

Summary
Advances in ‘omics technologies now allow for an unprecedented level of phenotyping for human diseases, including obesity, where individual responses to excess weight are heterogeneous and unpredictable. To aid the development of better understanding of these phenotypes, we performed a controlled longitudinal weight perturbation study combining multiple omics strategies (genomics, transcriptomics, multiple proteomics assays, metabolomics and microbiomics) during periods of weight gain and loss in humans. Results demonstrated that: a) Weight gain is associated with the activation of strong inflammatory and hypertrophic cardiomyopathy signatures in blood; b) Although weight loss reverses some changes, a number of signatures persist, indicative of long-term physiologic changes; c) We observed ‘omics signatures associated with insulin resistance that may serve as novel diagnostics; d) Specific biomolecules were highly individualized and stable in response to perturbations, potentially representing stable personalized markers. Most data are available open access and serve as a valuable resource for the community.
eTOC Blurb
Extensive multi-omic profiling of the blood and microbiomes of healthy and insulin resistant humans as they gain and lose weight reveals insights into the systemic impacts of weight gain.
Introduction
With 34% of the US adult population classified as obese (Flegal et al., 2010) and 3 million obesity-attributable deaths worldwide (Finucane et al., 2011), it is imperative that we gain a better understanding of the factors contributing to obesity-associated morbidities, especially cardiovascular disease (CVD), cancer, and type 2 diabetes mellitus (T2DM). These diseases are mediated in part by insulin resistance (IR) (Kahn et al., 2006). While IR is overall positively correlated with measures of adiposity, there is great heterogeneity in metabolic phenotypes between individuals who are similarly obese; as such our ability to predict these phenotypes is poor. One reason for this is that the onset of metabolic disease and related complications involves numerous pathways and complex interactions between metabolically active tissues such as fat, liver, and muscle (Kahn et al., 2006) (and most recently implicated, the microbiome (Janssen and Kersten, 2017)). As such, a more comprehensive molecular profiling approach may offer novel insights into the diversity of systemic responses to the increase in body fat stores.
While most human studies have simply examined plasma markers that occur in association with increasing adiposity, another way to approach this problem is to compare equally-obese individuals who are either IR or insulin sensitive (IS) to characterize differences between these metabolically-disparate groups. Many metabolic risk markers differ markedly as a function of IR/IS independent of adiposity (McLaughlin et al., 2002; McLaughlin et al., 2007; McLaughlin et al., 2006). Induction of obesity in mice via high fat diets has revealed important insights into the biological links between weight gain and metabolic disease. While inflammation and oxidative stress are clear players in mice, there are very few studies on determinants of insulin resistance/metabolic disease in response to weight gain and loss in humans, and causality is harder to determine given the relative difficulty in manipulating genes or proteins in human subjects.
The integration of multiple large-scale omics profiling technologies across biological fluids and tissues has recently been demonstrated to offer novel insights into disease development and progression, including T2DM (Chen et al., 2012; Hood et al., 2015; Lee et al., 2016; Price et al., 2017; Williams et al., 2016). In this context, we performed a controlled short-term weight gain and weight loss intervention in humans. Overweight to moderately-obese (body-to-mass index (BMI) 25–35 kg/m2) healthy, nondiabetic participants were selected to span a wide range of insulin resistance. This group was targeted since they are most at risk for T2DM, and by subjecting individuals of different insulin resistance profiles to identical weight perturbations, we sought to identify pathways involved in obesity-mediated insulin resistance. Furthermore, subjects already overweight require little additional weight gain to trigger stress responses in adipose tissue (McLaughlin et al., 2014) which contributes to systemic responses mediating disease states such as type 2 diabetes and cardiovascular disease (McLaughlin et al, 2016). Unlike many prior studies, the current human study entailed a perturbation in body weight such that changes with gain and loss could be evaluated and in particular, with insulin resistance, which was quantified by a gold-standard physiologic measure (modified insulin suppression test (Greenfield et al., 1981; Pei et al., 1994; Shen et al., 1970)). Multi-omics profiling including genomics, transcriptomics, proteomics, metabolomics from blood peripheral blood mononuclear cells (PBMCs), plasma and serum, and microbiomics from stool was performed over the course of the study to generate a wealth of personal longitudinal data, thereby enabling a detailed map of the individual molecular changes that occur in response to weight gain and weight loss in the context of insulin sensitivity and resistance. Our study revealed a number of important findings including: 1) modest weight gain in overweight humans was associated with the activation of inflammatory signatures in the blood, induction of markers and pathways for cardiovascular disease, and significant changes to the microbiome; 2) many of these changes affected IR participants differently from metabolically healthy controls (e.g. microbiome and metabolome differences); 3) the longitudinal nature of the designed perturbation results in statistical power increases that are equivalent to orders-of-magnitude larger cross sectional studies, an important consideration for the design of future large-scale (and costly) multi-omics studies.
Results
Overview of the multi-omics study
The overall goal of the study was twofold: A) assemble a comprehensive map of the molecular changes in humans (in circulating blood as well as the microbiome) that occur over the course of a carefully controlled weight gain and reversibility with weight loss; and B) determine whether IS and IR individuals who are matched for degree of obesity demonstrate unique biomolecular signatures and/or pathway activation during similar weight gain. Identifying specific molecules and/or pathways that characterize IR versus IS individuals may reveal the fundamental mechanisms by which obesity potentiates insulin resistance and associated diseases such as type 2 diabetes and cardiovascular disease. Participants were recruited as part of the current iteration of the Human Microbiome Project (iHMP) (Integrative, 2014), and ‘omic data are open access (http://hmp2.org/).
We sampled 23 carefully selected healthy participants with BMI 25–35 kg/m2 at baseline before perturbation and during periods of short-term weight gain followed by weight-loss, with samples drawn and metabolic measurements made at baseline (T1), peak weight (T2) and post weight loss (T3) (Fig. 1), and after three months of weight stability following return to baseline weight (T4). All subjects provided written informed consent and all evaluations and blood/stool samples were obtained in the Clinical and Translational Research Unit (CTRU) after an overnight fast. Metabolic phenotyping included quantification of insulin-mediated glucose uptake using the modified insulin-suppression test (Pei et al., 1994; Shen et al., 1970), which replaces endogenous insulin secretion with a controlled intravenous infusion of insulin and glucose such that at steady state all individuals will have the same insulin concentration, but different plasma glucose concentrations, which reflect the relative ability of insulin to dispose of a glucose load. In this test, which has been validated against the euglycemic clamp method (Greenfield et al., 1981), the higher the steady-state plasma glucose (SSPG) indicates relative resistance to insulin action. Thirteen fasted overweight IR participants (defined by SSPG ≥ 150 mg/dl (Yeni-Komshian et al., 2000), Table S1 and Materials and Methods); and ten BMI-matched healthy overweight IS participants completed the study. Three others dropped out after baseline tests and were not included in the analyses. Comprehensive anthropometric, clinical, and plasma measures of general health (waist circumference, blood pressure, hematocrit, cholesterol, triglycerides, liver function tests, creatinine, etc.) were also performed (Table S1).
Fig. 1.
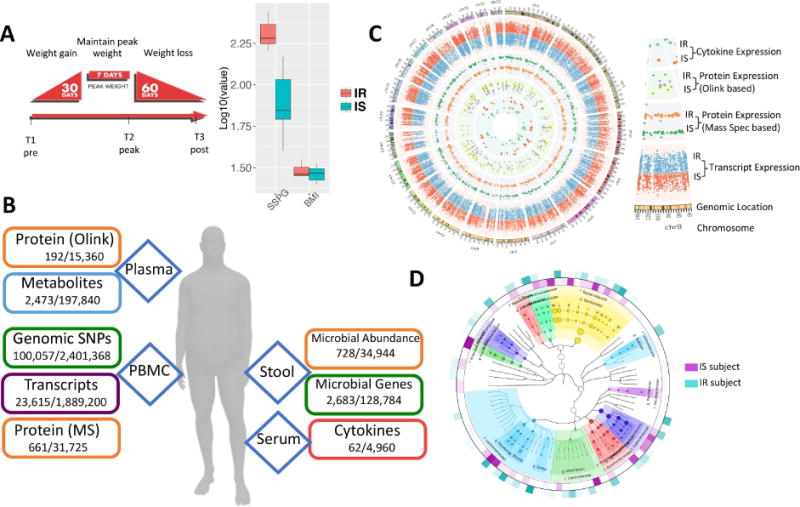
Overview of the multi-omic weight perturbation experiment. (A) Schematic of the weight gain and loss perturbation. The sampling timepoints (T1–T3) are indicated at the specific time in the perturbation when they occur. Inset: SSPG and BMI measurements for IS and IR subjects. (B) The types of ‘omics analyses performed are indicated along with the types of biological materials they are performed on. Listed below each data type are the number of analytes measured per timepoint for each individual, as well as the total number of analytes measured across the study. (C) Circos plot of multi-omic data points from selected assays. The transcriptome, both targeted and untargeted proteome and serum cytokine levels are plotted according to their genomic location as well as the average expression in IR and IS participants (see inset labels). (D) The stool microbiome phylogenetic tree is visualized by GraPhlAn for taxonomies present across all participants along with the respective relative abundance in IR and IS (outer layers).
Blood and stool were sampled from fasted subjects at baseline (T1, Fig. 1A); participants then underwent a controlled hypercaloric diet for a period of 30 days, with each participant supplementing their normal diet with high-caloric foods provided by the Stanford CTRU Research Kitchen (see Material and Methods) to achieve an average of 880 kcal per day as determined for each individual by resting metabolic caloric requirement and an activity factor (Material and Methods). At the end of the 30-day weight gain period, participants maintained a eucaloric diet for seven days, at which point a second fasted sampling of blood and stool was collected (T2, Fig. 1A). Participants gained an average of 2.8 kg over the course of the perturbation (Fig. S1). Each participant then underwent a caloric-restricted diet under nutritionist supervision (see Material and Methods) for a subsequent 60-day period designed to return each participant back to his/her initial baseline weight, at which point a third set of fasted samples of blood and stool were collected (T3, Fig. 1A). A subset of participants returned for a follow-up sampling approximately three months after the end of the perturbation (T4). A total of 90 timepoints were sampled. The large-scale multi-omics assays performed at all timepoints were: genomics (germline exome sequencing from whole blood, performed once for each individual), transcriptomics (RNA-seq from blood PBMCs performed over the timecourse), proteomics performed three different orthogonal ways (untargeted liquid chromatography - mass spectrometry (LC-MS) from blood PBMCs, 276 targeted protein assays from plasma (Proseek multiplex) and a 63-protein cytokine/chemokine/adipokine panel from serum), metabolomics (untargeted LC-MS from plasma using a pipeline we recently developed (Contrepois et al., 2015)) and microbiomics done using two different methods (16S and whole metagenome sequencing from stool) (Fig. 1B and C). In total, across all timepoints over 2 million unique datapoints were measured.
Differences in omics profiles and biochemical pathways between IR and IS participants
We first examined whether baseline differences were detectable in omics profiles between insulin sensitive and insulin resistant individuals; such differences may influence how individuals respond to the dietary perturbation. In order to identify any individuals that may harbor germline mutations in known metabolic disease susceptibility genes as well as enable accurate mapping of the different omics data, exome sequencing was performed on each subject followed by reconstruction of their personal exome which was used for improved transcriptome mapping. A detailed analysis of the genomic risk of the subjects is reported elsewhere (Rego et al., 2017), however we did not find any known high-risk variants for diabetes or insulin resistance (e.g. MODY mutations), thus allowing us to study insulin resistance in a population without elevated Mendelian genetic risk. As expected, the overall density of variants was consistent from participant to participant (Fig. S1).
At baseline, modest differences were evident between IR and IS groups. In blood PBMCs, significant transcripts differentially expressed between IR and IS (Fig 2A and Table S2) comprised a number of common significant functional groups including pathways associated with the cardiovascular system (FDR < 0.0001), angiogenesis (FDR < 0.04) and actin/myosin cell motility (FDR < 0.00001), as well as multiple pathways representing an immune/inflammatory response (FDR < 0.05) (Fig. S2 and Table S2). To explore whether these expression differences translated to the proteome level, we performed the same comparison in LC-MS proteomic profiles of the same PBMC population. Baseline analysis revealed three proteins (TC2N, DMTN and PKD1) that were different between IR and IS participants; the smaller number of differential proteins versus RNA is likely due to the size of the cohort and potentially higher variability of protein levels. Indeed, expanding our analysis to the pathway level, we observed significantly enriched pathways that closely mirrored those uncovered in the transcriptome as well as those that were unique to the proteome (fat cell differentiation, etc.) (Table S2). A combined ranked pathway analysis of transcriptome and proteome from PBMCs further confirmed the enrichment of these core pathways (Fig. 2B), as well as uncovered additional enriched pathways that spanned RNA and proteins (platelet/blood coagulation, plasminogen activation). Despite the increased immune/inflammatory responses observed here in IR participants, we note that all participants self-identified as healthy (no common colds etc.) at the time of sampling, so it was particularly striking that participants exhibited this phenotype at baseline.
Fig. 2.
