

ABSTRACT
The APH(2″)-Ia aminoglycoside resistance enzyme forms the C-terminal domain of the bifunctional AAC(6′)-Ie/APH(2″)-Ia enzyme and confers high-level resistance to natural 4,6-disubstituted aminoglycosides. In addition, reports have suggested that the enzyme can phosphorylate 4,5-disubstituted compounds and aminoglycosides with substitutions at the N1 position. Previously determined structures of the enzyme with bound aminoglycosides have not indicated how these noncanonical substrates may bind and be modified by the enzyme. We carried out crystallographic studies to directly observe the interactions of these compounds with the aminoglycoside binding site and to probe the means by which these noncanonical substrates interact with the enzyme. We find that APH(2″)-Ia maintains a preferred mode of binding aminoglycosides by using the conserved neamine rings when possible, with flexibility that allows it to accommodate additional rings. However, if this binding mode is made impossible because of additional substitutions to the standard 4,5- or 4,6-disubstituted aminoglycoside architecture, as in lividomycin A or the N1-substituted aminoglycosides, it is still possible for these aminoglycosides to bind to the antibiotic binding site by using alternate binding modes, which explains the low rates of noncanonical phosphorylation activities seen in enzyme assays. Furthermore, structural studies of a clinically observed arbekacin-resistant mutant of APH(2″)-Ia revealed an altered aminoglycoside binding site that can stabilize an alternative binding mode for N1-substituted aminoglycosides. This mutation may alter and expand the aminoglycoside resistance spectrum of the wild-type enzyme in response to newly developed aminoglycosides.
KEYWORDS: aminoglycoside, antibiotic resistance, kinase, phosphotransferase, substrate recognition
INTRODUCTION
Aminoglycosides are a class of antibiotics effective against many bacterial pathogens. However, widespread resistance to these compounds limits their clinical utility (1). Most commonly, resistance emerges through the action of enzymes that chemically modify these antibiotics, transferring chemical groups from donor molecules to an acceptor group on the antibiotic to render it inactive (2). Most aminoglycosides are based upon a neamine core, i.e., a central 2-deoxystreptamine (2-DOS) ring with an aminohexose sugar linked to 2-DOS at the 4-position (Fig. 1). These rings form the minimal active element of an aminoglycoside antibiotic (3), and two subclasses of aminoglycosides are formed from elaboration of this scaffold. Addition of rings to the 5- and 6-positions of 2-DOS differentiate aminoglycosides into the 4,5-disubstituted (ribostamycin, neomycin, and lividomycin) and 4,6-disusbstituted (kanamycin, gentamicin, and dibekacin) groups. The central importance of the neamine core has been illustrated through studies of the mode of binding to the ribosome: both classes of compound conserve the same interactions between the neamine core and the ribosome, while the additional rings are accommodated and form additional binding interactions (4).
FIG 1.

Aminoglycoside antibiotics discussed in this article. 4,5-Disubstituted and 4,6-disubstituted aminoglycosides are both built on a minimal active unit represented by the compound neamine. Addition of rings to the 5-position creates the 4,5-disubstituted compounds ribostamycin, neomycin, and lividomycin, while addition of a ring to the 6-position generates kanamycin and similar compounds. Dibekacin is produced by changing some chemical groups on the 4-linked ring of kanamycin, while the N1-substituted compounds amikacin and arbekacin are produced by addition of an (S)-2-hydroxy-4-aminobutyrate group to the N1 position of the 2-DOS ring.
Following the emergence of aminoglycoside-inactivating enzymes, semisynthetic aminoglycosides were developed that maintained antibiotic activity by maintaining ribosome binding while blocking binding to resistance enzymes through the addition of a bulky group (5). The most successful modification has been acylation of the N1 group of aminoglycosides with an (S)-2-hydroxyl-aminobutyrate (AHB) group, producing amikacin from kanamycin A (6) and arbekacin from dibekacin (7). Both these compounds have been subjects of recent reviews (8, 9). The AHB modification is still tolerated for ribosomal binding (10) but greatly reduces the binding of modified compounds to resistance enzymes, often effectively evading enzymatic aminoglycoside resistance.
The structure of an aminoglycoside dictates its ability to bind to and be modified by an antibiotic resistance enzyme. Shape complementarity, charge distribution, formation and breakage of hydrogen bonds, and changes in conformational entropy all play a role in the interactions of these compounds with resistance enzymes. As a result, some aminoglycoside resistance enzymes act upon both 4,5-disubstituted and 4,6-disubstituted compounds, while others are limited to only one group. Broad-spectrum enzymes that act upon both groups do so by binding to the conserved neamine core in a shared orientation while tolerating the 5- or 6-linked rings through various mechanisms (11–13).
A widespread resistance factor for aminoglycoside antibiotics is the bifunctional protein AAC(6′)-Ie/APH(2″)-Ia, which contains two aminoglycoside-modifying enzyme activities within a single polypeptide. These domains appear to be associated structurally (14, 15), but to date, the full-length enzyme has not had its structure unambiguously defined. The phosphotransferase domain of this enzyme, APH(2″)-Ia, can be studied in isolation and phosphorylates 4,6-disubstituted aminoglycosides on the 2″-OH group. This produces high-level clinical resistance to gentamicin and other 4,6-disubstituted aminoglycosides. The enzyme uses GTP as the phosphate donor (16) and magnesium ions to facilitate the transfer. The activity of this domain has been subjected to thorough enzymological characterization, including studies that have indicated that the enzyme is active toward N1-substituted (17) and 4,5-disubsituted (18) aminoglycosides. This occurs in addition to the activity toward prototypical 4,6-disubstituted substrates, such as gentamicin. These anomalous activities toward 4,5-disubstituted and N1-modified 4,6-disubstituted aminoglycosides are not easily explained by existing structural models.
