

ABSTRACT
LCB01-0371 is a novel oxazolidinone with broad-spectrum activity against Gram-positive pathogens in both in vitro studies and animal infection models. The objectives of this study were to evaluate its safety, tolerability, pharmacokinetics, and pharmacodynamics following single ascending doses. Single oral doses of 600 mg linezolid, a placebo, or LCB01-0371 of between 50 mg and 3,200 mg were tested in 69 healthy male subjects. Blood and urine were sampled, LCB01-0371 concentrations were measured, and the serum inhibitory and bactericidal titers of LCB01-0371 and linezolid were determined. LCB01-0371 was well tolerated up to 2,400 mg. The most common drug-related clinical and laboratory adverse events were nausea with or without vomiting, decreased neutrophil counts, and increased total bilirubin levels. The frequency of adverse events and drug-related adverse events was similar among the treatment groups. The systemic exposure was approximately dose proportional over the range of 50 mg to 800 mg, which includes the anticipated clinical dose. The mean clearance, renal clearance, and volume of distribution were significantly decreased at higher doses (above 800 mg). LCB01-0371 exhibited early bacteriostatic activity against all tested strains except for Streptococcus pneumoniae strains, and the potency of LCB01-0371 at 800 mg was similar to that of linezolid at the therapeutic dose (600 mg). However, LCB01-0371 had less bactericidal activity than linezolid. Taken together, LCB01-0371 was well tolerated, exhibited approximate dose proportionality within the anticipated clinically relevant dose range, and showed bacteriostatic and bactericidal activity comparable to that of linezolid. These results support the further clinical development of LCB01-0371. (This study has been registered at ClinicalTrials.gov under registration no. NCT01554995.)
KEYWORDS: LCB01-0371, pharmacokinetics, pharmacodynamics, safety, tolerability
INTRODUCTION
Oxazolidinone antibiotics are both effective and, in general, rather well tolerated (1, 2). Linezolid was the first oxazolidinone antibiotic approved for use in the United States and in many countries worldwide. Because of its strong activity against almost all Gram-positive microorganisms, including multidrug-resistant pathogens, such as methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE), oxazolidinone antibiotics have been widely used for the treatment of bacterial infections (1). Despite the success of linezolid, its use has been limited by the occurrence of bacterial resistance and the potential for adverse effects, including hematological abnormalities, neuropathy, and hyperlactatemia, when used over a prolonged course of therapy (2, 3, 4, 5). Thus, there is an unmet need for newer oxazolidinones that maintain the broad-spectrum activity but minimize the risk of use-limiting adverse effects.
LCB01-0371, a novel oxazolidinone, has recently been developed as an oral formulation. It differs from linezolid in that the C-5 methylacetamide chain and the C-4′ morpholine ring of the phenyl oxazolidinone pharmacophoric group (rings A and B) are replaced by a hydroxymethyl group and a cyclic amidrazone group, respectively (Fig. 1). LCB01-0371 has demonstrated broad-spectrum activity against Gram-positive pathogens in both in vitro studies and animal infection models. Notably, LCB01-0371 was more potent than linezolid against systemic infection in animals. LCB01-0371 does not significantly induce or inhibit the tested hepatic cytochrome P450 (CYP) isozymes. In metabolic studies, no major metabolite was formed, but several minor metabolites were observed. With respect to reversible myelosuppression, the most notable safety concern of linezolid, LCB01-0371 exhibited no adverse effect in a 4-week rat study with oral doses of up to 60 mg/kg of body weight/day, while linezolid exhibited no adverse effect in a study with the same study design with doses of up to 20 mg/kg/day (unpublished data). Assuming that LCB01-0371 and linezolid have similar bioavailabilities, LCB01-0371 is expected to be safer than linezolid according to the use-limiting adverse effects of linezolid.
FIG 1.
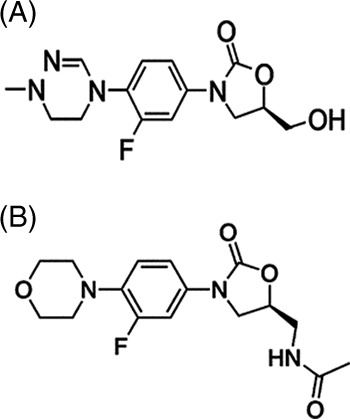
Chemical structure of LCB01-0371 ((R)-3-(3-fluoro-4-(1-methyl-5,6-dihydro-1,2,4-triazine-4(1H)-yl)penyl)-5-(hydroxymethyl) oxazolidin-2-one).
Due to the promising in vitro activity and favorable safety profile of LCB01-0371 in animals, we investigated its safety, tolerability, pharmacokinetics (PKs), and pharmacodynamics (PDs) in an ascending single-dose phase 1 study with healthy male volunteers (ClinicalTrials.gov registration no. NCT01554995).
RESULTS
Study population.
