

ABSTRACT
Simeprevir is a novel NS3/4A protease inhibitor (PI) of hepatitis C virus (HCV). The baseline polymorphism NS3-Q80K is frequently observed in genotype (GT) 1a HCV and often associated with treatment failure in simeprevir-containing regimens. We aimed to elucidate mechanisms of treatment failure due to NS3-Q80K. We included a Q80R mutation in our study and generated a series of Huh-7.5 cell lines, each of which harbored either wild-type GT 1a strain H77S.3 or the Q80K or Q80R variant. The cells were cultured with increasing concentrations of simeprevir, and NS3 domain sequences were determined. The mutations identified by sequence analyses were subsequently introduced into H77S.3. The sensitivity of each mutant to the NS3/4A PIs simeprevir, asunaprevir, grazoprevir, and paritaprevir was analyzed. We introduced the mutations into GT 1b strain N.2 and compared the sensitivity to simeprevir with that of GT 1a strain H77S.3. While simeprevir treatment selected mutations at residue D168, such as D168A/V in the wild-type virus, an additional mutation at residue R155, R155K, was selected in Q80K/R variants at simeprevir concentrations of <2.5 μM. Sensitivity analyses showed that simeprevir concentrations of <1 μM significantly boosted the replication of Q80K/R R155K variants. Interestingly, this boost was not observed with the other NS3/4A PIs or in Q80R R155Q/G/T/W variants or GT 1b isolates. The boosted replication of the Q80K+R155K variant by simeprevir could be related to treatment failure in simeprevir-containing antiviral treatments in GT 1a HCV-infected patients with the NS3-Q80K polymorphism. This result provides new insight into how resistance-associated variants can cause treatment failure.
KEYWORDS: direct-acting antivirals, drug resistance mechanisms, hepatitis C virus, simeprevir
INTRODUCTION
About 180 million people worldwide are infected with hepatitis C virus (HCV) (1), and approximately 704,000 people die each year from HCV-related liver diseases such as liver failure and hepatocellular carcinoma (2). Established HCV cell culture systems have facilitated the development of direct-acting antiviral reagents (DAAs) targeting the nonstructural protein 3/4A (NS3/4A) protease, the NS5A protein, and the NS5B polymerase. Recently, DAA combination regimens with dramatically increased sustained virologic responses (SVRs) have been reported; however, the preexistence and selection of resistance-associated variants (RAVs) can still result in treatment failure (3). Baseline RAVs and selected RAVs after treatment failure are clinically important, because given the limited number of DAA classes and the possibility of cross-resistance, they can also interfere with second-line DAA treatments. Therefore, it is crucial to clarify the mechanisms of the emergence and selection of RAVs.
Simeprevir is a novel macrocyclic NS3/4A protease inhibitor (PI). It was initially used with PEGylated interferon (PEG) and ribavirin (RBV), and since then, it has been used with other DAAs, such as NS5A or NS5B inhibitors. Although newer types of NS3/4A PIs, such as grazoprevir, paritaprevir, and glecaprevir, are available, the latest American Association for the Study of Liver Diseases (AASLD)-Infectious Diseases Society of America (IDSA) and European Association for Study of Liver (EASL) guidelines still recommend the use of simeprevir in combination with the NS5B inhibitor sofosbuvir as one of the treatment options for patients infected with HCV genotypes (GTs) 1 and 4 (4, 5).
When simeprevir was used with PEG and RBV, the Q80K polymorphism in the NS3 protease domain at baseline was found to be significantly related to treatment failure, particularly in HCV GT 1a-infected patients. For example, the SVR rate was lower in patients with the Q80K variant than in those without it (58% versus 84% in GT 1a-infected treatment-naive patients and 47% versus 79% in GT 1a-infected prior relapsers) (6, 7). Recently, combination regimens containing only DAAs without PEG, namely, interferon-free DAA treatments, have become the main antiviral treatments for HCV. Accordingly, simeprevir has been frequently used with sofosbuvir. Several clinical studies have demonstrated that this combination usually achieves SVR rates of over 90%. The impact of the Q80K polymorphism on the SVR with simeprevir-sofosbuvir combination treatment seems to be attenuated in comparison to that with treatment with simeprevir, PEG, and RBV. The COSMOS study, a phase II trial, evaluated the efficacy and safety of simeprevir plus sofosbuvir with or without RBV for 12 or 24 weeks in HCV GT 1-infected patients. Six patients, including four with the Q80K polymorphism at baseline, experienced viral relapse (8). Subsequently, two phase III trials were conducted to further evaluate the efficacy and safety of simeprevir plus sofosbuvir, namely, the OPTIMIST-1 and OPTIMIST-2 studies. The OPTIMIST-1 study included noncirrhotic HCV GT 1-infected patients, and the combination of simeprevir plus sofosbuvir was administered for 8 or 12 weeks. The OPTIMIST-2 study included cirrhotic GT 1-infected patients with simeprevir plus sofosbuvir administered for 12 weeks. The OPTIMIST-2 study showed that HCV GT 1a-infected patients without baseline Q80K polymorphisms had a higher SVR rate (92%) than did those with baseline Q80K polymorphisms (74%) (9). On the other hand, the OPTIMIST-1 study showed that the Q80K polymorphism at baseline was not related to treatment failure in HCV GT 1a-infected patients in the 12-week arm (SVR12) (96% with the Q80K polymorphism versus 97% without it); however, that study also revealed that the patients without the baseline Q80K polymorphism had a higher SVR12 rate (84%) than did those with the baseline Q80K polymorphism (73%) in the 8-week arm (10). Although there is disagreement about the importance of the Q80K polymorphism at baseline in the GT 1a HCV treatment described above, baseline detection of Q80K should be considered a relevant factor related to the failure of simeprevir-containing treatments.
The Q80K polymorphism is reported to be present in up to 47% of HCV GT 1a-infected patients (11–13), with the highest prevalence in North America (14). A recent study analyzing samples from clinical trials across the world showed that the Q80K polymorphism was detected in patients infected with GT 1a (36%), GT 1b (1.7%), GT 5a (100%), and GT 6a (100%), although there were many fewer samples of GTs 5a and 6a than of GTs 1a and 1b (15). The Q80K polymorphism has also been reported to be observed more frequently in patients coinfected with human immunodeficiency virus (HIV) than in those without coinfection (12). Therefore, it is crucial to clarify the mechanisms related to treatment failure in patients with the NS3-Q80K polymorphism.
The Q80K mutation, in addition to the Q80R mutation, is known to confer low levels of simeprevir resistance in vitro (16, 17); however, this low level of drug resistance caused by these mutations seems insufficient to explain the failures of simeprevir-containing treatments in patients with the Q80K baseline polymorphism. In this study, using HCV cell culture systems, we demonstrated that Q80K- or Q80R-harboring variants under drug pressure via simeprevir preferentially selected a second-site mutation at residue R155, R155K, at simeprevir concentrations of <2.5 μM, in the GT 1a NS3 protease backbone. We found that the replication capacity of the Q80K/R+R155K double variants was unexpectedly boosted by low-dose simeprevir and that the Q80K+R155K variant had the highest selective advantage in terms of replication capacity at around 0.75 to 1 μM simeprevir. This remarkable result could explain the reason for treatment failures among patients with the NS3-Q80K polymorphism of HCV GT 1a and provides new insight into the mechanism of treatment failure among resistance-associated variants.
RESULTS
Although the importance of identifying the NS3-Q80K polymorphism at baseline is clinically recognized, the mechanisms underlying the causes of treatment failure with this polymorphism remain unclear. In addition to the Q80K mutation, we also included the Q80R mutation in our study because it has also been reported to cause simeprevir resistance in vitro (16).
Selection of simeprevir-resistant variants from wild-type Q80K/R virus-replicating cells.
