

ABSTRACT
The incidence of invasive fungal infections has risen significantly in recent decades as medical interventions have become increasingly aggressive. These infections are extremely difficult to treat due to the extremely limited repertoire of systemic antifungals, the development of drug resistance, and the extent to which the patient's immune function is compromised. Even when the appropriate antifungal therapies are administered in a timely fashion, treatment failure is common, even in the absence of in vitro microbial resistance. In this study, we screened a small collection of FDA-approved oncolytic agents for compounds that impact the efficacy of the two most widely used classes of systemic antifungals against Candida albicans, Candida glabrata, and Aspergillus fumigatus. We have identified several drugs that enhance fungal growth in the presence of azole antifungals and examine the potential that these drugs directly affect fungal fitness, specifically antifungal susceptibility, and may be contributing to clinical treatment failure.
KEYWORDS: antagonism, antifungal treatment failure, azoles, echinocandins, induced resistance
INTRODUCTION
The global burden of invasive fungal infections (IFIs) has increased dramatically as the population of susceptible individuals continues to expand (1). Worryingly, the mortality rate for many IFIs exceeds 50%, despite the provision of appropriate antifungal agents. While the increasing incidence of antifungal drug resistance undoubtedly contributes to the frequency of treatment failure (2, 3), in vitro resistance is only observed in about one-third of such cases. While a variety of factors have been speculated to account for the remaining nonresponsive patients, including inadequate drug distribution or severity of immune dysfunction, little evidence has been provided in support of these arguments. We considered an additional explanation—the influence of other medications on the fungal pathogen itself. This is especially pertinent, given that individuals at the greatest risk of developing IFIs are usually receiving a multitude of drugs to treat a variety of underlying conditions (4). Furthermore, as eukaryotes, human and fungal cells share the same basic biology and signaling pathways. Accordingly, many drugs that induce a physiological response in humans are likely to induce a response in fungi. Yet the influence of most medications upon fungal physiology, antifungal susceptibility, a patient's response to antifungal therapy, and, in a broader sense, the outcome of infection, remains largely unknown.
Several approved drugs are known to enhance the efficacy of existing antifungal medications and may therefore provide a basis for adjunctive therapies (5). However, to date, there has been no systematic attempt to identify approved medications that promote survival of infectious fungi in the presence of antifungal drugs and may therefore undermine their clinical efficacy. Thus, while drug-drug interactions are a serious concern from the perspective of patient toxicity, the consequences of similar interactions on the fungal pathogen itself have not been widely appreciated. The purpose of this study was to determine the effect of approved oncology drugs on the efficacy of the two most important classes of antifungal pharmacotherapies—the azoles and the echinocandins. In so doing, we focused on three of the most prevalent fungal pathogens within this patient population, Candida albicans, Candida glabrata, and Aspergillus fumigatus (6, 7).
RESULTS
We screened a library of oncology drugs to identify any that interfere with the antifungal activity of the azoles or the echinocandins, under conditions that closely mimic those used in the Clinical and Laboratory Standards Institute (CLSI) protocol for determining MICs. Fungal cells were diluted into RPMI medium with suprainhibitory concentrations of the selected antifungal and dispensed into the wells of 96-well plates, with each well containing a single test compound, at a final concentration of 5 μM. Oncology drugs that increased fungal growth at least 2-fold versus the antifungal drug alone (control) were called hits. Of the 129 compounds in the library, 21 were identified as hits in at least one screen, indicating that this type of interaction may be far more common than originally anticipated.
Candida albicans.