A total of 69 healthy male volunteers were enrolled and completed all assessments. Of these, 48 received LCB01-0371, 15 received the placebo, and 6 received linezolid. The mean age was 26.24 years (range, 20 to 44 years), and the mean body weight was 69.06 kg (range, 53.2 to 86.45 kg).
Safety and tolerability.
There were no serious adverse events (AEs) or withdrawals due to AEs. Table 1 summarizes all AEs in the treatment groups by causality. In total, 24 AEs were reported by 19 of the 69 (27.5%) subjects, including 22 AEs in 17 of the 63 (27.0%) subjects who received either LCB01-0371 or the placebo. Of these, 20 AEs occurred in 15 of the 48 (31.25%) subjects who received LCB01-0371, and 2 AEs occurred in 2 of the 15 (13.3%) subjects who received the placebo. The remaining two AEs occurred in two of the six (33.3%) subjects who received linezolid.
TABLE 1.
Summary of AEs after oral administration of LCB01-0371
| Drug and dose (mg) | Drug-related AE | No. (%) of subjects with the following AE: |
Total no. of AEsa | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Decreased neutrophil count | Increased creatine kinase level | Increased total bilirubin level | Headache | Dizziness | Nausea | Vomiting | Hordeolum | Hypertriglyceridemia | Total | |||
| Placebo (n = 15) | Possibly | 0 (0.0) | 1 (6.7) | 0 (0.0) | 0 (0.0) | 1 (6.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (13.3) | 2 |
| LCB01-0371 | ||||||||||||
| 50 (n = 6) | Possibly | 1 (16.7) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (33.3) | 2 |
| 100 (n = 6) | Possibly | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 1 |
| 200 (n = 6) | Possibly | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 1 |
| No | 2 (33.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (33.3) | 2 | |
| 400 (n = 6) | Possibly | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 1 |
| 800 (n = 6) | Possibly | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 1 |
| 1,600 (n = 6) | Possibly | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (33.3) | 2 |
| Unlikely | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 1 (16.7) | 1 | |
| No | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 1 (16.7) | 1 | |
| 2,400 (n = 6) | Possibly | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 1 |
| 3,200 (n = 6) | Probably | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 1 (16.7) | 2 (33.3) | 2 (33.3) | 0 (0.0) | 0 (0.0) | 2 (33.3) | 6 |
| Possibly | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 1 | |
Some subjects reported the same adverse event more than once.
For LCB01-0371, the primary clinical AEs were nausea with or without vomiting. One subject in the 1,600-mg group and two subjects in the 3,200-mg group reported nausea. All incidences of nausea were reported within the first 2 h after dosing, and one of these incidences of nausea was accompanied by dizziness and headache. Two subjects reported headache approximately 24 h after dosing (one subject each in the 400- and 3,200-mg groups). Two subjects experienced dizziness. This was reported by a subject in the 3,200-mg group within 1 h after dosing and by one subject in the placebo group on the second day after dosing.
Some laboratory AEs occurred. These included decreases in neutrophil count, increases in total serum bilirubin levels, and increases in serum creatine kinase levels. One subject each in the 50- and 2,400-mg groups and three subjects in the 200-mg group experienced mild decreases in the neutrophil count, which were detected at 24 h after drug administration in three subjects and before dosing in two subjects. One subject each in the 100-, 800-, 1,600-, and 3,200-mg groups exhibited elevated total bilirubin levels, which were detected at 24 h after dosing. One subject in the 50-mg group and one subject in the placebo group had increased creatine kinase levels, which were detected at 24 h after dosing. No clinically significant abnormalities in vital signs or in electrocardiography (ECG) data were observed upon physical examination.
The AEs were mild in all groups up to the 2,400-mg group. In the 3,200-mg group, four moderate AEs occurred in two subjects (vomiting in one subject and dizziness, nausea, and vomiting in the other). All subjects except one recovered without treatment during the observation period. One subject took platycodon root and five other ingredients to treat a hordeolum, which the investigators felt was unlikely to have been drug related.
Nineteen of the AEs were considered doubtful to have been drug related, while 13 were thought to possibly have been drug related (the 3 reports of decreases in neutrophil count, the 2 reports of increases in the creatine kinase level, the 5 reports of increases in the total bilirubin level, and the 1 report of headache, dizziness, and nausea) and 6 were thought to probably have been drug related (1 report of headache, 1 occurrence of dizziness, 2 reports of nausea, and 2 occurrences of vomiting). Further dose escalation was stopped at 3,200 mg owing to the four moderate but likely drug-related AEs. No apparent differences in the numbers of total AEs or the numbers of drug-related AEs were observed across the treatment groups.
For linezolid, increases in total serum bilirubin levels and laceration at the right eyebrow were reported by two subjects. They were mild and were thought to possibly have been drug related and unlikely to have been drug related, respectively.
Pharmacokinetics.
Mean plasma concentration-time curves for each dose of LCB01-0371 are shown in Fig. 2. The values of the PK parameters of LCB01-0371, determined using a noncompartmental analysis, are summarized in Table 2. In general, LCB01-0371 was rapidly absorbed following oral administration, with the maximum concentration (Cmax) being attained within 1 h of dosing.