We introduced the Q80K and Q80R mutations individually into H77S.3/Blast5A, as described in our previous study (18), and designated these strains H77S.3/Blast5A-Q80K and H77S.3/Blast5A-Q80R, respectively. Briefly, we inserted a blasticidin S resistance-encoding gene into the NS5A region of GT 1a strain H77S.3 (see Fig. S1A in the supplemental material) and transfected this HCV RNA into Huh-7.5 cells to subsequently select blasticidin S-resistant cells in the presence of blasticidin S. All blasticidin S-selected cells should support HCV replication. We transfected H77S.3/Blast5A, H77S.3/Blast5A-Q80K, and H77S.3/Blast5A-Q80R RNAs into Huh-7.5 cells for subsequent selection with blasticidin S for 2 weeks. We confirmed the expression of the HCV core protein in all blasticidin S-selected cells by immunofluorescence (Fig. S1B). We also determined the sequence of the NS3 protease domain from HCV RNA in these selected cells. In the case of H77S.3/Blast5A RNA-transfected cells, no mutations were observed, while in H77S.3/Blast5A-Q80K/R RNA-transfected cells, the Q80K/R mutation was preserved, without the appearance of any other mutations (Fig. S1C).
These HCV-replicating cells were treated with or without simeprevir under continuous selection by blasticidin S. The initial concentration of simeprevir was 10 nM, which was almost the same as the 50% effective concentration (EC50), which was then gradually increased every two passages to 50 nM, 100 nM, 250 nM, and 500 nM (Fig. 1A). At first, cell death due to blasticidin S was observed, but it was not observed at the fourth passage or thereafter. The cells were split when they reached confluence. Total cellular RNA was extracted at each split to quantify the abundance of HCV RNA and determine the viral sequences.
FIG 1.

Simeprevir treatment of HCV-replicating cells and abundance of HCV RNA. (A) Schematic representation of simeprevir treatment in HCV-replicating cells. Huh-7.5 cells harboring H77S.3/Blast5A (wt), H77S.3/Blast5A-Q80K (Q80K), and H77S.3/Blast5A-Q80R (Q80R) viruses were treated with or without simeprevir under continuous selection by 10 μg/ml blasticidin S, and the cells were split when they reached confluence. The starting concentration of simeprevir was 10 nM, in addition to 0 nM, i.e., the control, which was then gradually increased at every second passage to 50 nM, 100 nM, 250 nM, and 500 nM. Total cellular RNA was extracted at each split to quantify the abundance of HCV RNA and determine the viral sequences. (B) HCV RNA levels during simeprevir treatment. The levels of HCV RNA and β-actin RNA in the total cellular RNA were measured by qRT-PCR, and the level of HCV RNA was normalized to that of β-actin. The normalized level of HCV RNA at the first time point in each passage in Huh-7.5 cells harboring the wt, Q80K, or Q80R virus is shown, and the error bars show the standard deviations from three independent measurements.
Regarding the abundance of HCV RNA measured by reverse transcription-quantitative PCR (qRT-PCR), in the case of the wild-type (wt) virus, the amount of HCV RNA in the simeprevir-treated cells continuously decreased until the third passage, and a low level was maintained, but it started to increase from the ninth passage. In the case of Q80K/R viruses, the amount of HCV RNA in the simeprevir-treated cells decreased until the fourth passage but started to increase from the fifth passage (Fig. 1B).
Population-based sequence analysis of simeprevir-resistant variants.
We performed sequence analysis on the NS3 protease domain of HCV RNA in cells treated with simeprevir. By a population-based sequence analysis, we found that mutations R155K, D168A, and D168V emerged in the wt virus from the sixth passage. While the abundance of the R155K mutation started to decrease from the ninth passage, mutations D168A and D168V became enriched until the last time point. We did not observe any other mutations during the entire course of simeprevir treatment, including at residue 80. In the case of experiments with the Q80K and Q80R viruses, both baseline mutations were preserved throughout the course of simeprevir treatment. The R155K mutation emerged from the seventh passage and persisted until the last time point. Interestingly, in contrast to the wt virus, there were no mutations observed at residue 168 (Fig. 2).
FIG 2.
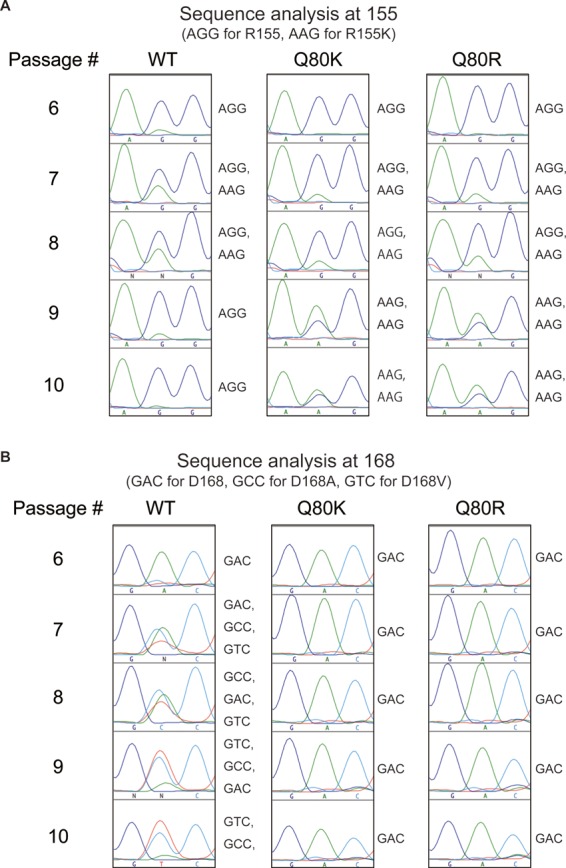
Population-based sequence analysis of the NS3 protease domain. The sequences of the entire NS3 protease domain in HCV RNA starting with wt, Q80K, and Q80R viruses were determined by a population-based analysis. (A) Sequence analysis of the 155th position between the 6th and 10th passages. (B) Sequence analysis of the 168th position between the 6th and 10th passages.
We also treated Q80K and Q80R variant-replicating cells with up to 5,000 nM simeprevir in a separate experiment and performed a population-based sequence analysis. Both baseline mutations Q80K and Q80R were preserved throughout the course of simeprevir treatment. The R155K mutation was continuously observed at simeprevir concentrations of up to 2,500 nM but disappeared at higher concentrations. Instead of the R155K mutation, the D168V mutation was observed (see Fig. S2 in the supplemental material).
Clonal sequence analysis of simeprevir-resistant variants.
We analyzed the last time point, at 1,000 nM simeprevir, of the simeprevir selection pressure experiment by clonal sequence analysis. We determined the sequences of 30 clones derived from the experiment with the wt virus and 35 clones in each case derived from the experiment with the Q80K and Q80R viruses. In the experiment with the wt virus, the most frequently observed variant was D168V, followed by D168A. Variants harboring D168 mutations such as D168A, D168V, and D168V+Y56H accounted for about 82.8% of the clones analyzed, whereas only 14.3% of the variants harbored R155 mutations such as R155K and R155K+T177A. In the experiment with the Q80K virus, the baseline mutation was preserved within all clones. The most frequently observed variant was Q80K+R155K, and variants harboring both Q80K and R155K mutations, such as Q80K+R155K and Q80K+S101P/V151A+R155K, accounted for 83.3% of the clones. On the other hand, variants harboring the D168 mutation Q80K+D168E constituted only 3.3% of the clones. In the experiment with the Q80R virus, the baseline mutation was preserved in 96.7% of the clones, whereas the Q80H mutation appeared in 3.3% of the clones. The Q80R+R155K variant was observed most frequently, and variants harboring both Q80R and R155K mutations, such as Q80R+R155K and Q80R+S55F/P88A/L127F+R155K, accounted for 69.9% of the clones. On the other hand, 16.7% of the variants harbored D168 mutations such as Q80R+D168E, Q80R+D168E, and Q80R+S102P+D168E (Table 1). The results showed that the mutation pattern selected by simeprevir treatment depended on the presence of baseline mutations at residue Q80. While treatment with simeprevir was likely to select D168A/V resistance mutations in the wt virus, it was more likely to select R155K mutations in the Q80K/R viruses.