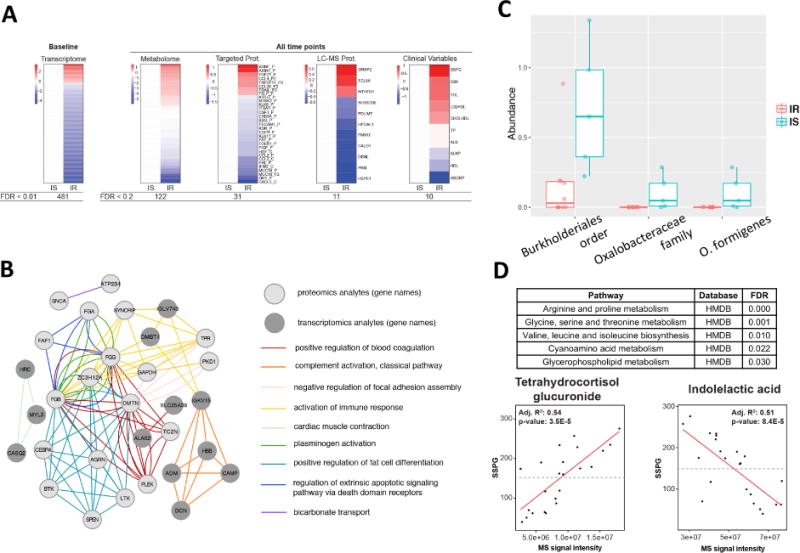
Differences between IR and IS participants at baseline. (A) Heatmap showing differences between IR and IS in baseline molecular abundance for each ‘omic type. Each analyte is normalized according to the average expression in IS and significant differences in the IR group are plotted (red=upregulated in IR, blue=downregulated in IR). (B) Pathways exhibiting significant transcriptomic and proteomics differences between IR and IS. The top Gene Ontology categories are presented and top transcripts and proteins are plotted in a network diagram showing pathway connections. (C) Differences in microbial abundance (%) between IR and IS by both 16S and shotgun metagenomic sequencing. (D) Regression analysis detailing association of multiple metabolites with clinical steady-state-plasma glucose (SSPG). MS signal intensity is plotted versus SSPG (mg/dl) for the selected metabolites indolelactic acid and tetrahydrocortisol glucuronide. Inset are the R2 and p-values for the selected comparisons.
Using both 16S and shotgun metagenomics of the stool microbiome, we observed significant differences between IR and IS participants in the abundance of the gram-negative proteobacterium Oxalobacter formigenes (p < 0.006, Fig. 2C, Table S2). Interestingly, whereas this bacterium was present at relatively high levels in IS participants, it was not detected in any of the IR participants’ samples. O. formigenes is particularly unique in that it processes oxalate, and absence of this bacterium is associated with increased risk of kidney stones (Duncan et al., 2002), which was linked to diabetes and insulin resistance (Chung et al., 2011; Daudon et al., 2006), and can be sensitive to high oxalate-containing foods such as almonds (Haaskjold et al., 2015). While we only observed significant IR-associations with one microbe at baseline, we hypothesized that this may be due to the small sample size of the cohort. As such, we expanded the analysis to include all timepoints, revealing a substantially different picture. Both 16S and shotgun metagenomics identified that bacteria of Alistipes genus under the Rikenellaceae family were more abundant in IS than IR subjects (p < 0.04). Alistipes were shown previously to associate with glucose regulation, diabetes and obesity (Brown et al., 2011; Ridaura et al., 2013; Serino et al., 2012). Our results demonstrate that many important biological molecules, pathways and microorganisms differ in IR and IS patients. Although some of these differences were known previously in diabetics or obese individuals relative to healthy people, our results demonstrate that these differences are already present in earlier stages/aspects of the disease.
Expanding the analysis to include all timepoints also revealed IR-associated signatures in other omic data types. In plasma, we observed an IR-associated proteomic signature spanning multiple targeted assays (Fig. 2A, Table S2). Among these include the folate receptor FOLR1 (FDR < 0.004), and the hormone prolactin (FDR < 0.007), and brain-derived neurotrophic factor (BDNF) (FDR < 0.03). BDNF has been shown to be one of the main signaling factors for appetite as well as heart function (Feng et al., 2015; Fulgenzi et al., 2015; Rosas-Vargas et al., 2011). The former is an interesting target as folate supplementation has been explored as a method to combat metabolic disease-associated cardiovascular disease (van Etten et al., 2002) and prolactin levels have been shown to be inversely associated with diabetes (Wang et al., 2013). Along with targeted plasma proteomic assays, clinical blood panels showed 10 analytes that were significantly different between IS and IR participants (Table S2, FDR < 0.2). Most of these clinical variables were associated with dysregulated lipid metabolism (i.e. triglycerides (TGL), lipoprotein LDL:HDL ratio, etc.) and inflammation (C-reactive protein (hs-CRP)), consistent with results from PBMCs and plasma (Fig. 2A).
Untargeted metabolomic profiling of plasma also led to the identification of 122 metabolites different between IR and IS participants (FDR < 0.2) (Fig. 2A, Table S2). While the SSPG cutoff for defining IR and IS has precedent clinically, we asked whether different thresholds for defining IR and IS led to the identification of different numbers of metabolites. We observed that changes to the SSPG cutoff (from 100 to 170) did not significantly change the number of differential metabolites in IR vs IS participants (Fig. S2) suggesting that there is some consistency in metabolic differences across a relatively wide range of insulin resistance measurements.
Pathway analysis revealed that amino acid as well as lipid metabolism were deregulated in IR individuals compared to healthy controls (FDR < 0.05) (Fig. 2). These observations are consistent with the observation that compromised insulin action is associated with altered intermediary metabolism of fats and amino acids (Adams, 2011). In particular, we found that the branched-chain amino acid (BCAA) biosynthetic pathway was deregulated in insulin resistant participants (FDR = 0.01) which is consistent with previous findings (Pedersen et al., 2016; Yoon, 2016). In addition to BCAAs, many more amino acids such as sulfur-containing amino acids (e.g. methionine) were found in higher levels in the plasma of insulin-resistant individuals (Fig. S2). Lipid metabolism was also altered with the increased level of many short- and medium-chain acylcarnitines, and the reduction of phospholipids and plasmalogens (Fig. S2). Accumulation of acylcarnitines is commonly observed in cases of insulin resistance and T2DM (Schooneman et al., 2013).
For highly significant metabolites, we also tested how well-correlated their plasma abundances were with clinical insulin resistance measures (e.g. SSPG), thus indicating their potential as a biomarker for IR. Indeed, tetrahydrocortisol glucuronide was strongly positively associated with baseline SSPG levels and indolelactic acid was negatively associated with SSPG levels (Fig. 2D). Interestingly, indolelactic acid has recently been associated with insulin resistance (Pedersen et al., 2016). Tetrahydrocortisol glucuronide is a modified version more water-soluble of tetrahydrocortisol and allow elimination from the body and/or ease its transportation around the body. To the best of our knowledge, this is the first report of an association between insulin resistance and tetrahydrocortisol glucuronide, and based on these data, both metabolites show promise as an IR biomarker.
We next attempted to use the metabolome to predict SSPG values. We performed binary classification using ensemble learning, as well as quantitative prediction using delta SSPG values based on delta metabolomics features. Using Random forest and AdaBoost classification, we achieved an accuracy rate of 87.5 % on our held-out test set (see STAR Methods). Predictive performance measure in terms of Precision, Recall, and F1 score were 87% for the Random forest method and 78% for AdaBoost (Fig. S2). The high accuracy and precision shows that the model was able to correctly predict the future SSPG trend by only looking at the difference between the current and future metabolomics features. From both methods, only one wrong prediction was made, which was a false positive (see confusion matrix, Fig. S2). For the regression model to compute ΔSSPG at time (tn), we used LASSO and Elastic Net regularized regression, with 10-fold cross validation. We achieved a low root-mean-square error (RMSE) of 27.5110 for the Elastic Net model. Regression curves (Fig. S2) show that the predicted ΔSSPG(tn) are very close to the actual values for most samples. In conclusion, metabolomics profiling has significant potential as a predictor for changes in insulin resistance.
Omics profiles and pathways associated with weight gain
Following characterization of group differences, we next examined the biomolecules and pathways that were specifically responsive to the 30-day weight gain perturbation (Fig. 3). Comparing omics profiles at peak weight to baseline profiles, we observed a number of significant changes across multiple omes. Specifically, at the transcriptome level, we observed 318 transcripts that were significantly differentially expressed between baseline and peak weight (FDR < 0.01, Table S3). As expected, this included an increase in expression for a number of genes associated with lipid metabolism such as lipoprotein lipase (LPL, FDR < 5 × 10−5). Pathway enrichment analysis showed that these genes comprised a number of common functional categories (Fig. 3B), including a large number of inflammatory response genes and pathways at peak weight; this may indicate significant stress response despite the relatively modest weight gain (average 2.4 kg). Importantly, we also observed an increase in expression of genes associated with dilated cardiomyopathy, potentially reflecting a molecular explanation for the association between weight gain and heart failure (Dela Cruz and Matthay, 2009).
Fig. 3.
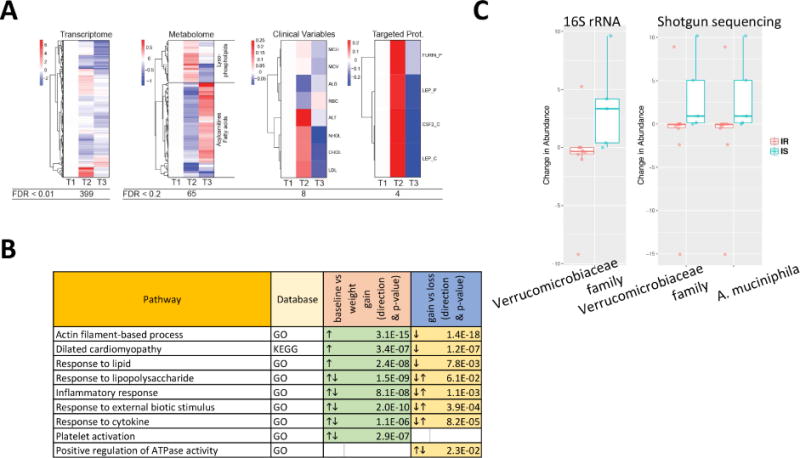
Multi-omic differences over the course of weight gain and loss perturbation experiment. (A) Heatmap showing analytes that vary in abundance in response to the weight gain and loss perturbation. (B) Pathways that are significantly different between baseline versus weight gain, and weight gain versus weight loss, respectively. (C) IR or IS-specific microbiome changes are shown for selected taxonomic units confirmed by both methods (16S and shotgun metagenomics).
At the microbiome level, 16S sequencing revealed a significant increase in microbes of the Verrucomicrobiaceae family in response to weight gain; using shotgun metagenomics we were able to further specify this response to the species Akkermansia muciniphila (p < 0.03; Fig. 3C and Table S3). Interestingly, this response was only observed in the insulin sensitive participants; this is of particular note in light of prior studies showing that A. muciniphilia confers a protective effect against insulin resistance in response to weight gain in animal models (Everard et al., 2013; Roopchand et al., 2015). Overall, these results indicate that there are substantial biological pathways that change during weight gain that affect our immune response, heart function, biochemistry and microbiome.
Omics profiles and pathways associated with weight loss
As short-term weight gain induced a number of significant blood-based and microbiome-based changes, we next determined whether these responses would persist or revert upon each participant’s return to baseline weight. We compared omics profiles assayed after weight loss to those of the previous peak weight sample and again observed a number of significant changes across multiple omes (Fig. 3). At the transcriptome level, 213 genes were significantly differentially expressed in weight loss versus peak weight (Fig. 3A and Table S3). Pathway enrichment analyses strikingly revealed that the majority of the significant pathways observed after weight gain reversed after subsequent weight loss (Fig. 3B); examples include the genes associated with lipid metabolism and inflammation. This reversal upon weight loss is of particular importance as it suggests that the negative effects of short-term weight gain can potentially be ameliorated by corrective action.
Mirroring the dynamic changes of RNA levels in PBMCs, most of the 65 plasma metabolites that changed upon weight gain reversed after weight loss (Fig. 3A and Table S3; FDR < 0.2). Interestingly, most of these molecules function in lipid metabolism, including acylcarnitines and fatty acids, showing that a subtle change in weight profoundly impacts lipid metabolic pathways. We specifically examined associations with BMI, and identified 133 metabolites that were significantly associated with changes in BMI (Table S3). Similar to the time points model, most changing molecules belong to lipid metabolism and include many acylcarnitines, fatty acids and lysophospholipids (Fig. S3), possibly reflecting decreased catabolism due to an increased calorie intake. We also had the opportunity to validate these results in a separate yet-unpublished weight gain cohort conducted in Sweden with blood LC-MS metabolomics generated using the Metabolon platform. We examined the subset of metabolites that were positively identified by both platforms and asked whether they behaved similarly across both cohorts. Of these, we found that 77% of weight-responsive metabolites were also enriched upon weight change in the second study (Table S4). Thus, despite differences in study populations and metabolomic analytical platforms, weight-responsive metabolites were reproducible across studies.
