

Abstract
Nylon hydrolase (NylC) is initially expressed as an inactive precursor (36 kDa). The precursor is cleaved autocatalytically at Asn266/Thr267 to generate an active enzyme composed of an α subunit (27 kDa) and a β subunit (9 kDa). Four αβ heterodimers (molecules A-D) form a doughnut-shaped quaternary structure. In this study, the thermostability of the parental NylC was altered by amino acid substitutions located at the A/D interface (D122G/H130Y/D36A/L137A) or the A/B interface (E263Q) and spanned a range of 47 °C. Considering structural, biophysical, and biochemical analyses, we discuss the structural basis of the stability of nylon hydrolase. From the analytical centrifugation data obtained regarding the various mutant enzymes, we conclude that the assembly of the monomeric units is dynamically altered by the mutations. Finally, we propose a model that can predict whether the fate of the nascent polypeptide will be correct subunit assembly, inappropriate protein-protein interactions causing aggregation, or intracellular degradation of the polypeptide.
Introduction
Protein-protein interactions leading to correct subunit assembly play important roles in protein stability or various biological functions, which could include the following: i) regulation of enzyme activities by the allosteric effect, ii) generation of an active center by utilizing several catalytic/substrate-binding residues located in different subunits, and iii) generation of a multifunctional enzyme by integrating several functions initially compartmentalized in different enzyme subunits into a single oligomeric enzyme. In addition, the stability of a monomeric protein is generally improved by the oligomerization of the protein1–3. An appropriate protein-protein interaction results in correct subunit assembly, whereas an inappropriate interaction causes the proteins to aggregate. The formation of insoluble protein aggregates and the aggregates themselves, as in the case of the amyloidogenesis of amyloid β peptides, can lead to various diseases, including diseases of the nervous system4,5. Therefore, it is important to analyze the molecular basis of protein assembly to discriminate between the correct oligomer formation required for a functional enzyme and incorrect associations leading to the aggregation of the monomeric units.
Nylon hydrolase (NylC) is one of the three enzymes responsible for the endo-type degradation of the by-products of nylon-6 manufacture (cyclic and linear oligomers of 6-aminohexanoate (Ahx) with a degree of polymerization greater than three) and various aliphatic nylons (e.g., nylon-6 and nylon-66)6–14. NylC is a member of the N-terminal nucleophile (N-tn) hydrolase family15–28 and has been found in Arthrobacter (pOAD2 plasmid-encoded NylC; NylCp2), Agromyces (NylCA), and Kocuria (NylCK)12–14. NylCA and NylCK have 5 and 15 substitutions, respectively, in their sequences of 355 amino acid residues compared with the sequence of NylCp2 (Fig. S1), and both have 10–20 °C higher thermostability than does NylCp213. The NylC enzymes are initially expressed as an inactive enzyme. However, the post-translational autocleavage of the nascent polypeptide between Asn266 and Thr267 generates an active enzyme with an α subunit (27 kDa) and a β subunit (9 kDa) (Figs 1 and 2)7,8. Based on the X-ray-crystallographic structure of NylCA, we reported that four αβ heterodimers (molecules A-D) form a doughnut-shaped quaternary structure9. In addition, we proposed that the nucleophilic N-terminal residue Thr267 in the β subunit functions as a catalytic residue for either autocleavage (from precursor to active enzyme) or substrate hydrolysis9. Moreover, the thermostability of NylCp2 (melting temperature (Tm) identified by circular dichroism (CD) analysis = 52 °C) can be cumulatively enhanced by the D122G/H130Y/D36A/E263Q quadruple mutations (Tm = 88 °C) (Figs 1c, S1, and Table 1)9.
Figure 1.

Structure of NylC. (a) Autocleavage of the NylC precursor between Asn266 and Thr267 generates an active enzyme composed of α and β subunits. (b) The quaternary structure of NylCp2 is shown as a ribbon model, with different colors highlighting the individual monomer molecules A (green), B (blue), C (red), and D (yellow). The positions of helix α1 in monomers A and D are marked by circles with double solid lines. (c) An enlarged view of monomer A and its interfaces with the adjacent monomers B and D in the NylCp2-G122Y130A36Q263 mutant enzyme is shown as a stereo diagram. Three mutated residues (Ala36, Gly122, and Tyr130) integrated into wild-type NylCp2 and seven additionally mutated residues (Ile75, Leu106, Ser141, Pro157, Ala179, Asn235, and Gln299) obtained by random mutagenesis are shown as space-filling models. The catalytic residue Thr267 (in the N-terminus of the β subunit) is shown in reddish purple. (d) The secondary structure of NylCp2-G122 mutant was illustrated by the program “ENDscript” (http://espript.ibcp.fr/ESPript/ENDscript/index.php). The amino acid sequence is shown using one-letter codes. The autocleaved site (N266/T267) and the D122G mutation are shown as a blue vertical arrow and a red triangle, respectively. Five α-helices (α1–α5) and five 310-helices (η1–η5) are shown as black coils. Twenty β-strands (β1–β20) are shown as horizontal black arrows. The seven loop regions (L1-L7) described in the text are shown in red. Due to poor electron density, the conformations of the amino acid residues marked by stars could not be determined. (e) Subunit structure of the NylCp2-G122 mutant shown as a ribbon model. The α-helices and β-strands are colored in green and orange, respectively. Catalytic residue Thr267 is shown in reddish purple.
Figure 2.

Genetic and mutational effects on the expression of the NylC. (a) The transcription, translation and autocleavage responsible for the expression of functional NylC are schematically shown. (b) Mutants obtained by PCR-induced random mutagenesis and site-directed mutagenesis were classified into four types. Type 1: obtained as active enzymes. Type 2: obtained as soluble precursors. Type 3: obtained as insoluble precursors (protein aggregation). Type 4: subjected to fragmentation during the cultivation/purification process (see Table 3).
Table 1.
Oligomeric states, thermostability and enzyme activity of wild-type NylC and mutant enzymes.
| Enzyme | Amino acid substitutions in NylCp2 sequence | Oligomeric state*1 | Subunit identified by SDS-PAGE | CD-melting analysis*2 Tm (°C) | Enzyme activity*3 (U mg−1) |
|---|---|---|---|---|---|
| NylCp2 | — | Trimer/Dimer (Monomer) |
α, β | 52 | 7.1 |
| NylCA | G111S D122G H130Y L137A V225M | Tetramer | α, β | 60 | 15.7 |
| NylCK | D36A A41V M50T I60V A62S G111S D122G H130Y L137A V225M T230G V231I V257L E263Q G354A | Tetramer (Monomer, Higher oligomer) |
α, β | 67 | 14.9 |
| NylCp2-Y130 | H130Y | Not tested | α, β | 63 | 0.85 |
| A137 | L137A | Dimer (Monomer, Higher oligomer) |
Precursor | 41 | < 0.1 |
| G122Y130 | D122G H130Y | Not tested | α, β | 81 | 7.1 |
| G122A137 | D122G L137A | Dimer or monomer (Trimer) | Mixture of α, β, precursor | Not tested | Not tested |
| G122Y130A36Q263 | D122G H130Y D36A E263Q | Tetramer (Monomer) | α, β | 88 | 6.8 |
*1Oligomeric states of NylC in aqueous solution were estimated from analytical centrifugation. Minor populations are shown in parentheses. For example, in wild-type NylCp2, trimeric and dimeric αβ heterodimers were found at equal amounts, while a small population was present as monomeric αβ heterodimer. In NylCp2-A137, uncleaved dimeric molecules share the dominant population.
*2The thermostability determined previously by CD melting analysis (ref.9) is shown.
*3The enzyme activity was assayed at 30 °C under standard assay conditions (4 mg ml−1 of the cyclic Ahx oligomer) (ref.9).
In this study, we isolated various mutants that affect protein stability, autoprocessing of the precursor to the active enzyme, and subunit assembly. The mutants obtained were classified into the following four types (Fig. 2): active enzymes with altered thermostability or enzyme activity (type 1); soluble precursors (type 2); insoluble precursors (protein aggregation) (type 3); and fragments proteolyzed during the cultivation/purification process (type 4). Based on X-ray-crystallographic analysis, we describe the structural basis of the stability of nylon hydrolase and the fate of the protein, which is directed along one of the following pathways: i) correct subunit assembly generating dimer/tetramer structures, ii) inappropriate protein-protein interactions causing aggregation, or iii) intracellular disintegration of the nascent NylC polypeptide.
Results and Discussion
Oligomeric states of NylC
To study the oligomeric states of nylon hydrolase, we purified three wild-type NylCs (NylCp2, NylCA, and NylCK) and several NylC mutants to homogeneity and analyzed the differences in the patterns of sedimentation using analytical ultracentrifugation (Fig. 3). As described below, the assembly of monomeric units (A-D) dynamically switched between the monomer, dimer (A/B or A/D), trimer, and tetramer (as well as higher oligomers), and the oligomeric states were affected by mutations, especially at the subunit interface (Fig. 4). In addition, amino acid substitutions at specific positions altered not only the protein stability but also the catalytic activity, as well as the processability to the active enzyme (Table 1).
