

ABSTRACT
Peptidoglycan (PG) and wall teichoic acid (WTA) are the major staphylococcal cell wall components, and WTA biosynthesis has recently been explored for drug development. Targocil is a novel agent that targets the TarG subunit of the WTA translocase (TarGH) that transports WTA across the membrane to the wall. Previously we showed that targocil treatment of a methicillin-susceptible Staphylococcus aureus strain led to a rapid shut down of cellular autolysis. Targocil II, which targets the TarH subunit of TarGH, also resulted in a drastic decrease in autolysis. Here, we address the mechanism of targocil-mediated decreased autolysis. The mechanism is WTA dependent since targocil treatment decreased autolysis in methicillin-resistant strains but not in a WTA-deficient mutant. Similar to cellular autolysis, autolysin-retaining crude cell walls isolated from targocil-treated cells had vastly decreased autolytic activity compared to those from untreated cells. Purified cell walls from control and targocil-treated cells, which lack autolytic activity, were similarly susceptible to lysozyme and lysostaphin and had similar O-acetyl contents, indicating that targocil treatment did not grossly alter PG structure and chemistry. Purified cell walls from targocil-treated cells were highly susceptible to autolysin extracts, supporting the notion that targocil treatment led to decreased autolysin in the crude cell walls. Quantitative real-time PCR analysis revealed that the decrease in autolysis in the targocil-exposed cells was not due to transcriptional repression of the autolysin genes atl, lytM, lytN, and sle1. Zymographic analysis of peptidoglycan hydrolase profiles showed a deficiency of cell surface autolysins in targocil-treated cells but higher activity in cell membrane fractions. Here, we propose that the untranslocated WTA molecules in the targocil-exposed cells sequester Atl at the membrane, resulting in significantly decreased autolysis.
KEYWORDS: Staphylococcus aureus, targocil, wall teichoic acid, autolysis, Atl
INTRODUCTION
Multiple antibiotic-resistant Staphylococcus aureus is a leading worldwide cause of infections causing significant morbidity and mortality (1). Several useful antistaphylococcal antibiotics target the cell wall, including methicillin (and other β-lactams), vancomycin, and daptomycin, but resistance has developed to all these agents, leading to methicillin-resistant S. aureus (MRSA), strains showing decreased susceptibility to vancomycin (VISA), and strains showing decreased susceptibility to daptomycin (here referred to as DRSA) (2–6). These agents affect biosynthesis of the major cell wall polymer peptidoglycan (PG), which is responsible for containing the internal turgor pressure of the cell. PG constitutes a highly dynamic structural framework and is maintained throughout the cell cycle by remodeling of the preexisting polymer resulting in overall biosynthesis that incorporates controlled localized dissolution. In order for PG to be remodeled and to be dissolved in a controlled fashion during cell division, S. aureus possesses multiple enzymes known as PG hydrolases or autolysins (7). It is a common finding that VISA and DRSA show decreased autolytic activity by which the cells build up the ability to tolerate higher concentrations of the antibiotics (8, 9). These cell wall-targeting antibiotics perturb PG biosynthesis, and strains susceptible to these antibiotics are typically lysed by their own autolysins and killed. Hence, in order to maintain the structural integrity of PG, S. aureus should be able to precisely regulate the autolysins temporally, as well as spatially (10). Although various autolysins have been discovered and characterized over the past several decades, our knowledge of the control of autolysin activity in S. aureus is still not complete.
PG and wall teichoic acid (WTA) are the major staphylococcal cell wall components, and biosynthesis of the latter polymer has recently been targeted for drug development (11). Targocil is a novel agent that interacts with TarG and inhibits the TarGH complex that translocates WTA across the membrane in S. aureus (12, 13). WTA, an anionic polysaccharide, binds cationic groups to maintain cell surface integrity and is required for proper cell growth and to colonize tissues during infection (11, 12, 14–16). WTA precursors are synthesized on a bactoprenyl-phosphate carrier lipid in the cytoplasm and are translocated through the membrane, where they are covalently linked to the PG in the cell wall with release of the carrier lipid (11). Knocking out the tarO gene that encodes the first enzyme in WTA biosynthesis in S. aureus renders the cells highly susceptible to autolysis (17–20). Because targocil blocks WTA translocation through the membrane (12), resulting in cell walls deprived of WTA (21), it might be expected that targocil-treated cells would be as susceptible to autolysis as the ΔtarO mutant. However, targocil exposure rapidly shuts down the rate of autolysis in S. aureus (22).
Because the synthesis of both PG monomers and WTA molecules consume available bactoprenyl-phosphate (11) and their biosyntheses are intricately connected (23), blocking of fully synthesized WTA at the membrane impacts the overall growth of S. aureus in targocil-containing medium. However, in relation to autolysis, the transcriptomic data for targocil-exposed S. aureus SH1000 revealed that the expression of autolysin genes was not affected (22). In this study, we undertook an investigation to understand the underlying mechanism of the reduced rate of autolysis in targocil-treated S. aureus MW2, a community-associated MRSA strain. We found that the major autolysin, Atl, had restricted translocation through the membrane in the targocil-exposed cells, and we propose that Atl is sequestered inside at the cytoplasmic membrane by unflipped WTA molecules.
RESULTS AND DISCUSSION
Targocil inhibits autolysis in S. aureus but not in a WTA-deficient mutant.