FIG 2.

Mean plasma concentration-time profiles of LCB01-0371 following a single oral dose of 50, 100, 200, 400, 800, 1,600, 2,400, or 3,200 mg.
TABLE 2.
PK variables for LCB01-0371 and linezolida
| Drug and dose (mg) | Cmax (ng/ml) | Tmax (h) | AUClast (ng · h/ml) | AUCinf (ng · h/ml) | t1/2 (h) | V/F (liters) | CL/F (liters/h) | fe (%)b | CLR (liters/h) |
|---|---|---|---|---|---|---|---|---|---|
| LCB01-0371 | |||||||||
| 50 | 525.5 (95.82) | 0.75 | 834.54 (66.91) | 874.21 (65.03) | 1.46 (0.11) | 121.18 (10.45) | 57.47 (4.42) | 6.97 (1.77) | 4.18 (1.07) |
| 100 | 1,323.83 (578.32) | 0.5 | 1,710.08 (562.36) | 1,753.31 (570.55) | 1.41 (0.18) | 123.93 (31.3) | 62.28 (20.31) | 5.92 (1.41) | 3.72 (1.21) |
| 200 | 1,651.67 (437.15) | 0.75 | 3,092.37 (580.14) | 3,153.18 (582.89) | 1.48 (0.3) | 136.36 (16.93) | 65.12 (11.02) | 4.61 (2.36) | 2.9 (1) |
| 400 | 3,460.00 (889.90) | 0.51 | 7,433.55 (1,328.23) | 7,483.02 (1,312.50) | 1.55 (0.10) | 122.46 (20.10) | 55.16 (11.83) | 5.35 (2) | 2.9 (0.95) |
| 800 | 9,516.67 (2,330.36) | 1 | 19,538.55 (3,553.11) | 19,669.64 (3,589.96) | 1.61 (0.15) | 97.21 (20.81) | 41.84 (7.80) | 4 (0.83) | 1.68 (0.43) |
| 1,600 | 21,650.00 (6,409.60) | 1 | 52,900.54 (11,626.03) | 53,105.34 (11,636.44) | 2.16 (1.14) | 94.96 (42.28) | 31.58 (8.10) | 6.08 (1.54) | 1.85 (0.32) |
| 2,400 | 27,450.00 (8,971.68) | 1 | 85,839.64 (27,533.38) | 86,179.46 (27,425.07) | 3.41 (1.10) | 153.79 (79.01) | 30.29 (9.72) | 4.16 (2.01) | 1.22 (0.55) |
| 3,200 | 38,900.00 (16,311.84) | 0.75 | 136,664.51 (43,212.08) | 136,827.24 (43,199.08) | 2.35 (0.69) | 85.57 (39.01) | 24.95 (6.02) | 8.23 (3.88) | 1.89 (0.4) |
| Linezolid, 600 | 12,519.46 (4,440.54) | 1.05 | 130,534.55 (26,887.19) | 134,252.63 (26,336.04) | 7.94 (0.95) | 52.22 (7.51) | 4.61 (0.88) |
Values for all parameters except Tmax are the mean (standard deviation). Values for Tmax are the median. Six subjects received each dose.
fe, the fraction of the drug excreted unchanged in the urine.
Graphical and statistical analysis of the dose proportionality of LCB01-0371 revealed that Cmax increased proportionally over the 50- to 3,200-mg dose range, whereas the area under the plasma concentration-time curve (AUC) from 0 h to the time of the last measurable concentration after dosing (AUClast) and the AUC from time zero to infinity (AUCinf) increased more than proportionally over this dose range (Fig. 3). Using the power model to assess dose proportionality, the 90% confidence interval (CI) for the slope of the parameters for the range of evaluated doses was 0.96 to 1.14 (6). The estimated slopes for the AUC values were completely outside the 90% CI, whereas the slope for Cmax was within the interval. However, analysis over smaller ranges revealed that the slopes for the AUC values were more dose proportional over the dose range of 50 to 800 mg (slopes for the AUClast, 1.0511 to 1.1946; slopes for the AUCinf, 1.037 to 1.1802). When the dose increased in ratios of 2 (100/50 mg), 4 (200/50 mg), 8 (400/50 mg), 16 (800/50 mg), 32 (1,600/50 mg), 48 (2,400/50 mg), or 64 (3,200/50 mg), the corresponding mean ratios for AUCinf were approximately 2.0, 3.6, 8.6, 22.5, 60.7, 98.6, and 156.5, respectively. The corresponding ratios for Cmax were 2.5, 3.1, 6.6, 18.1, 41.2, 52.2, and 74.0, respectively.
FIG 3.

Mean and 90% CI of Cmax (A), AUClast (B), and AUCinf (C) versus dose following a single oral dose of 50, 100, 200, 400, 800, 1,600, 2,400, or 3,200 mg. The points indicate individual values.