Figure 3.

Polyacrylamide gel electrophoresis and ultracentrifugation analysis of wild-type and mutant enzymes. (a) SDS-PAGE (17.5% gel) of purified NylC. Lane M, molecular size marker; Lane 1, NylCA; Lane 2, NylCK; Lane 3, NylCp2; Lane 4, NylCp2-K122; Lane 5, NylCp2-L122; Lane 6, NylCp2-R122; Lane 7, NylCp2-V122; Lane 8, NylCp2-Q122; Lane 9, NylCp2-N122; Lane 10, NylCp2-G122Y130A36Q263; Lane 11, NylCp2-A137; Lane 12, NylCp2-G122Y130; Lane 13, NylCp2-G122A137; Lane 14, NylCp2-G122; and Lane 15, NylCp2-Y130. (b) Native-PAGE (10% gel) of purified NylC. The wild-type and most NylC mutants exhibited single bands corresponding to the dimeric structure (shown as “D”). However, some mutants (NylCp2-A137 and NylCp2-G122A137) exhibited a faster-migrating band (presumed to be the monomer and shown as “M”). In contrast, the most thermostable mutant, G122Y130A36Q263, showed the slowest-migrating band (presumed to be tetrameric and shown as “T”). Lane 1, NylCA; Lane 2, NylCK; Lane 3, NylCp2; Lane 4, NylCp2-L122; Lane 5, NylCp2-N122; Lane 6, NylCp2-Q122; Lane 7, NylCp2-R122; Lane 8, NylCp2-K122; Lane 9, NylCp2-V122; Lane 10, NylCp2-G122; Lane 11, NylCp2-G122Y130A36Q263; Lane 12, NylCp2-G122Y130; Lane 13, NylCp2-A137; and Lane 14, NylCp2-G122A137. The grouping of portions cropped from different gels was made explicit using delineation with dividing white space. Full-length gels are included in a Supplementary Information file (Fig. S8). (c,d) Ultracentrifugation analysis. The subunit assembly of the wild-type enzymes (NylCp2, NylCA, and NylCK) and mutant enzymes derived from NylCp2 (G122Y130A36Q263, A137, and G122A137) was analyzed by sedimentation velocity (c) and sedimentation equilibrium methods (d). The estimated oligomeric states, which are the monomer (M), dimer (D), trimer (Tr), and tetramer (T), for each peak in (c) are marked. The deviations of the s values of the same oligomeric state suggest that the shape of the protein structure (spatial location of loop regions and bulkiness of protein) affects the sedimentation coefficient in aqueous solution. The wild-type NylCA gave a single peak (4.7 S) corresponding to a tetramer. The wild-type NylCK gave one major peak (4.3 S) and two minor peaks (7.5 S and 2.1 S), suggesting that the monomer, tetramer, and higher oligomers coexisted in equilibrium. The wild-type NylCp2 gave two major peaks [3.7 S (trimer) and 3.2 S (dimer)] and a minor peak (2.1 S) (monomer). The G122Y130A36Q263 mutant gave one major peak corresponding to 4.5 S (tetramer) and a minor peak (2.4 S) (monomer). The A137 mutant gave a major peak (2.7 S) (dimer) and two minor peaks [2.0 S (monomer) and 5.0 S] molecular species. The G122A137 mutant gave a major peak of 2.6 S (monomer or dimer) and a minor peak of 3.5 S (trimer).
Figure 4.

Subunit assembly and surface structure of NylC. (a) Model of the subunit assembly of NylC. The oligomeric states are dynamically altered depending on the structural changes caused by mutations. Typical association/dissociation reactions (reactions 1–8) are shown. Major assembly and dissociation reactions are assumed to be in equilibrium and are shown as solid lines (reactions 1 and 2), while the alternative equilibria shown as dashed lines may be possible. To simplify the designation of each monomer molecule in the quaternary structure, both the uncleaved precursor and the cleaved enzyme (αβ heterodimer) are expressed as molecules A-D. (b) Monomer (A), dimer (A/B and A/D), trimer (A/B/C), and tetramer structures are shown as surface models. Contacts at the A/B and C/D interfaces (3,121 Å2) were observed to be more extensive than those at the B/C and A/D interfaces (1,451 Å2). (c) The tetramer structure of the NylCp2-G122Y130A36Q263 mutant is shown as a space-filling model. The overall structures of monomer molecules A, B, C, and D are shown in light green, light blue, light pink, and light yellow, respectively. To highlight the catalytic residues and mutated sites, the catalytic nucleophile Thr267 (in the N-terminus of the β subunit in monomer A) and other presumed catalytic residues (Lys189 and Asn219) are shown in red, dark blue, and dark green, respectively. The mutated residues (Ala36, Gly122, Tyr130, and Leu137) in monomer A are shown in purple. The mutated residues (Ala36, Tyr130, and Leu137) in monomer D are shown in orange. The conformation of the Glu263 side chain in NylCp2-G122Y130A36Q263 could not be determined due to poor electron density. Pro259/Pro260, which is linked to the loop 5 region, is shown in green (monomer A) and magenta (monomer C). Arg328 and Arg330 in loop 7 are marked. (d) To show the structure of the A/D interface in the NylCp2-G122Y130A36Q263 mutant, the structure (space-filling model) is shown by omitting monomer D.
Wild-type NylCA produced a single peak in the sedimentation velocity [sedimentation coefficient at 20 °C (s20) = 4.7 S], indicating that the enzyme exhibited a narrow distribution in the oligomeric states (Fig. 3c). The average molecular weight (MAV) of NylCA was 130,928 based on sedimentation equilibrium analysis (Fig. 3d). Therefore, NylCA was mainly present as a tetramer of αβ-heterodimers in aqueous solutions (Table 1). In contrast, the MAV of wild-type NylCp2 was 85,602 based on sedimentation equilibrium analysis. This value was in good agreement with the molecular weight (93,000) estimated by gel-filtration chromatography11. These results suggest that the trimeric structure of αβ-heterodimers (3.7 S), as well as the monomeric (2.1 S) and dimeric (3.2 S) structures, was formed in the case of NylCp2. Wild-type NylCK and the most thermostable mutant, NylCp2-G122Y130A36Q263 (Tm = 88 °C), should exist mainly as a tetramer of αβ-heterodimers (MAV = 151,384 and 130,895, respectively) (Fig. 3d).
The MAV values of NylCp2-A137(60,258) and NylCp2-G122A137(67,397) obtained by equilibrium analysis were between the values corresponding to the monomer (36 kDa) and the dimer (72 kDa). The presence of the monomeric unit was also confirmed by native polyacrylamide gel electrophoresis (PAGE) analysis of the NylCp2-A137 and NylCp2-G122A137 mutants (Fig. 3b). SDS-PAGE analysis revealed that the NylCp2-A137 mutant was obtained as a precursor (type 2 mutation), whereas the more stable G122A137 mutant was obtained as a mixture of the precursor and the α- and β-subunits (Fig. 3a, lanes 11 and 13).
The oligomeric states of NylC enzymes should also depend on the enzyme concentration and environmental conditions to which the enzymes are exposed. Under the enzyme concentrations (0.34 mg ml−1) used for ultracentrifugation analysis, NylCp2 was in a monomer/dimer/trimer equilibrium, NylCA provided a homogeneous tetrameric structure, but NylCK produced higher oligomer and monomer molecules in addition to the major tetrameric structures (Fig. 3). In contrast, native PAGE analysis of wild-type NylCp2, NylCA, NylCK, and most mutants yielded a single band, presumed to be a dimer (Fig. 3b, lanes 1–3). These results suggest that the oligomeric states are more homogeneous in the gel matrix of polyacrylamide and that the tetrameric (NylCA and NylCK) and trimeric (NylCp2) molecules are likely to dissociate into dimeric structures.
The expected oligomeric states in aqueous solution, processability from the precursor to the active enzyme, thermostability, and enzyme activity obtained in the present experiments are summarized in Table 1. In this study, we demonstrate that the local alterations induced by mutations in certain cases generated additional contacts with the adjacent monomers, resulting in protein aggregation (type 3 mutation in Fig. 2). In contrast, if the structural alterations induced by mutations weakened the interactions required for protein assembly, the nascent monomeric polypeptides did not always fold and assemble to form stable oligomeric structures (type 4 mutation). The molecular basis of subunit assembly is discussed below on the basis of the three-dimensional structure (Fig. 4).
