

ABSTRACT
Nocardia spp. are Gram-positive opportunistic pathogens that affect largely immunocompromised patients, leading to serious pulmonary or systemic infections. Combination therapy using the folate biosynthesis pathway inhibitors trimethoprim (TMP) and sulfamethoxazole (SMX) is commonly used as an antimicrobial therapy. Not surprisingly, as antibiotic therapies for nocardiosis can extend for many months, resistance to TMP-SMX has emerged. Using experimental evolution, we surveyed the genetic basis of adaptation to TMP-SMX across 8 strains of Nocardia nova and 2 strains of Nocardia cyriacigeorgica. By employing both continuous experimental evolution to provide longitudinal information on the order of changes and characterization of resistant endpoint isolates, we observe changes that are consistent with modifications of two enzymes of the folate biosynthesis pathway: dihydrofolate reductase (DHFR) and dihydropteroate synthase (DHPS) (FolP), with the mutations often being clustered near the active site of the enzymes. While changes to DHFR and DHPS might be expected, we also noted that mutations in a previously undescribed homolog of DHPS (DHPS2 or FolP2) that was annotated as being “nonfunctional” were also sufficient to generate TMP-SMX resistance, which serves as a cautionary tale for the use of automated annotation by investigators and for the future discovery of drugs against this genus. Additionally, folP2 overlapped glucosyl-3-phosphoglycerate synthase. Remarkably, an adaptive frameshift mutation within the overlapping region resulted in a new in-frame fusion to the downstream gene to produce a potentially new bifunctional enzyme. How a single potentially bifunctional DHPS2 enzyme might confer resistance is unclear. However, it highlights the unexpected ways in which adaptive evolution finds novel solutions for selection.
KEYWORDS: Nocardia, experimental evolution, genome-wide analysis, trimethoprim-sulfamethoxazole resistance
INTRODUCTION
Nocardia species are Gram-positive, aerobic actinomycetes found ubiquitously in soil and water (1–3). Although they are usually considered opportunistic pathogens, affecting immunocompromised individuals, about one-third of nocardiosis patients are immunocompetent (4, 5). Laboratory identification of nocardiae is challenging and time-consuming because of their inconsistency in routine microbiological assays as well as their low growth rates, sometimes requiring up to several weeks on routine culture media (1, 6, 7). For these reasons, it has been suggested that diagnoses and the broader role of nocardiosis in human disease have been underestimated (8, 9). The incidence rate of Nocardia species isolation has increased in recent decades, but this increase has sometimes been attributed to advances in molecular techniques to identify and characterize nocardiae (10). Regardless of whether strains of Nocardia are emerging or have been historically underestimated, nocardial infections are being recognized as an emerging public health problem (11, 12).
Treatment of nocardial infections usually involves long-term antimicrobial therapy that can last up to a year, particularly for immunocompromised patients or patients with disseminated infection (1, 13). Combination therapy using the folate biosynthesis pathway inhibitors trimethoprim (TMP) and sulfamethoxazole (SMX) is most commonly used. TMP-SMX is also given as prophylaxis to immunocompromised patients, especially HIV patients, to protect against Pneumocystis jirovecii and nocardial infections (1, 14, 15). This long-term exposure to antibiotics raises the concern of bacteria becoming resistant to these drugs. In a 10-year retrospective evaluation conducted on 765 Nocardia isolates submitted to the Centers for Disease Control and Prevention (CDC) from 1995 to 2004, 42% were found to be resistant to TMP-SMX (16).
Resistance to TMP and SMX, alone or in combination, has been studied in other bacterial species, and mutations in two genes involved in the folate biosynthesis pathway, dihydrofolate reductase (DHFR) (FolA) and dihydropteroate synthase (DHPS) (FolP), have been associated with resistance (17–23). Additionally, resistant variants of these two genes have been identified on mobile elements or plasmids that can be transmitted from species to species (24–29). In a recent study to identify resistance determinants in Nocardia, targeted amplification of genes known to be involved in resistance was performed on 76 TMP-SMX-resistant clinical isolates (30). DHFR and DHPS variants carrying mutations that make them nonsusceptible to TMP-SMX were identified. While most of these genes were associated with integrons, the authors of that study suggested that an additional mechanism was needed to explain the strong TMP resistance seen in several isolates (30).
We have used a combination of experimental evolution and deep sequencing to survey the evolutionary trajectories conferring TMP-SMX resistance as broadly and comprehensively as possible. By starting with 10 diverse strains of Nocardia and two distinct experimental-evolution approaches, we identified both expected and unexpected pathways to resistance. Figure 1 depicts the folate biosynthesis pathway and mutations identified in this pathway leading to TMP-SMX resistance in Nocardia. Among the unexpected findings were a gene previously characterized as nonfunctional (folP2) that is capable of conferring resistance, new genes that interact with better-known alleles to increase resistance, and an unexpected gene fusion event that highlights the multitude of ways in which adaptive evolution can find new solutions to challenges during natural selection.
FIG 1.
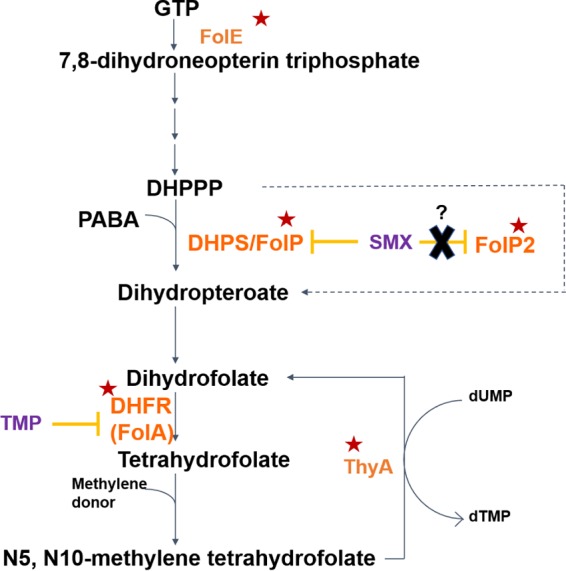
Folate biosynthesis pathway and mutations identified in TMP-SMX-adapted Nocardia strains. The first step in the pathway involves the conversion of GTP by GTP cyclohydrolase I (FolE) to 7,8-dihydroneopterin triphosphate, which is converted to 6-hydroxymethyl-7,8-dihydropterin pyrophosphate (DHPPP) by a series of reactions. Dihydropteroate synthase (DHPS or FolP), which is targeted by SMX, catalyzes the reaction of para-aminobenzoic acid with DHPPP to produce dihydropteroate. Dihydropteroate is converted to dihydrofolate (DHF), which is then reduced to tetrahydrofolate (THF) by dihydrofolate reductase (DHFR or FolA). TMP competes with DHF for DHFR binding. THF becomes methylenated and is used for 1-carbon metabolism. Thymidylate synthase (ThyA) can regenerate DHF from N5,N10-methylene tetrahydrofolate during deoxy-dTMP synthesis. Enzymes in this pathway encoded by genes mutated during TMP-SMX adaptation are identified by a red star. Surprisingly, mutations within a FolP homolog (FolP2), previously characterized and annotated as being “nonfunctional,” are sufficient to confer TMP-SMX resistance.
RESULTS AND DISCUSSION
Evolution of TMP-SMX resistance identifies changes within the folate synthesis pathway.
To survey the adaptive strategies leading to TMP-SMX resistance, two distinct experimental-evolution methods providing different selection environments for adaptation were used to evolve 10 pathogenic strains of Nocardia. Table S1 in the supplemental material provides details of the strains used, and Fig. S1 shows their phylogenetic relationship. Nine strains were evolved to TMP-SMX resistance by using a serial flask transfer approach to survey how the very different starting genomes (Fig. S1) might alter the accessible evolutionary trajectories leading to resistance. These populations also provide information on the reproducibility and potential stochasticity of the identified genetic changes. As a complementary strategy, one strain, Nocardia cyriacigeorgica MDA3349, was evolved as a continuous culture in a bioreactor to quantitatively assess the order and frequency of adaptive alleles as they arise within an evolving polymorphic population to reconstruct parsimonious evolutionary trajectories leading to resistance (31, 32).
MICs were determined for all the endpoint isolates obtained from all the adapted populations. Mutations in genes associated with the folate biosynthesis pathway were observed in resistant isolates. As shown in Table 1, particular mutations within a specific gene as well as the epistatic relationships among genes are critical to the level of resistance conferred. While all the flask transfer endpoint isolates were found to be resistant to TMP-SMX, isolates obtained from the bioreactor varied widely from completely susceptible to completely resistant.
TABLE 1.