Targeted proteomic assays (Luminex and Proseek) revealed 27 proteins (FDR < 0.2) associated with BMI change, with the adipokine leptin (p < 8×10−5) positively associated with BMI change (Fig. S3 and Table S3) with a substantial 30% increase in leptin levels per unit of BMI; this is consistent with previous cross-sectional studies finding increased leptin levels in obese subjects (Considine et al., 1996). Novel significant responses to changes in BMI included the immuno-modulating covertase furin (p < 8×10−5) (Pesu et al., 2008), as well as lipopolysaccharide induced TNF Factor (LITAF) (p < 4×10−5). The latter is of particular note considering that LPS-responsive immune/inflammatory pathways also exhibited a significant response in the PBMC transcriptome. As LITAF and LPS-responsive pathways are known to respond to endotoxins of microbial origin, this response may indicate crosstalk between the microbiome and the human host’s immune system in response to weight gain/loss. Consistent with this interpretation was the association of white blood cell counts with delta BMI. Also associated with delta BMI were non-HDL cholesterol (p < 0.03), LDL-cholesterol (p < 0.02) measured as part of the clinical panel (Table S3). In summary, these results indicate that most changes identified during weight gain reverse upon weight loss.
Uncovering trends in response to dietary change
While the prior analysis provides insight into biomolecules that vary along with changing weight/BMI, it is possible that more complex patterns may be evident across the perturbation. Specifically, we were interested in analytes that a) may have changed in response to weight gain but did not revert upon subsequent weight loss (and indicative of a long-term effect) or b) molecules that may not have changed upon weight gain but were only responsive to weight loss. To better understand the breadth of possible responses, we performed longitudinal pattern recognition analysis across blood-based analytes (transcriptome, proteome and metabolome) using fuzzy c-means clustering. From these data, we observed diverse response patterns to the perturbation (Fig. 4A).
Fig. 4.
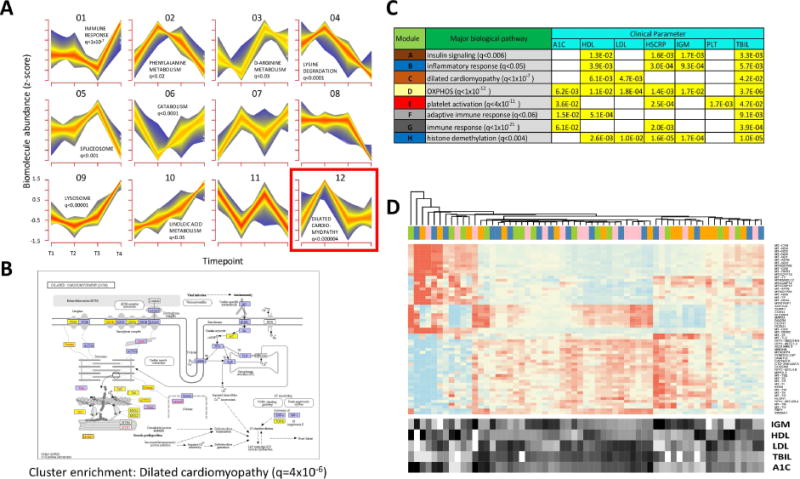
Multiparametric and trend analyses reveal novel responses to weight gain and loss. (A) Longitudinal pattern recognition using fuzzy c-means clustering across all host ‘omes. Data from the transcriptome, proteome, cytokines and metabolites were standardized to z-scores for each analyte and subjected to c-means clustering across all four timepoints. Each subplot shows a unique cluster and the trend for all analytes comprising the cluster. (B) KEGG pathway diagram for analytes implicated in dilated cardiomyopathy, a pathway that was significantly enriched in Cluster 12 (FDR < 0.000004). Elements highlighted in yellow indicate the pathway analytes that comprise Cluster 12. (C) Table showing biological pathway enrichment and association with clinical blood panel analytes for key gene co-expression clusters. (D) Gene expression heatmap for transcripts comprising the yellow module from Fig. S3. The expression for each gene is shown for all timepoints (T1, pink; T2, blue; T3, orange; T4, green.) along with the relative levels for each of the enriched clinical parameters (A1C, LDL, HDL, IGM and bilirubin (TBIL)).
From these analyses, a number of key patterns were evident. For the pattern observed in Cluster 12 (Fig. 4A), which comprised an initial increase in biomolecular abundance followed by a decrease back to baseline that persisted through the quarterly follow-up visit, and most closely resembled the weight gain/loss trajectory each participant exhibited (Fig. S1), we observed a highly significant enrichment for genes and proteins associated with risk for dilated cardiomyopathy (FDR < 5×10−6, KEGG enrichment via the DAVID algorithm (Huang da et al., 2009a, b)). As shown by the KEGG pathway diagram (Fig. 4B), this enrichment included a large number of members of this functional category spanning the cell surface receptors to the internal machinery. The overfeeding-induced changes in these analytes may indicate ill-effects of the short-term weight gain, such as increased coronary artery disease, however it is important to note that this biomolecular signature returned to baseline upon subsequent weight loss. Other intriguing responses included sets of biomolecules that were anticorrelated with weight gain. A cluster genes/proteins and metabolites comprising catabolic pathways decreased upon weight gain and returned to baseline upon subsequent weight loss (Fig. 4A, Cluster 6). A downregulation of catabolism is logical in an overfed state as the body may switch to a largely anabolic state with an overabundance of energy intake from food.
Of particular note was a cluster of biomolecules that increased upon weight gain but failed to return to baseline at the end of the weight loss period (Fig. S4). Enrichment testing showed that Cluster 2 comprised biomolecules associated with a variety of metabolic pathways including folate metabolism (p < 0.001), phenylalanine metabolism (p < 0.005) and branched-chain amino acid degradation (p < 0.03). Thus, these pathways have long-lasting effects after weight gain.
In order to identify biomolecular trends across the dataset in a more unbiased fashion, we performed weighted gene-coexpression analysis (Zhang and Horvath, 2005) across all conditions and timepoints (Fig. S4). From this analysis, a series of core gene expression patterns emerged, and the co-expressed genes were functionally related based on pathway enrichment. We next asked whether these common expression patterns were significantly associated with any of the clinical variables tested and thus may cause clinical phenotypes. A number of these significant associations emerged (Fig. 4C). For example, a gene module significantly enriched for mitochondrial genes involved in oxidative phosphorylation (FDR < 1×10−12) was significantly associated with multiple clinical parameters including glycated hemoglobin (HbA1C), C-reactive protein (hs-CRP), and LDL cholesterol (Fig. 4D); this may suggest a link between mitochondrial activity and diabetes/cardiovascular related issues, thus showing that novel associations can be gleaned from the unsupervised associations.
Associations between ‘omic measures and the microbiome
In order to better understand how the microbiome may interact with the changes we observed in human molecular physiology over the course of the dietary perturbation, we explored a) how microbes co-vary with each other across the entire dataset and b) how microbes co-vary with metabolites in the host. Some microbes showed a high degree of covariance in both IR and IS subjects over the course of the perturbation experiment; for example, the proportion of Bacteroides dorei was positively correlated with Alistipes putredinis (Fig. 5A, highlighted by squares). B. dorei has been shown to modulate immune responses in humans (Vatanen et al., 2016) and A. putredinis has been shown to be significantly responsive to dietary changes (David et al., 2014), thus painting a complex portrait of how diet-induced changes to the microbiome may relate to the immune changes we observe in the host. In addition to these similarities, some microbiota show strikingly opposite correlations depending on IR and IS context. For example, Eubacterium Hallii and Parabacteroides exhibit a strong positive correlation in IR participants across timepoints and a negative correlation in IS individuals. This is of particular interest, as Eubacterium Hallii has been shown to metabolize glucose and is a major contributor of short-chain fatty acids in the gut (Engels et al., 2016), whereas Parabacteroides has previously been shown to be directly associated with weight gain (Lecomte et al., 2015). It is intriguing to speculate that the unique gut microenvironment in IR versus IS individuals contributes to these differing responses to host dietary intervention and weight changes. Additional examples are Bacteroides vulgatus and Eubacterium eligens which exhibit a positive correlation in IR individuals and a negative correlation in IS individuals (Fig. 5A, highlighted by circles). This is of particular note as B. vulgatus has been shown to be a mediator between BCAA metabolism and insulin resistance (Pedersen et al., 2016), whereas E. eligens has been shown to respond significantly to dietary fiber (Chung et al., 2016). Furthermore, most pair-wise correlations were significant only in IR or IS individuals, again showing a potential difference where IR individuals may possess a unique microbiome that possibly contributes to the pathology or progression of their disease.
Fig. 5. Associations of analytes across IR and IS and across ‘omes.
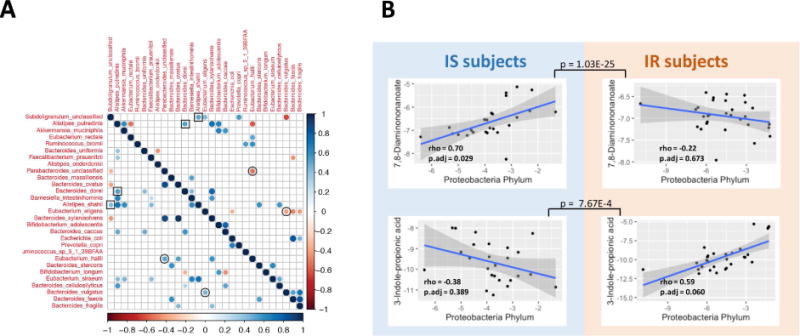
(A) Co-varying microbial species are plotted based on whether they are co- or inversely associated (blue or red, respectively), and whether this occurs in IR (upper quadrant) or IS (lower quadrant). (B) Co-variation of microbes and metabolites for IR and IS is plotted for selected associations. Inset are the Spearman’s rho and adjusted p-values after FDR correction for the selected associations. Also, adjusted p values by FDR are shown between IS and IR individuals for the interaction term of linear model describing different trends in two groups.
The contribution of microbial products to host metabolic signaling is one of the fundamental mechanisms underlying the host-microbiome interactions (Holmes et al., 2011). In order to better understand how these microbiome alterations are associated with host metabolism, we examined whether specific microbes exhibited correlation with host metabolites. Associations between microbial populations at the phylum level (with at least 1% abundance) and metabolite quantities revealed 26 cross-omics associations significantly different between IR and IS (p < 0.1 for the interaction term in the ANOVA model), of which 8 showed significant associations only in IR or IS individuals (Table S3). As an example of these, we found that the antioxidant 3-indolepropionic acid was positively associated with Proteobacteria, but specifically in the IR subjects. As a second example, N6-Trimethyllysine was significantly positively associated with the phylum Proteobacteria exclusively in the IS participants (Fig. 5B). It is worth noting that 3-Indolepropionic acid is a microbial fermentation product from tryptophan, and it can be a potential drug target for the management of insulin resistance (Khan et al., 2014). In the case of N6-Trimethyllysine, it is a precursor for L-carnitine biosynthesis, which improves glucose tolerance, increases total energy expenditure in obesity (Flanagan et al., 2010) and decreases cardiovascular risks (Koeth et al., 2013; Ussher et al., 2013). These results demonstrate that microbial differences exist between IR and IS participants and these are associated with differences in metabolism in the host.
Each individual has unique biomolecular profiles and molecules variation can come from distinct sources
Although much of the prior analyses focused on the similarities between the individuals across the dietary perturbation, with such a diverse set of multi-omic measurements we also had the unique opportunity to describe in greater detail the differences that exist among individuals both at steady-state and through a longitudinal perturbation. The extensive measurements across different people and perturbations allowed us to use ANOVA to decompose the observed variance for each analyte into components originating from inter-personal differences, the experimental perturbation (weight gain, weight loss, IR vs IS) and other parameters (technical noise and unknown sources). This was examined for individual molecules as well as the general type of molecule (RNA, cytokine, microbiome) (Figs. 6A and S5). Strikingly, we found that all molecular measurements were dominated by inter-personal variation, which accounted for more than 90% of the observed variance in some cases (e.g. cytokines). On the opposite side of the spectrum, proteomics and metabolomics measurements had a substantial unexplained component (30% and 35 %, respectively), highlighting the presence of unaccounted factors (e.g. food, exercise and other changing environmental factors) or a subject-specific reaction to the perturbation. We then examined how cytokine levels differed between individuals versus within multiple timepoints for an individual (Fig. 6B). To exclude the effects of the dietary perturbation for this exercise, we performed this analysis in the set of participants that provided follow-up timepoints three months after the end of the perturbation study (from our prior analyses we observed that the majority of the effects of the weight perturbation have dissipated at this follow-up timepoint (Fig. 4B)). Comparing the variation in cytokine levels between multiple ‘baselines’ in a single individual versus across individuals, we observed a striking difference: for almost all cytokines, the within-individual coefficient of variation was under 20%, while the variation across individuals was 40–60%. This shows that our baseline cytokine profiles are very unique to the individual, a point that has significant implications for one-size-fits-all clinical cytokine assays for the detection and/or monitoring of disease. We observed similar (but less dramatic) effects for the other ‘omes (Fig. S5).
Fig. 6. Personal variation of ‘omics data.