Mutations that affect the protein stability and autoprocessing
Among the various mutants obtained from wild-type NylCp2, which differ in thermostability, a single D122G mutation drastically improves the thermostability by 24 °C9. To extensively analyze the effect of amino acid substitution at position 122, we constructed 10 new mutants in which Asp122 of NylCp2 was replaced with other amino acids by site-directed mutagenesis. The wild-type and mutant enzymes exhibited the typical far-UV CD spectrum (200–250 nm) for non-denatured proteins at 25 °C, whereas for CD analysis at 95 °C, the enzymes exhibited the typical pattern for denatured proteins (Fig. S2). The values of Tm (melting point of the heat denaturation) and ΔH (change in enthalpy for the global unfolding of proteins) were determined by regression analysis using nonlinear least-squares fitting of the CD melting curve (Fig. S2c). The Tm values increased in the order of Asp122 (wild-type NylCp2) < Leu < Gln < Asn < Arg < Lys < Val < Gly and ranged from 52.9 °C to 75.1 °C (type 1 mutation in Fig. 2) (Table 2). The CD analyses were performed under more diluted enzyme concentrations (0.1 mg ml−1) than the ultracentrifugation analysis. However, the tendency of the thermostability obtained by the residual enzyme assay using a higher concentration (1 mg ml−1) was largely similar to the stability obtained by the CD analysis (Table 2 and Fig. S3). Therefore, we estimated the molecular basis underlying the protein stability on the basis of the structure (X-ray crystallography) and the oligomeric states (ultracentrifugation and native PAGE analysis).
Table 2.
Thermostability and enzyme activity of NylC mutant enzymes.
| Enzyme | Amino acid substitutions in NylCp2 sequence | Subunit identified by SDS-PAGE | CD-melting analysis*1 | Thermostability*2 (°C) by residual activity assay | Enzyme Activity*3 (U mg−1) | |
|---|---|---|---|---|---|---|
| Tm (°C) | ∆H (kJ mol−1) |
|||||
| NylCp2 | — | α, β | 52.9 ± 0.1 | −401 ± 15 | 42 | 7.1 |
| NylCp2-G122 | D122G | α, β | 75.1 ± 0.1 | −543 ± 12 | 73 | 6.3 |
| V122 | D122V | α, β | 74.7 ± 0.0 | −655 ± 14 | 72 | 6.5 |
| K122 | D122K | α, β | 70.8 ± 0.1 | −520 ± 13 | 65 | 7.5 |
| R122 | D122R | α, β | 69.5 ± 0.1 | −409 ± 8 | 65 | 7.1 |
| Q122 | D122Q | α, β | 64.3 ± 0.1 | −252 ± 4 | 61 | 6.1 |
| N122 | D122N | α, β | 66.7 ± 0.1 | −394 ± 11 | 60 | 7.5 |
| L122 | D122L | α, β | 60.2 ± 0.1 | −355 ± 7 | 55 | 5.7 |
| H122 | D122H | Enzyme is not expressed in cell. | ||||
| P122 | D122P | |||||
| W122 | D122W | |||||
*1Tm (melting point of the heat denaturation) and ΔH (change in enthalpy for the global unfolding of proteins) were determined from the CD melting curve by regression analysis using nonlinear least squares fitting.
*2Enzyme solutions were incubated at various temperatures for 30 min, and the residual enzyme activity was analyzed (Fig. S3).
*3The enzyme activity was assayed at 30 °C under standard assay conditions (4 mg ml−1 of the cyclic Ahx oligomer) (ref.9).
In the H122, W122, and P122 mutants of NylCp2, no protein bands corresponding to the active enzyme (27 kDa and 9 kDa) or the precursor (36 kDa) were detected by Western blot analysis, whereas low levels of NylC-antigenic protein were detected by a direct ELISA (Table 3 and Fig. S4). Our attempts to express mutant enzymes having a single D122H, D122W, or D122P substitution in NylCp2 were unsuccessful, even though we conducted three independent experiments. The elution profiles from ion-exchange column chromatography of the cell extracts of the E. coli clone showed no protein peaks corresponding to the NylC fractions (Fig. S5). We speculate that protein destabilization results in the denaturation of the protein even at the cultivation temperature. Consequently, the nascent mutant NylC polypeptide is cleaved into fragments by intracellular proteases (type 4 mutation in Fig. 2).
Table 3.
Immunological detection of NylC-antigenic protein and RT-PCR analysis.
| Mutant enzymes derived from NylCp2 |
Mutant phenotype*1 | Western blotting*2 | Direct ELISA*3 (µg ml−1) | RT-PCR*4 (%) | |
|---|---|---|---|---|---|
| Soluble | Insoluble | ||||
| P122 | Type 4 | — | — | 8.2 | 129 |
| H122 | Type 4 | — | — | 2.4 | 142 |
| W122 | Type 4 | — | — | 3.2 | 112 |
| G122Y130A36Q263 | Type 1 | 27 kDa, 9 kDa | — | 11.4 | 100 |
| G122Y130A36Q263-T179 | Type 4 | — | — | 8.1 | 121 |
| G122Y130A36Q263-T295 | Type 1 | 27 kDa, 9 kDa | — | 17.7 | 113 |
| G122Y130A36Q263-D299 | Type 4 | — | — | 4.4 | 154 |
| G122Y130A36Q263-E299 | Type 3 | — | 36 kDa | 11.7 | 106 |
| G122Y130A36Q263-V75 | Type 3 | — | 36 kDa | 1.1 | 120 |
| G122Y130A36Q263-R106 | Type 3 | — | 36 kDa | 0.2 | 118 |
| G122Y130A36Q263-L141 | Type 4 | — | — | 2.9 | 124 |
| G122Y130A36Q263-L157 | Type 3 | — | 36 kDa | 3.1 | 96 |
| G122Y130A36Q263-N191 | Type 1 | 27 kDa, 9 kDa | 14.3 | 121 | |
| G122Y130A36Q263-D235 | Type 3 | — | 36 kDa | 6.9 | 67 |
*1NylC mutants were classified into four types (see Fig. 2b). Type 1: obtained as active enzymes, Type 2: obtained as soluble precursors (e.g., NylCp2-A137; see Table 1). Type 3: obtained as insoluble precursors (protein aggregation). Type 4: subjected to fragmentation during the cultivation/purification process.
*2The cell extract (soluble fraction) and precipitates obtained by the centrifugation of sonicated cells (insoluble fraction) were boiled in SDS, and NylC-antigenic proteins were detected by Western blot analysis using anti-NylC antibody (Fig. S4).
*3The amount of NylC-antigenic protein was analyzed by direct ELISA using anti-NylC antibody. The antigenic protein was estimated by subtracting the background level (4.9 µg ml−1 for cell extracts harboring the vector pBluescript) from the data obtained for E. coli clones harboring the mutant nylC gene.
*4To check the expression of the mutant nylC gene, we prepared RNA samples from E. coli clones. Reverse transcription (RT)-PCR analysis revealed that PCR products corresponding to nylC-mRNA were present in all mutants.
X-ray-crystallographic analysis of wild-type NylCp2 and typical mutant enzymes
The crystal of NylCA contains fifteen monomeric molecules in a single unit cell9. Such a large number of monomers in one unit cell is not suitable for structural analysis at high resolution. Therefore, we screened and established a new crystallization condition, which generated crystals with only two molecules (corresponding to the A/B dimer) related by a non-crystallographic two-fold axis in an asymmetric unit (space group C2221)11. We performed X-ray-crystallographic studies of wild-type NylCp2 and the seven typical mutants at 1.05–2.00 Å resolution (Table S1). The four identical αβ heterodimers (molecules A-D) were mutually related by D2 symmetry (Fig. 1b). Each monomer molecule (A-D) of NylC contained five α-helices (α1–α5), five 310-helices (η1–η5), 20 β-strands (β1–β20), and 29 loop regions (Fig. 1d), generating a stacked αββα core structure (Fig. 1e). The overall structure of NylCp2 was almost identical to that of NylCA determined previously9 (Fig. S6) with a root mean square deviation (rmsd) of 0.552 Å. The rmsd of the main-chain atoms between NylCp2 and NylCA and between NylCp2 and NylCp2-G122Y130A36Q263 was generally less than 1 Å, but loop 1 (positions 15–34), loop 2 (positions 88–93), loop 3 (positions 125–133), α3 (positions 246–257), loop 5 (positions 258–269), and loop 7 (positions 319–333) displayed significant structural differences (Fig. S6). It should be noted that α1 and its adjacent loop 3 regions showed the largest structural difference between NylCA and NylCp2, whereas a difference of less than 2 Å was found between NylCA and NylCp2-G122Y130A36Q263 (Fig. S6b). The structural differences in the α1 and loop 3 regions that included position 122 seemed to correlate with the thermostability of the enzymes (see below). Large deviations around position 30 were due to the difference in crystal contacts. The loop 5 region, which was close to the autocleavage site, displayed high temperature factors, and consequently, the residues at positions 261–266 were invisible (Fig. S7).