List of mutations associated with the folate biosynthesis pathway identified in endpoint isolates of TMP-SMX-evolved Nocardia strainsa
| Strain | Isolate | MIC (mg/liter TMP-SMX) | Mutation(s) |
Descriptionb | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DHFR/FolA | DHPS/FolP | FolP2 | FolE | FtsH | GpgS | ThyA | ||||
| ATCC 33727 | 2-4 | >32-608 | Ile99Thr | Ser27Pro | 1-bp deletion at position −64 position | SNP at position −37, transcriptional regulator, TetR family; V560G sensory box/GGDEF family protein; d-alanyl–d-alanine carboxypeptidase/integral membrane efflux protein (position −88/−33); SNP at position +60, CAIB/BAIF family; 2-bp →CA (R209Q) at position 625, enoyl-CoA hydratase; 9 others | ||||
| MDA0897 | 1-5 | >32-608 | Met30Arg | SNP at position −16 | Siderophore biosynthesis nonribosomal peptide synthetase modules at bacillibactin synthetase component F (I10849L); 13 others | |||||
| MDA5529 | 2-5 | >32-608 | Phe26Ser | Gly210Glu | SNP at positions −65 and −12, GntR family; W31* sensor histidine kinase MtrB; R142M, enoyl-CoA hydratase; 5 others | |||||
| MDA3349 (c) | 1-4 | 16-304 | Phe36Leu | Val101Ala | C6→7 (gene fusion) | 2 others | ||||
| MDA0897 | 2-1 | 16-304 | Ile99Leu | Phe26Cys | Lys212Glu | N98K, methionine ABC transporter; 5 others | ||||
| MDA0897 | 2-3 | 16-304 | Ile99Leu | Phe26Cys | Lys212Glu | P99L and N98K, methionine ABC transporter; 3 others | ||||
| ATCC 33727 | 1-4 | 16-304 | Ile155Thr | Phe26Cys | Insertion at nucleotide 227 | Δ12-bp sensory box/GGDEF protein; 2-bp →CA (R209Q) at position 625, enoyl-CoA hydratase; 5 others | ||||
| ATCC 10905 | 1-1 | 16-304 | Ile155Thr | Phe26Cys | Thr116Pro | SNP at position −150 | S269L, sensor histidine kinase MtrB; K53E transcriptional regulator, TetR family; S183A, PstB; 7 others | |||
| ATCC 10905 | 1-2 | 16-304 | Ile155Thr | Phe26Cys | Thr116Pro | SNP at position −150 | S269L, sensor histidine kinase MtrB; 5 others | |||
| MDA3349 (c) | 1-33 | 16-304 | Phe36Leu | C6→7 (gene fusion) | Transcriptional regulator, MarR family, E90*; siderophore biosynthesis nonribosomal peptide synthetase modules at bacillibactin synthetase component F (A6732V); 2 others | |||||
| MDA3349 (c) | 1-38 | 16-304 | Phe36Leu | C6→7 (gene fusion) | Transcriptional regulator, MarR family, E90*; siderophore biosynthesis nonribosomal peptide synthetase modules at bacillibactin synthetase component F (A6732V); 2 others | |||||
| MDA3732 (c) | 1-1 | 16-304 | Phe36Leu | C6→7 (gene fusion) | TC→CA at position −63 | P253T, transcriptional regulator, AraC family | ||||
| ATCC 10905 | 2-1 | 16-304 | Phe26Cys | 2 others | ||||||
| ATCC 10905 | 2-2 | 16-304 | Phe26Ser, Glu164Asp | 6 others | ||||||
| MDA5529 | 1-4 | 16-304 | Phe26Leu | C6→5 | R142M, enoyl-CoA hydratase; 4 others | |||||
| ATCC 33727 | 1-2 | 16-304 | Phe26Cys | Insertion at nucleotide 227 | Δ12-bp sensory box/GGDEF protein; 2-bp → CA (R209Q) at position 625, enoyl-CoA hydratase; 6 others | |||||
| BAA2227 | 2-4 | 16-304 | Phe13Ser | Lys212Glu | SNP at position −27, transcriptional regulator, AraC family; 7 others | |||||
| MDA3139 | 2-1 | 16-304 | Phe13Leu | C6→7 (gene fusion) | SNP at position −61, Glu215Gln | +G at nucleotide 12, integral membrane protein (rhomboid family); L371P, methionine ABC transporter; SNP at position −175, enoyl-CoA hydratase; 14 others | ||||
| MDA3139 | 2-3 | 16-304 | Phe13Leu | C6→7 (gene fusion) | SNP at position −61, Glu215Gln | Siderophore biosynthesis nonribosomal peptide synthetase modules at bacillibactin synthetase component F (F1011S); L371P, methionine ABC transporter; 15 others | ||||
| MDA3349 (c) | 1-31 | 16-304 | Phe13Ser, Asp172Glu | 12-bp deletion | Asp770Asp | SNP at position −34, integral membrane protein (rhomboid family); 1 other | ||||
| MDA3397 | 2-3 | 16-304 | Phe13Cys | Arg243Cys | Δ144-bp transcription repair-coupling factor; *210E transcriptional regulator, TetR family; 7 others | |||||
| MDA3397 | 2-5 | 16-304 | Phe13Cys | Arg243Cys | 4 others | |||||
| MDA5529 | 2-1 | 16-304 | Phe26Ser | Gly210Glu | W31* sensor histidine kinase MtrB; R142M, enoyl-CoA hydratase; 6 others | |||||
| BAA2227 | 1-1 | 16-304 | Ile112Thr | d-Alanyl–d-alanine carboxypeptidase/integral membrane efflux protein (position −80/−41); F458Y, CAIB-BAIF family; 15 others | ||||||
| BAA2227 | 1-3 | 16-304 | Ile112Thr | Transcriptional regulator, MarR family, R136C; I36S, PstB; 13 others | ||||||
| MDA3349 (c) | 2-18 | 16-304 | C6→7 (gene fusion) | Asp770Asp | 1 other | |||||
| MDA3349 (c) | 2-69 | 16-304 | I131T, SNP at position −53 | 8 others | ||||||
| MDA3139 | 1-1 | 8-152 | Ile155Thr | Phe13Ser | Lys212Glu | Transcriptional regulator, MarR family, at position −30; Δ1 bp at position 9, transcription repair-coupling factor; 12 others | ||||
| MDA3139 | 1-5 | 8-152 | Ile155Thr | Phe13Ser | Lys212Glu | Asp473Gly | Transcriptional regulator, MarR family, at position −30; Δ1 bp at position 9, transcription repair-coupling factor; 12 others | |||
| MDA0666 | 1-2 | 8-152 | Ile30Thr | Phe13Cys | C5→6 | P34A, transcriptional regulator, ArsR family; insertion at position 632 and L429P, sensory box/GGDEF family protein; 12 others | ||||
| MDA0666 | 1-1 | 8-152 | Ile30Thr | Phe13Cys | SNP at position −135 | P34A, transcriptional regulator, ArsR family; insertion at position 632, sensory box/GGDEF family protein; 10 others | ||||
| MDA3349 (c) | 1-7 | 8-152 | Phe36Leu | C6→7 (gene fusion) | 5 others | |||||
| MDA3349 (c) | 1-1 | 8-152 | Phe36Leu | C6→7 (gene fusion) | 2 others | |||||
| MDA3732 (c) | 1-2 | 8-152 | Phe36Leu | C6→7 (gene fusion) | TC→CA at position −63 | P253T, transcriptional regulator, AraC family | ||||
| BAA2227 | 2-2 | 8-152 | Phe13Ser | Lys212Glu | SNP at position −27, transcriptional regulator, AraC family; 8 others | |||||
| MDA5529 | 1-5 | 8-152 | Phe26Leu | C6→5 | *798Trp | R142M, enoyl-CoA hydratase; 3 others | ||||
| MDA3349 (c) | 2-110 | 8-152 | Asp172Glu | Asp770Asp | SNP at positions −44 and −50, GntR family; 2 others | |||||
| MDA3349 (c) | 2-16 | 8-152 | Asp172Glu | Asp770Asp | 3 others | |||||
| MDA3349 (c) | 2-93 | 8-152 | Asp172Glu | Asp770Asp | 2 others | |||||
| MDA3349 (c) | 2-12 | 8-152 | Asp172Glu | Asp770Asp | 1 other | |||||
| MDA3349 (c) | 2-89 | 8-152 | Asp172Glu | Asp770Asp | 1 other | |||||
| MDA3349 (c) | 2-94 | 8-152 | Asp172Glu | Asp770Asp | 1 other | |||||
| MDA3349 (c) | 2-103 | 8-152 | Asp172Glu | Asp770Asp | 1 other | |||||
| MDA3349 (c) | 2-3 | 8-152 | C6→5, Arg82Gln | |||||||
| MDA3349 (c) | 2-13 | 8-152 | C6→7 (gene fusion) | Asp770Asp | 1 other | |||||
| MDA3349 (c) | 2-106 | 8-152 | C6→7 (gene fusion) | Asp770Asp | 2 others | |||||
| MDA3732 (c) | 2-1 | 8-152 | Asp770Asp | P253T, transcriptional regulator, AraC family; 2 others | ||||||
| MDA3732 (c) | 2-2 | 8-152 | Asp770Asp | SNP at position −39, integral membrane protein (rhomboid family); P253T, transcriptional regulator, AraC family; 3 others | ||||||
| MDA3349 (c) | 1-47 | 8-152 | Δ2-bp transcription repair-coupling factor | |||||||
| MDA3349 (c) | 1-48 | 4-76 | C6→7 (gene fusion) | 4 others | ||||||
| MDA3349 (c) | 1-14 | 4-76 | Arg262His | 1 other | ||||||
| MDA3349 (c) | 2-42 | 4-76 | Arg82Gln | Transcriptional regulator, MarR family, E73*; SNP at position −60, transcriptional regulator, ArsR family; T170P, transcriptional regulator, TetR family; 2 others | ||||||
| MDA3349 (c) | 1-19 | 4-76 | 12-bp deletion | Asp770Asp | SNP at position −34, integral membrane protein (rhomboid family); R454C, sensor histidine kinase MtrB; 2 others | |||||
| MDA3349 (c) | 2-65 | 1-19 | E84K, transcription repair-coupling factor | |||||||
| MDA3349 (c) | 2-66 | 1-19 | 4 others | |||||||
| MDA3349 (c) | 1-26 | 1-19 | 1 other | |||||||
| MDA3349 (c) | 1-44 | 1-19 | 1 other | |||||||
| MDA3349 (c) | 1-11 | 1-19 | 1 other | |||||||
Isolates are arranged in decreasing order of TMP-SMX MICs. MDA3349 isolates 1-47, 1-15, 1-43, 2-41, and 2-6 had MICs of 1-19 mg/liter TMP-SMX and had no mutations. Abbreviations: DHFR/FolA, dihydrofolate reductase; DHPS/FolP, dihydropteroate synthase; FolP2, dihydropteroate synthase homolog; FolE, GTP cyclohydrolase I; FtsH, cell division protein; GpgS, glucosyl-3-phosphoglycerate synthase; ThyA, thymidylate synthase; SNP, single nucleotide polymorphism. (c) represents Nocardia cyriacigeorgica strains. All other strains are Nocardia nova strains.