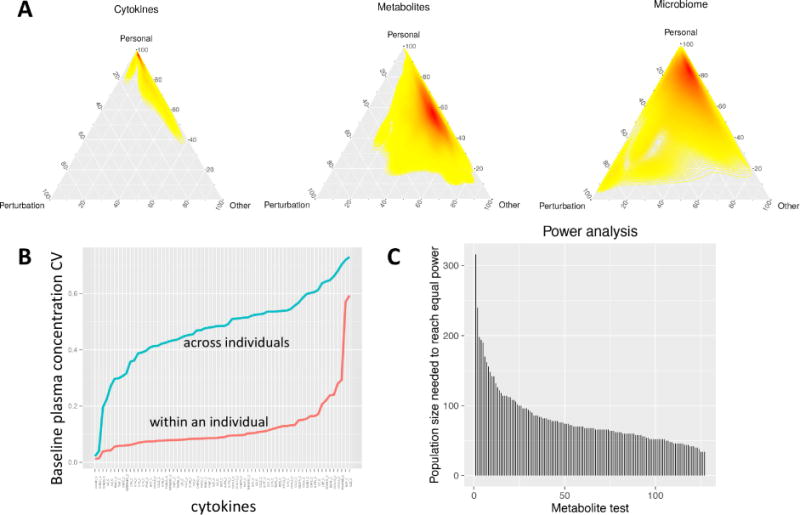
(A) Variance decomposition analysis of selected ‘omes (see Fig. S9 for others). The variance across all timepoints was deconvolved into experiment-dependent variation (i.e. due to the perturbation), personal variation (within an individual), or other types of variation (technical or unknown sources). The heatmap color (yellow to red) indicates the density of analytes at each particular coordinate. (B) Variation in cytokine/chemokine/adipokine abundance within participants versus across participants. The coefficient of variation for all measured Luminex immunoassays is plotted for across steady-state timepoints (T1 and T4) within an individual (red) and across individuals (blue). (C) Power comparison for longitudinal versus group wise study designs using metabolomics data as an example.
The power of longitudinal analyses
As personal variation proved to be one of the main sources of variation across these datasets, we next quantified to what degree the longitudinal study design (i.e. comparing each individual to his/herself across time) increased our ability to identify biomolecular responses to weight gain and loss. To do this, we performed a power analysis, using the metabolome data as an example (Fig. 6C). By comparing our analysis, which accounts for the personal baseline, to a regular, group comparison using a t-test, we can compute the population size required to detect the same effect size with the same statistical power as found in our study. For the 127 significant pairwise differences (p < 0.05) across 73 metabolites, we find that a cross-sectional setup would, on average, require 79 participants to reach the same power, with some analytes requiring 300 participants or more in a cross-sectional setup. Therefore, by correcting for the personal baseline of each analyte we were able to improve power in every single case, for some analytes quite dramatically. This is consistent with the fact that for 85.6 % of all metabolites we found significant (q-value < 0.2) differences evident in baseline samples that also persisted throughout the perturbation. These findings highlight the fact that each individual is biochemically unique and stresses the need for personalized analysis in medicine.
Discussion
Obesity and T2DM are progressive disorders, in which our key to a deep understanding of the etiology likely will come not from single observations of a limited number of analytes, but from deep analyses and longitudinal profiling. Here, we have made millions of measurements of humans and their microbiomes across a longitudinal perturbation and identified some striking patterns within and between subjects. First, by taking an integrated multi-omic approach (Fig. 1), we find marked differences between IR and IS individuals. In the fasted blood of IR individuals, we observed differential regulation of inflammatory/immune response pathways (Fig. 2A and B). This is consistent with prior literature (Festa et al., 2000; Mardi et al., 2005), however earlier studies employed only a few analytical markers, limiting the breadth of information that can be gleaned. Using deep multi-omic profiling, we show that this response includes dysregulation of a large number of factors that specifically function in an antimicrobial response (LTF, CAMP as well as various defensins), and was reflected in each of the different omics profiles (PBMC transcriptome, proteome and circulating cytokines).
In addition, many circulating metabolites were present at different levels in IR and IS participants belonging to amino acid (BCAAs, sulfur-containing amino acids) and lipid metabolism (acylcarnitines and ether-linked phosphatidylcholines) (Fig. 2A). BCAAs have been most consistently described in the literature as associated with insulin resistance. Additionally, indolelactic acid presented a very strong negative correlation with SSPG levels (Fig. 2D). This molecule is of particular interest as it is produced specifically by the microbiome in humans (Patten et al., 2013; Wikoff et al., 2009). Moreover, we show that metabolomics profiles can be used as an accurate predictor for changes in insulin resistance, and it will be of significant interest to apply this approach across significantly larger cohorts. It is possible that a metabolomic signature or a subset of metabolites could be used as a clinical assessment of the insulin sensitivity level in place of the expensive and time-consuming procedure that measures SSPG levels.
Concomitant with these observations is the differential relative abundance of several bacteria in the gut microbiome of IR compared to IS individuals, including gram-negative proteobacterium Oxalobacter formigenes and several Firmicutes species (e.g. Lachnospiraceae blautia and Lachnospiraceae dorea at >1% abundance level) (Fig. 2C, Table S2). Increases in the relative proportion of Firmicutes have been shown to be associated with obesity in numerous contexts (Ley et al., 2006; Turnbaugh et al., 2009; Turnbaugh et al., 2006), and in mice, there is evidence that low-grade inflammation associated with weight gain is at least partially due to the microbiome (Cani et al., 2008). While this association has been largely attributed to the metabolite lipopolysaccharide produced by gram-negative bacteria, it is interesting to note that increases in gram-positive Firmicutes correlate with increased inflammation in this study, raising the possibility of other non-LPS triggers of a low-level systemic immune response in overweight/obese humans. Interestingly, a prior study showed that Firmicutes abundance was reduced in patients with T2DM relative to healthy controls (Larsen et al., 2010), so it is possible that upon progression from a prediabetic state to outright diabetes, a remodeling of the microbiome occurs, again emphasizing the need for detailed longitudinal ‘omic monitoring of patients at risk for T2DM in order to better understand the events that precede the development of frank T2DM.
We also found extensive molecular changes after weight gain and weight loss. Notably the inflammation response was one of the major pathways induced upon weight gain; similar results have been found previously for fat cells (Nishimura et al., 2009). This dysregulation is evident at several different levels including transcriptome, proteome and cytokines (Figs. 3 and 4). These results suggest that a systemic inflammatory pathway is activated in response to short term weight gain, somewhat surprising given the modest weight gain induced here (~2–3 kg), however it is important to note that this response was largely reversed upon subsequent weight loss (Fig. 3).
Of particular interest from the global pathway analysis was the discovery that the dilated cardiomyopathy (DCM) pathway is activated upon weight gain (Fig. 4). Since heart conditions are associated with increased weight, these results provide a potential biochemical explanation for this response; indeed many of the associated biochemical pathways observed herein including inflammation and oxidative stress have been previously implicated as causative for DCM (Tiwari and Ndisang, 2014). Interestingly, we also observed activation of the platelet plug formation pathway which was also altered during diabetes onset (Chen et al., 2012). This pathway may be an indication of altered metabolism and possibly play a role in signaling and/or preparation for blood clotting response during loss of metabolic control.
It is important to note that not all of the responses we observed were consistent across IR and IS participants. In particular, for the microbiome, we observed that the microbe A. muciniphila was weight-gain responsive only in insulin sensitive participants (Fig. 3C). The abundance of this particular microbe in IR individuals did not change across perturbations, and was barely or not detectable in most IR individuals. While prior studies have also shown that gut colonization by A. muciniphila is associated with protection from metabolic disease in mice and humans (Dao et al., 2016; Everard et al., 2013; Roopchand et al., 2015), here we show in humans that A. muciniphila is responsive to even a modest weight gain and clearly differentiates between IR and IS participants. Additionally, we observed a subset of microbes synchronizing in response to the dietary perturbation but are different between IR and IS individuals (Fig. 5A). Furthermore, we discovered longitudinal associations between microbes and host metabolites that are significantly different between the two groups (Fig. 5B and 5C). Taken together, microbial composition and their metabolic activities may be one explanation as to why some individuals respond metabolically very poorly to weight gain while others do not; as such we hypothesize that ensuring the presence of certain classes of microbes in the gut could be a key target for diabetes prevention.
Our study also allowed us to investigate the relation between inter-individual variation and intra-individual variation over time and during a perturbation. Our analysis of variance components showed that for all -omics measurements, the inter-individual variation dominated the intra-individual variation and was the main explanatory factor for different analyte levels (inter-individual variation ranged from 93% (for cytokines) to 54% (for transcripts)). Our data highlight how personalized analysis can provide additional insight and improve statistical power several fold. Even within different types of analytes (metabolites, cytokines etc.), we observed that subsets exhibit highly personal variability while others showed common trends in response to the perturbation. In addition, the study participants have agreed to participate in long-term followup with regular sampling, thus it will eventually be possible to track individual trajectories of specific biomolecules over multi-year periods, further adding to our understanding of personal versus universal changes in biomolecular abundance.
While patterns emerged that implicate common pathways found in the PBMC transcriptome, plasma proteome and metabolome, and gut microbiome it is important to note that despite performing many high-throughput biochemical assays across multiple biospecimen types from the same patient, we are likely a long way off from a complete ‘omic representation of complex human biochemical systems. For example, the PBMC cell types profiled here represent only a fraction of the myriad cell types and tissues that actively secrete free proteins and metabolites into the plasma. Many of these tissues are inaccessible in a non-invasive fashion, limiting the breadth of ‘omic profiling in humans versus similar model organism studies. To this end, mechanistic models derived from mouse or other studies could be leveraged as a template for mapping quantitative human ‘omic data, potentially leading to more accurate predictive signatures for T2DM or other human diseases.
In addition to demonstrating the dynamics of extensive molecular changes during weight gain and loss, this study provides a unique resource for the scientific community. Nearly all of the data are publically available, enabling exploration of inter-omic relationships and alterations across a longitudinal perturbation. With the many levels of linked biological information available here, from genome, transcriptome, proteome, metabolome and microbiome, all open-access, we feel that this study may provide a valuable resource for the development and validation of bioinformatic tools and pipelines integrating disparate data types.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Michael P. Snyder (mpsnyder@stanford.edu).
Experimental Models and Subject Details
Participant recruitment and IRB consent
Participants provided informed written consent for the study under a research study protocol approved by the Stanford University Institutional Review Board. All participants were studied after an overnight fast at the Stanford Clinical and Translational Research Unit (CTRU).
Participants were recruited via placement of advertisements in local newspapers and radio stations seeking “healthy volunteers” for study on body weight changes and effect on metabolism. Screening in the CTRU entailed history and physical, anthropometric measurements, and fasting blood tests for exclusions including presence of diabetes defined as fasting blood glucose > 126 mg/dL, anemia defined as hematocrit < 30, renal disease defined as creatinine > 1.5, history of any cardiovascular, malignancy, chronic inflammatory, psychiatric disease, and history of any bariatric surgery or liposuction.
Eligible consented subjects underwent quantification of insulin mediated glucose uptake via the modified insulin suppression test as previously described and validated (Greenfield et al., 1981; Pei et al., 1994; Shen et al., 1970). Briefly, following an overnight fast, subjects were infused for 180 minutes with octreotide (0.27 Δμg/m2 min), insulin (25 mU/m2 min), and glucose (240 mg/m2 min). Blood was drawn at 10-minute intervals from 150 to 180 minutes of the infusion to measure plasma glucose (oximetric method) and insulin (radioimmunoassay) concentrations: the mean of these four values comprised the steady-state plasma glucose (SSPG) and insulin concentrations for each individual. At steady state, insulin concentrations (65 uU/mL) are similar in all subjects and the SSPG provides a direct measure of the relative ability of insulin to dispose of a glucose load: the higher the SSPG concentration, the more insulin-resistant the individual. While the SSPG is distributed continuously, for the purpose of this study, we defined IS as SSPG<120 mg/dL and IR as SSPG>150 (Yeni-Komshian et al., 2000), largely to provide separation between the two groups. Individuals with SSPG between 120 and 150 were excluded.
Subjects were then placed on a controlled weight gain diet for 30 days by adding an average of 880 calories/day in addition to their usual daily intake as previously described (McLaughlin et al, 2016). Exact caloric excess, administered by research dietitian in the form of snacks/beverages, with fixed macronutrient composition of 50% carbohydrate, 35% fat (<7% saturated), 15% protein, was calculated individually using the Harris Benedict Equation (Harris and Benedict, 1918) for each subject to attain weight gain of 0.8 kg per week (goal of 3.2 kg total). Subjects were not allowed to change their physical activity or change medications during the study. Weekly visits with study dietitian for weight checks, return of food diary, dispensation of snacks, and caloric adjustment if needed, ensured compliance. After weight gain for 28 days, subjects underwent 7 days of weight maintenance with eucaloric diet after which they were resampled (T2) to measure effect of increased body weight rather than caloric excess per se. subjects then underwent supervised weight loss for 6–9 weeks (average 60 days) such that they returned to their baseline weight. A three-week range was given to ensure all subjects could return to baseline weight. One subject was unable to return to baseline weight due to lack of interest or concern about the higher weight, and several highly motivated individuals lost weight to below baseline.