Three amino acid residues corresponding to the substitutions D122G, H130Y, and D36A (increasing the protein stability of NylCp2) and one amino acid residue corresponding to the substitution L137A (decreasing the protein stability) were located at the A/D interface, and the glutamate residue corresponding to the substitution E263Q was found at the A/B interface (Figs 1 and 4). Since there were no detectable contacts between monomers A and C, we focused on the effects of amino acid substitutions on the protein stability and structural alterations between monomers A, B, and D on the basis of the structure of monomer A.
Interaction between helices and the partial asymmetry caused by the mutations
At the A/D interface, helix α1 of monomer A was juxtaposed next to helix α1 of monomer D (Fig. 1b). However, the D2 symmetry was partly altered by mutations at position 122. For example, in wild-type NylCp2 (Asp122, Tm = 52.9 °C), the distance between position 112 (monomer A) and position 122 (monomer D) at Cα (designated as “a1”) was 7.04 Å (Fig. 5 and Table S2). In contrast, the distance between position 122 (monomer A) and position 112 (monomer D) (designated as “a2”) was considerably greater with a value of 8.43 Å. Plotting the protein stability (Tm value) estimated from the CD analyses against the distance between the two monomer molecules revealed that the protein stability exhibited an inverse correlation with the a2/a1 ratio (index of symmetry). In other words, the ratio changed from 0.979 to 1.197 from the wild-type (Asp122) to the mutant (Arg122, Lys122, Val122, and Gly122) enzyme, although this value should be equal to one for a perfectly symmetric structure (Fig. 5e). From the distance between positions 115 and 118 at the Cα of the α1 helices of the monomers A and D (designated “b1” and “b2”, respectively), the b2/b1 ratio was similarly calculated. Since the values (b2/b1 = 0.984–1.016) were closer to the theoretical value, the structural symmetry was better conserved at position 115/118. Thus, the partial asymmetry at the A/D interface inhibits tetramer formation but generates an irregular trimeric structure in aqueous solution, as shown in the proposed model (Fig. 4).
Figure 5.

Subunit interactions between monomers A and D. (a) The structure of helix α1 and its adjacent loop regions in monomers A and D is shown as a ribbon diagram. The structure of NylCp2-G122 (monomer A, green; monomer D, yellow) is superimposed on the structure of NylCp2 (monomer A, dark green; monomer D, orange). (b) The relationship between the α1 helices in monomers A and D is illustrated. a1, distance between position 112 (monomer A) and position 122 (monomer D) at Cα; a2, distance between position 122 (monomer A) and position 112 (monomer D); b1, distance between position 115 (monomer A) and position 118 (monomer D) at Cα; b2, distance between position 118 (monomer A) and position 115 (monomer D). (c,d). The structure of NylCp2 (c) and the NylCp2-G122 mutant (d) at the A/D monomer interface is shown as a stereo diagram. The side chains of Asp122, Gly122, and Lys159 (monomer A), as well as Glu115 and Tyr156 (monomer D), are shown as stick diagrams in 2Fo-Fc electron density maps. The contour level of the electron density map is 1σ. Possible hydrogen bonds and contacts between two atoms are indicated as dotted lines with the distances listed in Å. (e) The index of symmetry (a2/a1 and b2/b1) is plotted against the protein stability (melting temperature: Tm) estimated from CD analysis of NylCp2 (wild-type) and NylCp2-R122, K122, V122, and G122-mutants. (f) The distance from Lys159-NH3+ (located on helix α2 in monomer A) to Glu115-Oε1− (located on helix α1 in monomer D) is plotted against the Tm.
A clearer correlation was found between thermostability and the nearest distance between the side-chain atoms on helix α2 and helix α1. In wild-type NylCp2, Lys159-NH3+ (located on helix α2 in molecule A) was 2.87 Å away from Glu115-Oε1− (located on helix α1 in molecule D) (Fig. 5c and Table S3). In contrast, the distance was reduced to 2.65 Å in the thermostable Gly122 enzyme (Fig. 5d). Notably, this distance decreased with an almost linear correlation with increasing thermostability in the wild-type and mutant enzymes (Fig. 5f). The side-chain atoms of Lys122 existed as two conformers, with the distance of one conformer showing a linear relationship with thermostability. We suggest that the electrostatic effect between the two residues will enhance the subunit binding around Gly122 of helix α1. However, in NylCp2, the amino acid residue at position 122 was replaced with Asp, and the close proximity of the acidic residue Asp122 (close to Glu115) reduced this stabilization effect. Since the correlation between the Lys159-Glu115 distances and the enthalpy change (ΔH) for the unfolding of proteins was not strong (Table 2), the overall stabilization effects should be determined by the additional stabilization effect, probably by the interaction at the loop regions as described below.
In the case of NylCp2 mutants having single Asn122 (Tm = 66.7 °C), Gln122 (Tm = 64.3 °C), and Leu122 mutations (Tm = 60.2 °C), crystals were not obtained even under the crystallization conditions optimized for NylCp2. Therefore, it is likely that the local structural changes caused by the mutations altered the precise orientation of the enzyme molecules required for crystal formation.
Stabilization of loop regions by subunit interactions
His130 and Leu137 in NylCp2 were located in a loop between helix α1 and β-strand β7 (designated “loop 3”) (Fig. 1d). However, the structures of loop 1 and loop 3 containing His130 in NylCp2 were not built during X-ray-crystallographic analysis due to the poor electron density (Fig. 6a). Moreover, the temperature factors (index of thermal motion in a crystallographic structure) of loop regions 1–3 in NylCp2 were significantly larger than those of the corresponding regions in the thermostable NylC-G122Y130 mutant (Fig. S7). In the thermostable enzymes, loop 3 and η3 (in monomer A) was mutually stabilized by contacts with loop 1 and loop 4 in monomer D (Fig. 6). Moreover, a single L137A substitution in the η3 region of the native NylCp2 resulted in a decrease in thermostability by 11 °C. The L137A substitution resulted in a loss of the autocleavage function, which is required for converting the precursor to the active enzyme. The effect of this amino acid substitution on autocleavage is discussed below.
Figure 6.
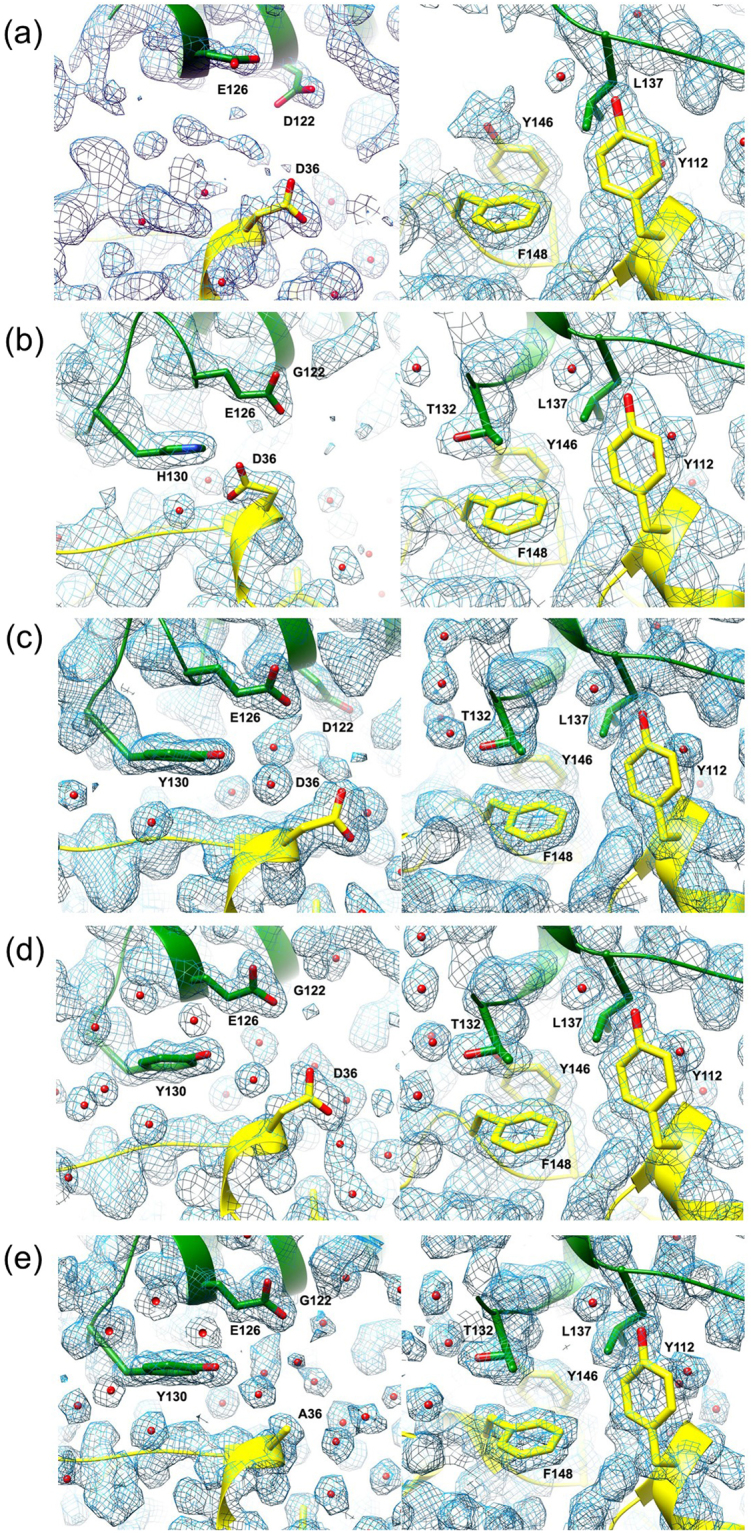
Structure of loop regions located at the A/D monomer interface and catalytic centers. The 2Fo-Fc electron density maps of NylCp2 (a), NylCp2-G122 (b), NylCp2-Y130 (c), NylCp2-G122Y130 (d), and NylCp2-G122Y130A36Q263 (e) are shown. Left panel: amino acid residues in monomer A (Asp/Gly122, Glu126, and His/Tyr130) and monomer D (Asp/Ala36) are shown as stick models. Right panel: amino acid residues in monomer A (Thr132 and Leu137) and monomer D (Tyr112, Tyr146, and Phe148) are shown as stick models. In the thermostable enzymes (c–e), “loop 3” and α3 (containing Tyr130, Thr132, and Leu137) (in monomer A) are mutually stabilized by contacts with “loop 1” and “loop 4” (containing Tyr146 and Phe148) in monomer D. Leu137 (in monomer A) has contacts with monomer D at Tyr146, Phe148, and Tyr112 (in helix α1), suggesting that hydrophobic interactions around Leu137 stabilize the protein structure. D36A substitution in NylCp2-G122Y130Q263 mutant (Tm = 84 °C) contributes to the increased stability, probably by the reduction of electrostatic repulsion between Asp36-COO− (monomer D) and Glu126-COO− (monomer A) (e).