APH(2″)-Ia crystallizes with four copies (chains A to D) in the crystal structure. Variation between these chains has allowed us to study the conformational changes that occur in the protein (19). Aminoglycosides can be introduced into crystals through crystallographic soaking experiments, allowing us to track the structural changes that take place upon introduction of the antibiotic to the protein. To probe the interactions of APH(2″)-Ia with aminoglycosides and to examine its activity toward nonstandard substrates, we determined structures of the APH(2″)-Ia enzyme following crystallographic soaking with the 4,5-disubstituted compounds ribostamycin and lividomycin A, the N1-substituted 4,6-disubstituted compounds amikacin and arbekacin, and arbekacin's parent compound, dibekacin. Using comparisons with previously determined aminoglycoside-bound structures, we report the most important structural interactions required to bind aminoglycosides, as well as alternate means by which aminoglycosides can bind the enzyme. These interactions also help to explain the enhanced resistance to arbekacin conferred by a clinical mutant (20), for which we also report structural data here.
RESULTS
The ribostamycin-bound crystal structure shows conservation of the neamine ring position but variability in the position of the 5-linked ring.
A crystal of APH(2″)-Ia grown with guanosine-β,γ-imidotriphosphate (GMPPNP) and a saturating concentration of magnesium was soaked with ribostamycin and the structure determined at 2.42-Å resolution. In three of four chains of the structure, the neamine rings are clearly resolved in the antibiotic binding site, and the 5-linked ribose ring is directed into the solvent-filled aminoglycoside binding cleft (Fig. 2a). In this structure, the neamine core binds in the same orientation as that in a higher-resolution structure previously determined with the compound (Protein Data Bank [PDB] entry 5IQD). However, an alternate position is resolved for the 5-linked ribose ring, which also shows some variability, indicating mobility (Fig. 2b). We refer to this conformation of ribostamycin as “ribose-up,” while the previously determined conformation is considered “ribose-down.”
FIG 2.
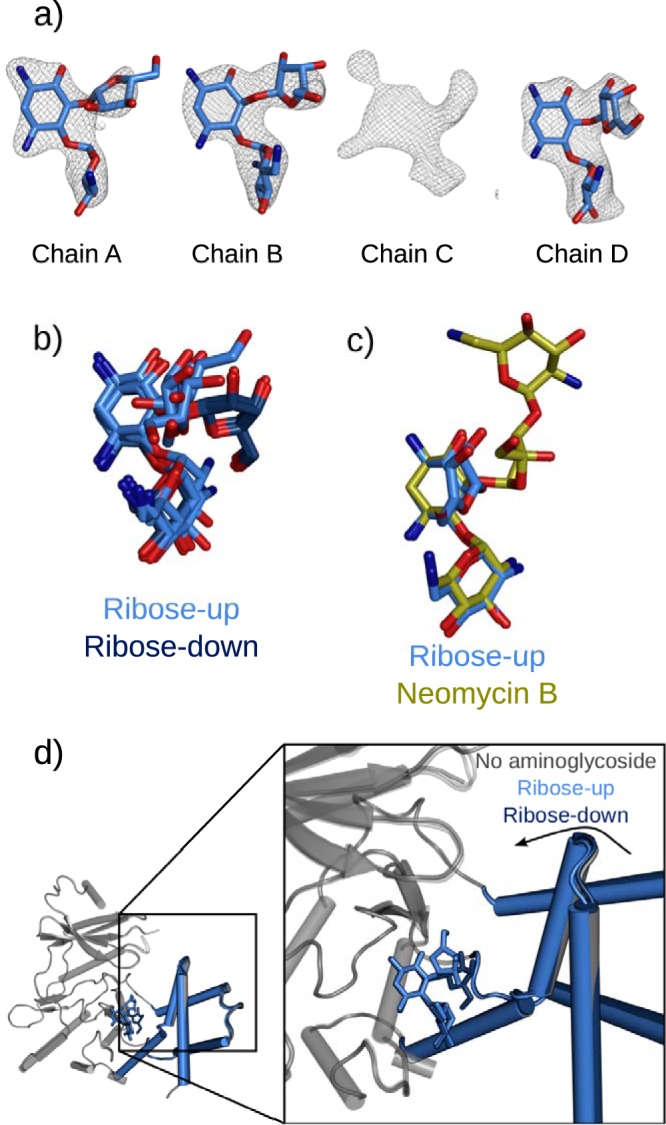
Alternate binding of ribostamycin to the aminoglycoside binding site of APH(2″)-Ia. (a) Difference electron densities following introduction of ribostamycin into APH-GMPPNP crystals. Ribostamycin can be modeled in chains A, B, and D, while chain C contains an ambiguous electron density that suggests multiple overlapping weak binding modes. Maps are Fo-Fc refined difference density maps contoured at a σ level of 2.5. (b) Superimposed ribose-up and ribose-down (PDB entry 5IQD) conformations aligned based upon the APH(2″)-Ia protein core, indicating the differential placement of the ribose ring in these bound forms. (c) Superimposition with bound neomycin (PDB entry 5IQE) indicates that the position of the ribose ring is also distinct from that of the equivalent ring of the larger compound. Molecules are superimposed using the core subdomain of the protein molecule, illustrating that the position of the neamine rings is unchanged in all of these structures. (d) Different structural conformations of APH(2″)-Ia on binding ribostamycin in the ribose-up and ribose-down conformations. In the ribose-up form, the enzyme can further close around the aminoglycoside, similar to when the enzyme is bound to kanamycin, gentamicin, or neomycin, while the ribose-down position holds the enzyme in a more open form.