For validation data obtained from a separate cohort conducted in Sweden, subjects were recruited by newspaper advertisements or by having participated in other studies in the laboratory. Inclusion criteria were general good health and no chronic medication. After initial careful phenotyping, including euglycemic clamps to measure degree of insulin sensitivity, 15 male individuals were placed on a hypercaloric diet aimed to increase their body weights by around 8% over a period of 6–8 wks. Phenotyping was then repeated after the weight gain and the volunteers were then helped by nutritionists and exercise plans to recover initial body weights.
Inclusion criteria were male sex, healthy and in general good health. Participants underwent clinical, radiological and metabolomic evaluation before and after a controlled weight gain of about 8% through a hypercaloric diet based on diet recall of each individual by a nutritionist. The 8% weight increase required about 6–8 weeks on the hypercaloric diet. Unbiased metabolomics was performed before and after the weight gain (Metabolon Inc.).
Lifestyle factors were evaluated through a questionnaire filled out at the laboratory. Body weight, height, waist and hip circumferences were recorded and BMI was calculated. The proportions of body fat and lean body mass were determined using bioelectrical impedance (single frequency, 50 kHz; Animeter, HTS, Odense, Denmark). Blood pressure was measured in a sitting position after a five minutes rest with a mercury sphygmomanometer.
To evaluate glucose tolerance status, fasting blood samples were drawn after 12 hours of fasting and were followed by an oral glucose tolerance test (OGTT) (75 g glucose orally). Samples for measurement of plasma glucose and serum insulin were drawn after 0, 30, 60 and 120 minutes.
At a separate examination and after 12 hours of fasting, an intravenous glucose tolerance test (IVGTT) was performed to determine the first and second phases of insulin secretion. A bolus of glucose (300 mg/kg in a 50% solution) was given within 30 seconds into the antecubital vein. Samples for the measurement of plasma glucose and insulin (arterialised venous blood) were drawn at −5, 0, 2, 4, 6, 8, 10, 20, 30, 40, 50 and 60 minutes. The acute and the late insulin responses, i.e. incremental area under the insulin curve, (AIR, 0–10 minutes; LIR, 10–60 minutes) were calculated using the trapezoidal method.
Fasting plasma insulin and fasting plasma glucose from the OGTT were used to calculate a HOMA-IR index using the formula HOMA-IR = (fasting plasma glucose × fasting plasma insulin)/22.5 (Matthews et al., 1985). The HOMA-IR value was used to assess insulin sensitivity in this study.
Plasma glucose was measured using standard laboratory methods (Department of Chemistry, Sahlgrenska University Hospital, Gothenburg, Sweden). Plasma insulin was measured at the University of Tübingen, Germany, by micro-particle enzyme immunoassay (Abbott Laboratories, Tokyo, Japan).
This latter study was approved by the local Ethical Committees at the Sahlgrenska Academy at the University of Gothenburg and was performed in agreement with the Declaration of Helsinki.
Method Details
Sample preparation - Blood samples
Blood was drawn from overnight-fasted participants at the indicated timepoints at the Stanford Clinical Translational Research Unit (CTRU). An aliquot of blood was incubated at room temperature to coagulate; clots were subsequently pelleted and the serum supernatant was pipetted off and immediately frozen at −80C. Blood from separate EDTA-tubes was immediately layered onto Ficoll media and spun via gradient centrifugation. The top layer plasma was pipetted off, aliquoted and immediately frozen at −80C. The PBMC layer was removed, counted via cell counter and aliquots of PBMCs were further pelleted and flash-frozen. For the subsequent multi-omic analyses, PBMCs were thawed on ice, and subsequently lysed and processed to DNA, RNA and protein fractions via Allprep Spin Columns (Qiagen) according to the manufacturer’s instructions and using the Qiashredder lysis option. Plasma analysis was performed on individual aliquots to prevent freeze-thaw cycles.
Sample preparation - Stool
Stool sampling was conducted according to the Human Microbiome Project – Core Microbiome Sampling Protocol A (hmpdacc.org). Once samples were received in the lab, they were subsequently stored in −80C until further processing.
Exome Sequencing
Briefly, DNA was isolated from blood via Gentra Puregene Kits (Qiagen) according to the manufacturer’s instructions. Exome sequencing was performed in a CLIA- and CAP-accredited facility utilizing the ACE Clinical Exome Test (Personalis) (Patwardhan et al., 2015). Variant calling was performed using an in-house developed automated pipeline (HugeSeq) (Lam et al., 2012).
RNA-seq
The transcriptome was evaluated by RNA sequencing (RNA-seq) (Wang et al., 2009) from bulk PBMCs. Ribosomal RNA was first removed and total RNA from each sample was converted into a cDNA sequencing library (using Illumina TruSeq Stranded Total RNA Gold kits according to the manufacturer’s instructions). Each resultant library was quantified via Agilent Bioanalyzer as well as Qubit Fluorometric quantification (ThermoFisher) using a dsDNA high sensitivity kit. Quantified, barcoded libraries were normalized and mixed at equimolar concentrations into a multiplexed sequencing library. The pooled library was quantified and loaded onto a single lane of an Illumina flowcell and sequenced on a HiSeq 2500.
Microbiome sequencing -16S
DNA extractions were performed following Human Microbiome Project – Core Microbiome Sampling Protocol A (HMP Protocol # 07-001, v12.0). Metagenomic DNA was isolated in a clean hood using the MOBIO PowerSoil DNA Extraction kit, with added proteinase K, followed by lysozyme and staphylolysin treatment. These digestion steps greatly improved the extraction of many difficult- to-lyse Gram-positive species.
For 16S (Bacterial) rRNA gene amplification, the V1 through V3 hyper-variable regions (V1–V3) of 16S were amplified from the metagenomic DNA using primers 27F and 534R (27F:5′-AGAGTTTGATCCTGGCTCAG-3′ and 534R: 5′-ATTACCGCGGCTGCTGG-3′). The oligonucleotides containing the 16S primer sequences also contain an adaptor sequence for the Illumina sequencing platform. A barcode sequence unique to each sample is embedded within each of the forward and reverse oligonucleotides used to create the amplicons (dual tags). The uniquely barcoded amplicons from multiple samples were pooled and sequenced on the Illumina MiSeq sequencing platform using a V3 2×300 sequencing protocol.
Microbiome sequencing – Metagenome shotgun
DNA extracted from stools were also subject to whole genome metagenomic shotgun sequencing (mWGS). The libraries were prepared following a standard protocol from Illumina, and at least 1Gb of 150 bp pair-end (PE) reads per sample were sequenced on a Illumina HiSeq or MiSeq instrument.
Untargeted metabolomics by LC-MS
Plasma samples were prepared and analyzed as previously described (Contrepois et al., 2015). Briefly, 400 10 μl of a solvent mixture of 1:1:1 acetone:acetonitrile:methanol was added to 100 μl of plasma, mixed for 15 min at 4°C and incubated for 2h at −20°C to allow protein precipitation. The solvent mixture contained seven internal standards to confirm extraction efficiency and evaluate LC-MS instrument performance. The supernatant was collected after centrifugation at 10,000 rpm for 10 min at 4°C and evaporated to dryness under nitrogen. The dry extracts were then reconstituted with 100 μl of a mixture of 1:1 methanol:water before analysis.
The metabolite extracts were analyzed in HILIC ESI(+) MS, HILIC ESI(−) MS, RPLC ESI(+) MS, RPLC ESI(−) MS using a Thermo Ultimate 3000 RSLC system coupled with a Thermo Q Exactive plus mass spectrometer. The Q Exactive plus was equipped with a HESI-II probe and operated in full MS scan mode. MS/MS data were acquired on quality control samples (QCs = equimolar mixture of all the samples comprised in the study). The source conditions were as follows: Spray Voltage = 3.4 kV (both ESI pos. and neg.), Vaporizer = 310°C, Capill ary temp. = 375°C, S-Lens RF level = 55.0, Sheath Gas = 45 for HILIC and 55 for RPLC, Auxiliary gas = 8 for HILIC and 15 for RPLC, Sweep Gas = 0. The acquisition settings were as follow: AGC (MS) = 3e6, AGC (MS2) = 1e5, Injection Time (MS) = 200 ms, Injection Time (MS2) = 50 ms, Mass Range = 70–1000 Da, Resolution MS = 70,000 (FWHM at m/z 200), Resolution MS2 = 35,000 (FWHM at m/z 200), Top-10 experiments, Isolation Window = 1.0 Da, Dynamic Exclusion = 14 for HILIC and 8s for RPLC, Normalized Collision Energy (NCE) = 25 and 35 for HILIC, 25 and 50 for RPLC. Between each batch, the source and the transfer capillary were cleaned and the mass spectrometer calibrated using an infusion of Pierce LTQ Velos ESI Positive Ion Calibration Solution or Pierce ESI Negative Ion Calibration Solution.
HILIC experiments were performed using a ZIC-HILIC column 2.1 × 100 mm, 3.5 μm, 200Å (Merck Millipore) and mobile phase solvents consisting of 10 mM ammonium acetate in 50/50 acetonitrile/water (A) and 10 mM ammonium acetate in 95/5 acetonitrile/water (B) (Contrepois et al., 2015). Metabolites were eluted from the columns at 0.5 mL/min using a 1–99% phase A gradient over 15 min. Before each injection, the column was equilibrated for 5 min with 1% phase A. Twelve QCs were injected at the beginning of the batch to equilibrate and condition the LC-MS system. The oven temperature was set at 40 °C, and the injection volume was 5 μL. RPLC experiments were performed using a Zorbax SBaq column 2.1 × 50 mm, 1.7 μm, 100Å (Agilent Technologies) and mobile phase solvents consisting of 0.06% acetic acid in water (A) and 0.06% acetic acid in methanol (B). Metabolites were eluted from the columns at 0.6 mL/min using a 1–99% phase B gradient over 9 min. Before each injection, the column was equilibrated for 5 min with 1% phase B. Five QCs were injected at the beginning of the batch to equilibrate and condition the LC-MS system. The oven temperature was set at 60°C, and the injection volume was 5 μL.
Additional metabolomics assays
For the independent cohort conducted in Sweden. non-targeted metabolomic profiling analysis was performed by Metabolon Inc. Samples were prepared using the automated MicroLab STAR® system from Hamilton Company. A recovery standard was added prior to the first step in the extraction process for quality control purposes. Sample preparation was conducted using aqueous methanol extraction process to remove the protein fraction while allowing maximum recovery of small molecules. The resulting extract was divided into four fractions: one for analysis by UPLC/MS/MS (positive mode), one for UPLC/MS/MS (negative mode), one for GC/MS, and one for backup. Samples were dried under nitrogen using a TurboVap® (Zymark) and resolubilized for the appropriate instrument either UPLC/MS/MS or GC/MS.
Untargeted Proteomics by LC-MS
Ethanol precipitated protein pellets from PBMC samples were resuspended in 110 uL of 100 mM ammonium bicarbonate (ABC) and 0.1% Octyl β-D-glucopyranoside (OG) and subjected to a sonicator probe for efficient resuspension. The Thermo Scientific Pierce Micro BCA Protein Assay Kit was then used to quantify protein levels. 1 mg of each resuspended protein sample then underwent denaturation in 900 uL of 8M Urea, 100mM ABC and 0.1% OG. The samples were once again sonicated with a sonicator probe. Each sample then underwent chemical reduction with 10 uL of 1M dithiolthreitol (DTT) and incubated at room temperature for 2 hours. 20 uL of 1M iodoacetamide (IAA) was then added in each sample for the alkylation step. The alkylation reactions were left to incubate in the dark at room temperature for 1 hour.
After fractionation, the unbound fraction from each sample was concentrated and buffer exchanged into 100 uL of 100 mM tetraethylammonium bromide (TEAB) using the Amicon Ultra-15 followed by the Amicon Ultra-4 centrifugal filter unit. Each sample was digested with 40 ng of Promega Sequencing Grade Trypsin in 100 mM TEAB solution. Samples were then labeled with 10-plex Thermo Scientific™ Tandem Mass Tag™ (TMT) Reagents, using instructions provided by the manufacturer. All the samples were divided into groups of nine and pooled together with a master reference sample to create 10-plexed samples.
Each TMT 10-plex sample underwent shotgun liquid chromatography-tandem mass spectrometry (LC-MS/MS) with the LTQ Orbitrap Elite™ Hybrid Ion Trap-Orbitrap Mass Spectrometer coupled with a Dionex RSLC 3000 Nano-HPLC. 15 μL of each sample was loaded onto a C18 trap column at 5 μL/min for 10 minutes. Peptides were then separated by a 25 cm C18 analytical column (Picofrit 75 μm ID, New Objective, packed in-house with MagicC18 AQ resin). Tryptic peptides were separated using a multi-step gradient at a flow rate of 0.6 μL/minute in which Buffer B (0.1% FA in acetonitrile) was increased from 0% (100% Buffer A, 0.1% FA in water) to 85% over 120 minutes. The column was re-equilibrated for 20 minutes at 98% Buffer A. Blank runs were performed between each sample. Samples were then ionized by electrospray ionization set to 2.25 kV with a capillary temperature of 200 °C. An initial MS1 scan over an m/z range of 400–1800 was performed, followed by 10 data-dependent higher energy collision-induced dissociation fragmentation (35 eV) events on the 10 most intense +2 or +3 ions from the MS1 spectrum over an acquisition time of 140 minutes.