Mutations listed in this column are those that occur in genes not directly involved in the folate biosynthesis pathway but are seen in more than 1 strain, suggesting that they may be adaptive. If a gene is affected within multiple isolates of the same strain, it is listed as “other.” See Data Sets S1 and S2 in the supplemental material for complete details of all mutations observed in these isolates.
Specific alleles associated with TMP-SMX resistance. (i) Dihydrofolate reductase (DHFR/FolA).
Dihydrofolate reductase catalyzes the reduction of dihydrofolate (DHF) to tetrahydrofolate in the folate biosynthesis pathway and is inhibited by TMP (Fig. 1). TMP has a high affinity for DHFR and binds proximally to the substrate binding site (Fig. 2) (33). Resistance to TMP has often been attributed to mutations in the DHFR substrate binding pocket and such mutations were identified in TMP-SMX-adapted Nocardia strains in this work (17, 24, 33, 34).
FIG 2.

Positions of adaptive DHFR mutations relative to the substrate and inhibitor binding pockets. Superimposition of DHF-bound DHFR (yellow sticks) from E. coli (PDB accession number 1RF7) and TMP-bound DHFR from M. tuberculosis (M. tb) (PDB accession number 1DG5) (orange sticks) shows that the inhibitor (TMP) binding site overlaps the substrate (DHF) binding pocket. DHFR adaptive mutations seen in Nocardia isolates and homologous residues in E. coli and M. tuberculosis DHFR proteins are shown in the inset table. Ile94, Phe31, and Leu/Gln28 in E. coli and M. tuberculosis proteins (corresponding to Ile99, Phe36, and Ile/Met30 in Nocardia, respectively) are involved directly in interactions with the substrate and/or inhibitor. The residues observed in the mutant Nocardia DHFR are indicated in red. Ile155 is located away from the substrate and inhibitor binding site of DHFR and is not shown. The image was generated by using PyMOL (PyMOL molecular graphics system, version 2.0.1; Schrödinger, LLC).
Adaptive mutations in DHFR were near the substrate binding pocket and the site of TMP binding (Fig. 2). Studies of other organisms have shown that mutations in DHFR can confer TMP-SMX resistance by decreasing the affinity of TMP for the enzyme while maintaining sufficient enzyme activity (18, 23, 35, 36). Our data suggest a similar biochemical mechanism, as in most cases, homologous positions near the substrate/inhibitor binding pockets are altered. Figure S2 in the supplemental material shows an alignment of DHFR protein sequences from Escherichia coli, Mycobacterium tuberculosis, and the 10 Nocardia strains used in this work. M. tuberculosis DHFR is the closest homolog (51% identity) for which there is a crystal structure available (PDB accession number 1DG5). Phe36 in N. cyriacigeorgica strains MDA3349 and MDA3732 corresponds to residue 31 in M. tuberculosis (37). In M. tuberculosis DHFR, Phe31 is within the drug binding pocket (Fig. 2) and makes van der Waals contacts with the pyrimidine of TMP. In E. coli, Phe31 is also involved directly in the interaction with the native substrate (Fig. 2) (38). Based on these structures, we hypothesize that the Phe36Leu mutation in Nocardia DHFR might alter the affinity for the inhibitor but still permit sufficient binding to dihydrofolate.
Ile94 in M. tuberculosis (aligning with Ile99 in MDA0897 and ATCC 33727) also makes van der Waals contacts with the pyrimidine of TMP, while its main-chain carbonyl group is involved in a potential hydrogen bond with TMP (Fig. 2) (37). Presumably, a change at position 94 may alter the local structure of the main chain such that the predicted H-bond cannot occur or the mutation induces changes within the local structure or dynamics of the structural pocket to disfavor the binding of the inhibitor. Likewise, mutations Ile99Leu and Ile99Thr in Nocardia could interfere with this interaction, weakening the binding of TMP to DHFR, making the enzyme TMP resistant.
Ile30 in MDA0666 (Ile30Thr) and Met30 in MDA0897 (Met30Arg) aligned with Leu28 in E. coli and comprises part of the binding pocket for DHF (Fig. 2). A Leu28Arg mutation in E. coli identified in a laboratory-adapted TMP-resistant strain was found to reduce TMP susceptibility (17, 39).
Ile155 in MDA3139 and ATCC 10905 corresponded to Leu153 in M. tuberculosis DHFR. Although this residue is not involved directly in substrate or inhibitor binding, a change from a nonpolar Leu or Ile residue to a polar Thr residue could also alter the protein structure to disfavor inhibitor binding or improve substrate binding while retaining sufficient enzyme activity. Taken together, the observed DHFR mutations in Nocardia along with what has been learned from other organisms provide a logical biochemical basis for resistance to TMP.
(ii) Dihydropteroate synthase (DHPS/FolP).
Dihydropteroate synthase catalyzes the condensation of 6-hydroxymethyl-7,8-dihydropterin pyrophosphate (DHPPP) with para-aminobenzoic acid (PABA) to form 7,8-dihydropteroate, which is subsequently converted to dihydrofolate in the bacterial folate biosynthesis pathway (Fig. 1) (24, 40, 41). SMX is a sulfonamide that has been shown to compete with PABA for binding to DHPS (41–43). In this study, mutations in DHPS affecting substrate and inhibitor binding were identified in TMP-SMX-resistant isolates.
Phe at position 13 in DHPS in Nocardia isolates MDA3349, MDA0666, BAA2227, MDA3139, and MDA3397 corresponded to Phe26 in isolates MDA0897, MDA5529, ATCC 10905, and ATCC 33727 (see Fig. S3 in the supplemental material). This highly conserved residue aligns with Phe31 in Neisseria meningitidis, Phe19 in M. tuberculosis, and Phe28 in E. coli and Yersinia pestis (41, 44–47). Nine of the 10 adapted strains developed a mutation at this position in DHPS (Table 1). In E. coli, a Phe28Leu mutation increases the MIC of sulfonamide 60-fold (48). Phe28 in Y. pestis is involved in interactions with the substrate PABA as well as the inhibitor SMX (Fig. 3), suggesting that substitutions at this position can alter sulfonamide susceptibility while also potentially impacting enzyme function.
FIG 3.