The ribose-up orientation of ribostamycin resembles that seen when APH(2″)-Ia binds neomycin, a 4,5-disubstituted compound with four linked rings (PBD entry 5IQE). The ribose-up conformation in both of these compounds projects the rings into the solvent-filled aminoglycoside binding cleft (Fig. 2c). In both cases, there is little contact between these rings and the APH(2″)-Ia protein. Almost all interactions are facilitated by the 2-DOS and 4-linked aminohexose rings, while the enzyme accommodates the additional rings in the spacious aminoglycoside binding cleft.
For chain C of this structure, the difference electron density indicates that a compound does interact with the aminoglycoside binding site, but it is not clearly defined enough to model the compound with a single orientation. The resulting electron density is explained instead by several alternative conformations instead of a single well-defined one. Similarly poor electron density was also seen for the equivalent chain of the neomycin-bound form of the enzyme (19), which was refined with high thermal factors reflecting flexibility. This copy of the APH(2″)-Ia protein also has the widest aminoglycoside binding cleft, and in this open conformation, ribostamycin cannot simultaneously contact both the core and helical subdomains. These interactions would facilitate the binding conformation seen in other chains of this structure and in structures soaked with other aminoglycosides. The open APH(2″)-Ia aminoglycoside binding site in this protein chain is fixed by crystal packing and facilitates alternative, weak binding of compounds to the aminoglycoside binding site.
This structure was determined from a crystal grown in 120 mM instead of 100 mM MgCl2 and in 10% instead of 8% glycerol. Changes in protein and ligand conformations between these two structures likely reflect these slightly different conditions, indicating that both are subject to subtle changes in the local chemical environment. Binding of ribostamycin in the ribose-down conformation corresponds to minimal structural changes to APH(2″)-Ia compared to the aminoglycoside-free structure. In the ribose-up conformation, the helical subdomain can close toward the active site of the enzyme in the same manner as that previously seen on introduction of neomycin, gentamicin, or kanamycin (Fig. 2d). This new ribostamycin-bound structure indicates that APH(2″)-Ia is capable of multiple aminoglycoside binding conformations, even within crystals prepared under very similar conditions. This flexibility builds on the intracrystal variation seen between copies of APH(2″)-Ia in a single crystal and the intercrystal variation seen when the protein is exposed to different ligands and mutations (19). Despite the conservation of neamine interactions, the enzyme maintains flexibility that allows it to accommodate the ligand in different orientations.
Lividomycin A is obstructed from canonical binding and interacts weakly with the APH(2″)-Ia site in multiple orientations.
Lividomycin A is the largest aminoglycoside, with 5 covalently linked rings (21). This compound was introduced to crystals of APH(2″)-Ia bound with GMPPNP in the same manner as that for ribostamycin and others (19). However, compared to these smaller aminoglycosides, lividomycin does not show a well-defined bound orientation in the enzyme active site. All four copies of the protein show electron density that reflects a ligand occupying the aminoglycoside binding site, but none is well defined enough to model a single unambiguous conformation. All instances of lividomycin indicate a weakly bound compound in multiple conformations.
As it was the largest aminoglycoside tested, poor binding of lividomycin may reflect the steric and entropic constraints of introducing this large compound into the aminoglycoside binding cleft. Lividomycin also lacks a 6′-amine group, instead carrying a 6′-hydroxyl group, which may also reduce the binding affinity of the neamine core. Despite the lack of a single well-defined bound conformation, we can use the electron density in the antibiotic binding site to infer some of the important interactions that must be taking place. The electron density produced by the introduction of lividomycin is different in each of four chains present in the protein crystal structure (Fig. 3a). This variation indicates that the means by which the aminoglycoside interacts depends considerably on the conformation of the protein, as seen with ribostamycin.
FIG 3.

Lividomycin interaction in the aminoglycoside binding site of APH(2″)-Ia. (a) Lividomycin-soaked APH(2″)-Ia crystals generate considerable difference electron densities in the binding site, which reflect a binding compound, although there are differences in density seen between all three chains, reflecting variability due to crystal packing on the respective protein chains. Maps are Fo-Fc refined aminoglycoside-free difference maps contoured at a σ level of 2.8. (b) Coincident models of lividomycin fragments best explain the electron density observed in chain D of the lividomycin-soaked crystals. (c) In the first of these fragments, neamine-based binding is maintained. (d) In the second case we observed, the rings which are unique to lividomycin instead interact in the neamine binding site.
In one of the four copies of the protein, there is sufficiently defined electron density to infer how lividomycin may bind in this site. The electron density in this chain can be explained by two superimposed fragments of lividomycin (Fig. 3b), each leaving two of the compound's five rings disordered and crystallographically invisible. Both superimposed conformations place the 5″-hydroxyl group near the catalytic center of the enzyme, suggesting that one of these orientations may be responsible for 5″-phosphorylation that has previously been observed for this compound (18). One of these binding modes (Fig. 3c) conserves the neamine-based interactions we have seen for other aminoglycosides, while the other conformation is inverted relative to this canonical binding mode (Fig. 3d). This alternate binding mode uses the distal rings of the compound to bind, including a ring found only in lividomycin, which may be why no other compound has exhibited similar behavior. Collectively, the density produced by introduction of lividomycin to APH(2″)-Ia-GMPPNP crystals indicates that lividomycin has multiple weak interaction modes in the APH(2″)-Ia antibiotic binding site. Some of these conformations may lead to productive phosphorylation and catalytic inactivation of the compound.
APH(2″)-Ia can bind the semisynthetic aminoglycosides dibekacin, amikacin, and arbekacin.