Plasma protein profiling using Proseek multiplex
Proteins were quantified from plasma at all time points using multiplex proximity extension assays (Proseek Multiplex, Olink Biosciences) according to the manufacturer’s instructions. In this study three panels, cardiovascular disease (CVD I 96x96), inflammation (Inflammation I 96×96) and oncology (Oncology I 96×96) were applied, and a total of 276 proteins were measured in the plasma samples. Briefly, in each well of 96-well plate 3 μL incubation solution containing with two incubation probes, protein target-specific antibodies conjugated with distinctive single-strand oligonucleotides, was mixed with 1 μL plasma sample. The mixture was incubated overnight at 4 °C, and then added with 96 μL extension solution containing extension enzyme and PCR reagents. The plate was then placed in a thermal cycler for the extension (50 °C, 20 min) and preamplification (9 5 °C 30 min, 17 cycles of 95 °C 30 sec, 54 °C 1 min and 60 °C 1 min). A 96.96 dynamic array IFC (Fluidigm) was prepared and primed according to the manufacturer’s instructions, 2.8 μL of the extension mix was mixed with 7.2 μL detection solution in a new 96-well plate. Next, 5 μL of the mix was loaded to the primed 96.96 Dynamic Array IFC (the right inlets), and 5 μL of each the 96 primer pairs was loaded to the other side of the 96.96 Dynamic Array IFC. The program for protein expression was run on a Fluidigm Biomark using the provided Proseek program (Olink BioSciences).
Luminex Assays
Levels of circulating cytokines in the blood were measured using a 63-plex Luminex antibody-conjugated bead capture assay (Affymetrix) that has been extensively characterized and benchmarked by the Stanford Human Immune Monitoring Center (HIMC). This assay was performed by the Human Immune Monitoring Center at Stanford University. Human 63-plex or Mouse 38-plex kits were purchased from eBiosciences/Affymetrix and used according to the manufacturer’s recommendations with modifications as described below. Briefly, beads were added to a 96 well plate and washed using a Biotek ELx405 washer. Samples were added to the plate containing the mixed antibody-linked beads and incubated at room temperature for 1 hour followed by overnight incubation at 4 °C with shaking. Cold and room temperature incubation steps were performed on an orbital shaker at 500–600 rpm. Following the overnight incubation plates were washed using a Biotek ELx405 washer and then biotinylated detection antibody added for 75 minutes at room temperature with shaking. The plate was washed as above and streptavidin-PE was added. After incubation for 30 minutes at room temperature a wash was performed as above and reading buffer was added to the wells. Each sample was measured in duplicate. Plates were read using a Luminex 200 instrument with a lower bound of 50 beads per sample per cytokine. Custom assay control beads by Radix Biosolutions are added to all wells.
Statistical Analyses
Power and variance calculations
To estimate power and sample sizes, we assumed an experiment on a set of patients with two conditions, before and after. From this, we wished to investigate the increase in power when we used paired tests vs unpaired tests. Our assumption is that there is inter-subject variance (personalized variance) and an effect with a size and variance. Thus, the formalized tests are as follows:
-
1 Unpaired test
In the case of a t-test of unequal variance we have the following test statistic:
where(1)
as calculated in a standard Welch’s test and N being the number of samples in each group (assuming each group has equal number of samples). -
2 Paired test
For a paired t test, we compute the following test statistic:
where
with n being the number of paired samples necessary and sΔ the sample standard deviation of the difference due to the effect. -
3 Power calculation
To compute the statistical power of the paired test, we compute the value of the cumulative distribution function under the alternative hypothesis HA for both sides:
where tn is the computed test statistics with a population size of n and Tα,n is the critical value from the Student t-distribution with n degrees of freedom for a given significance level α. For the power analysis, the power for the paired analysis is computed using the sample size from our analysis. Then, for increasing values of n, the test statistic for the unpaired test is computed until the power of the unpaired tests exceeds the power of the paired test. This value of n is then returned and reported as minimally necessary population size to achieve the same statistical power. Note that n for the unpaired test represents the number of subjects in one group, in the current study we had 3 groups (timepoints T1, T2 and T3) so the value is multiplied by 3.
Random Forest and AdaBoost classification
For these analyses, we proposed an automated prediction model based on delta metabolomics feature array which incorporates at least 2-time subsequent timepoints of SSPG measurement. The delta features (Δ) were computed by taking the 1st order derivatives of metabolomics predictor values of the consecutive time-points for every patient: Δi(tn) = metabolomicsi(tn) − metabolomicsi(tn−1), where metabolomicsi(tn) is the ith metabolomics predictor value at time n and metabolomicsi(tn−1) is value at the earlier timepoint. Using this technique, we created a “delta cohort” by computing the delta metabolomics feature array on the 23 patients, which gives us a total of 38 sample timepoints with 2472 features for each timepoint. We applied Z-score normalization to standardize the feature array to have a mean of zero and standard deviation of +/− 1. On top of the delta cohort, we adopted two different classes of supervised machine learning method to predict the temporal change in the SSPG value:
Binary classification using ensemble learning – We labeled the samples in the delta cohort into two groups based on SSPG increase or decrease compared to the earlier time point: (Group i) SSPG increase → SSPG(tn) ≥ SSPG(tn−1); (Group ii) SSPG decrease → SSPG(tx) < SSPG(tx−1). In order to propose generalizable model to discriminate the SSPG increase and SSPG decrease by analyzing the delta metabolomic feature array, we trained two popular ensemble learning models – Random Forest and AdaBoost. Finally, training and testing of the models were conducted using hold-out validation where 8 sample points (20% of the total cohort) were randomly chosen for testing. We utilized python scikit learn framework to implement the machine learning models. To handle high dimensionality of the metabolomics feature vector, the Random Forest was trained with 50 base estimators, 100 maximum tree depths, and entropy split criterion. AdaBoost was trained using 100 base estimators and learning rate of 0.1.
ΔSSPG computation at time (tn) using regression – We computed as: . We created a regression-based prediction model for computing the by analyzing delta metabolomics features. The model operates based on ElasticNet regularized regression that combines L1 penalties of Lasso and L2 penalties of Ridge to overcome the limitations of incorporating high dimensional features for relatively small number of samples.
RNA-seq data processing and analysis
For RNA-seq data analysis, raw transcripts were processed for adapter removal and low-quality base trimming using the Trimmomatic algorithm (Bolger et al., 2014). Depending on the analysis, reads were either mapped to personal genomes constructed from exome vcfs or the hg19 human reference genome using the STAR aligner (Dobin et al., 2012). Read counts after trimming and quality filtering and the fraction of features that were successfully mapped per sample are listed in Supplemental Table S5. Counts were assessed from STAR-aligned sam files for all ENSEMBL transcripts using the featureCounts function in the Subread package (Liao et al., 2014). Raw feature counts were normalized via the edgeR package and differentially expressed genes were calculated via negative-binomial exact test with false-discovery rate correction (Robinson et al., 2010).
Unsupervised co-expression module discovery was performed using weighted gene co-expression network analysis (WGCNA) (Zhang and Horvath, 2005). The scale-free topology overlap matrix was computed using the “signed” parameter and using an empirically-defined soft threshold power of 12, and co-expressing modules were defined from this network. For each identified module of co-expression biomolecules, representative eigengenes were calculated (WGCNA function ‘moduleEigengenes’) and correlations between module eigengenes and clinical parameters were calculated (corresponding p-values were adjusted for MHT using R function p.adjust (using the Benjamini-Hochberg setting).
Microbiome
The 16S rRNA gene is about 1.5kb, and includes nine variable regions that provide much of the sequence distinction between different taxa. Variable regions one through three are generally sufficient to identify taxa down to the genus level, and sometimes to the species level. Illumina’s software handles initial processing of all the raw sequencing data. One mismatch in primer and zero mismatch in barcodes were applied to assign read pairs to the appropriate sample within a pool of samples. Barcode and primers were removed from the reads. Reads were further processed by removing the sequences with low quality (average qual <35) and ambiguous bases (N’s). Chimeric amplicons were removed using UChime (Edgar et al., 2011), and amplicon sequences were clustered and Operational Taxonomic Units (OTU) picking by Usearch (Edgar, 2010) against GreenGenes database (May 2013 version) and final taxonomic assignment were performed using RDP-classifier (Wang et al., 2007). All details were executed using QIIME (Caporaso et al., 2010) with custom scripts. Alignment results and read counts for microbiome samples are listed in Supplemental Table S6.
For metagenomic data analysis, downstream processing of the mWGS reads included a) identification and masking human reads (using NCBI’s BMTagger, ftp://ftp.ncbi.nlm.nih.gov/pub/agarwala/bmtagger); b) removal of duplicated reads which are artifacts of sequencing process, c) trimming low quality bases and d) low-complexity screening (b–d were done through PRINSEQ (Schmieder and Edwards, 2011)). Reads trimmed to less than 60bp were removed and the remaining high quality reads were analyzed using MetaPhlAn2 (Segata et al., 2012; Truong et al., 2015) for strain-level taxonomic classification and HUMAnN2 (Abubucker et al., 2012) (http://huttenhower.sph.harvard.edu/humann2, v6.0) for functional reconstruction on the gene and pathway levels.
Abundance tables (taxonomy, genes and pathways) obtained above were further analyzed in R (version 3.0.1) by custom scripts. Specifically, pairwise comparisons on the abundances between IR and IS individuals and between timepoints were performed using Wilcoxon rank-sum test, and Spearman’s rank correlation coefficients were calculated for pairwise associations. Also, in the correlation analysis, a linear rank based regression model was employed to determine if trends were different between IR and IS groups (Rfit R package). As both 16S rRNA and whole genome shotgun sequencing (mWGS) methods have individual limitations (Poretsky et al., 2014; Shah et al., 2011), results were shown only for pairwise comparisons that were detected and significant by both methods. Among the intra- and inter-omic associations, mWGS data were used for species level analysis, and 16S for analysis on the phylum and genus levels to obtain the most accurate presentations.
Metabolomics
Samples for metabolomics were prepared and analyzed randomly. Data were analyzed using an in-house data analysis pipeline written in R (version 3.0.1). Metabolite features (characterized by a unique mass/charge ratio and retention time) were extracted, aligned and quantified with the “XCMS” package (version 1.39.4) after conversion of .RAW files to .mzXML using the ProteoWizard MS convert tool. Grouping and annotation were performed with the “CAMERA” package (version 1.16.0). Features from blanks were discarded. The signal drift with time was corrected by applying LOESS (Local Regression) normalization were each feature was independently corrected by fitting a LOESS curve to the MS signal measured in QCs injected repetitively along the batch. After normalization, samples from the same individual tend to cluster together (Figure S6). Metabolic features were putatively identified by matching the accurate masses (± 5 ppm) against a local database containing +60,000 entries (compilation of various public databases such as HMDB, FoodB, DrugBank). Discriminant metabolites were validated manually by comparing the retention time and/or fragmentation spectrum to a local or public spectral libraries. Pathway analysis was performed using the metabolites with a HMDB accession number in the web tool Metaboanalyst (Xia et al., 2015).
Untargeted Proteomics
Acquired data was converted and searched from the .raw files into peptide spectral matches (PSMs) using the Proteome Discoverer™ Software against the 2015 Human Reference Proteome and known contaminants from UniProt. The identifiers from Proteome Discoverer were mapped to Official Gene Symbols. Non-unique and modified peptides were filtered out before selection of top intensity unique PSMs in the reference channel for each 10-plex run. Ratios were then obtained by dividing the 9 sample channels with the reference channel and median centered. The intensities were then log2 transformed and then rolled into protein groups by taking the median log2 ratio of unique peptide groups in each run.
Multivariate data analysis
Data matrices from all omics (transcriptomics, MS-based proteomics, metabolomics, microbiome 16S data and WGS, Proseek Olink and Luminex cytokines) were merged into a common format. All data (excluding RNA-seq for reasons discussed below) was log-transformed and then a linear model was fitted for each individual analyte where the significance of the factors in the model were evaluated using ANOVA. For transcriptome, we used edgeR, a Bioconductor package specifically designed for modeling count data such as RNA-seq, and for improved variance estimation for overdispersed count data. We fit generalized linear models (GLMs) and performed ANOVA-like tests for main effects and interactions using the glmFit and glmLRT functions from the edgeR package (Robinson et al., 2010). The most basic model was used to identify analytes that differ between the two groups (insulin resistant and insulin sensitive):
Note that this model compares average differences between groups. In order to represent the experimental conditions accurately, a factor for the timepoint and an interaction term between the groups and timepoints was introduced which allows the IR and IS group to be different at each timepoint, thus allowing us to capture when the two groups react differently to the treatment. Note that this model was only used to estimate group differences, except for transcriptome, where we used it to estimate timepoint differences as well.
In order to estimate timepoint differences accurately, we used a model that allows each subject to have a different baseline level for each analyte:
By correcting for a personalized baseline, the model allows us to capture common differences between timepoints with higher power as people may start out at different baseline levels but move all in the same direction during perturbation - which would be hard to capture using averages only. It contains a term for the patient_id which estimates an average analyte level for each patient. It contains a term for the sample_timepoint which means the model estimates a different offset for each timepoint and it contains an interaction term which means that the model allows the IR and IS group have different relative analyte levels at each timepoint, thus allowing us to capture when the two groups react differently to the treatment. Note that by estimating a different offset for each patient, it is not possible any more to detect differences between the IR and IS groups which is why we used the first model for this purpose.