Eight compounds were identified as enhancing C. albicans growth in the presence of 1 μM fluconazole (∼8× MIC) (Table 1 and Fig. 1A). Strikingly, all but one of these drugs belong to one of two classes—specifically, topoisomerase or kinase inhibitors. A representative of each class causing the greatest restoration of fungal growth in the presence of fluconazole, idarubicin and ceritinib, as well as the unrelated microtubule inhibitor, cabazitaxel, were selected for follow-up analysis. Checkerboard assays were conducted to confirm and determine the extent of antifungal antagonism, as well as to determine the effective concentration range of each hit (Fig. 2). Ceritinib had a paradoxical effect, with a dose-dependent increase in antagonism up to 0.625 μM, at which point a 16-fold increase in resistance was observed, but at higher concentrations it enhanced fluconazole's antifungal activity. Idarubicin exhibited a dose-dependent increase in antifungal antagonism at concentrations ≥78 nM, inducing a 16-fold increase in fluconazole resistance at 5 μM. Cabazitaxel also antagonized fluconazole's activity at concentrations ≥0.625 μM, with 4-fold resistance observed at 2.5 μM. To determine if these interactions were specific to fluconazole, we performed checkerboard assays with two additional azole antifungals, itraconazole and voriconazole. While the extent of induced resistance and the specific concentrations at which the effects were observed varied, all three oncology agents tested also reduced the effectiveness of itraconazole and voriconazole (Fig. 3). Finally, we tested if a combination of these three agents would act in concert to further elevate fluconazole resistance; however, no additive effects were observed upon fluconazole resistance in the selected C. albicans strain (data not shown). Nonetheless, it remains possible that specific combinations of the other antagonistic oncology agents may induce resistance of greater magnitude than when provided alone. Using the same screening strategy, we did not identify any oncology agents that were able to rescue C. albicans growth in the presence of 500 nM caspofungin (∼8× MIC), indicating that the observed interactions are antifungal specific.
TABLE 1.
Oncology-related drugs that induce in vitro antifungal resistance in Candida and Aspergillus cellsa
| Organism | Antifungal (concentration) | Compound | Relative growth (×) | Z-score |
|---|---|---|---|---|
| C. albicans | None | None | 2.85 | 35.62 |
| Fluconazole (1 μM) | None | 1.00 | ||
| Tamoxifen | 2.54 | 31.29 | ||
| Epirubicin | 2.58 | 32.17 | ||
| Idarubicin | 3.92 | 59.4 | ||
| Nilotinib | 2.83 | 37.3 | ||
| Ceritinib | 3.2 | 44.72 | ||
| Daunorubicin | 2.45 | 29.52 | ||
| Cabazitaxel | 3.11 | 42.96 | ||
| Doxorubicin | 2.01 | 20.51 | ||
| C. glabrata | None | None | 4.85 | 62.52 |
| Fluconazole (100 μM) | None | 1.00 | ||
| Megestrol | 2.15 | 18.61 | ||
| Fluorouracil | 3.87 | 46.59 | ||
| Exemestane | 4.41 | 55.25 | ||
| Lomustine | 3.29 | 37.2 | ||
| Ixazomib | 2 | 16.27 | ||
| Regorafenib | 3.45 | 39.73 | ||
| Abiraterone | 4.64 | 59.04 | ||
| Daunorubicin | 3.23 | 36.12 | ||
| Dactinomycin | 4.55 | 57.6 | ||
| Romidepsin | 2.38 | 22.4 | ||
| Omacetaxine mepesuccinate | 4.32 | 53.81 | ||
| C. glabrata | None | None | 9.93 | 89.36 |
| Caspofungin (500 nM) | None | 1.00 | ||
| Tretinoin | 5.04 | 40.37 | ||
| A. fumigatus | None | None | ||
| Voriconazole (2 μg/ml) | None | |||
| Fluorouracil | ||||
| Thioguanine | ||||
| Floxuridine |
A small collection of FDA-approved oncolytic agents was tested for each compound's ability to antagonize antifungal activity against several common human fungal pathogens in 96-well format at a final test compound concentration of 5 μM. For C. albicans and C. glabrata wells, hits were defined as compounds that resulted in an OD600 of at least twice that of control wells treated with antifungal alone and are listed here with their relative growth and Z-scores. For A. fumigatus, hits were called based on visual inspection of the wells, due to the unreliable nature of OD reads with filamentous fungi.
FIG 1.