Positions of DHPS adaptive mutations relative to the substrate and inhibitor binding sites. Superimposition of Y. pestis DHPS structures bound to the substrate PABA (PDB accession number 3TYZ) and the inhibitor SMX (PDB accession number 3TZF) shows that Phe28 in Y. pestis (corresponding to Phe13/Phe26 in Nocardia [see the inset table]) is involved in the binding of both the substrate and inhibitor. The star in the table indicates that Phe13 and Phe26 align at the same position in the sequence alignment of different Nocardia DHPS sequences (see Fig. S3 in the supplemental material). Asp172 and Val101 in Nocardia correspond to Asp185 and Ile117 in Y. pestis DHPS, which interacts with the pterin substrate DHPPP (PDB accession number 3TZF) (41). Mutations in these residues were also seen in TMP-SMX-resistant Nocardia isolates (mutant residues shown in red text). The image was generated by using PyMOL (PyMOL molecular graphics system, version 2.0.1; Schrödinger, LLC).
Y. pestis DHPS residues corresponding to Val101 and Asp172 of Nocardia are in the pterin binding pocket of the enzyme interacting with the substrate DHPPP (Fig. 3) (41). Mutations such as the observed Asp172Glu and Val101Ala mutations that are involved in substrate binding might imply subtle changes in protein structure that prevent or reduce inhibitor binding while allowing the protein to function, perhaps with a reduced enzymatic efficiency. Ser27 in ATCC 33727 is known to be a conserved residue in DHPS, while Glu164 of DHPS from ATCC 10905 is not (42). Neither Ser27 nor Glu164 was proximal to the binding pocket, and thus, their role in substrate or inhibitor binding will need further biochemical and structural analyses.
Mutations identified in a DHPS homolog suggest a second path to TMP-SMX resistance.
All Nocardia strains used in this study have a DHPS homolog that is identified by automated annotation as “nonfunctional dihydropteroate synthase 2.” A surprising finding was the occurrence of mutations in the homolog of FolP, which was characterized as “nonfunctional” in Mycobacterium (49, 50). In this study, however, we identified multiple mutations in this gene in a variety of strains, including fully resistant isolates, with no accompanying mutations within genes well established in folate biosynthesis, such as folA or folP (Table 1). Other actinomycetes, such as Mycobacterium, Corynebacterium, and Rhodococcus, also have a second copy of a DHPS homolog.
Previous studies sought to identify the role of this additional copy of DHPS in Mycobacterium. A study of Mycobacterium leprae showed that while folP1 (DHPS gene) was active in this organism and could complement an E. coli folP knockout strain, the homolog folP2 could not (49). Studies performed on FolP1 and its homolog Rv1207 in M. tuberculosis showed that the purified FolP1 protein had the expected levels of DHPS enzyme activity but that Rv1207 did not, suggesting that the protein is inactive or at least unable to use DHPPP as a substrate (50). Superimposition of the Rv1207 and FolP1 structures showed that Leu180 of the canonical DHPS was positioned within the substrate binding pocket of FolP1 but was replaced by His220 in Rv1207. The substitution of Leu to His in Rv1207 suggested a potential steric clash within the reorganized substrate binding pocket, leading the authors of that study to suggest that the substrate could no longer bind within the homolog active site (see Fig. S4 in the supplemental material) (50). Furthermore, that study showed that the DHPS homolog of Mycobacterium bovis was not expressed during exponential growth, nutrient starvation, or anaerobiosis, while FolP1 was, and those authors concluded that the homolog was not a bona fide DHPS (50). Although it was demonstrated that this homolog did not encode a functional DHPS and was not expressed in M. tuberculosis and M. bovis (50), in Mycobacterium smegmatis, the deletion of folP2 caused an 8-fold decrease in the SMX MIC compared to that for the wild-type strain, suggesting a role for this gene in the action of SMX in some M. smegmatis strains (51). Nocardia strains adapted to TMP-SMX in this work showed numerous adaptive mutations in folP2 (Table 1), strongly suggesting that both Nocardia folP and folP2 could be targets for adaptive mutations leading to decreased sensitivity to SMX.
The DHPS2 point mutations in TMP-SMX-adapted Nocardia strains included Arg262 and Arg82 (MDA3349), Thr116 (ATCC 10905), and Ile112 and Lys212 (BAA2227) (Table 1 and Fig. 4). Sequence alignment showed that Arg262 in MDA3349 DHPS2 corresponded to Arg253 in M. tuberculosis DHPS and Arg287 in Rv1207 and was absolutely conserved among DHPS sequences (46). The guanidinium group of Arg253 in M. tuberculosis forms interactions with one face of the pterin ring of the substrate, and this residue is predicted to be involved in catalysis (46). Arg82 in MDA3349 FolP2 corresponded to Arg82 and Arg77 in Bacillus anthracis and Y. pestis DHPSs, respectively. This residue is involved in forming stabilizing interactions with Asp31, which is a part of the active site (41). Thr116 in ATCC 10905 FolP2 and Lys212 in BAA2227 FolP2 corresponded to highly conserved residues of FolP (Fig. 4). The role of the nonconserved residue corresponding to Ile112 in BAA2227 FolP2 was not known. A deletion of 12 nucleotides in a bioreactor-adapted isolate of MDA3349 led to the removal of 4 amino acids, amino acids 51 to 54, from FolP2 (highlighted in Fig. 4). These residues are not conserved among DHPS proteins and are not known to be involved in substrate or inhibitor binding. Contrary to what was observed for M. tuberculosis Rv1207 (50), mutations in FolP2 residues of TMP-SMX-adapted Nocardia strains that correspond to conserved as well as functionally important FolP residues suggest that this protein may be functional.
FIG 4.

Alignment of MDA3349 DHPS (FolP) and DHPS2 (FolP2). Blue and orange residues correspond to absolutely conserved and highly conserved residues, respectively (46). The two proteins share 36% sequence identity. Residues highlighted in yellow were found to be mutated in laboratory-adapted Nocardia strains (for mutations in strains other than MDA3349, the mutated residues of the other strains are shown using the numbering of MD3349). “*” represents conserved residues. “:” represents amino acids with strongly similar properties. “.” represents amino acids with weakly similar properties.
Alternatively, it may be possible that FolP2 effectively reduces the concentration of SMX in the cytoplasm by binding the inhibitor with a higher affinity than DHPS, thereby allowing folate biosynthesis, or that the adaptive mutations that we observed allow FolP2 to have the enzymatic activity of a DHPS or perhaps modify SMX. Notably, in some endpoint isolates of MDA3349 (isolates 2-3, 1-48, 1-14, and 2-42) and BAA2227 (isolates 1-1 and 1-3) shown in Table 1, the only mutations in the folate biosynthesis pathway are seen in FolP2, and based on their MICs, they have reached clinical levels of resistance. This provides further evidence for the importance of FolP2 in TMP-SMX resistance. Careful biochemical studies will be required to resolve the relationship of FolP and FolP2. What is clear is that mutations in a gene previously annotated as being nonfunctional can provide resistance to TMP-SMX.
A frameshift mutation that results in a combined single open reading frame for folP2 and the glucosyl-3-phosphoglycerate synthase gene (gpgS) appears important for TMP-SMX resistance.
An interesting finding was the common occurrence of frameshift mutations in folP2 during adaptation. folP2 overlapped the open reading frame of the downstream glucosyl-3-phosphoglycerate synthase gene (gpgS) by 4 nucleotides (Fig. 5). An insertion of a single cytosine near the 3′ end of folP2 caused a frameshift, bringing gpgS in frame with it and creating one long open reading frame potentially encoding a single fusion protein (Fig. 5). Since the mutations were located in runs of homopolynucleotides and detected by short-read whole-genome sequencing, we confirmed the mutations by Sanger sequencing. This mutation was observed in numerous endpoint isolates with high levels of resistance across different Nocardia strains (Table 1). The consequences of the gene fusion and its expression are currently being investigated.
FIG 5.
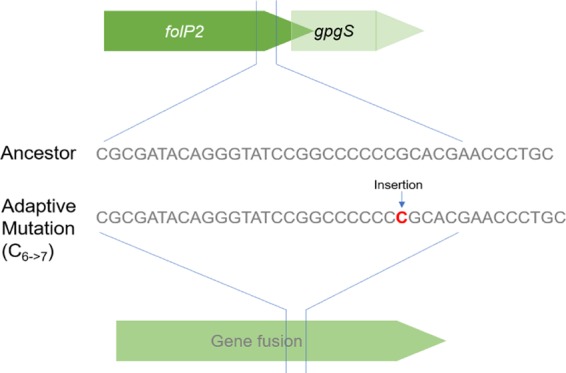
A frameshift mutation in folP2 leads to a gene fusion. The adjacent and overlapping genes folP2 and gpgS are brought in frame by the insertion of a single cytosine (C6→7) near the 3′ end of folP2 in TMP-SMX-adapted Nocardia strains.
Mutations in other genes may provide alternate pathways to TMP-SMX resistance. (i) GTP cyclohydrolase I (FolE).