Targocil is an antimicrobial agent that inhibits S. aureus WTA-translocase TarGH at the cell membrane and depletes WTA in cell wall (21) and, as a consequence, also inhibits the growth of various S. aureus strains, but not strains lacking WTA (11). Campbell et al. (22) showed that targocil exposure significantly inhibited the cellular autolysis of a methicillin-susceptible S. aureus strain SH1000. In this study, we extended the finding on the effects of targocil to autolysis of MRSA strains (COL, a hospital-associated strain; MW2, a community-associated strain) and observed that autolysis of the MRSA strains was similarly decreased (Fig. 1). Compared to the parent strain, the MW2ΔtarO strain lacking WTA was highly autolytic, and the cellular autolysis of the mutant was unaffected by targocil exposure. The MW2ΔtarO strain lacks WTA that otherwise inhibits autolysins (20, 24). Because targocil decreases the amount of WTA in the wall through inhibition of TarGH (11, 21), our observation suggests that the inhibition of autolysis is dependent on the inhibition of WTA translocation.
FIG 1.

Targocil inhibits autolysis of S. aureus strains irrespective of their methicillin susceptibilities but not in a strain lacking WTA. The cells were grown unexposed (−) or exposed (+) to targocil (5 μg/ml) for 1 h, harvested, and washed, and autolysis was measured. Error bars show standard deviations from the average percentages of the initial OD600 calculated from triplicate assays.
Targocil exposure decreases the autolysis of CCWs from strain MW2 but not from its WTA-deficient strain.
Since autolysins hydrolyze PG in the cell wall we prepared crude cell walls (CCWs), which retain autolytic activity (25), from the targocil-untreated and -treated strains to reveal alterations in cell wall properties due to targocil exposure. A lower concentration of targocil (0.4 μg/ml) than that used previously (5 μg/ml) that nevertheless noticeably inhibited growing cultures and shut down cellular autolysis was used to grow the larger culture volumes required for preparation of CCWs and assay them for autolysis. As shown in the Fig. 2, the autolytic patterns of the CCWs obtained from targocil-exposed MW2 cells was markedly reduced compared to those of the CCWs from unexposed MW2 cells. Further, there was no obvious difference in autolysis of CCWs from the targocil-unexposed and -exposed WTA-lacking mutant. Note that the CCW autolysis patterns were similar to those of their cellular autolysis (Fig. 1), which indicates that the effects in the cell walls represented the cellular effects of targocil exposure.
FIG 2.

Reduced rate of CCW autolysis of strain MW2 exposed to targocil but not its WTA-deficient mutant. Cells were grown in TSB without (−) or with 0.4 μg/ml (+) of targocil, CCWs were extracted, and autolysis was performed by resuspending them in phosphate buffer, followed by incubation at 37°C. The decrease in OD600 was monitored at intervals. Error bars show standard deviations from the average percentages of the initial OD580 calculated from triplicate assays.
Targocil exposure does not grossly alter PG structure and chemistry.
Since targocil exposure elicits the cell wall stress response (22), there could be alterations in cell wall structure and chemistry in targocil-exposed MW2 cells leading to a decreased rate of CCW autolysis and hence cellular autolysis. To determine whether a perturbation in PG might cause a decrease in autolysis, cell walls were purified from the MW2 strains unexposed or exposed to targocil and treated with lysozyme and lysostaphin. Lysozyme targets glycosidic bonds between sugar residues and lysostaphin digests pentaglycine cross-links between PG monomers in the cell wall. All purified cell walls (PCWs) were completely resistant to lysozyme digestion (see Fig. S1A in the supplemental material). The lysis kinetics of PCWs with lysostaphin were similar in each strain exposed or unexposed to targocil (see Fig. S1B in the supplemental material). Further, the levels of O-acetylation were similar in the walls from the targocil-treated and -untreated cells (see Fig. S1C in the supplemental material). These observations indicate that targocil exposure does not alter gross PG structure (e.g., increased cross-links) and/or chemistry (e.g., chemical modification of PG) leading to a decrease in cellular or CCW autolysis.
Targocil exposure decreases the amounts of autolysins in the cell wall.
WTA inhibits autolysin activity at the cell surface and, hence, WTA-deficient strains are highly susceptible to autolysis (20, 24). However, this raises the question of how the targocil-exposed S. aureus strains with depleted WTA in the cell wall (11, 21) experienced a rapid shutdown of autolysis. A decrease in the rate of autolysis in targocil-exposed cells could be due to a smaller amount of autolysins in the cell wall. To investigate this idea, equivalent amounts of PCWs from strain MW2 exposed and unexposed to targocil were treated with the same amount of autolysin extracts. PCWs do not have intrinsic autolysis activity because the autolysins were destroyed by boiling during PCW preparation. In contrast to the CCW autolysis patterns (Fig. 2), PCWs from the targocil-exposed MW2 cells were significantly more susceptible to lysis by autolysins compared to those from the unexposed cells (Fig. 3). Since targocil inhibits WTA translocase (11) and thus depletes WTA in the cell wall (21) that otherwise inhibits autolysins (20), the decreased WTA contents in the PCWs obtained from targocil-exposed cells might explain why the PCWs lysed faster than those from the unexposed cells. This observation supports the hypothesis that decreased cellular and CCW autolysis of the targocil-exposed cells might be due to the reduced amounts of autolysins therein. Hence, the CCWs from targocil-exposed MW2 cells might have significantly smaller amounts of autolysins and thus exhibit a lower rate of autolysis in spite of WTA depletion, which normally increases autolysis.
FIG 3.