The C-terminal region of the α subunit, including helix α3 and Pro260, flipped out toward the adjacent monomer at each subunit interface (Fig. 4c and d). Glu263 was located in the terminal region (loop 5) of the α subunit, which had a poor electron density distribution in the X-ray diffraction analysis of the wild-type and all NylC mutants. The Asp319-Val333 region (loop 7) was also flipped out towards the adjacent monomer. Thus, the loop 5 of monomer A was co-located with the loop 7 of monomer B at the surface of the tetramer structure (Fig. 4c and d). In the G122Y130A36Q263 mutant, the acidic amino acid residue Glu263 was replaced with a neutral Gln, suggesting that the alterations in the electrostatic environment induced by the E263Q mutation contributed to improving the subunit interactions. We confirmed that the E263Q substitution in G122Y130A36 (Tm = 84 °C) increased thermostability by 4 °C9.
We found that the V225M substitution, which was close to Gln299, decreased the expression and/or stability of NylCp2 in cells, whereas this mutation increased the stability of NylC-G122Y130 by 2 °C9. Therefore, the total stabilization effects depended on the combined interactions with the surrounding residues. Since Gln299 (in monomer A) was close to Arg296 (in monomer B), the replacement of the neutral amino acid Gln299 with acidic amino acids (Glu299 or Asp299) was expected to enhance the electrostatic interaction with the positively charged guanidium group of Arg296 at the A/B monomer interface. However, we found that the mutant enzyme with the Q299E substitution was aggregated (type 3 mutation), while the Q299D mutant enzyme was degraded at the cultivation stage (type 4 mutation) (Fig. 2b).
Cumulative mutational effects directing the successive fates of the enzyme
To analyze successive mutational effects on the most thermostable enzyme G122Y130A36Q263, we constructed various mutants by PCR-induced random mutagenesis. Of the 100 mutants isolated after random mutagenesis using the G122Y130A36Q263 enzyme, eight mutant enzymes with a single mutation were selected, and their properties were examined (Table 3). Only two mutants (H295T, D191N) were similar to the parental G122Y130A36Q263 mutant in the immunological analysis. However, the successive mutations caused protein aggregation (type 3 mutation) or intracellular degradation of the precursors in the cells (type 4 mutation), as described below.
Mutations causing protein aggregation
Western blot analysis of the NylCp2-G122Y130A36Q263-Glu299 mutant using a polyclonal NylC antibody and E. coli cells lysed by sonication was performed. The precursor (36 kDa) band was detected in the insoluble fraction, but no NylC-antigenic protein band was detected in the soluble fraction (Table 3 and Fig. S4). In addition, mutant enzymes with the I75V, L106R, P157L, or N235D single substitutions obtained by PCR-induced random mutagenesis exhibited a similar pattern in Western blot analysis (Table 3 and Fig. S4). Glu299 was located at the A/B monomer interface and on β-strand β19 in monomer A (Fig. 1). Asn235 was located at the A/B interface, which was close to Q299. In contrast, two mutations (I75V and L106R) were located on the central β-sheets and loop region, which were close to helix α1. Another mutation (P157L) was located on loop 4, which was also close to helix α2 (Fig. 1). Thus, the aggregation of the enzyme and the inability to autocleave the precursor were caused by mutations at various sites, rather than specific positions in the monomer structure.
Mutations causing intracellular degradation of the enzyme
The Asp299-mutant enzyme was thought to be degraded by intracellular proteases during cultivation of the E. coli clone, since the results obtained by immunological detection (Western blot analysis and sandwich ELISA) and RT-PCR analysis were very similar to the results for the NylCp2-His122, -Pro122, and -Trp122 enzymes (Table 2). The S141L or A179T substitutions in NylC-G122Y130A36Q263 caused similar phenotypic changes. S141 was located on β-strand β7 (Fig. 1). S141 was located close to A137, and the L137A substitution in NylCp2 showed pleiotropic effects on subunit assembly, autoprocessing, and thermostability (described below).
As stated above, amino acid substitution at a specific position (e.g., position 122 at the A/D interface and position 299 at the A/B interface) altered the local structure of the surrounding residues. Therefore, it was likely that these local structural alterations led to additional contacts with the adjacent monomers. We hypothesize that many monomer molecules were associated in an inappropriate orientation of monomers through these additional contacts, resulting in protein aggregation (type 3 phenotype). In contrast, if the structural alterations induced by mutations weakened the significant interactions required for protein assembly, the nascent unfolded monomeric polypeptides would be subjected to degradation by intracellular proteases (type 4 phenotype).
Effects of mutations on autocleavage
From the three-dimensional structure of NylCA and the results obtained from the other N-tn family enzymes15–28, we proposed that catalytic residue Thr267 is responsible for the autocleavage of the precursor and substrate hydrolysis9. We proposed that the auto-processing reaction is initiated by the nucleophilic attack of Thr267-Oγ on Asn266-Cα carbon, generating a tetrahedral intermediate. This intermediate rearranges into an ester intermediate (N-O acyl shift) that is subsequently hydrolyzed by an adjacent water molecule, producing the active enzyme9. The NylCp2-A137 mutant (type 2 mutant) was obtained as a precursor, whereas the more stable G122A137 mutant was obtained as a mixture of the precursor and α- and β-subunits (Fig. 3a, lanes 11 and 13). In the three-dimensional model, the α3 region (in monomer D) containing Phe134 and Leu137 is spatially close to Thr267 (processing site). The location of these structures around the catalytic center suggests that inappropriate positioning of the loop region caused by destabilization of the loop alters the initial triggering of the nucleophilic attack by Thr267 for the conversion of the precursor protein to the active enzyme. However, it should be noted that the inability of the autocleavage (type 3 phenotype) is caused by various mutations (I75V, L106R, P157L, N235D, and Q299E) in the NylCp2-G122Y130A36Q263 mutant, as described above (Table 3 and Fig. 1c).
Proposed model of subunit assembly
The complexity of the oligomeric states of NylC enzymes demonstrates that various association/dissociation steps are involved in the subunit assembly (Fig. 4). We estimated that the major subunit assembly generating the tetramer molecules proceeds via A/B dimers (by reactions 1 and 2) for the following reasons: (i) X-ray-crystallographic analyses of wild-type and mutant NylC enzymes revealed that the contact area of the A/B interface was approximately twice that of the A/D interface, suggesting that A/B dimer formation was more predominant than A/D dimer formation; (ii) the A/B dimer was actually identified in an asymmetric unit of the NylC crystals; (iii) the A/D dimer exhibited an irregular shape in which the terminal loop region responsible for contact with the adjacent monomer molecules was exposed to the solvent environment. This looped-out region should be amenable to intracellular proteolytic degradation. In this model, the C/D dimer is equivalent to the A/B dimer in the symmetrical relationship.