FolE catalyzes the first committed step in the folate biosynthesis pathway, converting GTP to dihydroneopterin triphosphate (52, 53). In some actinomycetes, such as Nocardia and Mycobacterium, folE is arranged in tandem with the cell division protein-encoding gene ftsH (Fig. 6). Downstream of folE is the gene encoding DHPS. In this work, several mutations in the putative promoter regions of ftsH and folE as well as within the folE gene were identified (Fig. 6 and Table 1). A synonymous mutation in ftsH was independently seen in two distinct TMP-SMX-adapted strains (Asp770Asp) (Table 1), MDA3349 and MDA3732. Although the mutation was located within ftsH, we speculate that its effect could be exerted on the downstream gene folE, since the mutation, 150 nucleotides upstream of the start codon of folE, may be within its promoter region. Increased copy numbers and the overexpression of folE, by itself or in combination with point mutations in folA or folP, have been reported to provide resistance to antifolates in Plasmodium falciparum and Streptococcus agalactiae by increasing the flux through the folate biosynthetic pathway (53, 54). A copy number variation was not identified for this region in the present study, but mutations in the promoter region as well as within the folE gene could indicate changes in the expression levels of genes in the operon or altered enzymatic activity that could potentially alter flux through the folate biosynthesis pathway, reducing the susceptibility of the organism to inhibitors of the pathway.
FIG 6.

Arrangement of ftsH and folE in Nocardia. GTP cyclohydrolase I, dihydropteroate synthase (DHPS), dihydroneopterin aldolase (DHNA), and hydroxymethyldihydropterin pyrophosphokinase (HPPK) are all involved in the folate biosynthesis pathway. Mutations identified in this region in the TMP-SMX-adapted Nocardia strains (listed in Table 1) are depicted with red starts (mutations within the DHPS gene, which have been discussed in Fig. 3, are shown here to demonstrate the extent of adaptive mutations in this region, making it a mutational hot spot).
(ii) Thymidylate synthase (ThyA).
In one endpoint isolate, 1-5, from MDA0897, a point mutation was observed 16 bp upstream of thyA, which is in an operon with folA (Table 1). This was accompanied by a Met30Arg mutation in FolA (FolAMet30Arg). As shown in Fig. 1, ThyA catalyzes the conversion of dUMP to dTMP using N5,N10-methylene tetrahydrofolate as the methyl group donor that is converted to dihydrofolate (55, 56). A mutation in the thyA promoter could modulate the expression of ThyA, altering flux through the folate biosynthetic pathway that may result from changes in FolA activity as a consequence of FolAMet30Arg.
(iii) Glucosyl-3-phosphoglycerate synthase (GpgS).
GpgS is involved in the transfer of glucose to 3-phosphoglycerate in M. tuberculosis, forming glucosyl-3-phosphoglycerate, an intermediate in the synthesis of the cytoplasmic methylated polysaccharide methylglucose lipopolysaccharide (MGLP), although its physiological role remains unclear (57, 58). Interestingly, resistant isolates of MDA3397 and MDA5529 had point mutations in gpgS resulting in amino acid substitutions Arg243Cys and Gly210Glu that correspond to Arg256 and Gly228, respectively, in M. tuberculosis GpgS. Both of these residues were absolutely conserved among GpgS proteins, and an M. tuberculosis Arg256Ala mutant protein created by site-directed mutagenesis had drastically reduced Km values for both of its substrates (57, 58). GpgS does not seem to have a direct role in folate biosynthesis, but its proximity to folP2 on the chromosome (Fig. 5) makes it a putative player in TMP-SMX resistance.
Another frameshift involving gpgS and folP2 was the deletion of a cytosine (C6→5 in Table 1) seen in MDA5529 (isolates 1-4 and 1-5) and MDA3349 (isolate 2-3), which also caused an extension of the folP2 reading frame but not in frame with gpgS. A third frameshift mutation involving a single nucleotide insertion at position 227 in the folP2 gene in ATCC 33727 isolates 1-2 and 1-4 introduced a premature stop codon in the gene, effectively removing the C-terminal 187 amino acids. In addition to potential coding changes, it is also possible that mutations in this region may alter an as-yet-unknown regulatory element.
In addition to the identification of genes known to confer TMP-SMX resistance, such as folA and folP, this comprehensive survey has allowed the identification of mutations in other members of the folate biosynthesis pathway as well as in a second copy of folP. As shown in Table 1, the particular mutations and their potential to have strong synergistic interactions are apparent. In MDA5529 2-5, DHPSF26S and GpgSG210E produce very high MICs, while in ATCC 33727 2-4, DHFRI99T, DHPSS27P, and an intergenic mutation upstream of GTP cyclohydrolase produce comparably high MICs. Not to be forgotten is that all the strains have unique mutations outside the folate pathway that could also be relevant to fitness (see Data Sets S1 and S2 in the supplemental material). Since the changes to genes outside the folate pathway were not consistent across multiple strains, it is difficult to assign a clear role. For example, some changes could be related more to adaptation to the resource conditions, such as a several changes observed in siderophores as well as in various transcriptional regulators that control functions such as cell division and metabolism (Table 1). Undoubtedly the genomic context matters as well (for instance, mutations affecting DHPS Phe13/26 were seen only in N. nova strains, while mutations affecting DHFR Phe36 and FtsH Asp770Asp were seen only in N. cyriacigeorgica strains), although the importance of the centrally essential genes identified in Table 1 extends across all the strains.
Experimental evolution of MDA3349 within a bioreactor reveals weaker selection and a potential role for biofilms and community structure in TMP-SMX resistance.
N. cyriacigeorgica MDA3349 was adapted to TMP-SMX in a bioreactor, which has been successfully used in studies of many other organisms adapting to a variety of drugs (31, 32, 59, 60). The continuous culture maintained in the vessel favors the formation of biofilms. All the Nocardia strains used in this work had a propensity to grow as aggregates in rich medium. In addition to the clumpy-growth phenotype, we observed that MDA3349 formed very thick biofilms on the glass and metal surfaces of the bioreactor, showing evidence of local spatial ecologies within the vessel, consistent with a polymorphic population (see Fig. S5 in the supplemental material).
An interesting observation was the morphological diversity of the bioreactor-adapted cells, which was in stark contrast to the fairly uniform morphologies of the flask transfer-adapted endpoint isolates (Fig. 7). While it is not unusual for bioreactor systems to show polymorphism, the extent of polymorphism was dramatic and is consistent with weaker selection than would be expected (61). The extent of polymorphism in this case was surprising, since the addition of an antibiotic usually provides stronger purifying selection, as was seen in our work with enterococci (32, 59). The presence of so many observable phenotypes was further borne out by the results of deep sequencing of the total DNA from the daily populations as well as the individual genomes of the 32 selected endpoint isolates. TMP-SMX MICs for the endpoint isolates were found to range from susceptible (≤2-38 mg/liter) to resistant (≥4-76 mg/liter) (Table 1). Ten isolates had MICs consistent with the sensitive ancestor despite the high concentration of the drug, suggesting that biofilms or aggregation may afford some protection from the bulk environment where the final drug concentration of the last 8 to 10 days was more than three times the MIC of the sensitive strains present at the end of the experiment. Colonies isolated from the biofilms collected at the end of adaptation showed similarly diverse morphologies. A relationship between colony morphology and MIC could not be established, and there was also no clear correlation between mutations in specific genes and their presence within the biofilm. The phenotypic diversity observed in the bioreactor suggests that Nocardia strains favor the formation of diverse communities within biofilms.
FIG 7.
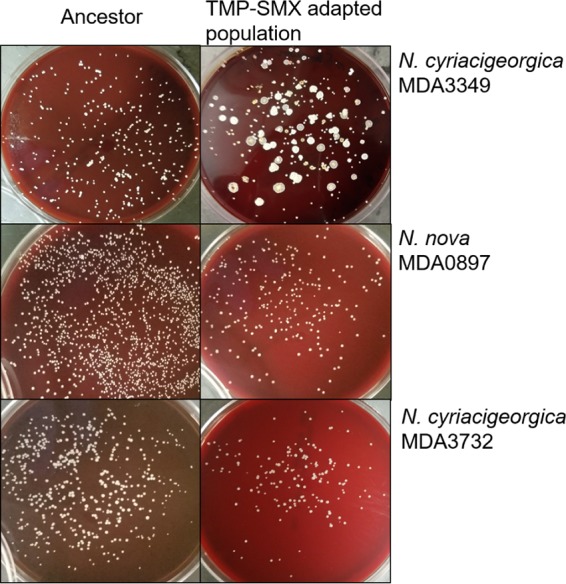
Morphologies of ancestor and TMP-SMX-adapted endpoint isolates. The strain adapted in the bioreactor, MDA3349, demonstrated dramatic morphological diversity upon adaptation to TMP-SMX, while flask transfer-adapted strains MDA0897 and MDA3732 looked uniform after adaptation. In contrast to the endpoint isolates from serial-transfer experiments, bioreactor isolates were frequently sensitive to TMP-SMX, suggesting an ability to persist within the well-established biofilms of the bioreactor.