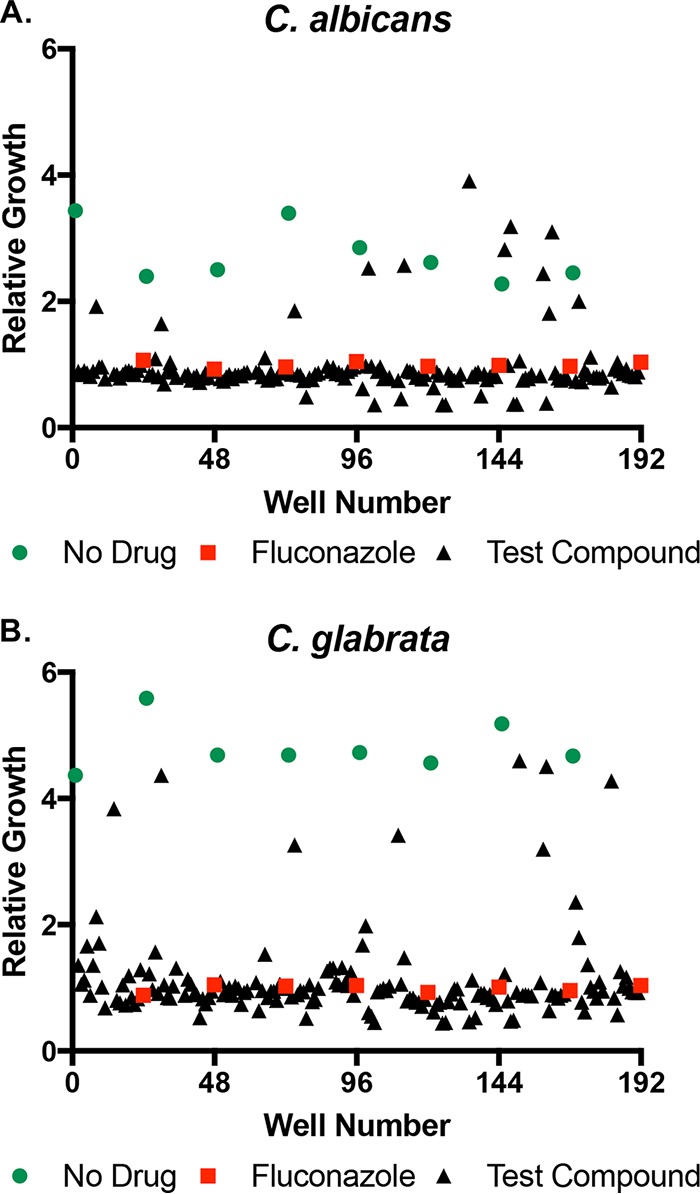
Identification of oncology agents that induce in vitro fluconazole resistance. (A) C. albicans strain SC5314 and (B) C. glabrata strain ATCC 2001 were grown in the presence of 1 and 100 μM fluconazole, respectively, in RPMI medium supplemented with a final concentration of 5 μM of each compound from the National Cancer Institute (NCI) oncology collection. After 24 h of incubation at 35°C, growth was measured as OD600 and normalized to the fluconazole alone controls (red squares). A second set of no-drug control wells had DMSO solvent alone (green circles).
FIG 2.
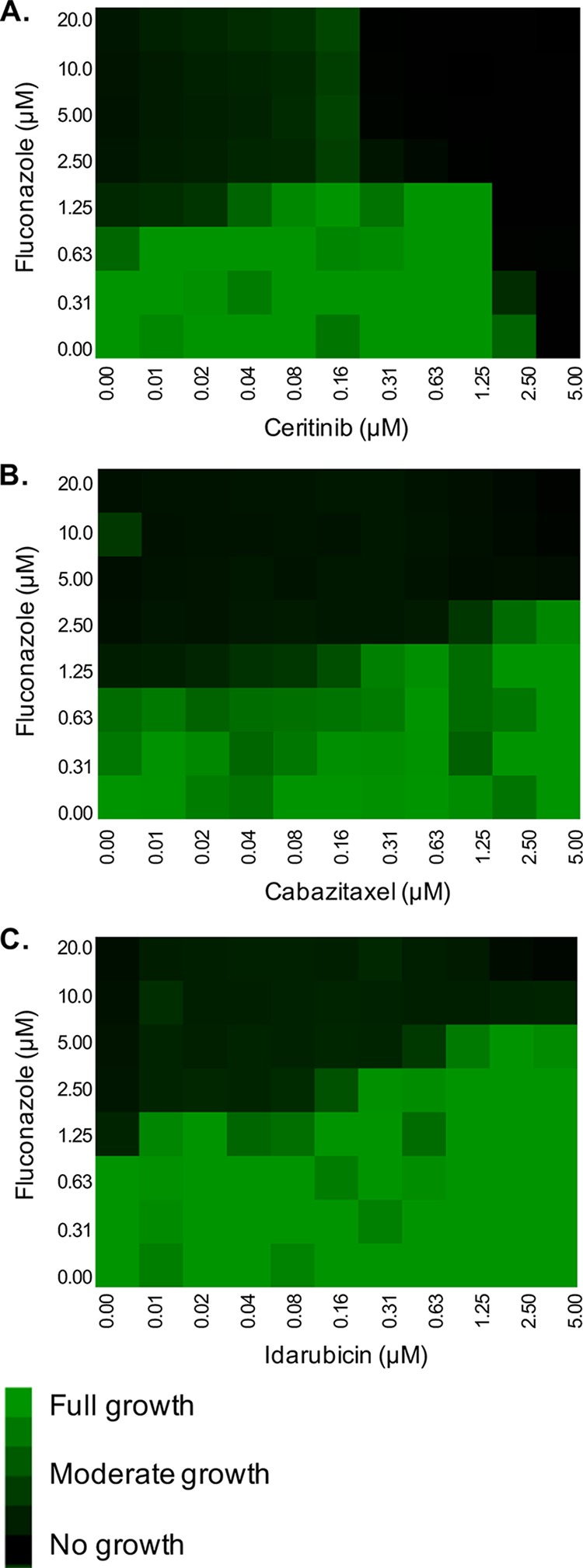
Oncology drugs reduce Candida albicans cell susceptibility to fluconazole. Checkerboard assays in RPMI were performed with C. albicans strain SC5314 across a range of fluconazole doses and (A) ceritinib, (B) cabazitaxel, and (C) idarubicin concentrations. After 24 h of incubation at 35°C, growth was quantified by OD600, normalized to the growth of the untreated control well, and presented as a heat map.