The median time to reach Cmax (Tmax) of LCB01-0371 ranged from approximately 0.5 to 1.0 h. The mean terminal-phase elimination half-life (t1/2) of LCB01-0371 was approximately 1.5 h over the dose range of 50 to 800 mg and increased to values ranging from 2.16 h (at 3,200 mg) to 3.41 h (at 1,600 mg) at higher doses. The mean apparent terminal volume of distribution (V/F) of LCB01-0371 ranged from 121.18 to 136.36 liters over the dose range of 50 to 400 mg and decreased to values ranging from 97.21 to 85.57 liters at all higher doses except the 2,400-mg dose level, where it was 153.79 liters. The apparent clearance (CL/F) of LCB01-0371 ranged from 55.16 to 65.12 liters/h over the dose range of 50 to 400 mg and decreased to values ranging from 41.84 to 24.95 liters/h at higher doses.
The majority of the drug recovered in urine was the parent drug. The percentage of LCB01-0371 recovered in urine ranged from 4.00% to 8.23% of the administered dose over the dose range of 50 to 3,200 mg. The renal clearance (CLR) of LCB01-0371 decreased slightly with increasing doses, from 4.61 liters/h for the 50-mg dose to 2.02 liters/h for the 3,200-mg dose (Table 2).
Pharmacodynamics.
The serum inhibitory titer (SIT) and serum bactericidal titer (SBT) were measured for three groups, the 400- and 800-mg LCB01-0371 group and the 600-mg linezolid group. The SIT and SBT ranges for the three groups against four different strains of bacteria (S. aureus ATCC 29213, Enterococcus faecalis ATCC 51299, MRSA ATCC 43300, and Streptococcus pneumoniae ATCC 49619) are listed in Table 3. Overall, there was no bacteriostatic or bactericidal effect in any subject at the trough time point (24 h). A bactericidal effect was observed at the peak time point (1 h) for some subjects that received 600 mg linezolid or 800 mg LCB01-0371, and on the basis of the median values, the bactericidal activity of 600 mg linezolid was slightly higher than that of 800 mg LCB01-0371. The SBT values for the linezolid group ranged from <2 to 4 against S. aureus, <2 to 8 against E. faecalis, and <2 to 8 against MRSA. The SBT values for the 800-mg LCB01-0371 group ranged from <2 to 4 against S. aureus and <2 to 2 against MRSA. As shown in Table 3, the SIT values at the peak time point against S. aureus ranged from <2 to 4 for the 400-mg LCB01-0371 group, 2 to 4 for the 800-mg LCB01-0371 group, and <2 to 8 for the 600-mg linezolid group. The SIT values at the peak time point against E. faecalis ranged from <2 to 2 for the 400-mg LCB01-0371 group, 2 to 4 for the 800-mg LCB01-0371 group, and 4 to 8 for the 600-mg linezolid group. The SIT values at the peak time point against MRSA ranged from <2 to 2 for the 400-mg LCB01-0371 group, 2 to 4 for the 800-mg LCB01-0371 group, and 4 to 8 for the 600-mg linezolid group. No inhibitory effect against S. pneumoniae was observed for either LCB01-0371 groups or the linezolid group.
TABLE 3.
SIT and SBT over time for LCB01-0371 and linezolid against Gram-positive pathogensa
| Organism | Drug | Dose (mg) | Titer range |
No. subjects with SIT of: |
No. subjects with SBT of: |
|||
|---|---|---|---|---|---|---|---|---|
| SIT | SBT | ≥2 | ≥4 | ≥2 | ≥4 | |||
| S. aureus ATCC 29213 | LCB01-0371 | 400 | <2–4 | <2 | 5 | 1 | 0 | 0 |
| LCB01-0371 | 800 | 2–4 | <2–4 | 6 | 1 | 2 | 1 | |
| Linezolid | 600 | <2–8 | <2–4 | 5 | 5 | 2 | 2 | |
| E. faecalis ATCC 51299 | LCB01-0371 | 400 | <2–2 | <2 | 5 | 0 | 0 | 0 |
| LCB01-0371 | 800 | 2–4 | <2 | 6 | 1 | 0 | 0 | |
| Linezolid | 600 | 4–8 | <2–8 | 6 | 6 | 4 | 4 | |
| MRSA ATCC 43300 | LCB01-0371 | 400 | <2–2 | <2 | 1 | 0 | 0 | 0 |
| LCB01-0371 | 800 | 2–4 | <2–2 | 6 | 1 | 1 | 0 | |
| Linezolid | 600 | 4–8 | <2–8 | 6 | 6 | 3 | 3 | |
| S. pneumoniae ATCC 49619 | LCB01-0371 | 400 | <2 | <2 | 0 | 0 | 0 | 0 |
| LCB01-0371 | 800 | <2 | <2 | 0 | 0 | 0 | 0 | |
| Linezolid | 600 | <2 | <2 | 0 | 0 | 0 | 0 | |
SIT, serum inhibitory titer; SBT, serum bactericidal titer. Values are the SIT and SBT at the peak time point (1 h after dosing). Six subjects received each dose.