Finally, we speculated that using BMI as a continuous predictor would potentially be more informative than taking sampling timepoints which do not capture the amount of weight gained or lost by individuals. We indeed see that (Figure S xxx delta_BMI.pdf) weight gain was not consistent across all individuals, even though all individuals gained weight and subsequently lost weight. By relating the analyte levels directly to the amount of weight gained, we wanted to obtain quantitative insights into changes associated with weight gain and loss:
For transcriptome data, we used the model:
After model fitting and ANOVA analysis, the resulting p-values were corrected using the Benjamini-Hochberg method and a cutoff of 0.2 was used for all data. The reduction in variance by each factor was used for the variance decomposition analysis.
For longitudinal pattern recognition analysis across the weight-gain/weight-loss perturbation we used the mfuzz soft-clustering algorithm (Futschik and Carlisle, 2005; Kumar and M, 2007). The mean expression/abundance was calculated for all biomolecules comprising the transcriptome, metabolome and proteome at each timepoint, and these values were subsequently standardized to z-scores (mfuzz::standardise). Soft clustering was run with fuzzifier parameter set to m=2.5. Cluster members at a low-stringency acore filter of 0.2 were used in subsequent enrichment analyses. Pathway enrichment analysis was conducted using multiple tools: DAVID for transcripts and proteins (Huang da et al., 2009b), and Metaboanalyst 3.0 for metabolites and transcripts (Xia and Wishart, 2016).
Data and Software Availability
Raw data included in this study are hosted on the NIH Human Microbiome 2 project site (http://www.hmp2.org). Data pertaining to this study are classified under the study ID “T2D” and datafiles can be retrieved by querying by subject ID and timepoint (Visit 1–4). Subject IDs included in this study are: ZJTKAE3, ZK112BX, ZK4CK8Y, ZKFV71L, ZKVR426, ZL63I8R, ZL9BTWF, ZLGD9M0, ZLPRB8E, ZLPZS0H, ZLTUJTN, ZM7JY3G, ZMBH10Z, ZMBVNFM, ZVGW5FI, ZVM4N7A, ZVTCAK9, ZW61YGW, ZWCZHHY, ZWFDEY0, ZWHMV5E, ZY1ZKJY, ZY7IW45. As LC-MS raw data consist of multiple samples isobarically tagged, they have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD007859″.
Supplementary Material
Table S2. Significant differences in transcripts, proteins, metabolites, microbes, clinical assays and pathways between IR and IS individuals. Related to Figure 2.
Individual analyte types are separated into unique tabs in the attached excel table. For RNA-seq, differentially expressed transcripts (Ensembl) at FDR < 0.01 are listed along with associated fold changes and p and q values for the test. For transcriptomic pathways, differential transcripts between IR and IS at FDR < 0.2 were used as input to the DAVID pathway analysis suite. Significant pathways from either KEGG or Gene Ontology biological process categories are listed. For proteomic pathways, proteomic data ranked by differential expression between IR and IS was used as input into GORILLA. Significant pathways are listed. For the microbiome, average abundance values from both 16S sequencing (in percentage) and whole genome shotgun sequencing (mWGS, in %) are listed for IR and IS individuals for bacteria that are significantly different (p < 0.05) between the group. Taxonomic units that are detected by both methods are bolded, with their prevalence indicated by columns labeled as “nonzeros”. For targeted Proseek multiplex assays, immunoassays were performed on plasma and the groupwise differences between IR and IS across all timepoints is shown. For Luminex cytokine/chemokine/adipokine assays, immunoassays were performed on plasma and the groupwise differences between IR and IS across all timepoints is shown. For clinical parameters, clinical blood panels were performed by the Stanford core lab, and significant analytes between IR and IS across all timepoints are shown. For metabolomics, LC-MS metabolomics differences between IR and IS (across all timepoints) are shown.
Table S3. Significant differences in transcripts, proteins, metabolites, metabolites and clinical assays across the weight gain/loss perturbation. Related to Figures 3, 4 and 5.
Individual analyte types are separated into unique tabs in the attached excel table. For RNA-seq, differentially expressed transcripts (Ensembl) at FDR < 0.01 are listed along with associated fold changes and p and q values for the test for both the differences between baseline and weight gain and weight gain vs weight loss. For the microbiome, average changes in abundance (T2 minus T1) from both 16S sequencing (in percentage) and whole genome shotgun sequencing (mWGS, in %) are listed for IR and IS individuals for bacteria that are significantly different (p < 0.05) between the group. Taxonomic units that are detected by both methods are bolded, with their prevalence indicated by columns labeled as “nonzeros”. For metabolomics data, differences across all timepoints are shown, as well as BMI-associated changes. For Proseek multiplex protein assays, all immunoassays were performed on plasma and associations with changes in BMI in the study are shown. For Luminex cytokine/chemokine/adipokine assays, the immunoassays were performed on plasma and associations with changes in BMI in the study are shown. Clinical blood panels were performed by the Stanford core lab, and significant associations with changes in BMI in the study are shown.
Highlights.
Analysis of insulin sensitive and resistant individuals across multiple ‘omes.
We reveal molecular changes in the human body after modest short-term weight gain.
Reversal of short-term weight gain eliminates negative molecular alterations.
Open-access multi-omic resource for further bioinformatics developments.
Acknowledgments
Our work was supported by grants from the NIH Common Fund Human Microbiome Project (HMP) (1U54DE02378901) (M.P.S., G.W., and T.M.), American Diabetes Association (grants 1-14-TS-28 and 1-11-CT-35) (T.M.), National Heart, Lung and Blood Institute (1T32HL098049) (B.D.P.), National Institute of Diabetes and Digestive and Kidney Diseases (1F32DK100072) (B.D.P.), as well as gifts from anonymous donors. H.L.R. was funded by SNSF (P2EZP3_162268) and is supported by EMBO (ALTF 854-2015). G.J.G.U is supported by Vetenskapsradet, Swedish Research Council (D0046401). The authors would like to thank the Stanford Human Immune Monitoring Center for performing immunoassays, the Stanford Center for Genomics and Personalized Medicine for sequencing services (grant 1S10OD020141-01), the Stanford Genetics Bioinformatics Service Center for computational and informatics support, The staff in the Stanford Clinical and Translational Research Unit, Lita M. Proctor, Jon LoTiempo and Salvador Secchi for HMP leadership and logistics as well as the dedicated research participants for their invaluable contributions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions:
Conceptualization, M.P.S., G.M.W. and T.L.M.; Methodology, M.P.S., G.M.W., T.L.M., E.S., B.D.P., W.Z., K.C., H.R., G.J.G.U., S.P., U.S. and A.M.; Investigation, B.D.P., W.Z., K.C., H.R., G.J.G.U., T.M., B.M.H., S.L., C.Y.Y., K.K., D.P., C.C., E.C., S.R., S.L., C.Z., J.W., M.R.S., L.L., A.M., U.S., S.P., and T.L.M.; Formal Analysis, H.R., W.Z., K.C., B.D.P., T.M., C.Y.Y., B.M.H., E.B., I.B., C.C., D.L.R., S.L., D.S., S.R., S.L., C.Z., M.R.S., L.L., A.M. and S.P.; Writing – Original Draft, B.D.P., W.Z., K.C., H.R., T.M. and C.Y.; Writing – Review & Editing, M.P.S., G.M.W., T.L.M., E.S., S.P., A.M., B.D.P., W.Z., K.C., H.R., G.J.G.U., C.Y.Y.; Visualization, C.A., H.R., W.Z., K.C., and B.D.P.; Funding Acquisition, M.P.S., G.M.W. and T.L.M.; Data Curation, D.S.; Supervision, M.P.S., G.M.W., T.L.M., E.S., S.P., U.S., A.M., D.L.R., and M.G.
Declaration of Interests:
M.P.S. is a founder and member of the science advisory board of Personalis and SensOmix and a science advisory board member of Genapsys and AxioMX.
References
- Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS computational biology. 2012;8:e1002358. doi: 10.1371/journal.pcbi.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams SH. Emerging perspectives on essential amino acid metabolism in obesity and the insulin-resistant state. Advances in nutrition. 2011;2:445–456. doi: 10.3945/an.111.000737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, Casella G, Drew JC, Ilonen J, Knip M, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One. 2011;6:e25792. doi: 10.1371/journal.pone.0025792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HY, Chen R, Miriami E, Karczewski KJ, Hariharan M, Dewey FE, et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. 2012;148:1293–1307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SD, Chen YK, Lin HC. Increased risk of diabetes in patients with urinary calculi: a 5-year followup study. The Journal of urology. 2011;186:1888–1893. doi: 10.1016/j.juro.2011.07.011. [DOI] [PubMed] [Google Scholar]
- Chung WS, Walker AW, Louis P, Parkhill J, Vermeiren J, Bosscher D, Duncan SH, Flint HJ. Modulation of the human gut microbiota by dietary fibres occurs at the species level. BMC biology. 2016;14:3. doi: 10.1186/s12915-015-0224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- Contrepois K, Jiang L, Snyder M. Optimized Analytical Procedures for the Untargeted Metabolomic Profiling of Human Urine and Plasma by Combining Hydrophilic Interaction (HILIC) and Reverse-Phase Liquid Chromatography (RPLC)-Mass Spectrometry. Mol Cell Proteomics. 2015;14:1684–1695. doi: 10.1074/mcp.M114.046508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao MC, Everard A, Aron-Wisnewsky J, Sokolovska N, Prifti E, Verger EO, Kayser BD, Levenez F, Chilloux J, Hoyles L, et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut. 2016;65:426–436. doi: 10.1136/gutjnl-2014-308778. [DOI] [PubMed] [Google Scholar]
- Daudon M, Traxer O, Conort P, Lacour B, Jungers P. Type 2 diabetes increases the risk for uric acid stones. Journal of the American Society of Nephrology: JASN. 2006;17:2026–2033. doi: 10.1681/ASN.2006030262. [DOI] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dela Cruz CS, Matthay RA. Role of obesity in cardiomyopathy and pulmonary hypertension. Clinics in chest medicine. 2009;30:509–523, ix. doi: 10.1016/j.ccm.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012 doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan SH, Richardson AJ, Kaul P, Holmes RP, Allison MJ, Stewart CS. Oxalobacter formigenes and its potential role in human health. Applied and environmental microbiology. 2002;68:3841–3847. doi: 10.1128/AEM.68.8.3841-3847.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels C, Ruscheweyh HJ, Beerenwinkel N, Lacroix C, Schwab C. The Common Gut Microbe Eubacterium hallii also Contributes to Intestinal Propionate Formation. Frontiers in microbiology. 2016;7:713. doi: 10.3389/fmicb.2016.00713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 2013;110:9066–9071. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng N, Huke S, Zhu G, Tocchetti CG, Shi S, Aiba T, Kaludercic N, Hoover DB, Beck SE, Mankowski JL, et al. Constitutive BDNF/TrkB signaling is required for normal cardiac contraction and relaxation. Proc Natl Acad Sci U S A. 2015;112:1880–1885. doi: 10.1073/pnas.1417949112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festa A, D’Agostino R, Jr, Howard G, Mykkanen L, Tracy RP, Haffner SM. Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS) Circulation. 2000;102:42–47. doi: 10.1161/01.cir.102.1.42. [DOI] [PubMed] [Google Scholar]
- Finucane MM, Stevens GA, Cowan MJ, Danaei G, Lin JK, Paciorek CJ, Singh GM, Gutierrez HR, Lu Y, Bahalim AN, et al. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet. 2011;377:557–567. doi: 10.1016/S0140-6736(10)62037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JL, Simmons PA, Vehige J, Willcox MD, Garrett Q. Role of carnitine in disease. Nutrition & metabolism. 2010;7:30. doi: 10.1186/1743-7075-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA: the journal of the American Medical Association. 2010;303:235–241. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- Fulgenzi G, Tomassoni-Ardori F, Babini L, Becker J, Barrick C, Puverel S, Tessarollo L. BDNF modulates heart contraction force and long-term homeostasis through truncated TrkB.T1 receptor activation. J Cell Biol. 2015;210:1003–1012. doi: 10.1083/jcb.201502100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futschik ME, Carlisle B. Noise-robust soft clustering of gene expression time-course data. Journal of bioinformatics and computational biology. 2005;3:965–988. doi: 10.1142/s0219720005001375. [DOI] [PubMed] [Google Scholar]
- Greenfield MS, Doberne L, Kraemer F, Tobey T, Reaven G. Assessment of insulin resistance with the insulin suppression test and the euglycemic clamp. Diabetes. 1981;30:387–392. doi: 10.2337/diab.30.5.387. [DOI] [PubMed] [Google Scholar]
- Haaskjold YL, Drotningsvik A, Leh S, Marti HP, Svarstad E. Renal Failure due to Excessive Intake of Almonds in the Absence of Oxalobacter formigenes. The American journal of medicine. 2015;128:e29–30. doi: 10.1016/j.amjmed.2015.07.010. [DOI] [PubMed] [Google Scholar]