TABLE 1.
Variants identified by clonal sequence analyses

Replication capacity of and production of infectious virus by simeprevir-resistant variants.
We assessed the replication capacities of variants such as the D168A, D168V, R155K, Q80K+R155K, and Q80R+R155K variants, which were selected with simeprevir treatment to constitute more than 11% of the clones in a clonal sequence analysis. We also included wt, Q80K, and Q80R viruses in this analysis. For this purpose, we used a full-length HCV genome that contained a Gaussia luciferase (GLuc)-encoding sequence between p7 and NS2 of genotype 1a strain H77S.3, as described in our previous study (19). In that study, we showed that the abundance of HCV RNA was correlated with GLuc activity. Since GLuc is secreted from the cells into the medium, we can easily monitor the replication capacity of a virus by using GLuc activity in the medium as an indicator. Because we previously created variants containing Q80R, R155K, D168A, and D168V mutations in the backbone of H77S.3/GLuc2A and reported their efficient replication (19), we newly inserted the Q80K, Q80K+R155K, and Q80R+R155K mutations into H77S.3/GLuc2A. After the transfection of these HCV RNAs into Huh-7.5 cells, we collected and replaced the media every 24 h until 96 h and measured the GLuc activity in the media (Fig. 3A). The replication capacities of the Q80K and Q80R variants were 1.26- and 0.67-fold higher than that of the wt virus, respectively. On the other hand, the replication capacities of the D168A, D168V, and R155K variants were 0.21-, 0.47-, and 0.32-fold higher than that of the wt virus, respectively. Interestingly, the addition of the R155K mutation to the Q80K/R mutation reduced the replication capacities of the Q80K/R variants to 0.63- and 0.10-fold higher than that of the wt virus, respectively. We also examined infectious-virus yields of these variants. For this purpose, the culture media collected from cells transfected with HCV RNAs of these variants in the backbone of H77S.3/GLuc2A were used. The collected culture media were then used to infect naive Huh-7.5 cells, and the ability to produce infectious virus was determined by a conventional focus-forming unit (FFU) assay 72 h after infection. The infectious-virus yield of the R155K, Q80K+R155K, and Q80R+R155K variants was significantly lower than that of the wt (Fig. 3B). We also compared the replication capacities of and the abilities to produce infectious virus by each variant. As shown in Fig. 3C, the replication capacity and the ability to produce infectious virus were well correlated in most variants, except for the Q80R+R155K variant. These results show that the Q80K+R155K and Q80R+R155K variants do not have an advantage in viral fitness measured by the replication capacity and infectious-virus yield in the absence of simeprevir.
FIG 3.
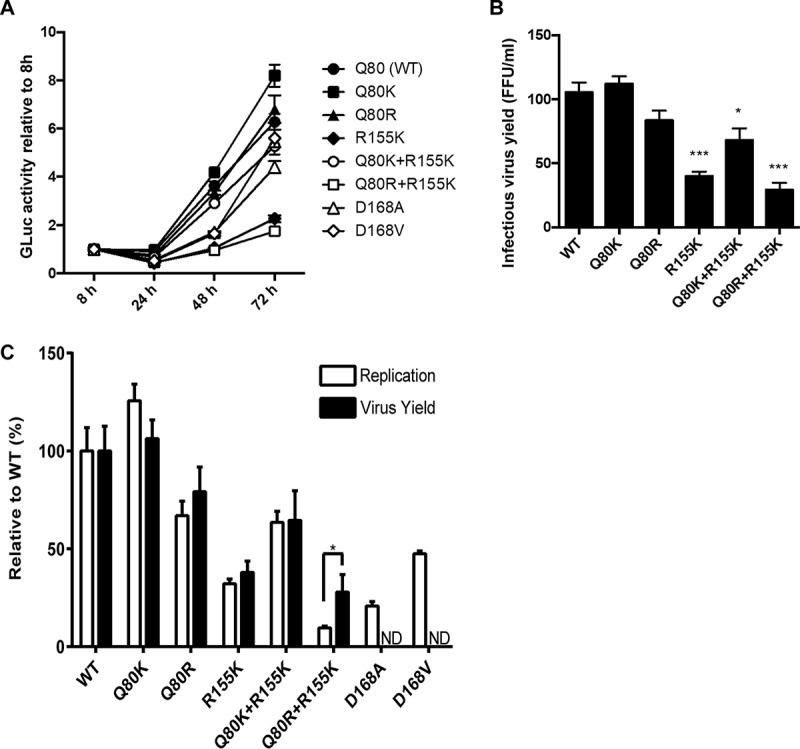
Replication capacities of and infectious-virus production by NS3 variants. Mutations such as Q80K, Q80R, Q80R, R155K, Q80K+R155K, Q80R+R155K, D168A, and D168V were introduced into H77S.3/GLuc2A, and the HCV RNAs were transfected into Huh-7.5 cells. The cell culture media were collected and replaced 8 h after transfection and then collected and replaced every 24 h thereafter until 72 h. The GLuc activity in the media collected was measured. (A) Replication capacity. Results for each variant were normalized to the 8-h GLuc activity, represent the means of data for triplicate samples, and are representative of data from multiple experiments. (B) Yield of infectious virus. The media collected 72 h after HCV RNA transfection were used immediately to infect naive Huh-7.5 cells. Seventy-two hours after infection, the numbers of infectious-virus foci were determined by an FFU assay. Data shown here represent the mean FFU per milliliter ± standard deviations from three independent experiments. (C) Impact of each mutation on the replication capacity and yield of infectious virus. The GLuc activity secreted by RNA-transfected cells at 72 h versus 8 h, as shown in panel A, was normalized to that of the wild type and plotted adjacent to the yield of infectious virus from each variant, similarly normalized to that of the wt. The difference between the relative capacities to replicate as RNA and to produce infectious virus is significant by Student's t test. ND, not determined; *, P < 0.05; ***, P < 0.001.
Sensitivity of each variant to simeprevir.
We then determined the EC50 values of simeprevir for the wt virus as well as for selected variants by using GLuc-containing HCV genomes. The HCV RNA of each variant was transfected into Huh-7.5 cells, and the cells were treated with simeprevir at concentrations in the range of 0 to 1,000 nM. As expected from data from previous reports (16, 19), all selected variants showed various degrees of resistance to simeprevir. The EC50s for the wt, Q80K, Q80R, and R155K viruses were 6 nM, 112 nM, 102 nM (fold change [FC] compared with the wt virus of 17), and 259 nM (FC of 43), respectively (Table 2). The EC50s for the D168A and D168V variants were >1,000 nM, and we did not observe any suppression of replication within the range of simeprevir concentrations tested. Unexpectedly, the replication capacities of the Q80K+R155K and Q80R+R155K variants increased with simeprevir treatment in a dose-dependent manner at concentrations of <1,000 nM and were 1.7- and 2.3-fold higher, respectively, with 1,000 nM simeprevir than without simeprevir. On the other hand, the replication capacity of the D168A or D168V variant was not altered by simeprevir in this concentration range (Fig. 4A). We also analyzed the sensitivities of viruses to simeprevir at a wider range of concentrations. The replication capacities of the Q80K+R155K and Q80R+R155K variants started to increase at 500 nM simeprevir, reached a maximum at 1,000 nM, and then began to decrease at 2,000 nM (Fig. 4B). At 10,000 nM simeprevir, the replication capacity of the D168A variant was severely inhibited, while that of the D168V variant was moderately inhibited (see Fig. S3 in the supplemental material). The EC50s were calculated for each variant and were 2,879 nM for the Q80K+R155K variant, 2,709 nM for the Q80R+R155K variant, 5,755 nM for the D168A variant, and 8,070 nM for the D168V variant (Table 2). When we examined the sensitivities of the Q80K/R+R155K variants to simeprevir in cell lines other than Huh-7.5 cells, such as FT3-7 cells and KH cells, simeprevir enhanced the replication of these variants at concentrations of up to 1,000 nM simeprevir in a dose-dependent manner, and replication then began to decrease at 2,000 nM simeprevir in both FT3-7 cells (Fig. S4A) and KH cells (Fig. S4B). Our results suggested that simeprevir boosted the replication capacities of the Q80K+R155K and Q80R+R155K double variants only at drug concentrations of <1,000 nM.