Amikacin, dibekacin, and arbekacin were all soaked into crystals of APH(2″)-Ia. Dibekacin elicited an excellent difference electron density, indicating that the compound was bound in the aminoglycoside binding site of the enzyme (Fig. 4a). This aminoglycoside is a modified kanamycin derivative created by removal of 3′- and 4′-hydroxyl groups from kanamycin B. The chemical differences between dibekacin and kanamycin A do not appear to have a negative impact on the binding of dibekacin to the enzyme, which binds in a conformation very similar to that of kanamycin (Fig. 4b).
FIG 4.

Semisynthetic aminoglycosides observed in the active site of APH(2″)-Ia crystals. All structures are superimposed by alignment of the core regions of APH(2″)-Ia, not by using any atoms from the ligands. (a) Dibekacin binds with unambiguous electron density in all four chains of the APH(2″)-Ia crystal structure. Electron densities are Fo-Fc difference electron densities, contoured at 2.8 σ and carved 2.25 Å from the ligand. (b) Superpositioning of dibekacin and kanamycin molecules in respective structures generated by soaking of the aminoglycoside indicate a nearly perfect coincidence of the structures. (c) Amikacin shows a positive difference electron density in two chains of the APH(2″)-Ia structure upon cocrystallization. Electron densities are Fo-Fc difference electron densities, contoured at 2.0 σ and carved 1.75 Å from the ligand. (d) Superimposition with kanamycin indicates divergent (chain A) and shared (chain D) binding modes in the aminoglycoside binding pocket. (e) Arbekacin shows a positive difference electron density in two chains of the APH(2″)-Ia structure upon cocrystallization. Electron densities are Fo-Fc difference electron densities, contoured at 2.0 σ and carved 1.75 Å from the ligand. (f) Superimposition with kanamycin indicates that arbekacin shares the neamine-based interactions with APH(2″)-Ia but places the 4-linked aminohexose in a different conformation. (g) In the S376N mutant of APH(2″)-Ia, clashes between the antibiotic and the mutant residue prevent the binding of aminoglycosides by use of the neamine rings. (h) The S376N mutant can still support the binding of amikacin via its alternative binding conformation.
In contrast to dibekacin's well-defined electron density, when the same experiment was carried out with amikacin and arbekacin, no difference density was observed. Dibekacin and kanamycin A soak readily and bind to the aminoglycoside binding site of the enzyme, but their N1-substituted counterparts, arbekacin and amikacin, do not, indicating that the N1 substitution blocks binding to the enzyme. The steric clash that results from this modification interferes with the neamine-based binding mode of the enzyme.
These soaking experiments were unsuccessful, but we made an important observation. Addition of most aminoglycosides to actively growing APH(2″)-Ia crystals halts crystal growth, which has always precluded the cocrystallization of APH(2″)-Ia with aminoglycosides. However, crystal growth continued after addition of amikacin, which indicated that cocrystallization with N1-substituted aminoglycosides might be possible. Crystals of APH(2″)-Ia prepared using a saturating concentration of magnesium, 3 mM GMPPNP, and 3 mM amikacin or arbekacin were grown and harvested, and full data sets were collected for these crystals.
In both cases, aminoglycosides could be modeled in two of four chains, with various levels of quality reflecting a variability in binding with the protein conformation. In amikacin cocrystals, the compound was seen in two distinct binding modes, modeled with 50% and 60% occupancy (Fig. 4c). In chain D, amikacin resembles unsubstituted kanamycin (Fig. 4d), with neamine rings in similar positions and the 6-linked ring near the active site. However, in this orientation, the AHB group adopts an unfavorable configuration to avoid generating clashes with the protein. In chain A of this structure, we identified a novel mode of binding to the aminoglycoside binding pocket. The aminoglycoside contacts residues near the active site but also a section of the antibiotic binding pocket that is not involved with binding other compounds. The neamine binding pocket that the enzyme uses to bind most compounds remains largely unoccupied. In this conformation, the 4- and 6-linked rings occupy distinct positions compared to those of the equivalent rings of kanamycin. However, this conformation still places the 2″-OH group in close proximity to the reactive center and the activated GMPPNP phosphate group, which means that modification of amikacin at the 2″ position remains possible despite this atypical binding mode.
In contrast to amikacin, arbekacin shows a well-defined conformation bound in one enzyme active site (Fig. 4e). Perhaps surprisingly, the compound still maintains most neamine-like interactions. The 2-DOS ring shifts slightly, and the N1-AHB group is directed away from the enzyme and into disordered solvent. In order to make room for this N1 modification, the 4-linked ring is twisted away from the active site (Fig. 4f), in a position where it rests against the helical subdomain of the enzyme. In this position, the ring cannot be modified, but it remains possible that when it is not restricted by the crystal lattice, the ring shifts to a position where it can productively be modified by the enzymatic center.
These structures illustrate that while N1-substituted aminoglycosides do not bind as easily as unsubstituted compounds in the antibiotic binding pocket of APH(2″)-Ia, they still show a capacity to interact with the enzyme active site. It is thus worth investigating how these compounds might interact with mutant enzymes that have improved activity toward these compounds.
The structure of S376N mutant APH(2″)-Ia suggests a possible means of increased modification of N1-substituted aminoglycosides.
The clinically identified S376N mutation of APH(2″)-Ia has been identified to elevate resistance to N1-substituted aminoglycosides but to eliminate modification of nonsubstituted compounds (20). We generated this mutant and purified it prior to determination of its structure (PDB entry 6CH4) to identify structural changes that take place that would facilitate modification of these compounds.