- Harris JA, Benedict FG. A Biometric Study of Human Basal Metabolism. Proc Natl Acad Sci U S A. 1918;4:370–373. doi: 10.1073/pnas.4.12.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E, Li JV, Athanasiou T, Ashrafian H, Nicholson JK. Understanding the role of gut microbiome-host metabolic signal disruption in health and disease. Trends Microbiol. 2011;19:349–359. doi: 10.1016/j.tim.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Hood L, Lovejoy JC, Price ND. Integrating big data and actionable health coaching to optimize wellness. BMC medicine. 2015;13:4. doi: 10.1186/s12916-014-0238-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009a;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009b;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Integrative HMPRNC. The Integrative Human Microbiome Project: dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell host & microbe. 2014;16:276–289. doi: 10.1016/j.chom.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen AW, Kersten S. Potential mediators linking gut bacteria to metabolic health: a critical view. The Journal of physiology. 2017;595:477–487. doi: 10.1113/JP272476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- Khan MT, Nieuwdorp M, Backhed F. Microbial modulation of insulin sensitivity. Cell metabolism. 2014;20:753–760. doi: 10.1016/j.cmet.2014.07.006. [DOI] [PubMed] [Google Scholar]
- Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar L, M EF. Mfuzz: a software package for soft clustering of microarray data. Bioinformation. 2007;2:5–7. doi: 10.6026/97320630002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam HY, Pan C, Clark MJ, Lacroute P, Chen R, Haraksingh R, O’Huallachain M, Gerstein MB, Kidd JM, Bustamante CD, et al. Detecting and annotating genetic variations using the HugeSeq pipeline. Nat Biotechnol. 2012;30:226–229. doi: 10.1038/nbt.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, Al-Soud WA, Sorensen SJ, Hansen LH, Jakobsen M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5:e9085. doi: 10.1371/journal.pone.0009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecomte V, Kaakoush NO, Maloney CA, Raipuria M, Huinao KD, Mitchell HM, Morris MJ. Changes in gut microbiota in rats fed a high fat diet correlate with obesity-associated metabolic parameters. PLoS One. 2015;10:e0126931. doi: 10.1371/journal.pone.0126931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Zhang C, Kilicarslan M, Piening BD, Bjornson E, Hallstrom BM, Groen AK, Ferrannini E, Laakso M, Snyder M, et al. Integrated Network Analysis Reveals an Association between Plasma Mannose Levels and Insulin Resistance. Cell metabolism. 2016;24:172–184. doi: 10.1016/j.cmet.2016.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Mardi T, Toker S, Melamed S, Shirom A, Zeltser D, Shapira I, Berliner S, Rogowski O. Increased erythropoiesis and subclinical inflammation as part of the metabolic syndrome. Diabetes research and clinical practice. 2005;69:249–255. doi: 10.1016/j.diabres.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- McLaughlin T, Abbasi F, Lamendola C, Liang L, Reaven G, Schaaf P, Reaven P. Differentiation between obesity and insulin resistance in the association with C-reactive protein. Circulation. 2002;106:2908–2912. doi: 10.1161/01.cir.0000041046.32962.86. [DOI] [PubMed] [Google Scholar]
- McLaughlin T, Abbasi F, Lamendola C, Reaven G. Heterogeneity in the prevalence of risk factors for cardiovascular disease and type 2 diabetes mellitus in obese individuals: effect of differences in insulin sensitivity. Archives of internal medicine. 2007;167:642–648. doi: 10.1001/archinte.167.7.642. [DOI] [PubMed] [Google Scholar]
- McLaughlin T, Lamendola C, Coghlan N, Liu TC, Lerner K, Sherman A, Cushman SW. Subcutaneous adipose cell size and distribution: relationship to insulin resistance and body fat. Obesity. 2014;22:673–680. doi: 10.1002/oby.20209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin T, Stuhlinger M, Lamendola C, Abbasi F, Bialek J, Reaven GM, Tsao PS. Plasma asymmetric dimethylarginine concentrations are elevated in obese insulin-resistant women and fall with weight loss. The Journal of clinical endocrinology and metabolism. 2006;91:1896–1900. doi: 10.1210/jc.2005-1441. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- Patten CL, Blakney AJ, Coulson TJ. Activity, distribution and function of indole-3-acetic acid biosynthetic pathways in bacteria. Crit Rev Microbiol. 2013;39:395–415. doi: 10.3109/1040841X.2012.716819. [DOI] [PubMed] [Google Scholar]
- Patwardhan A, Harris J, Leng N, Bartha G, Church DM, Luo S, Haudenschild C, Pratt M, Zook J, Salit M, et al. Achieving high-sensitivity for clinical applications using augmented exome sequencing. Genome medicine. 2015;7:71. doi: 10.1186/s13073-015-0197-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BA, Forslund K, Hildebrand F, Prifti E, Falony G, et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature. 2016;535:376–381. doi: 10.1038/nature18646. [DOI] [PubMed] [Google Scholar]
- Pei D, Jones CN, Bhargava R, Chen YD, Reaven GM. Evaluation of octreotide to assess insulin-mediated glucose disposal by the insulin suppression test. Diabetologia. 1994;37:843–845. doi: 10.1007/BF00404344. [DOI] [PubMed] [Google Scholar]
- Pesu M, Watford WT, Wei L, Xu L, Fuss I, Strober W, Andersson J, Shevach EM, Quezado M, Bouladoux N, et al. T-cell-expressed proprotein convertase furin is essential for maintenance of peripheral immune tolerance. Nature. 2008;455:246–250. doi: 10.1038/nature07210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poretsky R, Rodriguez RL, Luo C, Tsementzi D, Konstantinidis KT. Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS One. 2014;9:e93827. doi: 10.1371/journal.pone.0093827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price ND, Magis AT, Earls JC, Glusman G, Levy R, Lausted C, McDonald DT, Kusebauch U, Moss CL, Zhou Y, et al. A wellness study of 108 individuals using personal, dense, dynamic data clouds. Nat Biotechnol. 2017;35:747–756. doi: 10.1038/nbt.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego S, Dagan-Rosenfeld O, Zhou W, Sailani MR, Limcaoco P, Colbert E, Avina M, Wheeler J, Craig C, Salins D, et al. High Frequency Actionable Pathogenic Exome Mutations in an Average-Risk Cohort. bioRxiv. 2017 doi: 10.1101/mcs.a003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopchand DE, Carmody RN, Kuhn P, Moskal K, Rojas-Silva P, Turnbaugh PJ, Raskin I. Dietary Polyphenols Promote Growth of the Gut Bacterium Akkermansia muciniphila and Attenuate High-Fat Diet-Induced Metabolic Syndrome. Diabetes. 2015;64:2847–2858. doi: 10.2337/db14-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Vargas H, Martinez-Ezquerro JD, Bienvenu T. Brain-derived neurotrophic factor, food intake regulation, and obesity. Arch Med Res. 2011;42:482–494. doi: 10.1016/j.arcmed.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schooneman MG, Vaz FM, Houten SM, Soeters MR. Acylcarnitines: reflecting or inflicting insulin resistance? Diabetes. 2013;62:1–8. doi: 10.2337/db12-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nature methods. 2012;9:811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serino M, Luche E, Gres S, Baylac A, Berge M, Cenac C, Waget A, Klopp P, Iacovoni J, Klopp C, et al. Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut. 2012;61:543–553. doi: 10.1136/gutjnl-2011-301012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N, Tang H, Doak TG, Ye Y. Comparing bacterial communities inferred from 16S rRNA gene sequencing and shotgun metagenomics. Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing. 2011:165–176. doi: 10.1142/9789814335058_0018. [DOI] [PubMed] [Google Scholar]
- Shen SW, Reaven GM, Farquhar JW. Comparison of impedance to insulin-mediated glucose uptake in normal subjects and in subjects with latent diabetes. J Clin Invest. 1970;49:2151–2160. doi: 10.1172/JCI106433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari S, Ndisang JF. The role of obesity in cardiomyopathy and nephropathy. Current pharmaceutical design. 2014;20:1409–1417. doi: 10.2174/13816128113199990562. [DOI] [PubMed] [Google Scholar]
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nature methods. 2015;12:902–903. doi: 10.1038/nmeth.3589. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Ussher JR, Lopaschuk GD, Arduini A. Gut microbiota metabolism of L-carnitine and cardiovascular risk. Atherosclerosis. 2013;231:456–461. doi: 10.1016/j.atherosclerosis.2013.10.013. [DOI] [PubMed] [Google Scholar]
- van Etten RW, de Koning EJ, Verhaar MC, Gaillard CA, Rabelink TJ. Impaired NO-dependent vasodilation in patients with Type II (non-insulin-dependent) diabetes mellitus is restored by acute administration of folate. Diabetologia. 2002;45:1004–1010. doi: 10.1007/s00125-002-0862-1. [DOI] [PubMed] [Google Scholar]
- Vatanen T, Kostic AD, d’Hennezel E, Siljander H, Franzosa EA, Yassour M, Kolde R, Vlamakis H, Arthur TD, Hamalainen AM, et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell. 2016;165:842–853. doi: 10.1016/j.cell.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and environmental microbiology. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Lu J, Xu Y, Li M, Sun J, Zhang J, Xu B, Xu M, Chen Y, Bi Y, et al. Circulating prolactin associates with diabetes and impaired glucose regulation: a population-based study. Diabetes care. 2013;36:1974–1980. doi: 10.2337/dc12-1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A. 2009;106:3698–3703. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EG, Wu Y, Jha P, Dubuis S, Blattmann P, Argmann CA, Houten SM, Amariuta T, Wolski W, Zamboni N, et al. Systems proteomics of liver mitochondria function. Science. 2016;352:aad0189. doi: 10.1126/science.aad0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Wishart DS. Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Current protocols in bioinformatics/editoral board, Andreas D Baxevanis [et al] 2016;55:14 10 11–14 10 91. doi: 10.1002/cpbi.11. [DOI] [PubMed] [Google Scholar]
- Yeni-Komshian H, Carantoni M, Abbasi F, Reaven GM. Relationship between several surrogate estimates of insulin resistance and quantification of insulin-mediated glucose disposal in 490 healthy nondiabetic volunteers. Diabetes care. 2000;23:171–175. doi: 10.2337/diacare.23.2.171. [DOI] [PubMed] [Google Scholar]
- Yoon MS. The Emerging Role of Branched-Chain Amino Acids in Insulin Resistance and Metabolism. Nutrients. 2016;8 doi: 10.3390/nu8070405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Statistical applications in genetics and molecular biology. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Significant differences in transcripts, proteins, metabolites, microbes, clinical assays and pathways between IR and IS individuals. Related to Figure 2.
Individual analyte types are separated into unique tabs in the attached excel table. For RNA-seq, differentially expressed transcripts (Ensembl) at FDR < 0.01 are listed along with associated fold changes and p and q values for the test. For transcriptomic pathways, differential transcripts between IR and IS at FDR < 0.2 were used as input to the DAVID pathway analysis suite. Significant pathways from either KEGG or Gene Ontology biological process categories are listed. For proteomic pathways, proteomic data ranked by differential expression between IR and IS was used as input into GORILLA. Significant pathways are listed. For the microbiome, average abundance values from both 16S sequencing (in percentage) and whole genome shotgun sequencing (mWGS, in %) are listed for IR and IS individuals for bacteria that are significantly different (p < 0.05) between the group. Taxonomic units that are detected by both methods are bolded, with their prevalence indicated by columns labeled as “nonzeros”. For targeted Proseek multiplex assays, immunoassays were performed on plasma and the groupwise differences between IR and IS across all timepoints is shown. For Luminex cytokine/chemokine/adipokine assays, immunoassays were performed on plasma and the groupwise differences between IR and IS across all timepoints is shown. For clinical parameters, clinical blood panels were performed by the Stanford core lab, and significant analytes between IR and IS across all timepoints are shown. For metabolomics, LC-MS metabolomics differences between IR and IS (across all timepoints) are shown.
Table S3. Significant differences in transcripts, proteins, metabolites, metabolites and clinical assays across the weight gain/loss perturbation. Related to Figures 3, 4 and 5.
Individual analyte types are separated into unique tabs in the attached excel table. For RNA-seq, differentially expressed transcripts (Ensembl) at FDR < 0.01 are listed along with associated fold changes and p and q values for the test for both the differences between baseline and weight gain and weight gain vs weight loss. For the microbiome, average changes in abundance (T2 minus T1) from both 16S sequencing (in percentage) and whole genome shotgun sequencing (mWGS, in %) are listed for IR and IS individuals for bacteria that are significantly different (p < 0.05) between the group. Taxonomic units that are detected by both methods are bolded, with their prevalence indicated by columns labeled as “nonzeros”. For metabolomics data, differences across all timepoints are shown, as well as BMI-associated changes. For Proseek multiplex protein assays, all immunoassays were performed on plasma and associations with changes in BMI in the study are shown. For Luminex cytokine/chemokine/adipokine assays, the immunoassays were performed on plasma and associations with changes in BMI in the study are shown. Clinical blood panels were performed by the Stanford core lab, and significant associations with changes in BMI in the study are shown.