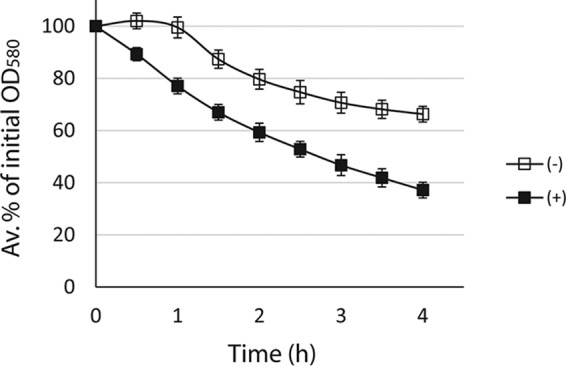
PCWs from targocil-exposed S. aureus are more susceptible to lysis by autolysins than those from targocil-unexposed cells. PCWs were prepared from S. aureus MW2 unexposed and exposed to targocil (0.4 μg/ml), resuspended to an OD580 ∼0.7 in equal amounts of secreted autolysin concentrate, and monitored for lysis by incubation at 37°C. The error bars indicate standard deviations from average percentages of the initial OD580 calculated from three experiments.
Targocil II treatment also inhibits cellular autolysis.
Recently, targocil II was discovered that inhibits TarH of S. aureus TarGH (26), whereas targocil targets TarG (12, 13). Strain MW2 and its WTA-deficient strain were exposed to 1 μg/ml of targocil II, and whole-cell autolysis was measured. As shown in Fig. 4, targocil II also inhibited autolysis in the strain MW2 but not in the WTA-lacking strain similarly to targocil. Targocil II inhibits TarH, the cytoplasmic ATPase component, by binding allosterically in the transmembrane domain in TarG (26), whereas targocil binds and inhibits the TarG component (12, 13). Thus, two agents that target TarGH in different locations both lead to decreased cellular autolysis.
FIG 4.

Targocil II also inhibits the autolysis of S. aureus. The cells were grown unexposed (−) or exposed (+) to targocil II (1 μg/ml) for 1 h, harvested, and washed, and autolysis was measured. Error bars show standard deviations from the average percentages of the initial OD600 calculated from triplicate assays.
Targocil exposure does not repress the genes that encode autolysins.
The decreased amount of autolysins in the cell surface of the targocil-exposed MW2 cells could be due to a downregulation of autolysin gene expression. Encoded by the atl gene, Atl is the major hydrolase responsible for cell division and cellular autolysis in S. aureus (27–31). The transcriptomic data of targocil-exposed S. aureus SH1000 revealed that targocil exposure had no effect on the expression of this or other autolysin genes (i.e., atl, lytM, lytN, and sle1) (22). However, the expression of some genes (arlR, sarA, agrA, clpC, and clpP) that could alter the expression of atl were affected. To confirm whether the decreased amount of autolysins on the cell surface of the targocil-exposed cells could be due to transcriptional repression of autolysin genes (viz., atl, lytM, lytN, and sle1) in S. aureus MW2 strain, quantitative real-time PCR (qRT-PCR) assay was performed to quantify the specific mRNAs in the targocil-exposed and -unexposed MW2 cells. As shown in Fig. 5, none of the autolysin genes was repressed with targocil exposure confirming that the decreased amount of autolysins in the cell wall of targocil-exposed S. aureus was unlikely due to transcriptional repression of the genes encoding autolysins.
FIG 5.

Targocil exposure of S. aureus does not repress the expression of autolysin genes. S. aureus MW2 was grown in TSB without or with targocil (5 μg/ml), total mRNA was extracted and converted to cDNA, and a qRT-PCR was performed to determine the level of expression of the known autolysin genes (viz., atl, lytM, lytN, and sle1). Error bars show standard deviations from average values from three independent biological replicates.
Targocil exposure causes internal sequestration of Atl.
The decreased amount of autolysins in the cell wall of targocil-exposed S. aureus MW2 could be due to obstruction of Atl translocation at the membrane. To compare the relative distribution of autolysins inside and outside the membrane, the enzymes in surface and membrane fractions were extracted from targocil-unexposed and -exposed cells and assayed by zymography. A zymogram is an SDS-PAGE gel containing Micrococcus luteus or S. aureus cells as a substrate for autolysin enzymes where the discrete bands of the lysed cells in the stained background represent autolysin activities.
The major autolysin in S. aureus Atl (138 kDa, also referred to as “pro-Atl”) has amidase (AM) and glucosaminidase (GL) domains (27–30) and is processed by proteolytic enzymes in the cell envelope, yielding separately active AM and GL enzymes (31–35). Note that multiple bands of each AM or GL are observed on the zymogram gels, indicating that the AM and GL enzymes are also processed into smaller molecules (8, 20, 27, 28, 36, 37).
As shown in Fig. 6, the activities of surface autolysins from targocil-exposed MW2 cells were much less than from the targocil-unexposed cells. On the other hand, the WTA-deficient strain had more easily extractable autolysins from the surface compared to the MW2 cells. Targocil exposure had no effect on the relative activities of the surface autolysins in the WTA-deficient strain, indicating that there was no obstruction in autolysin translocation by targocil exposure. Hence, the effect of targocil exposure on altering autolysis in the MW2 cell surface might be due to the untranslocated WTA in the cells.
FIG 6.

Atl is blocked at the inner membrane in targocil-exposed cells. Various autolysin fractions were electrophoresed in SDS-PAGE gels containing 0.2% M. luteus cells, washed, and renatured in renaturation buffer overnight before staining with methylene blue. The autolysins lysed M. luteus cells and thus appeared as discrete white bands on the gel that otherwise stained blue. Lanes: –, no treatment; +, treatment with targocil (5 μg/ml). The relative positions of the bands are indicated by molecular size in kilodaltons (numbers on the left side of the gel), as determined by Bose et al. (36).