Wild-type NylCA was largely present as a tetramer, as indicated by the crystallographic and ultracentrifugation analysis (single peak of 4.2 S in the sedimentation velocity). In contrast, NylCp2 existed at the monomer/dimer/trimer equilibrium in aqueous solution (2.1 S, 2.7 S, and 3.5 S). Thus, it was likely that the partial asymmetry at the A/D interface disrupted tetramer formation (reaction 2) but generated an irregular trimeric structure in aqueous solution by reaction 3 or by successive reactions (reaction 2 followed by reaction 8), although the enzyme was identified to be a tetramer by the crystallographic analysis described above.
Sedimentation velocity analysis of the NylCp2-G122Y130A36Q263 enzyme showed the major peak (4.5 S: tetramer) and the minor peak (2.4 S: monomer). The absence of a dimer molecule suggested that the equilibrium of reactions 1 and 2 were optimized to yield tetramers as the major product, leaving monomers as a minor population. Alternatively, the tetrameric structure was coordinately generated from monomers in a single step (reaction 4).
For wild-type NylCK, the minor peak corresponding to higher oligomers (7.5 S) was identified in addition to the major peak of tetramers (4.2 S) and the minor peak of monomers (2.1 S). This result may imply that further association of the molecules was induced by at least one of the eleven mutations that distinguished NylCK and NylCp2-G122Y130A36Q263 enzymes (Fig. S1).
In conclusion, we revealed that the observed protein stability and oligomeric states are determined by how the monomer unit constituting the oligomeric structure is stabilized due to the subunit interactions in the tetrameric structure. In addition, we demonstrated that the extent of relative stabilization effects at a wider A/B interface on narrower A/D interfaces drastically affects the correct subunit assembly and thermostability and that the amino acid substitutions at specific positions alter not only the protein stability but also the catalytic activity, oligomerization, and processability from the precursor to the active enzyme.
Methods
DNA preparation and mutagenesis
The plasmids pSKFC4 (NylCp2), pSKRC4 (NylCA), and pSKKC4 (NylCK) contained 1.1 kb genes flanked by BamHI and PstI restriction sites, which were cloned into the expression vector pBluescript II SK(+) (Stratagene, La Jolla, CA.)9. E. coli JM109 competent cells were prepared by the conventional CaCl2 method9 and stored at −80 °C for later use.
Site-directed mutagenesis was performed using a PrimeSTAR Mutagenesis Basal Kit (Takara Bio, Inc., Shiga, Japan) with the primers listed in Supplemental Table S4. The plasmids that contained the 1.1 kb fragments with mutated NylCp2 were isolated from transformed E. coli JM109 cells. DNA sequencing confirmed that the desired mutations were introduced into the wild-type nylCp2 sequence.
To construct a random mutant library from the nylC gene, pSKFC4–1 (having G122Y130A36Q263 mutations in pSKFC4) was initially digested with PstI and BamHI, and the linearized DNA was amplified by PCR in the presence of nucleotide analogues (2, 5, 10, 40, or 70 μM concentrations of both 8-oxo-2′-deoxyguanosine-5′-triphosphate (8-oxo-dGTP) and 6H,8H-3,4-dihydropyrimido(4,5-C)(1,2)oxazin-7-one-8-β-D-2′ -deoxy-ribofuranoside-5′-triphosphate (dPTP)] (Jena Bioscience GmbH, Jena, Germany) using two primers, FE-BamHI and RE-PstI (Table S4)29,30. The amplified 1.1 kb fragment containing the nylC gene was digested with BamHI and PstI and the 1.1 kb BamHI-PstI fragment was recovered. The plasmid pSKFC4 was also digested with BamHI and PstI, and a 2.6 kb fragment was recovered. To obtain the mutant library, the two fragments were combined by ligation followed by transformation into E. coli KP3998 using ampicillin resistance as the selection marker9.
Culture, enzyme assay and enzyme purification
The culture of E. coli clones and subsequent enzyme purification were performed as previously reported9,11. In the NylC activity assays, the enzyme solution (0.1 ml) was mixed with an Ahx-cyclic oligomer solution (0.9 ml) [4 mg ml−1 of Ahx-cyclic oligomer in 20 mM phosphate buffer (pH 7.3) containing 10% glycerol (buffer A)] and incubated at 30 °C (standard assay conditions). An increase in the concentration of the amino group was found using trinitrobenzene sulfonic acid (TNBS)9,11. Kinetic studies were performed under standard assay conditions, with the exception of the different Ahx-cyclic oligomer concentrations used. NylC encoded on the Arthrobacter plasmid pOAD2 (NylCp2) and its mutant enzymes were expressed in E. coli JM109 and purified by ammonium sulfate fractionation, anion-exchange column chromatography, and gel-filtration chromatography11.
Analytical centrifugation
The sedimentation velocity and sedimentation equilibrium of each NylC enzyme (0.34 mg ml−1) in phosphate buffer were examined at 20 °C using analytical ultracentrifugation. Centrifugation was performed at 40,000 rpm and 14,000 rpm for sedimentation velocity and equilibrium experiments, respectively, by monitoring the absorbance at 280 nm at intervals of 5 min. Analytical ultracentrifugation experiments were performed using a Beckman-Coulter Optima XL-A analytical ultracentrifuge (Fullerton, CA) after pre-centrifugation at 3,000 rpm for 5 min. Double-sector 12-mm-thick charcoal-epon centerpieces and matched quartz windows were used for all experiments. The experimental sedimentation coefficients were corrected to s20, w, the sedimentation coefficient expressed in terms of the standard state of water at 20 °C, with the van Holde-Weischet method using the software UltraScan 8.0 (www.ultrascan.uthscsa.edu). Frictional ratios and partial specific volumes were also obtained using the UltraScan software31.
Crystallographic analysis
To analyze the molecular basis of protein stabilization, comparative structural analysis of mutant enzymes that differ in thermostability is informative. Previously, based on the three-dimensional structure of NylCA determined at 2.0 Å resolution, we identified the structural interactions occurring at the subunit interfaces A/D (D122G, H130Y, and D36A) and A/B (E263Q). However, the crystals of NylCA obtained contain 15 monomer molecules in a large unit cell (space group I222); unit-cell parameters a = 155.86, b = 214.45, and c = 478.80 Å). The presence of a large number of monomers in a single asymmetric unit is unsuitable for further crystallographic analysis at higher resolutions. Recently, we reported a new crystallization condition of the NylCp2 protein to obtain a crystal that belonged to the space group C2221 with two molecules in an asymmetric unit (unit-cell parameters a = 70.84, b = 144.90, and c = 129.05 Å) (Table S1).
The crystals of NylCp2 and its mutants (G122, R122, K122, V122, Y130, G122Y130, and G122Y130A36Q263) were grown by the sitting-drop vapor-diffusion method with ammonium sulfate as a precipitant in 0.1 M HEPES buffer (pH 7.5) containing 0.2 M NaCl and 25% glycerol11. Placing the crystal in a cold nitrogen stream at 100 K, cryocooling was performed for data collection. Diffraction data sets were collected at SPring-8 (Hyogo, Japan) with a beamline BL41XU equipped with an ADSC Q315 CCD detector system. The following parameters were chosen for data collection: wavelength, 1.0000 Å; crystal-to-detector distance, 200 mm; oscillation range per image, 1.0°. Indexing, integration and scaling of reflections were performed using the HKL-2000 program package32. Diffraction data were collected from the crystal of NylCp2 at resolutions of 1.60 Å. The obtained crystal was spindle-shaped, with a C-centered orthorhombic space group C2221 and unit-cell parameters a = 70.84, b = 144.90, and c = 129.05 Å. Diffraction data were collected from the NylCp2 crystal at resolutions of 1.60 Å (Table S1). From NylC mutants, diffraction data were collected at resolutions of 2.00 Å (NylCp2-G122), 1.20 Å (NylCp2-R122), 1.10 Å (NylCp2-K122), 1.05 Å (NylCp2-V122), 1.90 Å (NylCp2-Y130), 1.39 Å (NylCp2-G122Y130), and 1.03 Å (NylCp2-G122Y130A36Q263).
The NylC structure was determined by the molecular replacement method using one protomer (monomer A) of the NylCA structure (PDB ID: 3AXG) as a template. Rotation and translation search for molecular replacement was performed by PHASER33 within the CCP4 program suite (Collaborative Computational Project, Number 4, 1994). There were two molecules in the asymmetric unit. The crystal packing analysis revealed that each of the NylCp2 monomers seems to extensively interact with the adjacent monomer related by the crystallographic two-fold axis.