Mutation of serine 376 to asparagine does not lead to substantial rearrangements in the aminoglycoside binding site. In fact, the addition of the larger asparagine residue in place of serine 376 creates an obstruction that prevents binding in the neamine binding pocket (Fig. 4g). As a result, any compounds that bind using the neamine rings are blocked from the antibiotic binding site of APH(2″)-Ia. However, this mutation remains compatible with one of the alternate binding modes of amikacin, which does not use this site to interact with the enzyme (Fig. 4h). If the S376N mutation favors this conformation, it may improve binding of N1-substituted compounds. This alternately bound antibiotic could then exploit a weak promiscuous activity of the enzyme on this alternate conformation, similar to that of 4,5-disubstituted compounds, to generate low levels of antibiotic modification and resistance.
DISCUSSION
APH(2″)-Ia conserves a ribosome mimic binding site to bind neamine rings.
The structures reported here recapitulate that for small, unobstructed aminoglycosides, the predominant mode of interaction with APH(2″)-Ia is a conserved binding pocket for the neamine rings. In addition to previously published structures with ribostamycin, neomycin, kanamycin, and gentamicin (19), these additional structures of ribostamycin and dibekacin continue to conserve a binding mode that preserves the interaction of these rings with the enzyme. These new structures reaffirm that besides binding these rings, the enzyme tolerates a great degree of conformational freedom in the 5- or 6-linked rings, even responding to slightly different crystal growth conditions.
Amikacin and arbekacin, which carry an N1-modified group, are mostly excluded from this mode of binding. However, under conditions that appear to have become accessible through cocrystallization, both compounds showed that they are capable of binding to at least one conformation of the enzyme. We previously observed that one protein chain of APH(2″)-Ia shows considerable freedom of motion within crystals, and this freedom appears to allow the adoption of conformations that can weakly facilitate binding of N1-substituted aminoglycosides.
The binding of these structurally distinct compounds by use of the neamine rings reflects a degree of target mimicry by the enzyme, as the neamine rings are used for binding to the ribosome (4). The neamine rings are the most important components of the antibiotic for its bacteriotoxic effect, so the enzyme has been selected toward the structural motif of the antibiotic that carries the most functional importance. A similar strategy is adopted by other broad-spectrum aminoglycoside resistance enzymes, which also conserve neamine-based binding to the compound but tolerate the variable rings through different structural and thermodynamic strategies. While APH(3′)-IIIa binds variable rings through loop rearrangement (12), AAC(6′)-Ib uses structured solvent molecules to compensate for differences in aminoglycoside structure (11), and APH(2″)-Ia uses a large cleft filled with disordered solvent. The lack of compensating interactions with the variable rings indicates that the binding of the neamine portion of the compound must be remarkably strong.
Binding of an aminoglycoside to APH(2″)-Ia in turn has effects on the protein conformation. An important hinge point in this motion appears to be the linkage between tyrosine 448, which stacks with the 4-linked aminohexose of the compound, and proline 449, which creates a large kink in the α9 helix of the helical subdomain. This hinge motion is linked to aminoglycoside binding and conformation, as evidenced by the different degrees of hinge movement with ribostamycin bound in the ribose-up and ribose-down conformations. This informs the design of new aminoglycosides, as antibiotic modifications that alter the structural changes of the enzyme may lead to aminoglycosides that are less effectively inactivated by wild-type APH(2″)-Ia.
Noncanonical aminoglycoside binding to APH(2″)-Ia.
APH(2″)-Ia binds the neamine rings of a compound if possible, and it accommodates binding of the rest of the molecule through its spacious cavity and structural changes. This seems to be true for all unobstructed aminoglycosides but is more difficult for large aminoglycosides or compounds with an N1 substitution. In the case of lividomycin, crystals soaked with the aminoglycoside still exhibited positive residual electron density in the aminoglycoside binding site, indicating weak binding interactions. This electron density hints at possible means by which the enzyme might phosphorylate this compound at noncanonical positions (Fig. 3a).
Daigle et al. (18) found that lividomycin is modified on the 5″-OH group of the ribose ring. We observed electron density consistent with placement of this group at the reactive center of the enzyme, most convincingly in a binding mode which uses the fifth, unique ring of lividomycin. In the same study, neomycin was modified on the 3′ and 3‴ sites, which is not compatible with this binding mode or a previously observed bound conformation of neomycin (19). Weak alternate binding modes may also exist for this compound, but they are not visible crystallographically because the neamine-based binding of the compound predominates. This promiscuity of binding and modification of 4,5-disubstituted compounds aligns with another early finding for this enzyme, i.e., that it can also phosphorylate structurally unrelated molecules, such as peptides (22), albeit at low rates.
The promiscuous binding and modification of aminoglycosides by APH(2″)-Ia echo the results of studies of unrelated aminoglycoside-modifying enzymes. Other resistance enzymes also show a defined regiospecificity but have leaky activity toward other compounds or toward binding in alternative conformations. AAC(1) (45), ANT(4′) (24), and Eis (46) can all modify substrates at multiple positions, indicating that they are capable of binding to compounds in multiple ways. APH(3′)-IIIa modifies 4,5-disubstituted compounds at the 5″ position as well as the canonical 3′ position (23). ANT(4′) shows broad specificity of aminoglycoside binding, thought to be an intrinsic feature that increases enzyme activity toward as many compounds as possible (24). The nomenclature of these enzymes restricts our categorization to single activities, but biochemical analysis indicates that they are less rigidly defined and more adaptable in their activities.
APH(2″)-Ia binding to nonnatural aminoglycoside substrates.
Unlike many antibiotics, amikacin and arbekacin are semisynthetic aminoglycosides which did not exist in the natural environment prior to their invention and development. As a result, it has been expected that antibiotic resistance enzymes will not have developed an effective means of biochemical resistance to these compounds. However, multiple enzymes have been observed to modify amikacin, albeit at lower rates than those against native substrates (25, 26).
