] [Google Scholar]
- 77.Cherny RA, et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron. 2001;30:665–76. doi: 10.1016/S0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 78.Lei P, et al. Clioquinol rescues Parkinsonism and dementia phenotypes of the tau knockout mouse. Neurobiol. Dis. 2015;81:168–75. doi: 10.1016/j.nbd.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 79.Cherny RA, et al. PBT2 Reduces Toxicity in a C. elegans Model of polyQ Aggregation and Extends Lifespan, Reduces Striatal Atrophy and Improves Motor Performance in the R6/2 Mouse Model of Huntington’s Disease. J Huntingtons Dis. 2012;1:211–219. doi: 10.3233/JHD-120029. [DOI] [PubMed] [Google Scholar]
- 80.Adlard PA, et al. Metal ionophore treatment restores dendritic spine density and synaptic protein levels in a mouse model of Alzheimer’s disease. PLoS One. 2011;6:e17669. doi: 10.1371/journal.pone.0017669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ritchie CW, et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch. Neurol. 2003;60:1685–1691. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 82.Faux NG, et al. PBT2 rapidly improves cognition in Alzheimer’s Disease: additional phase II analyses. J. Alzheimer’s Dis. 2010;20:509–516. doi: 10.3233/JAD-2010-1390. [DOI] [PubMed] [Google Scholar]
- 83.Huntington Study Group Reach, H. D. I. Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 14, 39–47 (2015). [DOI] [PubMed]
- 84.Sampson, E. L., Jenagaratnam, L. & McShane, R. Metal protein attenuating compounds for the treatment of Alzheimer’s dementia. in Cochrane Database of Systematic Reviews (ed. Sampson, E. L.) CD005380 (John Wiley & Sons, Ltd). 10.1002/14651858.CD005380.pub4 (2012).
- 85.Lannfelt L, et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008;7:779–786. doi: 10.1016/S1474-4422(08)70167-4. [DOI] [PubMed] [Google Scholar]
- 86.Adlard PA, et al. Metal chaperones prevent zinc-mediated cognitive decline. Neurobiol. Dis. 2015;81:196–202. doi: 10.1016/j.nbd.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 87.Adlard PA, et al. Rapid Restoration of Cognition in Alzheimer’s Transgenic Mice with 8-Hydroxy Quinoline Analogs Is Associated with Decreased Interstitial Aβ. Neuron. 2008;59:43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 88.Frazzini V, et al. Altered Kv2.1 functioning promotes increased excitability in hippocampal neurons of an Alzheimer’s disease mouse model. Cell Death Dis. 2016;7:e2100. doi: 10.1038/cddis.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bomba M, et al. Exenatide promotes cognitive enhancement and positive brain metabolic changes in PS1-KI mice but has no effects in 3xTg-AD animals. Cell Death Dis. 2013;4:e612. doi: 10.1038/cddis.2013.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Isopi E, et al. Pyruvate prevents the development of age-dependent cognitive deficits in a mouse model of Alzheimer’s disease without reducing amyloid and tau pathology. Neurobiol. Dis. 2015;81:214–224. doi: 10.1016/j.nbd.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 91.Frederickson CJ, Kasarskis EJ, Ringo D, Frederickson RE. A quinoline fluorescence method for visualizing and assaying the histochemically reactive zinc (bouton zinc) in the brain. J. Neurosci. Methods. 1987;20:91–103. doi: 10.1016/0165-0270(87)90042-2. [DOI] [PubMed] [Google Scholar]
- 92.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Granzotto A, Sensi SL. Intracellular zinc is a critical intermediate in the excitotoxic cascade. Neurobiol. Dis. 2015;81:25–37. doi: 10.1016/j.nbd.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 94.Bolte S, Cordelieres FP. A guided tour into subcellular colocalisation analysis in light microscopy. J. Microsc. 2006;224:13–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 95.Fiala JC. Reconstruct: a freeeditor for serial section microscopy. J. Microsc. 2005;218:52–61. doi: 10.1111/j.1365-2818.2005.01466.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data of the study are available from the corresponding author upon reasonable request.