FIG 3.

The antagonistic effect of several oncology agents is not fluconazole specific. Checkerboard assays in RPMI were performed with C. albicans strain SC5314 across a range of either itraconazole or voriconazole doses and (A and B) ceritinib, (C and D) cabazitaxel, and (E and F) idarubicin concentrations. After 24 h of incubation at 35°C, growth was quantified by OD600, normalized to the growth of the untreated control well, and presented as a heat map.
Candida glabrata.
Surprisingly, a total of 11 oncology drugs were identified as facilitating C. glabrata growth in the presence of 100 μM fluconazole (∼8× MIC), including one topoisomerase inhibitor and one kinase inhibitor (Table 1 and Fig. 1B). While the specific drugs inducing fluconazole resistance in C. glabrata were distinct from those identified for C. albicans, the common drug classes identified suggest that the underlying mechanisms are likely similar for many of the antagonistic interactions observed for either species. Additionally, several steroid-like compounds, namely, abiraterone, exemestane, and megestrol, were identified as antagonizing fluconazole's activity upon C. glabrata and were selected for follow-up (Fig. 4). We were not able to confirm the antagonistic interaction with megestrol, indicating that it was a false positive. However, exemestane induced a modest 2-fold increase in fluconazole MIC at concentrations of 2.5 to 5 μM, while abiraterone produced a 4-fold increase in MIC at concentrations as low as 0.156 μM. Tretinoin was the only agent identified in the caspofungin antagonism screen with C. glabrata; however, this interaction was not confirmed in follow-up experiments.
FIG 4.
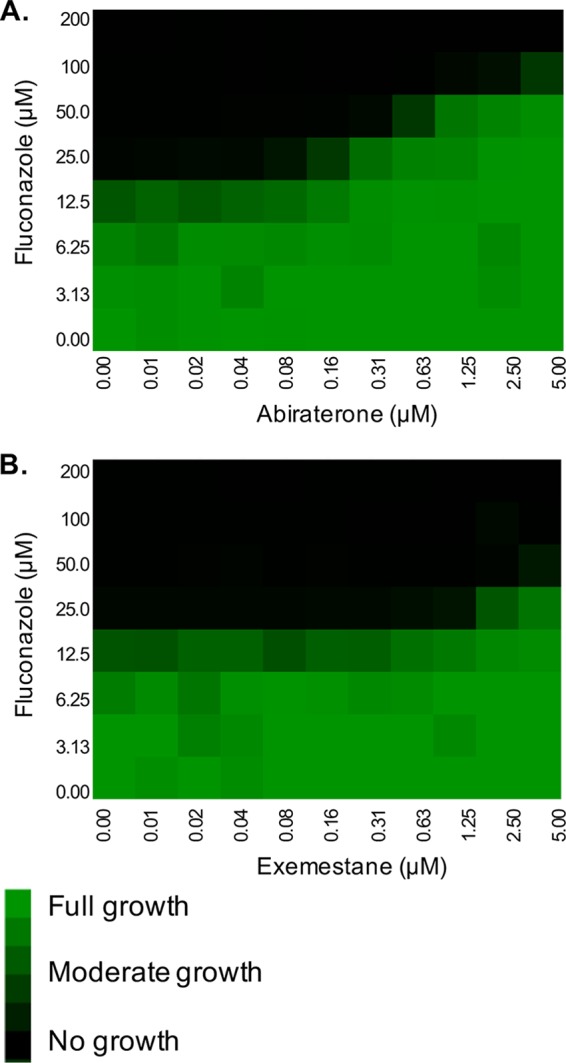
Oncology drugs reduce Candida glabrata cell susceptibility to fluconazole. Checkerboard assays in RPMI were performed with C. glabrata strain ATCC 2001 across a range of fluconazole doses and (A) abiraterone and (B) exemestane concentrations. After 24 h of incubation at 35°C, growth was quantified by OD600, normalized to the growth of the untreated control well, and presented as a heat map.