TABLE 2.
EC50 for each variant measured in the H77S.3/GLuc2A backbone
| Variant | EC50 (nM) | FC |
|---|---|---|
| Wild type | 6 | 1 |
| Q80K | 112 | 19 |
| Q80R | 102 | 17 |
| R155K | 259 | 43 |
| R155T | 85 | 14 |
| R155Q | 5 | 0.8 |
| R155G | 74 | 12 |
| R155W | 544 | 90.7 |
| D168A | 5,755 | 959 |
| D168V | 8,070 | 1,345 |
| Q80K+R155K | 2,879 | 480 |
| Q80R+R155K | 2,709 | 452 |
| Q80R+R155Q | 356 | 59 |
| Q80R+R155G | 2,677 | 446 |
| Q80R+R155T | 1,035 | 173 |
| Q80R+R155W | 4,110 | 685 |
FIG 4.

Sensitivities of NS3 variants to simeprevir. (A) wt HCV RNA or the HCV RNAs encoding NS3 variants, such as Q80K, Q80R, R155K, D168A, D168V, Q80K+R155K, or Q80R+R155K in H77S.3/GLuc2A, were transfected into Huh-7.5 cells. The medium was replaced with fresh medium containing only DMSO or simeprevir at 100, 200, 300, 400, 500, 750, and 1,000 nM after 24 h and at 24-h intervals thereafter. The secreted GLuc activity in the medium was determined at 72 h posttransfection. In each NS3 variant, the GLuc activity in simeprevir-treated cells was normalized to that in cells treated with DMSO only. Statistical significance was analyzed in comparison to 0 nM simeprevir by one-way ANOVA and Bonferroni's multiple-comparison tests. (B) HCV RNAs encoding the Q80K+R155K and Q80R+R155K variants in H77S.3/GLuc2A were transfected into Huh-7.5 cells. The medium was replaced with fresh medium containing only DMSO or simeprevir at 100, 500, 750, 1,000, 2,000, 3,000, 4,000, 5,000, and 10,000 nM at 24 h and at 24-h intervals thereafter. The secreted GLuc activity was determined at 72 h posttransfection. For each NS3 variant, the GLuc activity from simeprevir-treated cells was normalized to that of cells treated with DMSO only. Statistical significance was analyzed in comparison to 0 nM simeprevir by one-way ANOVA and Bonferroni's multiple-comparison tests. (C) HCV RNAs encoding the Q80K+R155K or Q80R+R155K variant in H77S.3 were transfected into Huh-7.5 cells. The medium was replaced with fresh medium containing only DMSO or simeprevir at 100, 250, 500, 750, 1,000, and 5,000 nM after 24 h. Total cell lysates were collected 96 h after transfection and then probed by Western blotting with anticore or β-actin antibody. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Replication boost of the Q80K/R+R155K double variants by simeprevir in HCV genomes without extra genes.
Because we used a GLuc-containing HCV genome, the insertion of additional genes into the HCV genome could potentially have caused this unexpected boost in the replication capacity. To exclude this possibility, we inserted the Q80K+R155K or Q80R+R155K mutations into a full-length H77S.3 genome that did not contain GLuc or foot-and-mouth disease virus 2A (FMDV2A) sequences. After the transfection of each variant RNA separately into Huh-7.5 cells, the cells were treated with simeprevir at concentrations of 100, 250, 500, 750, 1,000, and 5,000 nM, and the expression of the HCV core protein was analyzed by Western blotting. Similarly to the GLuc- and FMDV2A-containing HCV genomes, we found significantly enhanced expression of HCV core with simeprevir at concentrations in the range of 100 to 1,000 nM, whereas 5,000 nM simeprevir severely suppressed HCV core expression (Fig. 4C). This result demonstrated that the insertion of extra sequences did not cause unexpected boosts of the replication capacities of the Q80K+R155K and Q80R+R155K variants.
Relative replication capacities of each variant at various concentrations of simeprevir.
We compared the relative replication capacities of each variant at various concentrations of simeprevir in the range of 0 to 10,000 nM (Fig. 5). While the replication of the wt and the Q80K, Q80R, and R155K variants was reduced by simeprevir in a dose-dependent manner, the replication of the Q80K+R155K and Q80R+R155K variants increased with simeprevir concentrations of up to 1,000 nM and was reduced at higher concentrations in a dose-dependent manner. The replication of the D168A and D168V variants started to be reduced due to high drug resistance with 2,500 nM and 5,000 nM simeprevir, respectively. Importantly, in the range of 750 to 1,000 nM simeprevir, the replication capacity of the Q80K+R155K variant was higher than that of the wt in the absence of simeprevir. In the case of the Q80R+R155K variant, even though replication increased at simeprevir concentrations of up to 1,000 nM, the replication capacity never exceeded that of the wt in the absence of simeprevir. This result suggests that the Q80K+R155K variant would have the greatest advantage in replication capacity among the simeprevir-resistant variants in the range of 750 to 1,000 nM simeprevir.
FIG 5.
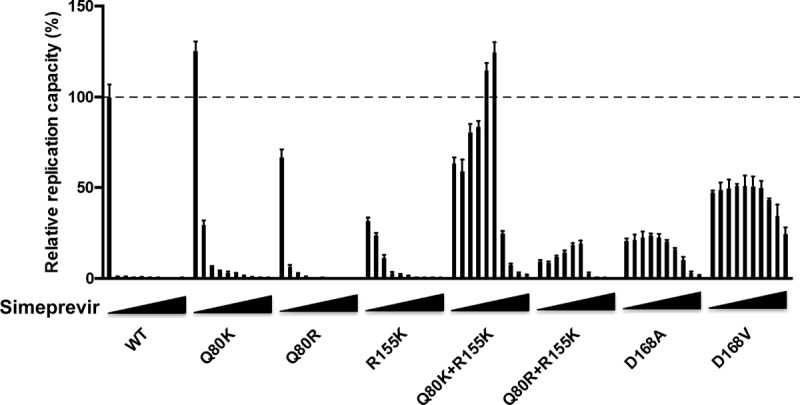
Relative replication capacities of each variant at various concentrations of simeprevir. wt HCV RNA or HCV RNA encoding NS3 variants, such as Q80K, Q80R, R155K, Q80K+R155K, Q80R+R155K, D168A, or D168V in H77S.3/GLuc2A, was transfected into Huh-7.5 cells. The medium was collected 8 h after transfection and replaced with fresh medium containing only DMSO or simeprevir at 100, 250, 500, 750, 1,000, 2,500, 5,000, 7,500, and 10,000 nM at 24-h intervals thereafter until 96 h. The secreted GLuc activity in the medium 8 h and 96 h after transfection was determined, and the GLuc activity at 96 h was normalized to that at 8 h for each variant. The normalized GLuc activity was further normalized to that of the wt without simeprevir, set to be 100%. Error bars show the standard deviations from three independent transfections.
Impact of mutations at residue 155 other than R155K on sensitivity to simeprevir.