DISCUSSION
In this study, single escalating doses of 50, 100, 200, 400, 800, 1,600, 2,400, and 3,200 mg LCB01-0371 were evaluated in healthy male volunteers (age range, 20 to 44 years). Further dose escalation was stopped at 3,200 mg owing to the occurrence of probable drug-related AEs. Thus, the 2,400-mg dose is defined as the maximum tolerated dose for LCB01-0371 in humans.
There were no severe or life-threatening AEs related to LCB01-0371. The most common drug-related clinical and laboratory AEs were nausea with or without vomiting, decreased neutrophil counts, and increased total bilirubin levels. There were no drug- or dose-related increases in the incidence of the reported AEs. The safety of a single oral dose of LCB01-0371 was further demonstrated by the lack of any significant effect on vital signs, physical examination findings, or ECG data. These safety and tolerability findings are comparable to those from single-dose studies of the clinically available oxazolidinones linezolid and tedizolid (7–10).
While oxazolidinones are generally well tolerated, one concern with this class of drugs is reversible myelosuppression, especially thrombocytopenia. This is well documented with linezolid, primarily in treatment courses longer than 14 days (11–14). Therefore, a study with a multiple-dosing period longer than 14 days will be required to evaluate the hematological safety profile of LCB01-0371, although hematological effects were not reported in this study, except for the slightly decreased neutrophil counts.
With respect to PKs, following oral administration, LCB01-0371 rapidly achieved its Cmax within 1 h and was eliminated with a dose-dependent t1/2 (approximately 1.5 to 3.41 h), showing absorption faster than that of linezolid and tedizolid and elimination equivalent to or more rapid than that of linezolid (7–10). The Cmax was linear and dose proportional over the range of 50 to 3,200 mg. Although the AUC increased more than proportionally with dose over the corresponding dose range, the deviation from dose proportionality over the anticipated clinically relevant dose range (50 to 800 mg) was almost within the criterion interval suggested by Smith et al. (6). The reason for the lack of proportionality was not investigated in this study, but it could have been caused by saturation of both an elimination pathway and an absorption pathway. Notably, considering that both CL/F and V/F tend to decrease with increasing dose, it is likely due to a nonlinear absorption mechanism, such as capacity-limited transport in the gastrointestinal tract or saturable first-pass metabolism. Additionally, considering a dose-dependent t1/2, it is also likely due to a nonlinear elimination mechanism.
The V/F of LCB01-0371 was similar to the values reported for tedizolid (8, 9) and exceeded the total body volume. The nonlinearity of CL/F and CLR was observed with increasing doses, consistent with the nonproportionality of the AUC. There was a wide variability in clearance, which can be accounted for primarily by the variability in non-CLR, because CLR exhibited little variability between subjects.
The urinary recovery reported here is much lower than that in previous nonclinical studies, in which 66.13% of the total study drug was recovered in the urine after oral administration of [14C]LCB01-0371 to male rats. Furthermore, the exposure of LCB01-0367 and LCB01-0352, major metabolites from in vitro studies, in both plasma and urine was very low in this study (data not shown). Considering these conflicting results between clinical and nonclinical studies, extensive metabolic profiling studies and quantitative whole-body autoradiography using radiolabeled drug in humans are needed to determine the clearance pathway and tissue distribution of LCB01-0371.
In this study, similarly to linezolid at the clinically therapeutic dose (600 mg), LCB01-0371 at the 400- and 800-mg doses showed early (1 h) bacteriostatic activity in serum against test strains of S. aureus, E. faecalis, and MRSA but did not show prolonged activity at 24 h or activity against S. pneumoniae at either time point. The SIT values for all three organisms generally increased with the dose level and correlated with the concentration of drug in the sample. A bactericidal effect was exhibited in some subjects that received 600 mg linezolid or 800 mg LCB01-0371, and linezolid at 600 mg was more potent than LCB01-0371 at 800 mg. In a previous study, Jeong et al. reported that LCB01-0371 had antimicrobial activity equivalent to or better than that of linezolid against these strains in vitro (15). When taking into account these in vitro activities and the results of our PK studies, the lower bioavailability, higher absorption rate, higher elimination rate, or higher serum protein binding for LCB01-0371 than for linezolid might be the cause of the lack of bactericidal activity of LCB01-0371 in this study. Taken together, the results of the serum bactericidal test suggest that LCB01-0371 should be given at a dose higher than 800 mg to achieve an antimicrobial activity similar to or greater than that of linezolid at the therapeutic dose. However, because the serum bactericidal test has the limitation that it does not predict either bacteriologic success or the clinical outcome (16), further study for the validation of these results, including the lack of antimicrobial activity against S. pneumoniae, will be needed. Additionally, given that LCB01-0371 and linezolid harbor similar antimicrobial activities, an at least twice-daily regimen should be considered for LCB01-0371, because the elimination half-life of LCB01-0371 was similar to or shorter than that of linezolid across a range of doses, and the recommended dosage of linezolid (given orally or intravenously) is 600 mg every 12 h (17).