Aspergillus fumigatus.
Three antimetabolite compounds—floxuridine, fluorouracil, and thioguanine—were identified as enabling A. fumigatus growth in the presence of 2 μg/ml voriconazole (∼4× MIC). Floxuridine is a prodrug that is rapidly converted into fluorouracil; it is therefore likely these agents act via the same mechanism. Both fluorouracil and thioguanine were tested in checkerboard assays and confirmed to enhance A. fumigatus growth in the presence of voriconazole in a dose-dependent manner (Fig. 5). While fluorouracil treatment enhanced fungal growth starting at 39 nM, it only caused a 2-fold increase in the voriconazole MIC at the highest concentration tested (5 μM). Thioguanine enhanced fungal growth in the presence of voriconazole at concentrations as low as 19 nM; however, it did not shift the MIC at any concentration tested. When this collection was screened for compounds that support A. fumigatus growth in the presence of 1 μM caspofungin (∼4× minimum effective concentration), no hits were identified, further supporting that the observed interactions are antifungal specific.
FIG 5.
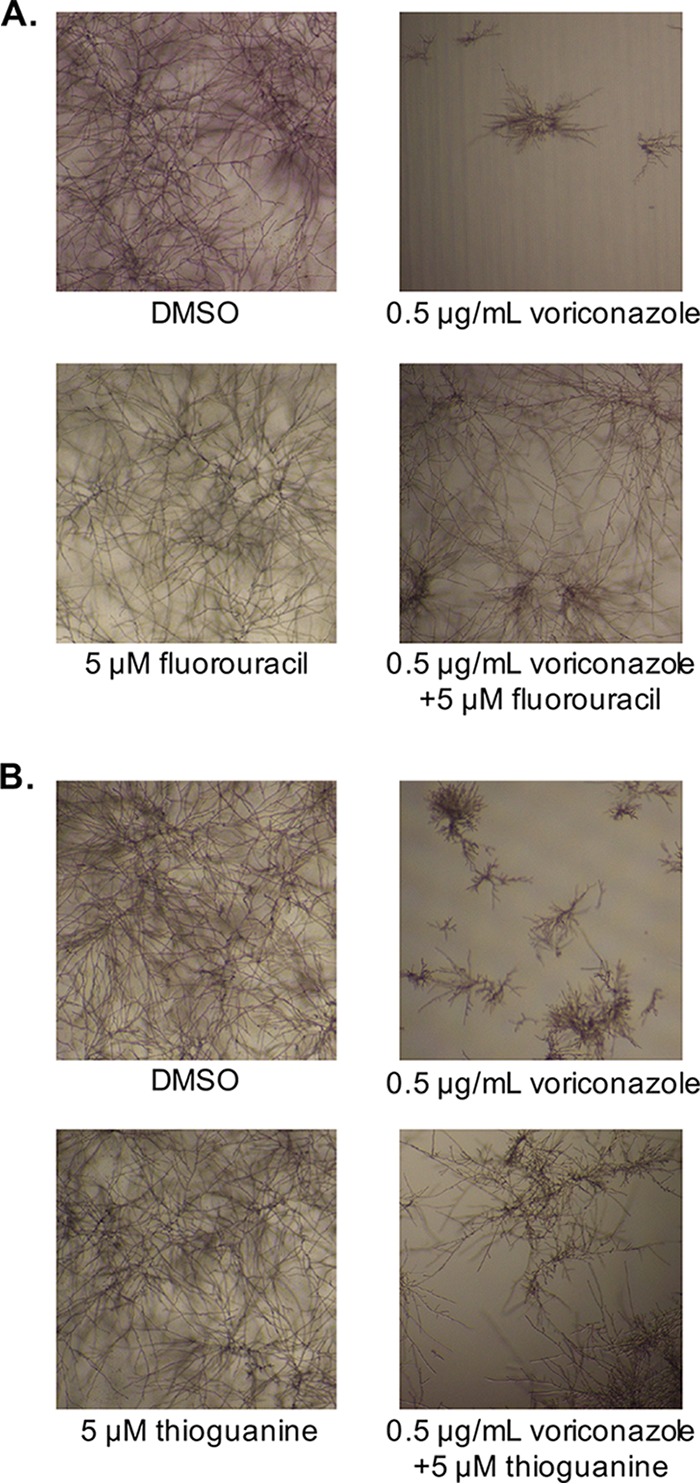
Fluorouracil and thioguanine enhance Aspergillus fumigatus growth in the presence of voriconazole. Checkerboard assays were performed with A. fumigatus strain Af293, using voriconazole in combination with either (A) fluorouracil or (B) thioguanine. Plates were incubated at 37°C for 48 h and then visually inspected and imaged. Representative images of the no-drug control, voriconazole alone, test compound alone, and voriconazole with test compound wells are shown.