Rigid-body refinement was performed using the coordinates of the initial model to fit the unit cell of the NylCA crystal followed by positional and B-factor refinement with the program REFMAC34. With several cycles of manual model rebuilding by Coot35, the R-factor and R-free values were determined to be, respectively, 16.1% and 19.2% (for NylCp2); 17.3% and 22.6% (for NylCp2-G122); 15.9% and 20.1% (for NylCp2-Y130); 17.1% and 20.0% (for NylCp2-G122Y130); 12.2% and 14.3% (for NylCp2-K122); 12.2% and 14.3% (for NylCp2-R122); 13.3% and 15.2% (for NylCp2-V122); and 11.6% and 13.4% (for NylCp2-G122Y130A36Q263). Ramachandran analysis showed 636 residues (96.1%) in favored regions, 36 residues (3.9%) in allowed regions, and 0 residues (0.0%) in outliers for NylCp2. No residue was found in the outlying regions.
The structures of the NylC mutants were determined by the molecular replacement method using one protomer (monomer A) of the NylCp2 structure as a template, and subsequently, rigid-body refinements were performed. The results of the crystal structure analysis are summarized in Table S1.
The atomic coordinates and structural factors for NylCp2 (PDB ID code: 5XYG); NylCp2-G122 (PDB ID code: 5XYO); NylCp2-R122 (PDB ID code: 5XYP); NylCp2-K122 (PDB ID code: 5XYQ); NylCp2-V122 (PDB ID code: 5XYS); NylCp2-Y130 (PDB ID code: 5XYT); NylCp2-G122Y130 (PDB ID code: 5Y0L); NylCp2-G122Y130A36Q263 (PDB ID code: 5Y0M) have been deposited in the Protein Data Bank (http://www.rcsb.org/).
Figures illustrating the three-dimensional structural models were generated using the programs MolFeat (ver. 4.0, FiatLux Co., Tokyo, Japan) and Chimera36.
CD measurements and thermal stability analyses
CD spectra at far-UV wavelengths (200–250 nm) were measured using a spectropolarimeter (Jasco, model J-720WI, Tokyo, Japan). A cuvette with a path length of 1 mm was used for far UV CD measurements. The results are expressed as the mean residue molar ellipticity, [θ], which is defined as [θ] = 100 (θobs − θback) l−1 c−1, where θobs is the observed ellipticity in degrees, θback is the observed ellipticity in degrees in the absence of enzyme (or background), c is the molar concentration of the residue, and l is the length of the light path (in centimeters). The temperature was kept at 25 or 95 °C with a Jasco PTC-348WI Peltier system. For thermal transition curve experiments, the temperature was increased from 25 to 95 °C with a Jasco PTC-348WI Peltier system at 1 °C min−1 by monitoring the CD signal at 220 nm. The protein concentration used was 0.1 mg ml−1.
Tm and ΔH were determined by regression analysis using nonlinear least-squares fitting of data to a sigmoidal equation under the assumption of a two-state transition between the folded and heat-denatured states37. We performed thermodynamic analyses, although the thermal unfolding of proteins was not confirmed. The following equation describes the signal intensity monitored by CD (the observed ellipticity in degrees).
where θobs is the signal intensity monitored by CD; the pre- and post-unfolding baselines are described by a + bT and c + dT, respectively; T, Tm, and R indicate the temperature, the midpoint temperature of denaturation, and the gas constant, respectively; the change in enthalpy for the global unfolding of proteins is represented by ΔH; and the change in heat capacity is shown by ΔCp.
Quantitative reverse-transcription-PCR (qRT-PCR)
Total RNA was extracted from the cells using a PureLinkTM RNA Mini Kit (Thermo Fisher Scientific, Inc., Waltham, U.S.A.) according to the manufacturer’s instructions, and qRT-PCR analysis was performed using a One Step SYBR® PrimeScript™ RT-PCR Kit II (Takara Bio, Inc. Shiga, Japan), a Bio-Rad MiniOpticon Real-Time PCR system (Bio-Rad, Inc., Hercules, U.S.A.) and Bio-Rad’s Real-Time PCR system (MiniOpticon, Bio-Rad Japan, Tokyo, Japan). Two primers (FE-BamHI and RE-PstI) were used for cDNA synthesis and amplification of the nylC gene (Table S5). A typical reaction mixture for qRT-PCR contained template RNA (50 ng), 2x One Step SYBR RT-PCR Buffer 4 (Takara Bio, Inc.) (12.5 µl), PrimeScript One Step Enzyme Mix 2 (Takara Bio, Inc.) (1.0 µl), each primer (0.4 µM), and RNase-free H2O (up to 25 µl). The thermal program employed was as follows: initial denaturation at 42 °C for 5 s, 95 °C for 10 s, 40 cycles of denaturation at 95 °C for 5 s, annealing and extension at 60 °C for 30 s, and the melting-curve step. The gene expression level was evaluated as the relative amounts of a specific RNA calculated from the qRT-PCR data using the 2−ΔΔCT method38.
Immunological detection of NylC antigenic protein
Antibody preparation
Polyclonal antibody against purified NylCp2-G122Y130A36Q263 was ordered from Eurofins Genomics (Tokyo, Japan). Briefly, anti-NylC serum was prepared by subcutaneous inoculation of rabbits with the antigen emulsified in Freund’s complete adjuvant. Rabbits were bled after four injections (0.2–0.3 mg antigen per injection, total 0.9 mg).
Western blot analysis
After E. coli cells were lysed by sonication and centrifuged (10,000 × g for 5 min), the cell extracts (soluble fraction) and precipitates (insoluble fraction) were obtained. These samples were boiled in SDS solution and fractionated by SDS-PAGE (17.5%). Protein bands were detected by Coomassie brilliant blue staining. For immunological detection, the proteins contained in the gels were electrophoretically transferred to Immobilon PVDF Transfer Membranes (Pore Size: 0.45 µm, Millipore) using a blotting instrument (Atto, model AE-7500). NylC-antigenic proteins were detected by immunoassay using polyclonal antibody against purified NylCp2-G122Y130A36Q263 and goat anti-rabbit IgG H&L (HRP) (Abcam, Cambridge, U.K.)39.
Sandwich ELISA
Standard ELISA was conducted by the conventional method with modifications. Briefly, 100 µl of the anti-NylC serum (10 µg ml−1) was dispensed into each well of a 96-well microtiter plate (MaxiSorp, Nunc), and the plate was left overnight in a refrigerator for immobilization. After removing the solution and washing, blocking treatment was performed, and the amount of NylC antigenic protein was quantified using a Protein Detector ELISA Kit (SeraCare Life Science, Massachusetts, U.S.A.). The samples were automatically analyzed by measuring the absorbance at 405 nm using a Crocodile Mini-Workstation (Titertek-Berthold, Pforzheim, Germany). Standard NylC solutions were prepared using purified NylCp2-G122Y130A36Q263 enzyme (up to 10 µg l−1) by diluting the stock solution with the coating solution. The amount of the NylC-antigenic protein was estimated from the calibration curve.
Electronic supplementary material
Acknowledgements
This work was supported in part by a grant-in-aid for scientific research (Japan Society for Promotion of Science, Nos 26289317 and 16K144931). This study was part of the “High-Quality Protein Crystal Growth Experiment on KIBO” promoted by JAXA (Japan Aerospace Exploration Agency). Russian spacecraft Progress and/or Soyuz provided by the Russian Federal Space Agency were used for space transportation. A part of the space crystallization technology had been developed by the European Space Agency and the University of Granada. The synchrotron radiation experiments were performed at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal Nos 2012A6721, 2012B6721, 2014A6925, and 2014B6925) and at PhotonFactory (Proposal No. 2014G188).
Author Contributions
S.N. designed, analyzed and interpreted all of the experiments and wrote the manuscript. N.S. performed the X-ray crystallographic analysis and helped to write the manuscript. Y.-H.L. performed the ultracentrifugation and CD analysis and helped to write the manuscript. I.T., R.K., K.N., and Y.T. performed the biochemical experiments and analyzed the data. D.K., M.T., Y.G., and Y.H. planned the experiments and analyzed the data.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-27860-w.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Seiji Negoro, Email: negoro@eng.u-hyogo.ac.jp.
Naoki Shibata, Email: shibach@sci.u-hyogo.ac.jp.
Young-Ho Lee, Email: mr0505@protein.osaka-u.ac.jp.