In conclusion, oral administration of a single dose of LCB01-0371 was generally safe and well tolerated in healthy male subjects at doses of up to 2,400 mg. The PK and pharmacodynamic profile of LCB01-0371 supports its further clinical development.
MATERIALS AND METHODS
Study drug.
The study drugs were LCB01-0371 (50, 100, 200, 400, 800, 1,600, 2,400, and 3,200 mg), placebo, and linezolid (600 mg), provided as tablets. The placebo formulation was identical in appearance to the active LCB01-0371 formulation.
Study population.
The study population consisted of healthy male subjects aged 20 to 45 years with body mass indexes of 19 to 27 kg/m2. For recruitment, the subjects underwent standard safety assessments during a prestudy screening. Subjects who reported the use of prescription medicine within the previous 14 days or over-the-counter drugs within the previous 7 days were excluded from the study. Consumption of grapefruit or grapefruit juice, caffeine, alcohol, and cigarettes was not permitted during the study. Written informed consent was obtained from each individual participating in the study prior to their enrollment.
Study design.
The study was a randomized, double-blind, placebo-controlled, single-dose escalation study involving eight sequential doses of LCB01-0371 (50, 100, 200, 400, 800, 1,600, 2,400, and 3,200 mg). In addition to the administration of LCB01-0371 to various cohorts, to compare the pharmacodynamics of LCB01-0371 and linezolid, linezolid was administered to six subjects at a single dose of 600 mg. The initial dose of LCB01-0371 selected for this study was 50 mg. This dose was calculated by deriving the human-equivalent dose (HED) from the lowest no-observed-adverse-effect level (NOAEL) found in preclinical 1-month repeat-dose toxicity studies and dividing the HED by the adjusted safety factor of 7. This starting dose was thought to allow judicious and efficient dose escalation to levels well beyond the anticipated therapeutic range. Cohorts of healthy male subjects were randomly assigned to receive tablets of LCB01-0371 (six subjects per dose group) or placebo (two subjects per dose group). A prestudy screening, including medical history, physical examination, and laboratory tests, was performed within 28 days prior to admission to the study site. Eligible subjects were admitted on the day prior to dosing (day −1). On the dosing day of the study (day 1), after fasting for 10 h, the subjects were orally administered LCB01-0371, the placebo, or linezolid with 240 ml water. The subjects continued to fast for an additional 4 h but were able to freely drink water after 2 h. During the admission period, blood and urine samples were collected for PK, pharmacodynamic, and safety evaluations before proceeding to the next cohort for the administration of the higher dose. The study was conducted at a single center in South Korea in full accordance with the principles of the Declaration of Helsinki and all its amendments. The protocol and all materials provided to the subjects were reviewed and approved by the Ministry of Food and Drug Safety of South Korea (Investigational New Drug application [IND] no. 11870) and the Institutional Review Board (IRB) of Seoul Asan Medical Center, South Korea.
Safety evaluation.
For all the cohorts that received LCB01-0371 or linezolid, records of all adverse events (AEs), vital signs, electrocardiography (ECG) data, and laboratory parameters were collected during each subject's stay in the clinic. Changes in vital signs, ECG data, and clinical laboratory parameters from the baseline (measured on day −1 or day 1 before dosing) were determined and evaluated for clinical relevance.
Sample collection.
Blood samples for PK analysis were collected within 0.5 h prior to dosing and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24 (day 2), and 48 h (day 3) postadministration. The blood samples for PK analysis were drawn into heparinized tubes and processed to obtain plasma. Additional blood samples were collected from all subjects in the 400- and 800-mg LCB01-0371 groups and the 600-mg linezolid group prior to dosing and at 1 and 24 h after dosing for pharmacodynamic analyses, i.e., measurement of the serum inhibitory titer (SIT) and serum bacterial titer (SBT). For each sample, the plasma or serum was aliquoted and stored at −70°C until further analysis. Urine samples for measurement of the LCB01-0371 concentration were collected into polyethylene containers over the following time intervals: predose (within 1 h prior to dosing) and 0 to 4, 4 to 8, 8 to 12, 12 to 24, and 24 to 48 h postadministration. During each collection period, the contents were stored in a refrigerator. At the end of each collection period, the volume was recorded and urine aliquots were stored at −70°C until further analysis.
Sample pharmacokinetic analysis.