The same has also been true of APH(2″)-Ia. Ever since the initial discovery and characterization of APH(2″)-Ia, the phosphorylation of amikacin by APH(2″)-Ia has been observed (27–29). Despite this activity, strains carrying these resistance factors are active at a low enough rate that they are typically considered amikacin susceptible. The cocrystallization results we report here are commensurate with a low level of modification, as the binding of amikacin is weak and not consistent with efficient modification of the antibiotic. This is also consistent with high KM values measured for N1-substituted compounds in APH(2″)-Ia phosphotransfer assays (16).
The mode of aminoglycoside binding we observe in chain A of the amikacin cocrystal structure completely forgoes interactions with the 2-DOS binding pocket of the enzyme. This suspends the 6-linked aminohexose ring in the center of the enzyme cleft. The lack of contact with the conserved binding pocket may help to explain the observation of a clinical point mutant of this enzyme, the S376N mutant, which is linked to elevated rates of arbekacin modification in clinical strains (20). In this strain, resistance to arbekacin is elevated, while that to gentamicin is reduced, as seen through MIC measurements. S376 is normally involved in binding of the neamine rings of natural 4,5-disubstituted and 4,6-disubstituted compounds. The S376N mutation exchanges this serine for a larger asparagine residue, which introduces a clash with any bound aminoglycosides. This mutation obviously disrupts normal aminoglycoside binding by placing a larger residue into the aminoglycoside binding pocket. However, the enzyme also modifies arbekacin more effectively than the wild-type enzyme does. Based on the structural analysis of this mutant and the bound N1-substituted aminoglycosides in the binding site, we suggest that this mutation promotes the adoption of an alternate binding mode for N1-substituted aminoglycosides, which elevates the rate at which they are modified. This leads to elevated arbekacin resistance by promoting this weak background activity of the enzyme.
Emergence of new activities in aminoglycoside-modifying enzymes.
In this study, we have highlighted structural evidence for promiscuous aminoglycoside binding and inactivating activities of APH(2″)-Ia. The enzyme has maintained an ancestral mode of antibiotic binding but also possesses the capacity for adaptation toward the binding of aminoglycosides for which it has not been optimized catalytically. This is important as we consider the role of this resistance factor in the greater context of all aminoglycoside-modifying enzymes (30).
An open and ongoing question in the study of antibiotic resistance enzymes is whether any given enzyme is an ancient, catalytically dedicated antibiotic resistance factor or the result of a de novo emergence of function. APH(2″)-Ia appears to be a dedicated resistance factor, optimized toward phosphorylation of 4,6-disubstituted aminoglycosides at the 2″ site, but these studies indicate that it may still be possible for de novo features to emerge in the enzyme.
Resistance to N1-substituted aminoglycosides by phosphorylation reflects the potential for emergence of a new antibiotic-phosphorylating activity in the enzyme. The interactions of APH(2″)-Ia with lividomycin and with N1-substituted semisynthetic compounds indicate a foothold of weak binding that may lead to phosphorylation at low rates. With sufficient selective pressure, the enzyme may adapt and elaborate upon this activity, resulting in new, potentially clinically important antibiotic resistance. This echoes the study of Holbrook and Garneau-Tsodikova on the expansion of the other domain of this bifunctional enzyme through mutation and truncation that leads to increased acetylation of arbekacin (31). As in their work, we observed that the mutated protein does not undergo changes that lead to a substantive difference in mechanism but simply exhibits weak-affinity alternative binding modes. While we do not observe any evidence to suggest alternative folding as posited by these researchers, an effect that we do recapitulate is that the modest chemical changes between amikacin and arbekacin result in substantial differences in their ability to bind and be modified by the resistance enzymes.
The development of new function in enzymes remains an area of considerable study in evolutionary biochemistry. A leading model for the development of new function in enzymes involves the amplification of genes that express low-level promiscuous activities, which can then develop mutations that alter and refine the new activity (32). This model is consistent with observed patterns of expression of N1-aminoglycoside modification by APH(2″)-Ia. Some resistance mutations greatly increase the expression of the gene (33), and in one case, an amplified and mutated gene was observed alongside the wild type, as a paralog whose function had drifted.
Enzymes with promiscuous activity are common, as enzymes typically evolve to be “good enough” at their selected function and not perfect to the exclusion of other activities (34). Promiscuous detoxification activity may be advantageous on encountering a novel antibiotic, so the enzymes may in fact have some promiscuous activity by selection, not just as a by-product of their primary role. In addition, weak background activities can provide a starting point upon which an enzyme can elaborate and optimize its catalytic activity toward new functions.
Like the unrelated acetyltransferase enzyme AAC(6′)-Ib, which has expanded its aminoglycoside-modifying range (35) and in one case even developed resistance to another class of antibiotics altogether (36), APH(2″)-Ia may be a scaffold upon which mutation produces new antibiotic-deactivating activities. In vitro studies have probed for the development of resistance to N1-substituted aminoglycosides in other APH(2″) enzymes (37, 38). As with the S376N mutant, in both cases the contribution of mutant residues to elevated N1-substituted aminoglycoside resistance is not easily rationalized. Further studies probing the structure and dynamics of aminoglycoside binding to these enzymes will be greatly informative in elucidating the mechanisms by which these mutations expand the substrate range of the enzyme.
The S376N mutation of APH(2″)-Ia may be the first step in the development of a new, highly active amikacin-modifying activity of APH(2″)-Ia. While this mutation does not make APH(2″)-Ia highly active toward N1-substituted compounds, it has enough activity to have clinically manifest effects (20). It is likely that additional mutations would improve this activity enough to create a much more effective resistance factor for N1-substituted aminoglycosides. Further exploration of the promiscuity in binding of APH(2″)-Ia and its mutants is necessary to anticipate and counter emergent antibiotic resistance activities.