To understand whether an altered distribution of autolysins in the membranes of targocil-exposed cells was due to untranslocated WTA molecules, the membrane fractions from MW2 cells exposed and unexposed to targocil were also analyzed. As shown in Fig. 6 (right panel), the intensity of the pro-Atl band in the membrane fraction from the targocil-exposed MW2 cells was much brighter than that from the unexposed cells, indicating that targocil exposure somehow blocked autolysin translocation at the membrane in the MW2 cells.
Since the major autolysin Atl is involved in cellular autolysis, an atl knockout strain was created in the MW2 background and studied to demonstrate whether the major autolysin (Atl) was affected by targocil exposure. The loss of the atl gene in the mutant was confirmed by whole-cell autolysis showing a markedly reduced rate of autolysis (see Fig. S2 in the supplemental material) and by scanning electron microscopy showing tightly attached cell clusters (see Fig. S3 in the supplemental material). Subsequently, the surface autolysins (Fig. 6, left panel) of the MW2Δatl strain exposed and unexposed to targocil were analyzed. There was no autolysin activity on the gel for both targocil-unexposed and -exposed MW2Δatl cells, indicating that the mutant was deficient in Atl activities and that the bands for MW2 and its WTA-deficient strains were specific to their Atl and the Atl-processed molecules.
Although the structure and enzymatic activities of Atl have been extensively characterized (27–30, 35–37), how this enzyme is translocated across the membrane is not known. Atl has a calculated isoelectric point of ∼9.6 (calculated using an online software [https://web.expasy.org/compute_pi/]), and negatively charged lipoteichoic acid (polyglycerophosphate) with a similar molecular structure to WTA (polyribitol phosphate) was shown to bind autolysins (38). These facts have led us speculate that the untranslocated WTA molecules in targocil-exposed cells could bind pro-Atl inside the membrane and block its translocation, resulting in decreased cellular autolysis in wild-type cells. However, in the WTA-lacking mutant the pro-Atl molecules could be translocated unrestricted across the membrane into the cell wall, resulting in an unaltered rate of autolysis. WTA is synthesized at the division septum in S. aureus and is required to properly localize penicillin-binding protein 4, a secondary transpeptidase that creates extensive PG cross-linking (17). Since the expression, localization, and activity of Atl should be precisely regulated to avoid unwanted autolysis, we speculate that WTA is an important factor in guiding Atl to the septum in a dividing S. aureus cell. Taken together, our data support the idea that WTA is an important spatial regulator of Atl activity, a crucial enzyme involved in cell division and autolysis.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains, plasmids, and primers used in this study are listed in Tables 1, 2, and S1, respectively. S. aureus strains were grown in tryptic soy broth (TSB; Becton-Dickinson Diagnostic Systems) by incubation with shaking at 200 rpm at 37°C. Escherichia coli Top10 cells were grown in Luria-Bertani broth (Becton-Dickinson Diagnostic Systems).
TABLE 1.
Bacterial strains used in the study
| Strain | Relevant characteristics | Source or reference |
|---|---|---|
| S. aureus COL | Hospital-associated MRSA, wild-type strain | 47 |
| S. aureus MW2 | Community-associated MRSA, wild-type strain, an USA400 | 48 |
| S. aureus MW2ΔtarO | Contain a premature stop codon through a TAC-to-TAG mutation at position 243 in tarO | 23 |
| S. aureus MW2Δatl::kanR | atl knockout of S. aureus MW2 strain with kanamycin cassette in place of atl in the chromosome | This study |
| E. coli Top10 | hsdR mcrA lacZΔM15 endA1 recA1 | Invitrogen |
TABLE 2.
Plasmids used in the study
| Plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| pJET1.2/blunt | rep(pMB1), blaR eco47IR, PlacUV5 | Thermo Fisher Scientific |
| U-pJET | pJET1.2/blunt with 1 kb upstream of >atl gene of S. aureus MW2 | This study |
| D-pJET | pJET1.2/blunt with 1 kb downstream of atl gene of S. aureus MW2 | This study |
| UD-pJET | pJET1.2/blunt with upstream and downstream of atl gene of S. aureus MW2 | This study |
| UKD-pJET | pJET1.2/blunt with kanamycin cassette in between of the upstream and downstream of atl gene of S. aureus MW2 | This study |
| pMAD | ermC blaR; oripBR322; pclpB bgaB, oripE194ts | 43 |
| UKD-pMAD | pMAD with the UKD fragment insert | This study |
Whole-cell autolysis of targocil-exposed cells.
Whole-cell autolysis assay was carried out as described previously (22). Briefly, cells grown overnight were diluted 100-fold in 20 ml of TSB in two 100-ml flasks for each strain and then grown to an optical density at 600 nm (OD600) of ∼0.3. Targocil (5 μg/ml) was added to one flask, and both were further incubated for 1 h. The cells were harvested at 2,700 × g for 10 min at 4°C, washed twice with cold water, and resuspended in 0.05 M Tris-HCl buffer (pH 7.2) with 0.05% Triton X-100 to an OD600 of 0.6. The suspensions were incubated at 37°C, and the OD600 was measured at intervals.
Preparation of CCWs retaining autolytic activity.
CCWs were prepared as described previously (39–41). Briefly, 1 liter of TSB was inoculated with 20 ml of overnight-grown strains without or with 0.4 μg/ml of targocil and incubated until the OD600 reached to 0.6 to 0.7. The cells were harvested at 13,800 × g for 10 min at 4°C, washed two times with cold 0.9% (wt/vol) NaCl, and resuspended in 50 ml of 0.9% NaCl. Each cell suspension was mixed with 50 ml of cold 0.1-mm glass beads (Biospec Products, Inc.). Using a Bead Beater, the cells were broken mechanically on an ice bath for five cycles of 2 min of beating and 2 min of rest. Each lysate was filtered through a sintered glass filter and washed with cold water. The filtrates were centrifuged, and the pellets were washed to get CCWs.