References
- 1.Tomko RJ, Jr., Hochstrasser M. Molecular architecture and assembly of the eukaryotic proteasome. Annu Rev Biochem. 2013;82:415–445. doi: 10.1146/annurev-biochem-060410-150257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Timucin E, Sezerman OU. Zinc modulates self-assembly of Bacillus thermocatenulatus lipase. Biochemistry. 2015;54:3901–3910. doi: 10.1021/acs.biochem.5b00200. [DOI] [PubMed] [Google Scholar]
- 3.Senger M, Stripp ST, Soboh B. Proteolytic cleavage orchestrates cofactor insertion and protein assembly in [NiFe]-hydrogenase biosynthesis. J Biol Chem. 2017;292:11670–11681. doi: 10.1074/jbc.M117.788125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlissel G, Krzyzanowski MK, Caudron F, Barral Y, Rine J. Aggregation of the Whi3 protein, not loss of heterochromatin, causes sterility in old yeast cells. Science. 2017;355:1184–1187. doi: 10.1126/science.aaj2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nguyen P, Derreumaux P. Understanding amyloid fibril nucleation and aβ oligomer/drug interactions from computer simulations. Acc Chem Res. 2014;47:603–611. doi: 10.1021/ar4002075. [DOI] [PubMed] [Google Scholar]
- 6.Negoro S. Biodegradation of nylon oligomers. Appl. Microbiol. Biotechnol. 2000;54:461–466. doi: 10.1007/s002530000434. [DOI] [PubMed] [Google Scholar]
- 7.Negoro S, Kakudo S, Urabe I, Okada H. A new nylon oligomer degradation gene (nylC) on plasmid pOAD2 from Flavobacterium sp. J. Bacteriol. 1992;174:7948–7953. doi: 10.1128/jb.174.24.7948-7953.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kakudo S, Negoro S, Urabe I, Okada H. Nylon oligomer degradation gene, nylC on plasmid pOAD2 from a Flavobacterium strain encodes endo-type 6-aminohexanoate oligomer hydrolase: purification and characterization of the nylC gene product. Appl. Environ. Microbiol. 1993;59:3978–3980. doi: 10.1128/aem.59.11.3978-3980.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Negoro S, et al. Three-dimensional structure of nylon hydrolase and mechanism of nylon-6 hydrolysis. J. Biol. Chem. 2012;287:5079–5090. doi: 10.1074/jbc.M111.321992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagai K, et al. Enzymatic hydrolysis of nylons: quantification of the reaction rate of nylon hydrolase for thin-layered nylons. Appl Microbiol Biotechnol. 2014;98:8751–8761. doi: 10.1007/s00253-014-5885-2. [DOI] [PubMed] [Google Scholar]
- 11.Nagai K, et al. Shibata N.. Crystallization and X-ray diffraction analysis of nylon hydrolase (NylC) from Arthrobacter sp. KI72. Acta Cryst. 2013;F69:1151–1154. doi: 10.1107/S1744309113024263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kato K, et al. A plasmid encoding enzymes for nylon oligomer degradation: nucleotide sequence and analysis of pOAD2. Microbiol. 1995;141:2585–2590. doi: 10.1099/13500872-141-10-2585. [DOI] [PubMed] [Google Scholar]
- 13.Yasuhira K, et al. 6-Aminohexanoate oligomer hydrolases from the alkalophilic bacteria Agromyces sp. strain KY5R and Kocuria sp. strain KY2. Appl. Environ. Microbiol. 2007;73:7099–7102. doi: 10.1128/AEM.00777-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yasuhira K, Uedo Y, Takeo M, Kato D, Negoro S. Genetic organization of nylon-oligomer-degrading enzymes from an alkalophilic bacterium Agromyces sp. KY5R. J. Biosci. Bioeng. 2007;104:521–524. doi: 10.1263/jbb.104.521. [DOI] [PubMed] [Google Scholar]
- 15.Oinonen C, Rouvinen J. Structural comparison of Ntn-hydrolases. Protein Sci. 2000;9:2329–2337. doi: 10.1110/ps.9.12.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bompard-Gilles C, et al. A new variant of the Ntn hydrolase fold revealed by the crystal structure of L-aminopeptidase D-Ala-esterase/amidase from Ochrobactrum anthropic. Structure. 2000;8:153–162. doi: 10.1016/S0969-2126(00)00091-5. [DOI] [PubMed] [Google Scholar]
- 17.Elkins JM, Kershaw NJ, Schofield CJ. X-ray crystal structure of ornithine acetyltransferase from the clavulanic acid biosynthesis gene cluster. Biochem J. 2005;385:565–573. doi: 10.1042/BJ20040814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sankaranarayanan R, et al. The molecular structure of ornithine acetyltransferase from Mycobacterium tuberculosis bound to ornithine, a competitive inhibitor. J. Mol. Biol. 2010;397:979–990. doi: 10.1016/j.jmb.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 19.Okada T, Suzuki H, Wada K, Kumagai H, Fukuyama K. Crystal structures of γ-glutamyltranspeptidase from Escherichia coli, a key enzyme in glutathione metabolism, and its reaction intermediate. Proc. Natl. Acad. Sci. USA. 2006;103:6471–6476. doi: 10.1073/pnas.0511020103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boanca G, Sand A, Barycki JJ. Uncoupling the enzymatic and autoprocessing activities of Helicobacter pylori gamma-glutamyltranspeptidase. J. Biol. Chem. 2006;281:19029–19037. doi: 10.1074/jbc.M603381200. [DOI] [PubMed] [Google Scholar]
- 21.Morrow AL, Williams K, Sand A, Boanca G, Barycki JJ. Characterization of Helicobacter pylori gamma-glutamyltranspeptidase reveals the molecular basis for substrate specificity and a critical role for the tyrosine 433-containing loop in catalysis. Biochemistry. 2007;46:13407–13414. doi: 10.1021/bi701599e. [DOI] [PubMed] [Google Scholar]
- 22.Guo HC, Xu Q, Buckley D, Crystal GuanC. structures of Flavobacterium glycosylasparaginase. An N-terminal nucleophile hydrolase activated by intramolecular proteolysis. J. Biol. Chem. 1998;273:20205–20212. doi: 10.1074/jbc.273.32.20205. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Guo H-C. Crystallographic snapshot of a productive glycosylasparaginase-substrate complex. J. Mol. Biol. 2007;366:82–92. doi: 10.1016/j.jmb.2006.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saarela J, Oinonen C, Jalanko A, Rouvinen J, Peltonen L. Autoproteolytic activation of human aspartylglucosaminidase. Biochem. J. 2004;378:363–371. doi: 10.1042/bj20031496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michalska K, Brzezinski K, Jaskolski M. Crystal structure of isoaspartyl aminopeptidase in complex with L-aspartate. J. Biol. Chem. 2005;280:28484–28491. doi: 10.1074/jbc.M504501200. [DOI] [PubMed] [Google Scholar]
- 26.Kim Y, Yoon K, Khang Y, Turley S, Hol WG. The 2.0 Å crystal structure of cephalosporin acylase. Structure. 2000;8:1059–1068. doi: 10.1016/S0969-2126(00)00505-0. [DOI] [PubMed] [Google Scholar]
- 27.Michalska K, Bujacz G, Jaskolski M. Crystal structure of plant asparaginase. J. Mol. Biol. 2006;360:105–116. doi: 10.1016/j.jmb.2006.04.066. [DOI] [PubMed] [Google Scholar]
- 28.Michalska K, Hernandez-Santoyo A, Jaskolski M. The mechanism of autocatalytic activation of plant-type L-asparaginases. J. Biol. Chem. 2008;283:13388–13397. doi: 10.1074/jbc.M800746200. [DOI] [PubMed] [Google Scholar]
- 29.Zaccolo M, Williams DM, Brown DM, Gherardi E. An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J Mol Biol. 1996;255:589–603. doi: 10.1006/jmbi.1996.0049. [DOI] [PubMed] [Google Scholar]
- 30.Rasila TS, Pajunen MI, Savilahti H. Critical evaluation of random mutagenesis by error-prone polymerase chain reaction protocols, Escherichia coli mutator strain, and hydroxylamine treatment. Anal Biochem. 2009;388:71–80. doi: 10.1016/j.ab.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 31.Demeler B. UltraScan A Comprehensive data analysis software package for analytical ultracentrifugation experiments. Modern Analytical Ultracentrifugation: Techniques and Methods. D. J. Scott, S. E. Harding and A. J. Rowe. Eds Royal Society of Chemistry (UK) 210–229 (2005).
- 32.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.McCoy AJ, et al. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 36.Pettersen EF, et al. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 37.Kim JY, et al. Non-covalent forces tune the electron transfer complex between ferredoxin and sulfite reductase to optimize enzymatic activity. Biochemical J. 2016;473:3837–3854. doi: 10.1042/BCJ20160658. [DOI] [PubMed] [Google Scholar]
- 38.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 39.Negoro S, et al. Construction of hybrid genes of 6-aminohexanoic acid-oligomer hydrolase and its analogous enzyme. Estimation of the intramolecular regions important for the enzyme evolution. J Biol Chem. 1984;259:13648–13651. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.