In addition to the R155K mutation, several other mutations at residue R155, such as R155T/G/Q/W, have been reported to confer drug resistance to NS3/4A PIs (19). To test the sensitivity of the R155T/G/Q/W variants to simeprevir, we introduced these mutations into H77S.3/GLuc2A and determined the EC50s of simeprevir for these variants. The R155Q variant did not show simeprevir resistance; however, the other variants, R155T, R155G, and R155W, did (Table 2). Next, we examined whether the replication capacities of the variants with Q80R+R155T/G/Q/W could also be boosted by simeprevir as in the case of the Q80R+R155K variant. For this purpose, we introduced a Q80R mutation into H77S.3/GLuc genomes with R155T/G/Q/W mutations and tested the sensitivities of these double variants to simeprevir at concentrations of 100, 250, 500, 750, 1,000, and 5,000 nM. While simeprevir enhanced the replication capacity of the Q80R+R155K variant in the concentration range of 100 to 1,000 nM, it suppressed the replication capacity of the Q80R+R155Q variant but did not alter the capacities of the Q80R+R155T/G/W variants at the same concentrations (Fig. 6 and Table 2). Our data suggest that only R155K, as a Q80K/R second-site mutation, is associated with an enhanced replication capacity in the presence of simeprevir, whereas other R155 mutations, such as R155T/G/Q/W, do not boost replication as Q80K/R second-site mutations.
FIG 6.

Simeprevir sensitivities of the NS3 variants containing Q80R with various amino acid mutations at position 155. HCV RNAs encoding NS3 variants, such as Q80R+R155K, Q80R+R155T, Q80R+R155G, Q80R+R155W, or Q80R+R155Q in H77S.3/GLuc2A, were transfected into Huh-7.5 cells. The medium was replaced with fresh medium containing only DMSO or simeprevir at 100, 250, 500, 750, 1,000, and 5,000 nM after 24 h and at 24-h intervals thereafter. The secreted GLuc activity in the medium was determined at 72 h posttransfection. For each NS3 variant, the GLuc activity in simeprevir-treated cells was normalized to that in cells treated with DMSO only.
Sensitivity of NS3 variants to other NS3/4A PIs.
We also examined whether other NS3/4A PIs, asunaprevir, paritaprevir, and grazoprevir, could boost the replication capacities of the Q80K/R+R155K variants. Because the Q80K variant was reported to cause low-level resistance to asunaprevir and simeprevir (20) but not to paritaprevir and grazoprevir, we expected that asunaprevir could boost the replication capacities of the Q80K/R+R155K variants. However, the replication capacities of the Q80K/R+R155K variants were efficiently suppressed by asunaprevir; no boost of the replication capacity was observed with any of the novel NS3/4A PIs tested at concentrations of 0.1, 10, 100, and 1,000 nM (Fig. 7A to C). We also examined the sensitivities of the Q80K/R and D168A/V variants to these NS3/4A PIs, and an efficient suppression of the replication capacity in a dose-dependent manner was found, but there was no boost in the replication capacity (see Fig. S5 to S7 and Table S1 in the supplemental material).
FIG 7.

Sensitivities of the Q80K/R+R155K variants to PIs other than simeprevir. wt HCV RNA or the HCV RNA encoding the Q80K+R155K or Q80R+R155K variant in H77S.3/GLuc2A was transfected into Huh-7.5 cells. The medium was replaced with fresh medium containing only DMSO, asunaprevir (A), grazoprevir (B), or paritaprevir (C) at 0.1, 5, 10, 500, or 1,000 nM after 24 h and at 24-h intervals thereafter. The GLuc activity in the medium was determined at 72 h posttransfection. With each NS3/4A PI, the GLuc activity in simeprevir-treated cells was normalized to that in cells treated with DMSO only.
Effects of simeprevir on sensitivity in GT 1b HCV.
We next investigated whether simeprevir could boost the replication capacities of the Q80K/R+R155K variant and variants of HCV GT 1b. For this purpose, Q80K, Q80R, Q80K+R155K, and Q80R+R155K mutations were introduced individually into GT 1b strain N.2/GLuc2A (21), which contained a GLuc-encoding sequence between p7 and NS2, followed by an FMDV2A-encoding sequence. The Q80K/R mutations conferred simeprevir resistance, with EC50s of 22.8 nM and 23.9 nM, respectively (see Table S2 in the supplemental material). Although both the Q80K+R155K and Q80R+R155K variants also showed simeprevir resistance (see Table S2 in the supplemental material) comparable to that of GT 1a, simeprevir did not boost the replication capacity, as observed for GT 1a, of either of the variants at concentrations of <1,000 nM (Fig. 8). Hence, the boost in the replication capacities of the Q80K+R155K and Q80R+R155K double variants is not likely to be universal among all HCV genotypes.
FIG 8.
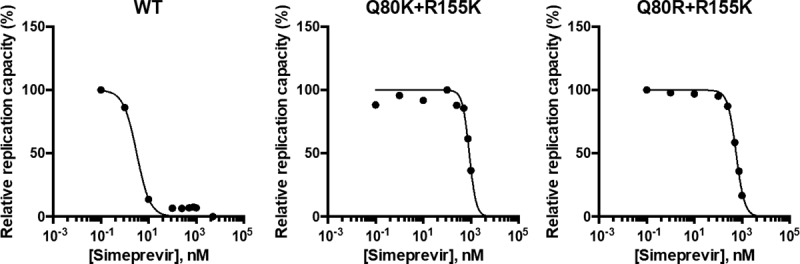
Sensitivities of the Q80K/R+R155K variants in GT 1b strain N.2 to simeprevir. wt HCV RNA and HCV RNA encoding the Q80K+R155K or Q80R+R155K variant in N.2/GLuc2A was transfected into Huh-7.5 cells. The medium was replaced with fresh medium containing only DMSO or simeprevir at 0.1, 1, 10, 100, 250,500, 750, 1,000, or 5,000 nM at 24 h and at 24-h intervals thereafter. The GLuc activity in the medium was determined at 72 h posttransfection. For each NS3 variant, the GLuc activity in simeprevir-treated cells was normalized to that in cells treated with DMSO only.
DISCUSSION
Lenz et al. (22) performed HCV sequence analysis on patients who failed to achieve SVRs with simeprevir, PEG, and RBV from baseline to the time of treatment failure in phase IIb/III clinical studies. In that study, the R155K mutation was the most frequently observed mutation (83.7%), and mutations at residue 168 were observed in only 12.2% of the patients who had the NS3-Q80K polymorphism at baseline. Although the R155K mutation was also observed most frequently (41.0%) in the case of patients who did not have the Q80K polymorphism at baseline, this frequency was much lower than that in patients who had the Q80K polymorphism (83.7%). Instead of the R155K mutation, mutations at D168 were frequently observed in patients who did not have the Q80K polymorphism at baseline; mutations at D168 without Q80K/R or R155K were observed at a rate of 21.3%, and those at D168 with Q80K/R or R155K were observed at a rate of 25.0%. The Q80K/R+R155K mutations were observed in just 6.6% of the patients without the Q80K polymorphism at baseline.
In our study, it was found that simeprevir treatment of the wt virus selected an R155K mutation at lower concentrations and that D168A/V mutations became dominant, where the R155K mutation was not observed at higher concentrations. Mutations at residue 80, such as Q80K/R, were not observed throughout simeprevir treatment. On the other hand, simeprevir treatment against Q80K/R viruses selected another mutation, R155K, with the Q80K/R mutations being preserved, and mutations at D168 were less frequent than with simeprevir treatment of the wt virus at concentrations of <2,500 nM (Fig. 2 and Table 1). At concentrations of >2,500 nM, the R155K mutation disappeared, and the D168V mutation was observed. This is quite reasonable from the relative replication capacities of the Q80K+R155K and Q80R+R155K variants at various concentrations of simeprevir; at concentrations of >2,500 nM, both variants could not replicate very well (Fig. 5). The pattern of the development of simeprevir-resistant variants at concentrations of <2,500 nM simeprevir in a cell culture system was quite similar to that observed in humans. These data suggest that the Q80K/R polymorphism at baseline should define the emerging pattern of simeprevir-resistant variants.