Determination of CCW autolysis.
The CCWs were resuspended in 0.05 M KH2PO4-K2HPO4 buffer (pH 7.2) to an OD580 of 0.4 to 0.6, followed by incubation at 37°C, and the OD580 was read at intervals as described previously (8, 25).
Preparation of PCWs.
Freshly prepared CCWs were resuspended in 2% SDS solution, boiled for 30 min, harvested at 13,800 × g for 10 min at 20°C, and washed with cold water to remove the SDS. The pellets were resuspended in 50 mM Tris-Cl (pH 6.8) with 5 mM MgCl2 and 5 μg/ml (each) of DNase (Sigma) and RNase (Sigma) and incubated for 2 h at 37°C with gentle shaking, and then trypsin (300 μg/ml; Sigma) was added, followed by incubation overnight. The cell wall suspensions were harvested and washed to get PCWs. The PCWs were lyophilized and stored at −20°C until used.
PCW lysis with lysozyme, lysostaphin, and autolysin extracts.
PCW preparations were assayed for lysis kinetics with chicken egg white lysozyme (Sigma), lysostaphin (Sigma), and autolysins extracted from S. aureus MW2 cells as described previously (8). Briefly, the PCWs were resuspended in 50 mM Tris-Cl (pH 7.5) to an OD580 of 0.5 to 0.6, and lysozyme (100 μg/ml) was added, followed by incubation at 37°C. The OD580 was measured at intervals. The lysis kinetics with lysostaphin (5 μg/ml) were similarly determined at OD620. Autolysins secreted in the overnight-incubated S. aureus MW2 culture medium were concentrated using an Amicon Ultra-15 centrifugal filter device (Millipore). Equal volumes of the extracted autolysins was added in the PCW suspensions to an OD580 of 0.5 to 0.6, followed by incubation at 37°C, and the OD580 was measured at intervals.
Determination of O-acetylation.
O-acetyl residues were released from the PCWs as described by Bera et al. (41). Briefly, 5-mg portions of the PCWs were suspended in 200 μl of 80 mM NaOH, incubated at 30°C for 3 h, and neutralized with 100 μl of 80 mM H2SO4. The suspensions were centrifuged at 13,800 × g at 4°C for 10 min, and the acetate content of the supernatants was determined from a calibration curve of acetate standards using an acetic acid assay kit (K-ACET; Megazyme International, Wicklow, Ireland).
Analysis of PG hydrolase enzymes of different cell fractions.
Overnight-grown cells were diluted 1:100 in 100 ml of TSB and incubated to an OD600 of 0.3, targocil (5 μg/ml) was added, and the cultures were incubated for 1 h. The cells were centrifuged, washed twice with cold 0.9% NaCl, and resuspended in 1.5 ml of 0.9% NaCl. Surface autolysins were extracted as described previously (42). Briefly, an equal cell mass of the strains in 2% SDS in 1× phosphate-buffered saline was incubated at room temperature for 1 h and centrifuged to recover the supernatants containing surface autolysins. Membranes were isolated from 500-ml portions of targocil-exposed and -unexposed cultures as described previously (39, 40). The cells were broken with 0.1-mm glass beads, and the unbroken cells were removed by filtration using sintered glass filters and by centrifugation at 13,800 × g at 4°C for 10 min. The membrane fractions were collected from the supernatants by centrifugation at 38,000 × g for 30 min at 4°C and resuspension of the pellets in Tris-HCl buffer (pH 7.2). The total protein in the membrane fractions was quantified by the Bradford method.
Zymography.
The relative activities of surface and cell membrane autolysins of the strains were assayed by zymography as described previously (8). Equal amounts of autolysin preparations from targocil-exposed and -unexposed strains (viz., strains MW2, MW2Δatl, and MW2ΔtarO) were subjected to electrophoresis in a 12% SDS-polyacrylamide gel containing 2 mg/ml M. luteus cells (Sigma-Aldrich). The gels were incubated in renaturation buffer (25 mM Tris-Cl [pH 8.0] and 1% Triton X-100) overnight, immersed in staining solution (1% methylene blue and 0.01% KOH) for 10 min, and destained to observe clear bands representing PG hydrolases.
Determination of autolysin genes expression by qRT-PCR.
The effect of targocil exposure on the expression of the S. aureus MW2 autolysin genes (viz., atl, lytM, lytN, and sle1) was determined by qRT-PCR. Briefly, overnight-grown cultures of strain MW2 were diluted 100-fold in two 250-ml flasks containing 50 ml of TSB each and grown to an OD600 of 0.3. Targocil (5 μg/ml) was added to one culture marked as “treated” that was allowed to incubate for 1 h, along with the untreated culture with dimethyl sulfoxide as a control. Cells were harvested, and the total RNA was extracted and purified using an RNeasy minikit (Qiagen). cDNA was prepared from 300 ng of each RNA using a high-capacity RNA-to-cDNA kit (Applied Biosystems). qRT-PCRs were conducted using a DyNAmo Flash SYGR Green kit (Thermo Scientific) with gene-specific primers (P1F/P1R for atl, P2F/P2R for lytM, P3F/P3R for lytN, P4F/P4R for sle1, and P5F/P5R for 16S-rRNA; Table 2). PCR was run using the ABI 7300 real-time PCR system. The reactions were run in triplicates for each set, and the experiment was repeated with three biological replicates to get the average values of threshold cycles (CT). The levels of expression of the autolysin genes in targocil-exposed cells over unexposed ones were calculated by the comparative CT method using the 16S rRNA gene as a reference.