Conclusions.
APH(2″)-Ia preferentially binds 4,5-disubstituted and 4,6-disubstituted aminoglycosides by using the neamine rings in a conserved pocket. Additional rings linked to this core neamine element are accommodated in various orientations in the spacious binding cleft of the enzyme. In cases where this means of binding is obstructed, the enzyme can bind compounds in alternative binding modes, though weakly. Some of these alternative binding modes may facilitate low levels of phosphorylation on additional sites of the aminoglycoside, such as the 5″ position of lividomycin.
Structures of the enzyme cocrystallized with the N1-substituted aminoglycosides amikacin and arbekacin indicate that these nonnatural aminoglycosides can still be bound by the enzyme and explain the low-level background phosphorylation at the 2″ position. These binding interactions may provide a means for the enzyme to develop mutations that shift the resistance spectrum toward new compounds, such as the S376N mutant with elevated arbekacin phosphorylation. Further mutations may also alter or expand the resistance profile conferred by APH(2″)-Ia, emphasizing the importance of tracking the sequences of resistance factors, not solely their presence in pathogenic bacteria.
MATERIALS AND METHODS
Protein production and purification.
The APH(2″)-Ia enzyme was produced and purified as previously described (19). To briefly summarize, the 305-residue, 35-kDa protein domain was produced in Escherichia coli by use of a pET-22b vector in autoinduction medium (39). Cells were harvested by centrifugation, flash frozen, and stored at −20°C until needed. A pellet corresponding to 125 ml of culture was resuspended in 40 ml 25 mM HEPES, pH 7.5, and 25 mM NaCl. These cells were subjected to ultrasonication followed by centrifugation to remove cell debris. The clarified lysate was passed through a 0.22-μm syringe filter and loaded onto a kanamycin-agarose resin prepared by incubating Affi-Gel 15 (Bio-Rad) with kanamycin A (Sigma-Aldrich). Elution from this column in a gradient of 25 to 500 mM NaCl recovered the APH(2″)-Ia enzyme as tracked by SDS-PAGE.
Fractions of eluate containing the APH(2″)-Ia enzyme were subjected to size exclusion chromatography on a Superdex 75 26/300 column (GE Healthcare) preequilibrated with 5% (wt/vol) glycerol and 10 mM HEPES, pH 7.5. The eluate from this kanamycin-agarose column was concentrated to 5 ml in an Amicon concentrator (Millipore) with a 30-kDa cutoff. The concentrated sample was loaded onto the size exclusion column, with most contaminating material eluted in the void volume, and a peak corresponding to the APH(2″)-Ia enzyme was confirmed to contain the enzyme by SDS-PAGE. This protein was concentrated in a new Amicon concentrator to 12 to 15 mg/ml, flash frozen, and stored at −80°C until ready for use.
The protein used to generate crystals bound with dibekacin and arbekacin was also further purified using an intermediate purification step on a ReSource Q resin (GE Healthcare), wherein the kanamycin-agarose eluate was exchanged into 25 mM NaCl and loaded onto the ion-exchange column, followed by elution in a 25 mM to 1 M gradient of NaCl. The protein eluted in a single peak from this column and was further purified by size exclusion chromatography as with previous protein preparations. This step removed additional impurities, simplifying the crystallization process, but the protein recovered was otherwise equivalent to that purified without this step.
Generation and purification of the S376N mutant of APH(2″)-Ia.
The S376N mutant of APH(2″)-Ia was generated using site-directed mutagenesis. Primers containing the sequences 5′-GTGTTTATGCCATAATGATTTTAATTGTAATCATCTATTGTTAGATGGC and 5′-GCCATCTAACAATAGATGATTACAATTAAAATCATTATGGCATAAACAC (the mutation is shown in italics) were purchased from BioCorp, Inc. (Montreal). These primers were prepared in molecular biology-grade water at a concentration of 0.1 ng/μl. Diluted primers were used to prepare an amplification reaction mixture in 50 μl with 0.05 ng/μl template DNA, i.e., the pET-22b-APH(2″)-Ia expression plasmid. This reaction mixture was prepared with a 100 μM concentration of each deoxynucleoside triphosphate (dNTP) and the PfuX7 enzyme (40), which was prepared in buffers described by Nørholm by using nickel affinity chromatography from an overnight autoinduction culture (39). The reaction was run for 30 cycles of alternating heat in a thermocycler, starting with 2 min of denaturation at 95°C and 15 min of annealing and extension at 72°C. On completion of this protocol, the amplification product was easily visible on an agarose gel and was transformed into competent E. coli DH5α cells and grown on ampicillin-agar. Single colonies were grown in Luria-Bertani broth, and the plasmids were extracted and purified by alkaline lysis. Plasmids were sequenced at the Genome Québec Innovation Centre to confirm the successful generation of mutant plasmids.
The S376N mutant enzyme was prepared and purified by the same protocol as that used for wild-type APH(2″)-Ia. While the protein was purified using the same affinity resin and buffers, the yield was considerably lower than those of the wild-type protein and other mutants, which is likely linked to the active site mutation that altered the binding site of the enzyme. This mutation reduced the ability of the protein to bind in the first aminoglycoside affinity step in the purification protocol.
Crystallization and processing of APH(2″)-Ia crystals.
Crystals of APH(2″)-Ia with soaked aminoglycosides were grown by use of similar procedures, with minor modifications as the crystal growth protocol was refined. The ribostamycin-soaked crystal was prepared by growing crystals of APH-GMPPNP in 100 mM HEPES, pH 7.5, 120 mM MgCl2, 10% polyethylene glycol (PEG) 3350, and 10% glycerol in a 1-μl-plus-1-μl hanging-drop crystallization experiment (41). Crystals formed spontaneously in the drop amid a considerable amount of precipitate. One millimolar ribostamycin sulfate was prepared in reservoir solution, and 1 μl of this ligand solution was added to the drop containing protein crystals. Following 24 h for soaking and equilibration, a crystal was mounted and flash-frozen for diffraction.