NS3 has both protease and RNA helicase enzymatic activities, which are known to play important roles in the propagation of HCV by promoting viral RNA synthesis, polyprotein processing, and infectious-virus assembly (19, 23–26). Both NS3 domains modulate their respective functions via an allosteric site within the domain interfaces (23). Q80 is located in this domain interface at the S2 pocket of the protease active site with hydrogen bonds to the guanidinium group of R155 that stabilize a salt bridge between R155 and D168 (27). As parts of the protease-helicase allosteric site, NS3 domain-domain interaction sites, and the interdomain linker in genotype 1a (N. Doncheva et al., unpublished data) (see Fig. S8A to S8C in the supplemental material), the structure topology of Q80 and R155 indicates a putative role for both residues in the assembly of infectious virus particles. These results indicate that mutations at both Q80 and R155, such as Q80K+R155K and Q80R+R155K, could cooperatively impact virus fitness. In the present study, while the replication capacities of the Q80K and Q80R variants were 126% and 67% higher than that of the wt virus, respectively, those of the variants harboring the additional R155K mutations were 63% and 10% higher than that of the wt virus, respectively. This result is somewhat different from those of a previous study; Lenz et al. (16) examined the replication capacities of the Q80K+R155K and Q80R+R155K double variants by inserting the mutations into a GT 1b subgenomic replicon and found that the replication capacities were 66% and 157% higher than that of the wt replicon, respectively. This could be due to the difference in GTs and HCV genomes used: we used GT 1a and full-length HCV genomes, while the previous study used GT 1b and subgenomic HCV genomes. Our results demonstrated that the addition of the R155K mutation to the Q80K/R variants reduced the replication capacities of the Q80K/R variants.
Interestingly, simeprevir boosted the replication capacities of the Q80K/R+R155K variants by about 1.5- and 2-fold compared to those of each variant in the absence of simeprevir, respectively, but only at concentrations of <1,000 nM (Fig. 4A). On the other hand, simeprevir inhibited the replication capacities of these variants at concentrations of >1,000 nM (Fig. 4B and C). Importantly, in the range of 750 to 1,000 nM simeprevir, the replication capacity of the Q80K+R155K variant was higher than that of the wt in the absence of simeprevir (Fig. 5). This result suggests that the Q80K+R155K variant would have the greatest advantage in replication capacity among the simeprevir-resistant variants in the range of 750 to 1,000 nM simeprevir. The boost in the replication capacities of the Q80K/R+R155K variants by simeprevir depended on amino acid substitutions at R155, since substitutions at R155 other than R155K, such as R155T/G/Q/W, did not increase the replication capacity with simeprevir treatment (Fig. 6). This boost was observed for GT 1a strain H77S.3 only; simeprevir did not boost the replication capacities of the Q80K/R+R155K variants in GT 1b strain N.2 at concentrations of <1,000 nM (Fig. 8). The boost was also specific to simeprevir among NS3/4A PIs, since other PIs, such as asunaprevir, grazoprevir, and paritaprevir, inhibited the replication of the Q80K/R+R155K variants without boosting at concentrations of <1,000 nM (Fig. 7).
The capacity of a PI to boost replication is an unexpected finding and mechanistically difficult to explain; however, the boost of the replication capacity by simeprevir was observed not only in Huh-7.5 cells but also in other cell lines, FT3-7 cells and KH cells (see Fig. S4 in the supplemental material), showing that this unexpected boost seems to be a relevant and universal phenomenon. So far, we can exclude a negative impact of simeprevir on cell division and/or viability within the concentration range tested, and simeprevir was stable during the whole experiment because it did not precipitate, even at the highest concentrations (data not shown). Other considerations are, for example, potential off-target effects due to the binding of the PI with high affinity at binding sites outside the protease active site, stabilizing the viral replication machinery without a negative impact on the processing of the viral polyprotein. NS3 is a multifunctional protein with highly interdependent protease and helicase activities that are involved in polyprotein processing, RNA synthesis, and the assembly of infectious virus particles. The binding of a small-molecule inhibitor, such as simeprevir, to the protease domain active site can effectively interfere with viral replication at different steps in the viral life cycle. Given our observation of the replication boost by simeprevir only at low concentrations in the Q80K/R+R155K variants and observations that Q80 affects the protease fold (our unpublished data), it seems possible that simeprevir, by unspecific binding (off target) to the double mutant protease, could stabilize a protein conformation that is involved essentially in one or multiple steps of the viral replication cycle, whereas simeprevir at high concentrations still effectively suppresses replication due to binding to the protease active site.
We should consider the concentration of simeprevir in humans because simeprevir can enhance the replication of the Q80R/K+R155K variants only at concentrations of <1,000 nM. The pharmacokinetic profile showed that the maximum concentration (Cmax) and minimum concentration (Cmin) in the plasma of HCV-infected patients after the administration of 150 mg simeprevir and 400 mg sofosbuvir for 14 days were 5.6 μM (standard deviation, 5.2 μM) and 1.3 μM (standard deviation, 1.7 μM) (28), respectively. The average Cmax and Cmin values in that report were generally >1 μM, when simeprevir inhibited the replication of the Q80K/R+R155K variants in vitro; however, the standard deviations were large, suggesting that the concentrations of simeprevir could differ among patients administered the same dose of simeprevir and that the concentrations of simeprevir could be around 1 μM in the plasma. We should further consider the concentration of simeprevir in hepatocytes infected by HCV. According to the drug information data (highlights of prescribing information for simeprevir from the FDA, December 2013 [available from https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/205123s001lbl.pdf]), the ratio of the distribution to liver versus blood is 29 to 1 in rats, suggesting that the concentration of simeprevir in the liver could be much higher than that in plasma. However, after simeprevir enters hepatocytes, it is metabolized; therefore, it is impossible to know the exact concentration of simeprevir in hepatocytes. In fact, there have been no available data from humans on the concentration of simeprevir in hepatocytes. Although the simeprevir concentration in hepatocytes might be higher than the EC50 for the Q80K+R155K variant, we would consider that simeprevir could enhance the replication capacity of the Q80K+R155K variant in some patients whose simeprevir concentrations in hepatocytes are relatively low due to individual differences in the absorption or metabolism of simeprevir. In other words, a higher dose of simeprevir could overcome treatment failures even if the Q80K polymorphism exists at baseline, because higher concentrations can suppress the replication of the Q80K+R155K variant.
In this study, we demonstrated that the Q80K- and Q80R-harboring variants under drug pressure with simeprevir preferentially selected a second-site mutation at residue R155, R155K, at concentrations of simeprevir of <2.5 μM in the GT 1a NS3 protease backbone. We found that the replication capacities of the Q80K/R+R155K double variants were unexpectedly boosted by concentrations of simeprevir of <1 μM, and the replication capacity of the Q80K+R155K variant at 0.75 to 1 μM simeprevir was higher than that of the wt in the absence of simeprevir. This remarkable result indicates the possible reasons for the failure of simeprevir-containing antiviral treatments in patients with the NS3-Q80K polymorphism and implies a remedy to overcome treatment failure in these patients. This result also provides a new insight into the potential cause of treatment failure with resistance-associated variants.
MATERIALS AND METHODS
Reagents.
Simeprevir and asunaprevir were purchased from Funakoshi Co., Ltd. (Tokyo, Japan), and grazoprevir and paritaprevir were purchased from Cosmo Bio (Tokyo, Japan). All reagents were dissolved in dimethyl sulfoxide (DMSO). The final dilutions contained 0.5% DMSO.
Cells.
The Huh-7.5 cell line, a Huh-7 derivative, was grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, penicillin, streptomycin, l-glutamine, and nonessential amino acids.