Construction of atl knockout strains.
An atl knockout mutant of S. aureus MW2 was constructed by allelic replacement of the atl gene with a kanamycin cassette, as described previously (43, 44). Briefly, 1 kb of upstream (U) and downstream (D) sequences of the atl gene was amplified from S. aureus MW2 genomic DNA using the primer sets P6F/P6R and P7F/P7R, respectively, ligated together, amplified as UD fragment by primer set P6F/P7R, and cloned into pJET. A kanamycin resistance (Kanr) cassette (amplified with the primer set P8F/P8R) was inserted into the plasmid to obtain UKD-pJET, and the UKD fragment was moved into a shuttle vector pMAD to obtain UKD-pMAD. The construct was purified from E. coli Top10 transformants and electroporated into S. aureus RN4220 using a Bio-Rad gene pulser. The plasmid was recovered from a RN4220 transformant and electroporated into the competent S. aureus MW2 strain. The plasmid was integrated into the chromosome of the MW2 cells by a first homologous recombination and replaced atl with the Kanr cassette by a second homologous recombination event to produce the atl knockout strain (MW2Δatl). The loss of blue color in the purified atl knockout strain indicated the loss of the plasmid that was released from the chromosome after the second homologous recombination event. The knockout of the atl gene was confirmed by (i) PCR using the primers P9F and P9R to demonstrate the absence of atl gene in the chromosome of S. aureus MW2, (ii) autolysis assays showing a loss of autolysis in the strains, (iii) the formation of characteristic clumps of the cell suspension sitting undisturbed for a few hours, as described elsewhere (18), and (iv) scanning electron microscopy (see below) to visualize cells tightly attached to each other.
Scanning electron microscopy.
S. aureus MW2, targocil-exposed MW2, MW2Δatl, and MW2ΔtarO strains were prepared for scanning electron microscopy as described previously (45, 46). Briefly, the cells were fixed by 2% glutaraldehyde on poly-l-lysine-coated glass coverslips, postfixed with 1% osmium tetroxide, dehydrated with an ethanol series, and dried to a critical control point. The cells were visualized under a Quanta 450 tungsten scanning electron microscope (FEI).
Targocil II.
Targocil II was purchased from Aurora Fine Chemicals LLC, San Diego, CA.
Supplementary Material
ACKNOWLEDGMENT
This study was supported in part by grant AI1083214 from the National Institutes of Health to S.W.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00323-18.
REFERENCES
- 1.Gould IM. 2005. The clinical significance of methicillin-resistant Staphylococcus aureus. J Hosp Infect 61:277–282. doi: 10.1016/j.jhin.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 2.Sieradzki K, Roberts RB, Haber SW, Tomasz A. 1999. The development of vancomycin resistance in a patient with methicillin-resistant Staphylococcus aureus infection. N Engl J Med 340:517–523. doi: 10.1056/NEJM199902183400704. [DOI] [PubMed] [Google Scholar]
- 3.Sieradzki K, Leski T, Dick J, Borio L, Tomasz A. 2003. Evolution of a vancomycin-intermediate Staphylococcus aureus strain in vivo: multiple changes in the antibiotic resistance phenotypes of a single lineage of methicillin-resistant S. aureus under the impact of antibiotics administered for chemotherapy. J Clin Microbiol 41:1687–1693. doi: 10.1128/JCM.41.4.1687-1693.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cosgrove SE, Carroll KC, Perl TM. 2004. Staphylococcus aureus with reduced susceptibility to vancomycin. Clin Infect Dis 39:539–545. doi: 10.1086/422458. [DOI] [PubMed] [Google Scholar]
- 5.Marty FM, Yeh W, Wennersten CB, Venkataraman L, Albano E, Alyea EP, Gold HS, Baden LR, Pillai SK. 2006. Emergence of a clinical daptomycin-resistant Staphylococcus aureus isolate during treatment of methicillin-resistant Staphylococcus aureus bacteremia and osteomyelitis. J Clin Microbiol 44:595–597. doi: 10.1128/JCM.44.2.595-597.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Julian K, Kosowska-Shick K, Whitener C, Roos M, Labischinski H, Rubio A, Parent L, Ednie L, Koeth L, Bogdanovich T, Appelbaum PC. 2007. Characterization of a daptomycin-nonsusceptible vancomycin-intermediate Staphylococcus aureus strain in a patient with endocarditis. Antimicrob Agents Chemother 51:3445–3448. doi: 10.1128/AAC.00559-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monteiro JM, Fernandes PB, Vaz F, Pereira AR, Tavares AC, Ferreira MT, Pereira PM, Veiga H, Kuru E, VanNieuwenhze M, Brun YV, Pinho MG. 2015. Cell shape dynamics during the staphylococcal cell cycle. Nat Commun 6:8055. doi: 10.1038/ncomms9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koehl JL, Muthaiyan A, Jayaswal RK, Ehlert K, Labischinski H, Wilkinson BJ. 2004. Cell wall composition and decreased autolytic activity and lysostaphin susceptibility of glycopeptide-intermediate Staphylococcus aureus. Antimicrob Agents Chemother 48:3749–3757. doi: 10.1128/AAC.48.10.3749-3757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dhand A, Sakoulas G. 2012. Reduced vancomycin susceptibility among clinical Staphylococcus aureus isolates (“the MIC Creep”): implications for therapy. F1000 Med Rep 4:1–11. doi: 10.3410/M4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uehara T, Bernhardt TG. 2011. More than just lysins: peptidoglycan hydrolases tailor the cell wall. Curr Opin Microbiol 14:698–703. doi: 10.1016/j.mib.