Reconstruction of representative timelines of genetic adaptation of Nocardia to SMP-TMX.
By consolidating whole-genome sequencing data of endpoint isolates and metagenomic populations, a timeline was constructed to identify successful evolutionary trajectories affecting the folate biosynthesis pathway (Fig. 8). From the order of mutations in bioreactor runs 1 and 2, it was evident that the first adaptive mutations appeared in both runs as soon as the drug concentration exceeded the MIC (1-19 mg/liter). Initially, the frameshift in folP2 that resulted in a fused open reading frame with gpgS was observed. Also evident in both runs was the synonymous mutation in ftsH, which affected the putative promoter region of the operon encoding DHPS. In both runs, this mutation appeared when the MICs for the populations were at 5 to 95 mg/liter TMP-SMX and was evidence for parallel evolution occurring in the two runs, making these genes likely drivers toward resistance. In addition, it was also observed that in run 2, three different lineages acquired the same synonymous mutation in ftsH. Surprisingly, mutations in DHFR and DHPS, which were so common in the serial-transfer experiments, did not reach frequencies higher than the 5% cutoff but could be observed in the endpoint isolates of the bioreactor populations. A consistent increase or decrease in the abundance of mutant alleles associated with adaptation was not observed in this case. This is highly unusual and has not been observed in our previous studies with other organisms (32, 60). The frameshift in folP2 was seen at a frequency of 34.5% at the end of run 1 and at a frequency of 17.2% at the end of run 2, while the synonymous mutation in ftsH was seen at frequencies of 8.3% and 7.2% in runs 1 and 2, respectively. A mutation in DHFR appeared on the very last day of adaptation in run 1 and was detected at only a 12.9% abundance in the final population. This further supported the notion that a large portion of the population in the bioreactor was not adapting to the selective pressure imposed by the drug. Besides the mutations shown in Fig. 8, both the populations had many other mutations that appeared sporadically over time (see Data Sets S1 and S2 in the supplemental material). A possible explanation for this could be the large amount of biofilm formed in the vessel (Fig. S5). Previous studies have shown that diversity in a population is strongly dependent on spatial structure, and simulated biofilm populations exhibit about 10-fold greater diversity than planktonic populations (62, 63). While diversity in the outermost, newly formed layers of the biofilm may be replication dependent, older cells persisting in the nutrient-starved, inner layers of the biofilm have a greater opportunity to accumulate nonreplicative mutations (62). Despite the observed diversity, resistant isolates from the bioreactor as well as the flask transfer experiments showed similar mutations in the folate biosynthesis pathway genes, further supporting the idea that these genes are the ones that are involved in resistance.
FIG 8.

Evolutionary trajectories and linkages of mutations in selected endpoint isolates (labeled in gray boxes) involved in TMP-SMX resistance. The x axis shows the time (in days) that it took for the mutation to arise in the bioreactor population during run 1 (a) and run 2 (b). The location of the mutation indicates the day on which it was first seen in the population at a >5% frequency, and the thicknesses of the arrows represent the abundance of the mutation in the population on the day on which it arose. Dashed arrows are for mutations that were not seen in any population sample at a ≥5% frequency but that were observed in endpoint isolates. It should be noted that this is not a comprehensive summary of all mutations occurring in the populations. This figure shows only trajectories of endpoint isolates with mutations in genes implicated in resistance to TMP-SMX. See Data Sets S1 and S2 in the supplemental material for genotypes of the daily polymorphic bioreactor samples and endpoint isolates.
The use of two different approaches to adapt Nocardia to TMP-SMX provides us with some insight into the way in which this organism grows and forms communities. One of the essential technical differences between these experiments is that within flask transfer experiments, the populations favored are the planktonic and nonadhering/aggregating lineages, since these populations are more likely to be transferred into a new flask with fresh media. In the bioreactor, the selection is inverted, and biofilm-forming lineages are favored. Although the endpoint isolates obtained by the two approaches showed consistency in the types of adaptive mutations acquired, the final evolved populations differed in composition. In many ways, the bioreactor may represent an environment that is more relevant to the clinical setting, wherein adherence to host tissues or medical implants can provide an advantage for the organism. The formation of such strong community structures may mean that treatment of nocardiosis may prove especially challenging, as drug exposure, even at high levels, may be unable to resolve a well-entrenched community of Nocardia.
MATERIALS AND METHODS
Strains and growth conditions.
Three strains of Nocardia nova were obtained from the ATCC, and seven strains, five of N. nova and two of N. cyriacigeorgica, were isolates from cancer patients at the MD Anderson Cancer Center, Houston, TX (see Table S1 in the supplemental material) (2). Strains were streaked out onto blood agar plates from frozen stocks (20% [vol/vol] glycerol stock) and incubated at 37°C until growth was observed (typically 2 to 7 days). Liquid cultures were started by picking 1 colony from the blood agar plate and resuspending it in Luria-Bertani (LB) broth (catalog number B214906; Becton Dickinson, NJ, USA), followed by shaking at 225 rpm at 37°C for ∼2 to 7 days. MIC tests showed that all strains were susceptible to TMP-SMX (Table 1). The phylogenetic relationship between these strains is shown in Text S1 and Fig. S1 in the supplemental material.
Experimental evolution by serial flask transfer.
Nine Nocardia strains were adapted to TMP-SMX by serial flask transfer (Table S1), as described in Text S2 in the supplemental material.
Bioreactor experimental evolution.
The bioreactor was set up as described previously, with some modifications (see Text S2 in the supplemental material) (31).
Phenotypic characterization of endpoint isolates.
Endpoint isolates were characterized based on their appearance on blood agar plates and growth in LB broth. MIC tests were done to characterize endpoint isolates based on their susceptibility to TMP-SMX (see Text S2 in the supplemental material).
Genomic DNA isolation, whole-genome sequencing, and analysis.
Genomic DNA was isolated from cell pellets for all the bioreactor samples as well as the endpoint isolates from flask transfer- and bioreactor-evolved populations, followed by whole-genome sequencing, genome assembly, and mutation identification, as described in Text S2 in the supplemental material.
Accession number(s).
Whole-genome sequencing data generated in this work have been deposited in the Sequence Read Archive (SRA) database under BioProject accession number PRJNA432572. The de novo-assembled genome sequences (Whole Genome Shotgun project) for 9 ancestor strains have been deposited in the DDBJ/ENA/GenBank database under accession numbers PSYZ00000000, PSZA00000000, PSZB00000000, PSZC00000000, PSZD00000000, PSZE00000000, PSZF00000000, PYHS00000000, and PYHT00000000. The complete genome of MDA3349 is available under GenBank accession numbers CP026745 to CP026746.
Supplementary Material
ACKNOWLEDGMENTS
We thank Luay Nakhleh for providing us with tools and resources for computational analysis.
H.M. and Y.S. were involved in drafting the manuscript. H.M., J.W., and A.P. were involved in performing experiments. H.M., J.W., and Y.S. were involved in data analysis and interpretation. R.A.L.E. constructed the phylogenetic tree. X.H. provided 7 strains used in this work and helped with critical analysis of the manuscript. All the authors contributed to the final manuscript.
This work is supported by grants from the Defense Threat Reduction Agency (HDTRA1-15-1-0069) to Y.S. and the National Science Foundation (DMS-1547433) to Luay Nakhleh (principal investigator) and R.A.L.E. (trainee). The content of the information does not necessarily reflect the position or the policy of the federal government, and no official endorsement should be inferred.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00364-18.