Plasmids.
pH77S.3 was derived from pH77S (19), a molecular clone of GT 1a HCV that contains an additional cell culture-adaptive mutation in E2 and a reversion of Q41R to wild-type Q41 in NS3. To easily monitor replications, the GLuc-encoding sequence, fused at its C terminus to the FMDV2A autoprotease, was inserted between p7 and NS2 of pH77S.3, yielding pH77S.3/GLuc2A (19). We used plasmids encoding several NS3 variants, as described in our previous study (19). NS3 mutations such as R155G/T/Q/W, Q80K+R155K, and Q80R+R155K were inserted into pH77S.3/GLuc2A, and the Q80K+R155K and Q80R+R155K mutations were also inserted into pH77S.3 by applying the same strategy as the one used in our previous study (19). NS3 mutations such as Q80K, Q80R, Q80K+R155K, and Q80R+R155K were inserted into pN.2/GLuc2A (21), which encodes a molecular clone of GT 1b, N.2, with some additional mutations, including Q41R in the NS3 protease domain, in the original clone, N (29).
RNA transcription and transfection.
HCV RNA was synthesized with the T7 RiboMAX Express large-scale RNA production system (Promega, Madison, WI) after linearization of the plasmids with XbaI. Following treatment with RNase-free DNase to remove the template DNA, RNA was purified by using the RNeasy minikit (Qiagen, Hilden, Germany). RNA transfection was carried out with the TransIT mRNA transfection kit (Mirus Bio LLC, Madison, WI), according to the manufacturer's protocol.
Luciferase activity assay.
Following RNA transfection, cell culture supernatant fluids were collected, and fresh medium was added at 24-h intervals. The secreted GLuc activity in 50-μl aliquots of the supernatants was measured by using the GLuc assay kit (New England BioLabs, Ipswich, MA), according to the manufacturer's protocol. The luminescent signal was measured on a GloMax-Multi+Microplate multimode reader with Instinct (Promega).
Antiviral activity assays.
Wild-type and mutant viral RNAs were transfected as described above. The medium was replaced with fresh medium containing serial dilutions of the antiviral compounds after 24 h and at 24-h intervals thereafter. The secreted GLuc activity was determined at 72 h posttransfection, as described above. The concentration of each compound required to reduce the amount of secreted GLuc activity by 50% (antiviral EC50) was determined by using a three-parameter Hill equation (SigmaPlot 12.5; Systat Software, Inc., Chicago, IL).
Sequence analysis of HCV RNAs.
Total RNA was extracted from the cells by using the RNeasy minikit (Qiagen) according to the manufacturer's instructions, and cDNA was synthesized with a high-capacity cDNA reverse transcription kit (Applied Biosystems, Carlsbad, CA), using random primers. Population-based sequence analysis and clonal sequence analysis of the NS3 protease (amino acids 1 to 180) were performed as described in our previous study (18).
Western blotting.
Western blotting was performed as described previously (30, 31). The expression of the HCV core protein and that of β-actin were evaluated with a mouse monoclonal antibody (C7-50) (catalog number MA1-080; Thermo Fisher Scientific, Inc., Waltham, MA) and a rabbit β-actin antibody (catalog number 4967; Cell Signaling Technology, Danvers, MA), respectively.
Focus-forming unit assay.
Huh-7.5 cells were seeded into 48-well plates at a density of 4.0 × 104 cells/well 24 h prior to inoculation with 100 μl of virus-containing medium. The cells were maintained at 37°C in a 5% CO2 environment and fed with 300 μl medium 24 h later. Following 48 h of additional incubation, the cells were fixed in methanol-acetone (1:1) at room temperature for 9 min and stained with the C7-50 monoclonal antibody to the HCV core protein (1:300). After extensive washing, the cells were stained with Alexa Fluor 568-conjugated anti-mouse IgG antibodies. A cluster of infected cells staining for the core antigen was considered to constitute a single infectious focus-forming unit (FFU); virus titers are reported as FFU per milliliter.
Statistics.
The results are expressed as means ± standard deviations. Significance was tested by Student's test, one-way analysis of variance (ANOVA), or Bonferroni's multiple-comparison tests, and a P value of <0.05 was considered statistically significant. Statistical analysis was performed by using GraphPad Prism7 (GraphPad Software, Inc., La Jolla, CA).
Supplementary Material
ACKNOWLEDGMENTS
We thank Takashi Shimizu for his critical reading of the manuscript.
This work was partially supported by JSPS KAKENHI grant JP15K08492 (grant-in-aid for scientific research [C] to Tetsuro Shimakami); Joint Research Projects and Seminars with Germany, the Japan Society for the Promotion of Science (to Tetsuro Shimakami); and Core-to-Core Program, B, Asia-Africa Science Platforms, the Japan Society for the Promotion of Science (to Shuichi Kaneko).
We have no conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02601-17.
REFERENCES
- 1.Gower E, Estes C, Blach S, Razavi-Shearer K, Razavi H. 2014. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol 61:S45–S57. doi: 10.1016/j.jhep.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 2.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, et al. . 2012. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sarrazin C. 2016. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J Hepatol 64:486–504. doi: 10.1016/j.jhep.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 4.AASLD/IDSA HCV Guidance Panel. 2015. Hepatitis C guidance: AASLD-IDSA recommendations for testing, managing, and treating adults infected with hepatitis C virus. Hepatology 62:932–954. doi: 10.1002/hep.27950. [DOI] [PubMed] [Google Scholar]
- 5.European Association for Study of Liver. 2015. EASL recommendations on treatment of hepatitis C 2015. J Hepatol 63:199–236. doi: 10.1016/j.jhep.2015.03.025. [DOI] [PubMed] [Google Scholar]
- 6.Forns X, Lawitz E, Zeuzem S, Gane E, Bronowicki JP, Andreone P, Horban A, Brown A, Peeters M, Lenz O, Ouwerkerk-Mahadevan S, Scott J, De La Rosa G, Kalmeijer R, Sinha R, Beumont-Mauviel M. 2014. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 146:1669.e3–1679.e3. doi: 10.1053/j.gastro.2014.02.051. [DOI] [PubMed] [Google Scholar]
- 7.Jacobson IM, Dore GJ, Foster GR, Fried MW, Radu M, Rafalsky VV, Moroz L, Craxi A, Peeters M, Lenz O, Ouwerkerk-Mahadevan S, De La Rosa G, Kalmeijer R, Scott J, Sinha R, Beumont-Mauviel M. 2014. Simeprevir with pegylated interferon alfa 2a plus ribavirin in treatment-naive patients with chronic hepatitis C virus genotype 1 infection (QUEST-1): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet 384:403–413. doi: 10.1016/S0140-6736(14)60494-3. [DOI] [PubMed] [Google Scholar]
- 8.Lawitz E, Sulkowski MS, Ghalib R, Rodriguez-Torres M, Younossi ZM, Corregidor A, DeJesus E, Pearlman B, Rabinovitz M, Gitlin N, Lim JK, Pockros PJ, Scott JD, Fevery B, Lambrecht T, Ouwerkerk-Mahadevan S, Callewaert K, Symonds WT, Picchio G, Lindsay KL, Beumont M, Jacobson IM. 2014. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: the COSMOS randomised study. Lancet 384:1756–1765. doi: 10.1016/S0140-6736(14)61036-9. [DOI] [PubMed] [Google Scholar]