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swoboda JG, Campbell J, Meredith TC, Walker S. 2010. Wall teichoic acid function, biosynthesis, and inhibition. ChemBioChem 11:35–45. doi: 10.1002/cbic.200900557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee K, Campbell J, Swoboda JG, Cunny GD, Walker S. 2010. Development of improved inhibitors of wall teichoic acid biosynthesis with potent activity against Staphylococcus aureus. Bioorganic Med Chem Lett 20:1767–1770. doi: 10.1016/j.bmcl.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swoboda JG, Meredith TC, Campbell J, Brown S, Suzuki T, Bollenbach T, Malhowski AJ, Kishony R, Michael S, Walker S, Gilmore MS, Walker S. 2010. Discovery of a small molecule that blocks wall teichoic acid biosynthesis in Staphylococcus aureus. ACS Chem Biol 4:875–883. doi: 10.1021/cb900151k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown S, Zhang YH, Walker S. 2008. A revised pathway proposed for Staphylococcus aureus wall teichoic acid biosynthesis based on in vitro reconstitution of the intracellular steps. Chem Biol 15:12–21. doi: 10.1016/j.chembiol.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meredith TC, Swoboda JG, Walker S. 2008. Late-stage polyribitol phosphate wall teichoic acid biosynthesis in Staphylococcus aureus. J Bacteriol 190:3046–3056. doi: 10.1128/JB.01880-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki T, Swoboda JG, Campbell J, Walker S, Gilmore MS. 2011. In vitro antimicrobial activity of wall teichoic acid biosynthesis inhibitors against Staphylococcus aureus isolates. Antimicrob Agents Chemother 55:767–774. doi: 10.1128/AAC.00879-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atilano ML, Pereira PM, Yates J, Reed P, Veiga H, Pinho MG, Filipe SR. 2010. Teichoic acids are temporal and spatial regulators of peptidoglycan cross-linking in Staphylococcus aureus. Proc Natl Acad Sci U S A 107:18991–18996. doi: 10.1073/pnas.1004304107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biswas R, Voggu L, Simon UK, Hentschel P, Thumm G, Götz F. 2006. Activity of the major staphylococcal autolysin Atl. FEMS Microbiol Lett 259:260–268. doi: 10.1111/j.1574-6968.2006.00281.x. [DOI] [PubMed] [Google Scholar]
- 19.Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Götz F. 1999. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem 274:8405–8410. doi: 10.1074/jbc.274.13.8405. [DOI] [PubMed] [Google Scholar]
- 20.Schlag M, Biswas R, Krismer B, Kohler T, Zoll S, Yu W, Schwarz H, Peschel A, Götz F. 2010. Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol Microbiol 75:864–873. doi: 10.1111/j.1365-2958.2009.07007.x. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Gill CJ, Lee SH, Mann P, Zuck P, Meredith TC, Murgolo N, She X, Kales S, Liang L, Liu J, Wu J, Santa Maria J, Su J, Pan J, Hailey J, McGuinness D, Tan CM, Flattery A, Walker S, Black T, Roemer T. 2013. Discovery of wall teichoic acid inhibitors as potential anti-MRSA beta-lactam combination agents. Chem Biol 20:272–284. doi: 10.1016/j.chembiol.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell J, Singh AK, Swoboda JG, Gilmore MS, Wilkinson BJ, Walker S. 2012. An antibiotic that inhibits a late step in wall teichoic acid biosynthesis induces the cell wall stress stimulon in Staphylococcus aureus. Antimicrob Agents Chemother 56:1810–1820. doi: 10.1128/AAC.05938-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campbell J, Singh AK, Santa Maria JP, Kim Y, Brown S, Swoboda JG, Mylonakis E, Wilkinson BJ, Walker S. 2011. Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem Biol 6:106–116. doi: 10.1021/cb100269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biswas R, Martinez RE, Göhring N, Schlag M, Josten M, Xia G, Hegler F, Gekeler C, Gleske AK, Götz F, Sahl HG, Kappler A, Peschel A. 2012. Proton-binding capacity of Staphylococcus aureus wall teichoic acid and its role in controlling autolysin activity. PLoS One 7:e41415. doi: 10.1371/journal.pone.0041415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qoronfleh MW, Wilkinson BJ. 1986. Effects of growth of methicillin-resistant and -susceptible Staphylococcus aureus in the presence of beta-lactams on peptidoglycan structure and susceptibility to lytic enzymes. Antimicrob Agents Chemother 29:250–257. doi: 10.1128/AAC.29.2.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matano LM, Morris HG, Hesser AR, Martin SES, Lee W, Owens TW, Laney E, Nakaminami H, Hooper D, Meredith TC, Walker S. 2017. Antibiotic that inhibits the ATPase activity of an ATP-binding cassette transporter by binding to a remote extracellular site. J Am Chem Soc 139:10597–10600. doi: 10.1021/jacs.7b04726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foster SJ. 1995. Molecular characterization and functional analysis of the major autolysin of Staphylococcus aureus 8325/4. J Bacteriol 177:5723–5725. doi: 10.1128/jb.177.19.5723-5725.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oshida T, Sugai M, Komatsuzawa H, Hong YM, Suginaka H, Tomasz a. 1995. A Staphylococcus aureus autolysin that has an N-acetylmuramoyl-l-alanine amidase domain and an endo-β-N-acetylglucosaminidase domain: cloning, sequence analysis, and characterization. Proc Natl Acad Sci U S A 92:285–289. doi: 10.1073/pnas.92.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sugai M, Komatsuzawa H, Akiyama T, Hong YM, Oshida T, Miyake Y, Yamaguchi T, Suginaka H. 1995. Identification of endo-β-N-acetylglucosaminidase and N-acetylmuramyl-l-alanine amidase as cluster-dispersing enzymes in Staphylococcus aureus. J Bacteriol 177:1491–1496. doi: 10.1128/jb.177.6.1491-1496.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Götz F, Heilmann C, Stehle T. 2014. Functional and structural analysis of the major amidase (Atl) in Staphylococcus. Int J Med Microbiol 304:156–163. doi: 10.1016/j.ijmm.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 31.Pinho MG, Kjos M, Veening J-W. 2013. How to get (a) round: mechanisms controlling growth and division of coccoid bacteria. Nat Rev Microbiol 11:601–614. doi: 10.1038/nrmicro3088. [DOI] [PubMed] [Google Scholar]