LCB01-0371 was extracted using the solid-phase extraction method and quantified by a validated high-performance liquid chromatography coupled to tandem mass spectrometry (HPLC-MS/MS) method with multiple-reaction monitoring. Separation was achieved using a Shiseido MG column (2.0 by 150 mm; particle size, 3 μm) with gradient elution. Both human plasma and urine calibration standards were used to quantify LCB01-0371 in the human quality control samples and unknown specimens. Sample concentrations were determined by back-calculation using weighted linear (1/x2) regression of a calibration curve generated from spiked matrix standards. The limits of quantification were set on the basis of standard acceptability criteria of the analyte/matrix peak ratio and the assay accuracy and precision. A 50-μl plasma aliquot was used for analysis of LCB01-0371. The normal range of quantification of LCB01-0371 in plasma was 10 to 10,000 ng/ml. The interrun and intrarun relative percent error and percent coefficient of variation for LCB01-0371 were −2.42 to 3.55% and 0.35 to 5.84%, respectively, over the linear range. A 100-μl sample was used for urine analysis. The normal range of quantification of LCB01-0371 in urine was 4 to 4,000 ng/ml. The interrun and intrarun relative percent error and percent coefficient of variation for LCB01-0371 were −9.69 to 5.20% and 1.26 to 9.04%, respectively, over the linear range.
Pharmacokinetics and statistical analysis.
Noncompartmental analyses of the concentration-time profile of LCB01-0371 in individual plasma samples were performed using the ncar package (version 0.3.7) of the open source data analysis program R. The following PK variables were derived for each subject: the maximum concentration (Cmax) and the time to reach Cmax (Tmax), the area under the plasma concentration-time curve from 0 h to the time of the last measurable concentration after dosing (AUClast), the terminal-phase elimination half-life (t1/2), the cumulative amount of drug excreted into the urine (Ae), the apparent clearance (CL/F), the apparent terminal volume of distribution (V/F), and the renal clearance (CLR). Plasma concentrations below the lower limit of quantification were set to zero if they occurred prior to the peak and were omitted from the PK analysis otherwise. Urine concentrations below the lower limit of quantification were set to zero. Cmax and Tmax were determined from visual inspection of individual plasma concentration-time profiles. The terminal-phase elimination rate constant was determined using linear least-squares regression of the terminal phase of the log plasma concentration-time profiles. t1/2 was calculated as ln(2)/(terminal-phase elimination rate constant). AUClast was calculated using the linear trapezoidal rule when the concentration was increasing or the logarithmic trapezoidal rule when the concentration was decreasing. The AUC from time zero to infinity (AUCinf) was calculated as the sum of AUClast and Clast/λz, where Clast is the last measurable drug concentration and λz is the elimination rate constant derived from the slope of the linear regression line of the apparent terminal linear portion of the log concentration-time curve over a minimum of the last three data points. CL/F and V/F were calculated as dose/AUCinf and dose/(AUCinf · λz), respectively. CLR was calculated as Ae divided by AUC over the same collection interval.
Dose proportionality was assessed by evaluating the PK parameters (i.e., Cmax, AUCinf, and AUClast) using the power model. The linear relationships between the log-transformed PK parameters and the natural log of the dose were tested using the criterion suggested by Smith et al. (6). A point estimate and 90% confidence interval (CI) were produced for the population mean slope. Approximate dose proportionality was established if the 90% CI for the slope was completely contained within the range of 0.96 to 1.14. The parameters that were not dose proportional over the range of 50 mg to 3,200 mg were investigated over several smaller ranges, e.g., 50 mg to 800 mg, to determine the dose-proportional range.
Pharmacodynamic analysis. (i) Bacterial strains.
The serum inhibitory and bactericidal titers of LCB01-0371 and linezolid were determined against the following strains: S. aureus ATCC 29213, E. faecalis ATCC 51299, MRSA ATCC 43300, and S. pneumoniae ATCC 49619.
(ii) Serum inhibitory activities and bactericidal titers.
Serum inhibitory activities and bacterial titers were determined by microdilution using cation-adjusted Mueller-Hinton broth, as recommended by the Clinical and Laboratory Standards Institute. An inoculum of 105 CFU per well was incubated for 24 h. Each determination was performed in duplicate. Wells with no visible growth and the first well to show growth were subcultured in supplemented Mueller-Hinton II agar and incubated for an additional 2 days prior to counting of the colonies. Isolates were tested against serum collected from all subjects in the 400- and 800-mg LCB01-0371 groups and the 600-mg linezolid group predosing and 1 h and 24 h after dosing. The bacteriostatic titer in serum was determined as the highest dilution with no visible growth, and the bactericidal titer in serum was determined as the highest dilution of serum yielding 99.9% killing. The median and geometric mean of the bacteriostatic and bactericidal titers at each time point were calculated.
ACKNOWLEDGMENTS
This study was funded by LegoChem Biosciences (Daejeon, South Korea) and was also supported by the Korea Drug Development Fund (KDDF), funded by the Ministry of Science and ICT, Ministry of Trade, Industry, and Energy, and Ministry of Health and Welfare (grant no. KDDF-201112-02, Republic of Korea), and by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI07C0001).
We thank Joon Seo Lim from the Scientific Publications Team at Asan Medical Center for his editorial assistance in preparing the manuscript.