DISCUSSION
While there have been reports of antagonism occurring with specific combinations of antifungal drugs (8, 9) including between flucytosine and fluconazole against C. glabrata (10), this is, to the best of our knowledge, the first study to systematically assess the influence of approved medications upon the activity of antifungal drugs. Our results indicate that this phenomenon may be more common than previously appreciated and may contribute to currently unexplained clinical treatment failure, especially in specific patient cohorts. The number of oncology drugs that negatively impact the antifungal activity of azoles was particularly surprising, especially considering the dearth of interactions with echinocandins. There are likely several factors contributing to this disparity that relate to the characteristics of target enzymes, as well as to the physiological consequences of their inhibition. For example, fluconazole is fungistatic against Candida spp., and thus fungal cells have an opportunity to mount a drug-induced adaptive response that enables growth to resume. In contrast, the echinocandins are fungicidal, which may restrict the opportunity to mount an adaptive response of sufficient magnitude to promote survival and proliferation. The distinct cellular location of the target enzymes may also be pertinent. The azole target enzyme, lanosterol demethylase (Erg11p) is intracellular, and thus mechanisms or responses resulting in decreased antifungal drug uptake, membrane permeability, or enhanced efflux can confer resistance. In contrast, the echinocandins target β1-3 glucan synthase, which is exposed at the cell surface and therefore not affected by efflux or cell permeability issues.
These findings raise several questions that require urgent attention: First, are the antagonistic interactions observed of clinical relevance or merely artifacts of in vitro culture? Moreover, can these interactions help explain the large number of treatment failures occurring in patients with IFIs that are not accounted for by heritable resistance of the infecting fungus? Investigation of these questions will necessitate determining if the antagonistic drugs are able to undermine antifungal efficacy at pharmacologically relevant concentrations, the use of appropriate animal models of infection, and an analysis of patient data. Initial studies indicate that all antagonistic agents confirmed herein exert an effect within an order of magnitude of plasma concentrations reported in the product information for these agents, supporting the notion that these interactions may have clinical relevance. Second, aside from oncology-related drugs, are there other pharmacotherapies that interfere with antifungal efficacy? Third, what are the underlying mechanisms of the observed antagonism? Azole resistance in C. albicans can be conferred by elevated expression of drug efflux pumps belonging to the major facilitator and ABC transporter super families, as well as elevated expression of the target enzyme itself, Erg11p (11). It is likely that some of these agents are acting through induction of these mechanisms. For example, the expression of several ergosterol biosynthetic genes has been shown to be responsive to various steroids (12). In addition, the transcription factors that regulate efflux pump expression are known to bind to and be activated by a variety of xenobiotics (13). For example, the antifungal flucytosine, which is metabolized into fluorouracil, has been shown to enhance efflux pump expression in C. glabrata in a Pdr1p-dependent manner (14). Some negative interactions may stem from drug-induced activation of stress responses that promote fungal survival upon antifungal challenge. It is also possible that some interactions result from direct chemical interaction or reaction leading to inactivation of the antifungal drug. Future work should focus on determining which mechanisms predominate and to what extent that varies across and within the classes of agents identified. Fourth, can drug-induced antifungal resistance act in concert with genetically encoded mechanisms to exacerbate resistance and/or tolerance? The latter point is highly important, as clinically relevant levels of resistance to the azole antifungals in C. albicans usually depend upon a combination of mechanisms (15). Fifth, do similar antagonistic drug interactions occur with additional infectious fungal species or, more broadly, with other pathogenic microbes, including protozoan parasites and bacteria? While we may expect fewer pharmacotherapies to interact with prokaryotes due to the greater evolutionary divergence between bacterial pathogens and their mammalian hosts, many protein classes are well conserved across all forms of life. Thus, the potential exists for drugs targeted at human proteins to engage bacterial homologs or by unrelated mechanisms to alter bacterial physiology to promote survival in the presence of antibacterial agents. Improving our understanding of how widespread the phenomenon of drug-induced antimicrobial resistance is, as well as identifying the underlying mechanisms, will be crucial to optimize therapeutic selection and ultimately improve patient outcomes.