REFERENCES
- 1.McNeil MM, Brown JM. 1994. The medically important aerobic actinomycetes: epidemiology and microbiology. Clin Microbiol Rev 7:357–417. doi: 10.1128/CMR.7.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang HL, Seo YH, LaSala PR, Tarrand JJ, Han XY. 2014. Nocardiosis in 132 patients with cancer: microbiological and clinical analyses. Am J Clin Pathol 142:513–523. doi: 10.1309/AJCPW84AFTUWMHYU. [DOI] [PubMed] [Google Scholar]
- 3.Al Akhrass F, Hachem R, Mohamed JA, Tarrand J, Kontoyiannis DP, Chandra J, Ghannoum M, Haydoura S, Chaftari AM, Raad I. 2011. Central venous catheter-associated Nocardia bacteremia in cancer patients. Emerg Infect Dis 17:1651–1658. doi: 10.3201/eid1709.101810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson JW. 2012. Nocardiosis: updates and clinical overview. Mayo Clin Proc 87:403–407. doi: 10.1016/j.mayocp.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coussement J, Lebeaux D, Van Delden C, Guillot H, Freund R, Marbus S, Melica G, Van Wijngaerden E, Douvry B, Van Laecke S, Vuotto F, Tricot L, Fernandez-Ruiz M, Dantal J, Hirzel C, Jais J-P, Rodriguez-Nava V, Lortholary O, Jacobs F. 2016. Nocardia infection in solid organ transplant recipients: a multicenter European case-control study. Clin Infect Dis 63:338–345. doi: 10.1093/cid/ciw241. [DOI] [PubMed] [Google Scholar]
- 6.Schlaberg R, Huard RC, Della-Latta P. 2008. Nocardia cyriacigeorgica, an emerging pathogen in the United States. J Clin Microbiol 46:265–273. doi: 10.1128/JCM.00937-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munoz J, Mirelis B, Aragon LM, Gutierrez N, Sanchez F, Espanol M, Esparcia O, Gurgui M, Domingo P, Coll P. 2007. Clinical and microbiological features of nocardiosis 1997–2003. J Med Microbiol 56:545–550. doi: 10.1099/jmm.0.46774-0. [DOI] [PubMed] [Google Scholar]
- 8.Abreu C, Rocha-Pereira N, Sarmento A, Magro F. 2015. Nocardia infections among immunomodulated inflammatory bowel disease patients: a review. World J Gastroenterol 21:6491–6498. doi: 10.3748/wjg.v21.i21.6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cattaneo C, Antoniazzi F, Caira M, Castagnola C, Delia M, Tumbarello M, Rossi G, Pagano L. 2013. Nocardia spp infections among hematological patients: results of a retrospective multicenter study. Int J Infect Dis 17:e610–e614. doi: 10.1016/j.ijid.2013.01.013. [DOI] [PubMed] [Google Scholar]
- 10.Hashemi-Shahraki A, Heidarieh P, Bostanabad SZ, Hashemzadeh M, Feizabadi MM, Schraufnagel D, Mirsaeidi M. 2015. Genetic diversity and antimicrobial susceptibility of Nocardia species among patients with nocardiosis. Sci Rep 5:17862. doi: 10.1038/srep17862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trichet E, Cohen-Bacrie S, Conrath J, Drancourt M, Hoffart L. 2011. Nocardia transvalensis keratitis: an emerging pathology among travelers returning from Asia. BMC Infect Dis 11:296. doi: 10.1186/1471-2334-11-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Apostolou A, Bolcen SJ, Dave V, Jani N, Lasker BA, Tan CG, Montana B, Brown JM, Genese CA. 2012. Nocardia cyriacigeorgica infections attributable to unlicensed cosmetic procedures—an emerging public health problem? Clin Infect Dis 55:251–253. doi: 10.1093/cid/cis341. [DOI] [PubMed] [Google Scholar]
- 13.Torres HA, Reddy BT, Raad II, Tarrand J, Bodey GP, Hanna HA, Rolston KVI, Kontoyiannis DP. 2002. Nocardiosis in cancer patients. Medicine 81:388–397. doi: 10.1097/00005792-200209000-00004. [DOI] [PubMed] [Google Scholar]
- 14.Minero MV, Marın M, Cercenado E, Rabadan PM, Bouza E, Munoz P. 2009. Nocardiosis at the turn of the century. Medicine 88:250–261. doi: 10.1097/MD.0b013e3181afa1c8. [DOI] [PubMed] [Google Scholar]
- 15.Lerner PI. 1996. Nocardiosis. Clin Infect Dis 22:891–905. doi: 10.1093/clinids/22.6.891. [DOI] [PubMed] [Google Scholar]
- 16.Uhde KB, Pathak S, McCullum I Jr, Jannat-Khah DP, Shadomy SV, Dykewicz CA, Clark TA, Smith TL, Brown JM. 2010. Antimicrobial-resistant Nocardia isolates, United States, 1995–2004. Clin Infect Dis 51:1445–1448. doi: 10.1086/657399. [DOI] [PubMed] [Google Scholar]
- 17.Toprak E, Veres A, Michel J-B, Chait R, Hartl DL, Kishony R. 2012. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat Genet 44:101–105. doi: 10.1038/ng.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abdizadeh H, Tamer YT, Acar O, Toprak E, Atilgan AR, Atilgan C. 2017. Increased substrate affinity in the Escherichia coli L28R dihydrofolate reductase mutant causes trimethoprim resistance. Phys Chem Chem Phys 19:11416–11428. doi: 10.1039/C7CP01458A. [DOI] [PubMed] [Google Scholar]
- 19.Vickers AA, Potter NJ, Fishwick CWG, Chopra I, Neill AJO. 2009. Analysis of mutational resistance to trimethoprim in Staphylococcus aureus by genetic and structural modelling techniques. J Antimicrob Chemother 63:1112–1117. doi: 10.1093/jac/dkp090. [DOI] [PubMed] [Google Scholar]
- 20.Padayachee T, Klugman KP. 1999. Novel expansions of the gene encoding dihydropteroate synthase in trimethoprim-sulfamethoxazole-resistant Streptococcus pneumoniae. Antimicrob Agents Chemother 43:2225–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang L, Crothers K, Atzori C, Benfield T, Miller R, Rabodonirina M, Helweg-Larsen J. 2004. Dihydropteroate synthase gene mutations in Pneumocystis and sulfa resistance. Emerg Infect Dis 10:1721–1728. doi: 10.3201/eid1010.030994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmitz F-J, Perdikouli M, Beeck A, Verhoef J, Fluit AC. 2001. Resistance to trimethoprim-sulfamethoxazole and modifications in genes coding for dihydrofolate reductase and dihydropteroate synthase in European Streptococcus pneumoniae isolates. J Antimicrob Chemother 48:935–936. doi: 10.1093/jac/48.6.935. [DOI] [PubMed] [Google Scholar]
- 23.Watson M, Liu J, Ollis D. 2007. Directed evolution of trimethoprim resistance in Escherichia coli. FEBS J 274:2661–2671. doi: 10.1111/j.1742-4658.2007.05801.x. [DOI] [PubMed] [Google Scholar]
- 24.Huovinen P. 2001. Resistance to trimethoprim-sulfamethoxazole. Clin Infect Dis 32:1608–1614. doi: 10.1086/320532. [DOI] [PubMed] [Google Scholar]
- 25.Shin HW, Lim J, Kim S, Kim J, Kwon GC, Koo SH. 2015. Characterization of trimethoprim-sulfamethoxazole resistance genes and their relatedness to class 1 integron and insertion sequence common region in Gram-negative bacilli. J Microbiol Biotechnol 25:137–142. doi: 10.4014/jmb.1409.09041. [DOI] [PubMed] [Google Scholar]
- 26.Coque TM, Singh KV, Weinstock GM, Murray BE. 1999. Characterization of dihydrofolate reductase genes from trimethoprim-susceptible and trimethoprim-resistant strains of Enterococcus faecalis. Antimicrob Agents Chemother 43:141–147. doi: 10.1093/jac/43.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kadlec K, Schwarz S. 2009. Identification of a novel trimethoprim resistance gene, dfrK, in a methicillin-resistant Staphylococcus aureus ST398 strain and its physical linkage to the tetracycline resistance gene tet(L). Antimicrob Agents Chemother 53:776–778. doi: 10.1128/AAC.01128-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JC, Oh JY, Cho JW, Park JC, Kim JM, Seol SY, Cho DT. 2001. The prevalence of trimethoprim-resistance-conferring dihydrofolate reductase genes in urinary isolates of Escherichia coli in Korea. J Antimicrob Chemother 47:599–604. doi: 10.1093/jac/47.5.599. [DOI] [PubMed] [Google Scholar]