REFERENCES
- 1.Perry CM, Jarvis B. 2001. Linezolid: a review of its use in the management of serious gram-positive infections. Drugs 61:525–551. doi: 10.2165/00003495-200161040-00008. [DOI] [PubMed] [Google Scholar]
- 2.Vinh DC, Rubinstein E. 2009. Linezolid: a review of safety and tolerability. J Infect 59(Suppl 1):S59–S74. doi: 10.1016/S0163-4453(09)60009-8. [DOI] [PubMed] [Google Scholar]
- 3.Gonzales RD, Schreckenberger PC, Graham MB, Kelkar S, DenBesten K, Quinn JP. 2001. Infections due to vancomycin-resistant Enterococcus faecium resistant to linezolid. Lancet 357:1179. doi: 10.1016/S0140-6736(00)04376-2. [DOI] [PubMed] [Google Scholar]
- 4.Herrero IA, Issa NC, Patel R. 2002. Nosocomial spread of linezolid-resistant, vancomycin-resistant Enterococcus faecium. N Engl J Med 346:867–869. doi: 10.1056/NEJM200203143461121. [DOI] [PubMed] [Google Scholar]
- 5.Tsiodras S, Gold HS, Sakoulas G, Eliopoulos GM, Wennersten C, Venkataraman L, Moellering RC, Ferraro MJ. 2001. Linezolid resistance in a clinical isolate of Staphylococcus aureus. Lancet 358:207–208. doi: 10.1016/S0140-6736(01)05410-1. [DOI] [PubMed] [Google Scholar]
- 6.Smith BP, Vandenhende FR, DeSante KA, Farid NA, Welch PA, Callaghan JT, Forgue ST. 2000. Confidence interval criteria for assessment of dose proportionality. Pharm Res 17:1278–1283. doi: 10.1023/A:1026451721686. [DOI] [PubMed] [Google Scholar]
- 7.Burkhardt O, Borner K, von der Hoh N, Koppe P, Pletz MW, Nord CE, Lode H. 2002. Single- and multiple-dose pharmacokinetics of linezolid and co-amoxiclav in healthy human volunteers. J Antimicrob Chemother 50:707–712. doi: 10.1093/jac/dkf163. [DOI] [PubMed] [Google Scholar]
- 8.Flanagan S, Fang E, Munoz KA, Minassian SL, Prokocimer PG. 2014. Single- and multiple-dose pharmacokinetics and absolute bioavailability of tedizolid. Pharmacotherapy 34:891–900. doi: 10.1002/phar.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flanagan SD, Bien PA, Munoz KA, Minassian SL, Prokocimer PG. 2014. Pharmacokinetics of tedizolid following oral administration: single and multiple dose, effect of food, and comparison of two solid forms of the prodrug. Pharmacotherapy 34:240–250. doi: 10.1002/phar.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stalker DJ, Jungbluth GL, Hopkins NK, Batts DH. 2003. Pharmacokinetics and tolerance of single- and multiple-dose oral or intravenous linezolid, an oxazolidinone antibiotic, in healthy volunteers. J Antimicrob Chemother 51:1239–1246. doi: 10.1093/jac/dkg180. [DOI] [PubMed] [Google Scholar]
- 11.Gerson SL, Kaplan SL, Bruss JB, Le V, Arellano FM, Hafkin B, Kuter DJ. 2002. Hematologic effects of linezolid: summary of clinical experience. Antimicrob Agents Chemother 46:2723–2726. doi: 10.1128/AAC.46.8.2723-2726.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Birmingham MC, Rayner CR, Meagher AK, Flavin SM, Batts DH, Schentag JJ. 2003. Linezolid for the treatment of multidrug-resistant, gram-positive infections: experience from a compassionate-use program. Clin Infect Dis 36:159–168. doi: 10.1086/345744. [DOI] [PubMed] [Google Scholar]
- 13.Lodise TP, Bidell MR, Flanagan SD, Zasowski EJ, Minassian SL, Prokocimer P. 2016. Characterization of the haematological profile of 21 days of tedizolid in healthy subjects. J Antimicrob Chemother 71:2553–2558. doi: 10.1093/jac/dkw206. [DOI] [PubMed] [Google Scholar]
- 14.Waldrep TW, Skiest DJ. 2002. Linezolid-induced anemia and thrombocytopenia. Pharmacotherapy 22:109–112. doi: 10.1592/phco.22.1.109.33504. [DOI] [PubMed] [Google Scholar]
- 15.Jeong JW, Jung SJ, Lee HH, Kim YZ, Park TK, Cho YL, Chae SE, Baek SY, Woo SH, Lee HS, Kwak JH. 2010. In vitro and in vivo activities of LCB01-0371, a new oxazolidinone. Antimicrob Agents Chemother 54:5359–5362. doi: 10.1128/AAC.00723-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ackerman BH, Dello Buono FA. 1996. In vitro testing of antibiotics. Pharmacotherapy 16:201–217. [PubMed] [Google Scholar]
- 17.Bouza E, Munoz P. 2001. Linezolid: pharmacokinetic characteristics and clinical studies. Clin Microbiol Infect 7(Suppl 4):S75–S82. [DOI] [PubMed] [Google Scholar]