MATERIALS AND METHODS
Strains.
Candida albicans strain SC5314 (16), Candida glabrata strain ATCC 2001 (17), and Aspergillus fumigatus strain Af293 (18) were used throughout this study.
Antifungal susceptibility testing.
All susceptibility testing was performed in accordance with the Clinical Laboratory and Standards Institute (CLSI) broth microdilution protocols (M27-A3) (19), using RPMI 1640 medium buffered with morpholinepropanesulfonic acid (MOPS) and pH adjusted to 7.0, except where specifically noted otherwise.
Compound library.
A collection of 129 FDA-approved oncology agents was provided by the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, which is part of the National Institutes of Health (NIH).
Screening.
The wells of the 96-well flat-bottom assay plates were seeded with 1 μl of the 1 mM stock solutions of each library compound in dimethyl sulfoxide (DMSO) or DMSO alone. Approximately 1,000 cells of either Candida species from an overnight culture were added to each well in 199 μl of RPMI 1640 medium containing the indicated concentrations of fluconazole or caspofungin and then incubated as described in the CLSI protocol. After 24 or 48 h, cells were manually resuspended before the optical density at 600 nm (OD600) was measured using a microplate reader. For A. fumigatus, 20,000 conidia were inoculated per ml of the RPMI medium and germinated at 37°C with shaking at 250 rpm for 4 h before the indicated antifungal drugs were added. Subsequently, 199 μl of the culture was dispensed into the assay wells. Due to the limitations of OD600 with filamentous fungi, all determinations were made by visual inspection. In all wells, the final DMSO concentration was 0.55%.
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award R01AI099080 (awarded to G.E.P.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. J.R.F. and W.G. are supported by NIH grant R01 AI106925 to J.R.F.
We also thank the National Cancer Institute (NCI), part of the National Institutes of Health, for providing the library of oncology-related drugs through the Open Chemical Repository Developmental Therapeutics Program.
REFERENCES
- 1.Richardson MD. 2005. Changing patterns and trends in systemic fungal infections. J Antimicrob Chemother 56(Suppl 1):i5–i11. doi: 10.1093/jac/dki218. [DOI] [PubMed] [Google Scholar]
- 2.Rex JH, Pfaller MA, Galgiani JN, Bartlett MS, Espinel-Ingroff A, Ghannoum MA, Lancaster M, Odds FC, Rinaldi MG, Walsh TJ, Barry AL. 1997. Development of interpretive breakpoints for antifungal susceptibility testing: conceptual framework and analysis of in vitro-in vivo correlation data for fluconazole, itraconazole, and Candida infections. Subcommittee on Antifungal Susceptibility Testing of the National Committee for Clinical Laboratory Standards. Clin Infect Dis 24:235–247. [DOI] [PubMed] [Google Scholar]
- 3.Sanglard D, Odds FC. 2002. Resistance of Candida species to antifungal agents: molecular mechanisms and clinical consequences. Lancet Infect Dis 2:73–85. doi: 10.1016/S1473-3099(02)00181-0. [DOI] [PubMed] [Google Scholar]
- 4.Badiee P, Hashemizadeh Z. 2014. Opportunistic invasive fungal infections: diagnosis and clinical management. Indian J Med Res 139:195–204. [PMC free article] [PubMed] [Google Scholar]
- 5.Robbins N, Spitzer M, Yu T, Cerone Robert P, Averette Anna K, Bahn Y-S, Heitman J, Sheppard Donald C, Tyers M, Wright Gerard D. 2015. An antifungal combination matrix identifies a rich pool of adjuvant molecules that enhance drug activity against diverse fungal pathogens. Cell Rep 13:1481–1492. doi: 10.1016/j.celrep.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pappas PG. 2006. Invasive candidiasis. Infect Dis Clin North Am 20:485–506. doi: 10.1016/j.idc.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 7.Maschmeyer G, Haas A. 2008. The epidemiology and treatment of infections in cancer patients. Int J Antimicrob Agents 31:193–197. doi: 10.1016/j.ijantimicag.