- 29.Antunes P, Machado J, Sousa JC, Peixe L. 2005. Dissemination of sulfonamide resistance genes (sul1, sul2, sul3) in Portuguese Salmonella enterica strains and relation with integrons. Antimicrob Agents Chemother 49:836–839. doi: 10.1128/AAC.49.2.836-839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valdezate S, Garrido N, Carrasco G, Villalon P, Medina-Pascual MJ, Saez-Nieto JA. 2015. Resistance gene pool to co-trimoxazole in non-susceptible Nocardia strains. Front Microbiol 6:376. doi: 10.3389/fmicb.2015.00376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehta HH, Prater AG, Shamoo Y. 2018. Using experimental evolution to identify druggable targets that could inhibit the evolution of antimicrobial resistance. J Antibiot (Tokyo) 71:279–286. doi: 10.1038/ja.2017.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beabout K, Hammerstrom TG, Wang TT, Bhatty M, Christie PJ, Saxer G, Shamoo Y. 2015. Rampant parasexuality evolves in a hospital pathogen during antibiotic selection. Mol Biol Evol 32:2585–2597. doi: 10.1093/molbev/msv133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huovinen P. 1987. Trimethoprim resistance. Antimicrob Agents Chemother 31:1451–1456. doi: 10.1128/AAC.31.10.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huovinen P, Sundstrom L, Swedberg G, Skold O. 1995. Trimethoprim and sulfonamide resistance. Antimicrob Agents Chemother 39:279–289. doi: 10.1128/AAC.39.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Young H-K, Skurray RA, Amyes SGB. 1987. Plasmid-mediated trimethoprim-resistance in Staphylococcus aureus. Characterization of the first Gram-positive plasmid dihydrofolate reductase (type S1). Biochem J 243:309–312. doi: 10.1042/bj2430309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Queener SF, Cody V, Pace J, Torkelson P, Gangjee A. 2013. Trimethoprim resistance of dihydrofolate reductase variants from clinical isolates of Pneumocystis jirovecii. Antimicrob Agents Chemother 57:4990–4998. doi: 10.1128/AAC.01161-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li R, Sirawaraporn R, Chitnumsub P, Sirawaraporn W, Wooden J, Athappilly F, Turley S, Hol WGJ. 2000. Three-dimensional structure of M. tuberculosis dihydrofolate reductase reveals opportunities for the design of novel tuberculosis drugs. J Mol Biol 295:307–323. doi: 10.1006/jmbi.1999.3328. [DOI] [PubMed] [Google Scholar]
- 38.Sawaya MR, Kraut J. 1997. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry 36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 39.Palmer AC, Toprak E, Baym M, Kim S, Veres A, Bershtein S, Kishony R. 2015. Delayed commitment to evolutionary fate in antibiotic resistance fitness landscapes. Nat Commun 6:7385. doi: 10.1038/ncomms8385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sköld O. 2000. Sulfonamide resistance: mechanisms and trends. Drug Resist Updat 3:155–160. doi: 10.1054/drup.2000.0146. [DOI] [PubMed] [Google Scholar]
- 41.Yun MK, Wu Y, Li Z, Zhao Y, Waddell BM, Ferreira AM, Lee RE, Bashford D, White SW. 2012. Catalysis and sulfa drug resistance in dihydropteroate synthase. Science 335:1110–1114. doi: 10.1126/science.1214641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Babaoglu K, Qi J, Lee RE, White SW. 2004. Crystal structure of 7,8-dihydropteroate synthase from Bacillus anthracis: mechanism and novel inhibitor design. Structure 12:1705–1717. doi: 10.1016/j.str.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 43.Palmer AC, Kishony R. 2014. Opposing effects of target overexpression reveal drug mechanisms. Nat Commun 5:4296. doi: 10.1038/ncomms5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fermer C, Kristiansen B-E, Skold O, Swedberg G. 1995. Sulfonamide resistance in Neisseria meningitidis as defined by site-directed mutagenesis could have its origin in other species. J Bacteriol 177:4669–4675. doi: 10.1128/jb.177.16.4669-4675.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Achari A, Somers DO, Champness JN, Bryant PK, Rosemond J, Stammers DK. 1997. Crystal structure of the anti-bacterial sulfonamide drug target dihydropteroate synthase. Nat Struct Biol 4:490–497. doi: 10.1038/nsb0697-490. [DOI] [PubMed] [Google Scholar]
- 46.Baca AM, Sirawaraporn R, Turley S, Sirawaraporn W, Hol WGJ. 2000. Crystal structure of Mycobacterium tuberculosis 6-hydroxymethyl-7,8-dihydropteroate synthase in complex with pterin monophosphate: new insight into the enzymatic mechanism and sulfa-drug action. J Mol Biol 302:1193–1212. doi: 10.1006/jmbi.2000.4094. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Q, Lambert G, Liao D, Kim H, Robin K, Tung CK, Pourmand N, Austin RH. 2011. Acceleration of emergence of bacterial antibiotic resistance in connected microenvironments. Science 333:1764–1767. doi: 10.1126/science.1208747. [DOI] [PubMed] [Google Scholar]
- 48.Dallas WS, Gowen JE, Ray PH, Cox MJ, Dev IK. 1992. Cloning, sequencing, and enhanced expression of the dihydropteroate synthase gene of Escherichia coli MC4100. J Bacteriol 174:5961–5970. doi: 10.1128/jb.174.18.5961-5970.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams DL, Spring L, Harris E, Roche P, Gillis TP. 2000. Dihydropteroate synthase of Mycobacterium leprae and dapsone resistance. Antimicrob Agents Chemother 44:1530–1537. doi: 10.1128/AAC.44.6.1530-1537.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gengenbacher M, Xu T, Niyomrattanakit P, Spraggon G, Dick T. 2008. Biochemical and structural characterization of the putative dihydropteroate synthase ortholog Rv1207 of Mycobacterium tuberculosis. FEMS Microbiol Lett 287:128–135. doi: 10.1111/j.1574-6968.2008.01302.x. [DOI] [PubMed] [Google Scholar]
- 51.Liu T, Wang B, Guo J, Zhou Y, Julius M, Njire M, Cao Y, Wu T, Liu Z, Wang C, Xu Y, Zhang T. 2015. Role of folP1 and folP2 genes in the action of sulfamethoxazole and trimethoprim against mycobacteria. J Microbiol Biotechnol 25:1559–1567. doi: 10.4014/jmb.1503.03053. [DOI] [PubMed] [Google Scholar]
- 52.Grawert T, Fischer M, Bacher A. 2013. Structures and reaction mechanisms of GTP cyclohydrolases. IUBMB Life 65:310–322. doi: 10.1002/iub.1153. [DOI] [PubMed] [Google Scholar]
- 53.Heinberg A, Kirkman L. 2015. The molecular basis of antifolate resistance in Plasmodium falciparum: looking beyond point mutations. Ann N Y Acad Sci 1342:10–18. doi: 10.1111/nyas.12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heinberg A, Siu E, Stern C, Lawrence EA, Ferdig MT, Deitsch KW, Kirkman LA. 2013. Direct evidence of the adaptive role of copy number variation on antifolate susceptibility in Plasmodium falciparum. Mol Microbiol 88:702–712. doi: 10.1111/mmi.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Belfort M, Maley G, Pedersen-Lane J, Maley F. 1983. Primary structure of the Escherichia coli thyA gene and its thymidylate synthase product. Proc Natl Acad Sci U S A 80:4914–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Howell EE, Foster PG, Foster LM. 1988. Construction of a dihydrofolate reductase-deficient mutant of Escherichia coli by gene replacement. J Bacteriol 170:3040–3045. doi: 10.1128/jb.170.7.3040-3045.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumar G, Guan S, Frantom PA. 2014. Biochemical characterization of the retaining glycosyltransferase glucosyl-3-phosphoglycerate synthase from Mycobacterium tuberculosis. Arch Biochem Biophys 564:120–127. doi: 10.1016/j.abb.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 58.Pereira PJB, Empadinhas N, Albuquerque L, Sa-Moura B, da Costa MS, Macedo-Ribeiro S. 2008. Mycobacterium tuberculosis glucosyl-3-phosphoglycerate synthase: structure of a key enzyme in methylglucose lipopolysaccharide biosynthesis. PLoS One 3:e3748. doi: 10.1371/journal.pone.0003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miller C, Kong J, Tran TT, Arias CA, Saxer G, Shamoo Y. 2013. Adaptation of Enterococcus faecalis to daptomycin reveals an ordered progression to resistance. Antimicrob Agents Chemother 57:5373–5383. doi: 10.1128/AAC.01473-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hammerstrom TG, Beabout K, Clements TP, Saxer G, Shamoo Y. 2015. Acinetobacter baumannii repeatedly evolves a hypermutator phenotype in response to tigecycline that effectively surveys evolutionary trajectories to resistance. PLoS One 10:e0140489. doi: 10.1371/journal.pone.0140489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saxer G, Krepps MD, Merkley ED, Ansong C, Kaiser BLD, Valovska M-T, Ristic N, Yeh PT, Prakash VP, Leiser OP, Nakhleh L, Gibbons HS, Kreuzer HW, Shamoo Y. 2014. Mutations in global regulators lead to metabolic selection during adaptation to complex environments. PLoS Genet 10:e1004872. doi: 10.1371/journal.pgen.1004872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eastman JM, Harmon LJ, La HJ, Joyce P, Forney LJ. 2011. The onion model, a simple neutral model for the evolution of diversity in bacterial biofilms. J Evol Biol 24:2496–2504. doi: 10.1111/j.1420-9101.2011.02377.x. [DOI] [PubMed] [Google Scholar]
- 63.Ponciano JM, La HJ, Joyce P, Forney LJ. 2009. Evolution of diversity in spatially structured Escherichia coli populations. Appl Environ Microbiol 75:6047–6054. doi: 10.1128/AEM.00063-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.