- 32.Baba T, Schneewind O. 1998. Targeting of muralytic enzymes to the cell division site of Gram-positive bacteria: repeat domains direct autolysin to the equatorial surface ring of Staphylococcus aureus. EMBO J 17:4639–4646. doi: 10.1093/emboj/17.16.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen C, Krishnan V, Macon K, Manne K, Narayana SVL, Schneewind O. 2013. Secreted proteases control autolysin-mediated biofilm growth of Staphylococcus aureus. J Biol Chem 288:29440–29452. doi: 10.1074/jbc.M113.502039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujimoto DF, Bayles KW. 1998. Opposing roles of the Staphylococcus aureus virulence regulators, Agr and Sar, in Triton X-100- and penicillin-induced autolysis. J Bacteriol 180:3724–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komatsuzawa H, Sugai M, Nakashima S, Yamada S, Matsumoto A, Oshida T, Suginaka H. 1997. Subcellular localization of the major autolysin, ATL and its processed proteins in Staphylococcus aureus. Microbiol Immunol 41:469–479. doi: 10.1111/j.1348-0421.1997.tb01880.x. [DOI] [PubMed] [Google Scholar]
- 36.Bose JL, Lehman MK, Fey PD, Bayles KW. 2012. Contribution of the Staphylococcus aureus Atl AM and GL murein hydrolase activities in cell division, autolysis, and biofilm formation. PLoS One 7:e42244. doi: 10.1371/journal.pone.0042244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh VK. 2014. High level expression and purification of Atl, the major autolytic protein of Staphylococcus aureus. Int J Microbiol 2014:615965. doi: 10.1155/2014/615965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bierbaum G, Sahl HG. 1985. Induction of autolysis of staphylococci by the basic peptide antibiotics Pep 5 and nisin and their influence on the activity of autolytic enzymes. Arch Microbiol 141:249–254. doi: 10.1007/BF00408067. [DOI] [PubMed] [Google Scholar]
- 39.Peterson PK, Wilkinson BJ, Kim Y, Schmeling D, Douglas SD, Quie PG, Verhoef J. 1978. The key role of peptidoglycan in the opsonization of Staphylococcus aureus. J Clin Invest 61:597–609. doi: 10.1172/JCI108971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilkinson BJ, Kim Y, Peterson PK, Quie PG, Michael AF. 1978. Activation of complement by cell surface components of Staphylococcus aureus. Infect Immun 20:388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bera A, Herbert S, Jakob A, Vollmer W, Götz F. 2005. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol Microbiol 55:778–787. [DOI] [PubMed] [Google Scholar]
- 42.Gross M, Cramton SE, Götz F, Peschel A. 2001. Key role of teichoic acid net charge in Staphylococcus aureus colonization of artificial surfaces. Infect Immun 69:3423–3426. doi: 10.1128/IAI.69.5.3423-3426.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arnaud M, Chastanet A, De M. 2004. New vector for efficient allelic replacement in naturally Gram-positive bacteria. Appl Environ Microbiol 70:6887–6891. doi: 10.1128/AEM.70.11.6887-6891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Löfblom J, Kronqvist N, Uhlén M, Ståhl S, Wernérus H. 2007. Optimization of electroporation-mediated transformation: Staphylococcus carnosus as model organism. J Appl Microbiol 102:736–747. doi: 10.1111/j.1365-2672.2006.03127.x. [DOI] [PubMed] [Google Scholar]
- 45.Hartmann M, Berditsch M, Hawecker J, Ardakani MF, Gerthsen D, Ulrich AS. 2010. Damage of the bacterial cell envelope by antimicrobial peptides gramicidin S and PGLa as revealed by transmission and scanning electron microscopy. Antimicrob Agents Chemother 54:3132–3142. doi: 10.1128/AAC.00124-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frankel MB, Hendrickx APA, Missiakas DM, Schneewind O. 2011. LytN, a murein hydrolase in the cross-wall compartment of Staphylococcus aureus, is involved in proper bacterial growth and envelope assembly. J Biol Chem 286:32593–32605. doi: 10.1074/jbc.M111.258863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gill SR, Fouts DE, Archer GL, Mongodin EF, DeBoy RT, Ravel J, Paulsen IT, Kolonay JF, Brinkac L, Beanan M, Dodson RJ, Daugherty SC, Madupu R, Angiuoli SV, Durkin AS, Haft DH, Vamathevan J, Khouri H, Utterback T, Lee C, Dimitrov G, Jiang L, Qin H, Weidman J, Tran K, Kang K, Hance IR, Nelson KE, Fraser CM. 2005. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol 187:2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki KI, Oguchi A, Nagai Y, Iwama N, Asano K, Naimi T, Kuroda H, Cui L, Yamamoto K, Hiramatsu K. 2002. Genome and virulence determinants of high-virulence community-acquired MRSA. Lancet 359:1819–1827. doi: 10.1016/S0140-6736(02)08713-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.