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 8.Liu N-N, Köhler JR. 2016. Antagonism of fluconazole and a proton pump inhibitor against Candida albicans. Antimicrob Agents Chemother 60:1145–1147. doi: 10.1128/AAC.02043-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cokol M, Weinstein Zohar B, Yilancioglu K, Tasan M, Doak A, Cansever D, Mutlu B, Li S, Rodriguez-Esteban R, Akhmedov M, Guvenek A, Cokol M, Cetiner S, Giaever G, Iossifov I, Nislow C, Shoichet B, Roth Frederick P. 2014. Large-scale identification and analysis of suppressive drug interactions. Chem Biol 21:541–551. doi: 10.1016/j.chembiol.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alves IA, Bandeira LA, Mario DAN, Denardi LB, Neves LV, Santurio JM, Alves SH. 2012. Effects of antifungal agents alone and in combination against Candida glabrata strains susceptible or resistant to fluconazole. Mycopathologia 174:215–221. doi: 10.1007/s11046-012-9538-7. [DOI] [PubMed] [Google Scholar]
- 11.Whaley SG, Berkow EL, Rybak JM, Nishimoto AT, Barker KS, Rogers PD. 2017. Azole antifungal resistance in Candida albicans and emerging non-albicans Candida species. Front Microbiol 7:2173. doi: 10.3389/fmicb.2016.02173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banerjee D, Lelandais G, Shukla S, Mukhopadhyay G, Jacq C, Devaux F, Prasad R. 2008. Responses of pathogenic and nonpathogenic yeast species to steroids reveal the functioning and evolution of multidrug resistance transcriptional networks. Eukaryot Cell 7:68–77. doi: 10.1128/EC.00256-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thakur JK, Arthanari H, Yang F, Pan S-J, Fan X, Breger J, Frueh DP, Gulshan K, Li DK, Mylonakis E, Struhl K, Moye-Rowley WS, Cormack BP, Wagner G, Näär AM. 2008. A nuclear receptor-like pathway regulating multidrug resistance in fungi. Nature 452:604. doi: 10.1038/nature06836. [DOI] [PubMed] [Google Scholar]
- 14.Steier Z, Vermitsky J-P, Toner G, Gygax SE, Edlind T, Katiyar S. 2013. Flucytosine antagonism of azole activity versus Candida glabrata: role of transcription factor Pdr1 and multidrug transporter Cdr1. Antimicrob Agents Chemother 57:5543–5547. doi: 10.1128/AAC.02394-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rogers PD, Barker KS. 2003. Genome-wide expression profile analysis reveals coordinately regulated genes associated with stepwise acquisition of azole resistance in Candida albicans clinical isolates. Antimicrob Agents Chemother 47:1220–1227. doi: 10.1128/AAC.47.4.1220-1227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Odds FC, Brown AJP, Gow NAR. 2004. Candida albicans genome sequence: a platform for genomics in the absence of genetics. Genome Biol 5:230–230. doi: 10.1186/gb-2004-5-7-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koszul R, Malpertuy A, Frangeul L, Bouchier C, Wincker P, Thierry A, Duthoy S, Ferris S, Hennequin C, Dujon B. 2003. The complete mitochondrial genome sequence of the pathogenic yeast Candida (Torulopsis) glabrata. FEBS Lett 534:39–48. doi: 10.1016/S0014-5793(02)03749-3. [DOI] [PubMed] [Google Scholar]
- 18.Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, Arroyo J, Berriman M, Abe K, Archer DB, Bermejo C, Bennett J, Bowyer P, Chen D, Collins M, Coulsen R, Davies R, Dyer PS, Farman M, Fedorova N, Fedorova N, Feldblyum TV, Fischer R, Fosker N, Fraser A, García JL, García MJ, Goble A, Goldman GH, Gomi K, Griffith-Jones S, Gwilliam R, Haas B, Haas H, Harris D, Horiuchi H, Huang J, Humphray S, Jiménez J, Keller N, Khouri H, Kitamoto K, Kobayashi T, Konzack S, Kulkarni R, Kumagai T, Lafton A, Latgé J-P, Li W, Lord A, Lu C, et al. . 2005. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438:1151. doi: 10.1038/nature04332. [DOI] [PubMed] [Google Scholar]
- 19.Clinical and Laboratory Standards Institute. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard, 3rd ed CLSI document M27-A3 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]