- 9.Lawitz E, Matusow G, DeJesus E, Yoshida EM, Felizarta F, Ghalib R, Godofsky E, Herring RW, Poleynard G, Sheikh A, Tobias H, Kugelmas M, Kalmeijer R, Peeters M, Lenz O, Fevery B, De La Rosa G, Scott J, Sinha R, Witek J. 2016. Simeprevir plus sofosbuvir in patients with chronic hepatitis C virus genotype 1 infection and cirrhosis: a phase 3 study (OPTIMIST-2). Hepatology 64:360–369. doi: 10.1002/hep.28422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwo P, Gitlin N, Nahass R, Bernstein D, Etzkorn K, Rojter S, Schiff E, Davis M, Ruane P, Younes Z, Kalmeijer R, Sinha R, Peeters M, Lenz O, Fevery B, De La Rosa G, Scott J, Witek J. 2016. Simeprevir plus sofosbuvir (12 and 8 weeks) in hepatitis C virus genotype 1-infected patients without cirrhosis: OPTIMIST-1, a phase 3, randomized study. Hepatology 64:370–380. doi: 10.1002/hep.28467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarrazin C, Dvory-Sobol H, Svarovskaia ES, Doehle BP, Pang PS, Chuang SM, Ma J, Ding X, Afdhal NH, Kowdley KV, Gane EJ, Lawitz E, Brainard DM, McHutchison JG, Miller MD, Mo H. 2016. Prevalence of resistance-associated substitutions in HCV NS5A, NS5B, or NS3 and outcomes of treatment with ledipasvir and sofosbuvir. Gastroenterology 151:501.e1–512.e1. doi: 10.1053/j.gastro.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 12.Jimenez-Sousa MA, Gutierrez-Rivas M, Alvaro-Meca A, Garcia-Alvarez M, Harrigan PR, Fedele CG, Briz V, Vazquez-Moron S, Resino S. 2016. NS3 resistance-associated variants (RAVs) in patients infected with HCV genotype 1a in Spain. PLoS One 11:e0163197. doi: 10.1371/journal.pone.0163197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarrazin C, Lathouwers E, Peeters M, Daems B, Buelens A, Witek J, Wyckmans Y, Fevery B, Verbinnen T, Ghys A, Schlag M, Baldini A, De Meyer S, Lenz O. 2015. Prevalence of the hepatitis C virus NS3 polymorphism Q80K in genotype 1 patients in the European region. Antiviral Res 116:10–16. doi: 10.1016/j.antiviral.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Bae A, Sun SC, Qi X, Chen X, Ku K, Worth A, Wong KA, Harris J, Miller MD, Mo H. 2010. Susceptibility of treatment-naive hepatitis C virus (HCV) clinical isolates to HCV protease inhibitors. Antimicrob Agents Chemother 54:5288–5297. doi: 10.1128/AAC.00777-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Welzel TM, Bhardwaj N, Hedskog C, Chodavarapu K, Camus G, McNally J, Brainard D, Miller MD, Mo H, Svarovskaia E, Jacobson I, Zeuzem S, Agarwal K. 24 March 2017. Global epidemiology of HCV subtypes and resistance-associated substitutions evaluated by sequencing-based subtype analyses. J Hepatol doi: 10.1016/j.jhep.2017.03.014. [DOI] [PubMed] [Google Scholar]
- 16.Lenz O, Verbinnen T, Lin TI, Vijgen L, Cummings MD, Lindberg J, Berke JM, Dehertogh P, Fransen E, Scholliers A, Vermeiren K, Ivens T, Raboisson P, Edlund M, Storm S, Vrang L, de Kock H, Fanning GC, Simmen KA. 2010. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrob Agents Chemother 54:1878–1887. doi: 10.1128/AAC.01452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verbinnen T, Fevery B, Vijgen L, Jacobs T, De Meyer S, Lenz O. 2015. In vitro activity of simeprevir against hepatitis C virus genotype 1 clinical isolates and its correlation with NS3 sequence and site-directed mutants. Antimicrob Agents Chemother 59:7548–7557. doi: 10.1128/AAC.01444-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu F, Shimakami T, Murai K, Shirasaki T, Funaki M, Honda M, Murakami S, Yi M, Tang H, Kaneko S. 2016. Efficient suppression of hepatitis C virus replication by combination treatment with miR-122 antagonism and direct-acting antivirals in cell culture systems. Sci Rep 6:30939. doi: 10.1038/srep30939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimakami T, Welsch C, Yamane D, McGivern DR, Yi M, Zeuzem S, Lemon SM. 2011. Protease inhibitor-resistant hepatitis C virus mutants with reduced fitness from impaired production of infectious virus. Gastroenterology 140:667–675. doi: 10.1053/j.gastro.2010.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McPhee F, Friborg J, Levine S, Chen C, Falk P, Yu F, Hernandez D, Lee MS, Chaniewski S, Sheaffer AK, Pasquinelli C. 2012. Resistance analysis of the hepatitis C virus NS3 protease inhibitor asunaprevir. Antimicrob Agents Chemother 56:3670–3681. doi: 10.1128/AAC.00308-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamane D, McGivern DR, Wauthier E, Yi M, Madden VJ, Welsch C, Antes I, Wen Y, Chugh PE, McGee CE, Widman DG, Misumi I, Bandyopadhyay S, Kim S, Shimakami T, Oikawa T, Whitmire JK, Heise MT, Dittmer DP, Kao CC, Pitson SM, Merrill AH Jr, Reid LM, Lemon SM. 2014. Regulation of the hepatitis C virus RNA replicase by endogenous lipid peroxidation. Nat Med 20:927–935. doi: 10.1038/nm.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lenz O, Verbinnen T, Fevery B, Tambuyzer L, Vijgen L, Peeters M, Buelens A, Ceulemans H, Beumont M, Picchio G, De Meyer S. 2015. Virology analyses of HCV isolates from genotype 1-infected patients treated with simeprevir plus peginterferon/ribavirin in phase IIb/III studies. J Hepatol 62:1008–1014. doi: 10.1016/j.jhep.2014.11.032. [DOI] [PubMed] [Google Scholar]
- 23.McGivern DR, Masaki T, Lovell W, Hamlett C, Saalau-Bethell S, Graham B. 2015. Protease inhibitors block multiple functions of the NS3/4A protease-helicase during the hepatitis C virus life cycle. J Virol 89:5362–5370. doi: 10.1128/JVI.03188-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma Y, Yates J, Liang Y, Lemon SM, Yi M. 2008. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J Virol 82:7624–7639. doi: 10.1128/JVI.00724-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 26.Bartenschlager R, Lohmann V, Penin F. 2013. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat Rev Microbiol 11:482–496. doi: 10.1038/nrmicro3046. [DOI] [PubMed] [Google Scholar]
- 27.Cummings MD, Lindberg J, Lin TI, de Kock H, Lenz O, Lilja E, Fellander S, Baraznenok V, Nystrom S, Nilsson M, Vrang L, Edlund M, Rosenquist A, Samuelsson B, Raboisson P, Simmen K. 2010. Induced-fit binding of the macrocyclic noncovalent inhibitor TMC435 to its HCV NS3/NS4A protease target. Angew Chem Int Ed Engl 49:1652–1655. doi: 10.1002/anie.200906696. [DOI] [PubMed] [Google Scholar]
- 28.Bourgeois S, Horsmans Y, Nevens F, van Vlierberghe H, Moreno C, Beumont M, Vijgen L, van Eygen V, Luo D, Hillewaert V, Van Remoortere P, van de Logt J, Ouwerkerk-Mahadevan S. 2 October 2017. Pharmacokinetic interactions between simeprevir and ledipasvir in treatment-naive hepatitis C virus genotype 1-infected patients without cirrhosis treated with a simeprevir/sofosbuvir/ledipasvir regimen. Antimicrob Agents Chemother doi: 10.1128/AAC.01217-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beard MR, Abell G, Honda M, Carroll A, Gartland M, Clarke B, Suzuki K, Lanford R, Sangar DV, Lemon SM. 1999. An infectious molecular clone of a Japanese genotype 1b hepatitis C virus. Hepatology 30:316–324. doi: 10.1002/hep.510300137. [DOI] [PubMed] [Google Scholar]
- 30.Shirasaki T, Honda M, Mizuno H, Shimakami T, Okada H, Sakai Y, Murakami S, Wakita T, Kaneko S. 2010. La protein required for internal ribosome entry site-directed translation is a potential therapeutic target for hepatitis C virus replication. J Infect Dis 202:75–85. doi: 10.1086/653081. [DOI] [PubMed] [Google Scholar]
- 31.Shirasaki T, Honda M, Shimakami T, Horii R, Yamashita T, Sakai Y, Sakai A, Okada H, Watanabe R, Murakami S, Yi M, Lemon SM, Kaneko S. 2013. MicroRNA-27a regulates lipid metabolism and inhibits hepatitis C virus replication in human hepatoma cells. J Virol 87:5270–5286. doi: 10.1128/JVI.03022-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.