The lividomycin-soaked crystal was prepared by cocrystallizing the enzyme with GMPPNP in 100 mM HEPES, pH 7.5, 100 mM MgCl2, 10% PEG 3350, and 8% glycerol. Preincubation of the enzyme with GMPPNP (3 mM) and magnesium chloride (6 mM) and mixture of this protein-ligand mix 2:1 with a reservoir solution of 10% PEG 3350, 8% glycerol, 100 mM MgCl2, and 50 mM HEPES, pH 7.5, were performed overnight. This solution had a considerable amount of precipitate and was recovered, filtered through a 0.1-μm membrane, assembled in a new hanging-drop crystallization apparatus, and streak seeded to nucleate new crystal growth. Following the growth of APH-GMPPNP crystals, lividomycin was prepared at 2 mM in mother liquor solution and introduced to the protein drop. The crystal was equilibrated for 3 days prior to harvesting, flash cooling, and screening for X-ray diffraction analysis.
The dibekacin-soaked crystal of APH(2″)-Ia-GMPPNP was also produced by the same means, but it did not require a preincubation step because the protein used to generate the parent crystals had undergone an additional purification step. The APH(2″)-Ia protein was mixed 1:1 with a reservoir solution of 10% PEG 3350, 8% glycerol, 100 mM MgCl2, and 50 mM HEPES, pH 7.5, and streak seeded to nucleate crystals. Following crystal growth, 2 mM dibekacin sulfate in mother liquor was added and allowed to equilibrate with the crystal before mounting, cryo-cooling, and diffraction analysis.
Soaking experiments with amikacin indicated that N1-substituted compounds did not halt crystal growth and thus might be added directly to the crystallization solutions. Cocrystallization of the enzyme with amikacin was carried out by mixing the protein at ∼13 mg/ml with 3 mM GMPPNP, 6 mM MgCl2, and 3 mM amikacin sulfate and setting up the tray by the same means as that for the GMPPNP-bound form. Crystals of this enzyme grew over a week and could be harvested directly, flash cooled, and mounted for diffraction analysis. The same process was repeated to recover crystals with bound arbekacin.
Lastly, the S376N mutant of APH(2″)-Ia was crystallized by mixing of the purified mutant protein 1:1 with reservoir solution, seeding, and crystal growth over ∼2 weeks. A crystal of this APH(2″)-Ia S376N-GMPPNP complex was harvested and cryo-cooled and diffraction data collected, although the longer crystal growth time led to some degradation of the GMPPNP ligand in this sample, resulting in reduced electron density for the triphosphate group in this crystal.
Data collection, model building, and analysis.
Data sets for all six crystal structures were collected at the CMCF diffraction facility at the Canadian Light Source in Saskatoon, Canada. All were collected at cryogenic temperatures and 0.9795-Å-wavelength radiation. Complete data sets for all six crystals were collected to resolutions between 2.2 to 2.6, and the data were integrated using iMOSFLM (42). These data were merged and scaled using AIMLESS (43) before proceeding to refinement in REFMAC5 (43) and model building in Coot (44). Structures were all determined by Fourier synthesis, using a reduced GMPPNP-bound structure of APH(2″)-Ia (PDB entry 5IQA) as the starting model, followed by manual building of flexible loops and ligands. Restraints for lividomycin, amikacin, dibekacin, and arbekacin were generated using GRADE (Global Phasing, Ltd.). A common set of translation-libration-screw refinement (TLS) groups was defined for all structures and refined in parallel. Data collection and structural statistics are provided in Tables 1 and 2, respectively.
TABLE 1.
Data collection statistics for APH(2″)-Ia and APH(2″)-Ia S376N data setsa
| Parameter | Value or description |
|||||
|---|---|---|---|---|---|---|
| WT APH-GMPPNP-ribostamycin (alternate) | WT APH-GMPPNP- lividomycin | WT APH-GMPPNP- dibekacin | WT APH-GMPPNP- amikacin | WT APH-GMPPNP- arbekacin | APH-S376N-GMPPNP | |
| PDB ID | 6C5U | 6CEY | 6CAV | 6CGD | 6CGG | 6CH4 |
| Data collection statistics | ||||||
| Space group | P21 | P21 | P21 | P21 | P21 | P21 |
| a, b, c (Å) | 90.2, 100.3, 94.0 | 90.2, 100.2, 94.0 | 89.9, 98.3, 93.2 | 90.1, 100.2, 94.0 | 90.3, 99.8, 93.5 | 90.4, 99.9, 93.2 |
| α, β, γ (°) | 90, 105.3, 90 | 90, 105.2, 90 | 90, 105.2, 90 | 90, 105.1, 90 | 90, 105.4, 90 | 90, 105.2, 90 |
| Resolution (Å) | 90.71–2.42 (2.48–2.42) | 59.07–2.40 (2.46–2.40) | 98.34–2.60 (2.68–2.60) | 50.11–2.20 (2.24–2.20) | 99.78–2.40 (2.46–2.40) | 58.89–2.30 (2.35–2.30) |
| CC1/2 | 0.998 (0.173) | 0.988 (0.287) | 0.978 (0.358) | 0.991 (0.557) | 0.992 (0.383) | 0.987 (0.434) |
| Rmerge | 0.079 (1.543) | 0.110 (1.345) | 0.162 (1.072) | 0.115 (1.022) | 0.115 (1.024) | 0.133 (0.965) |
| I/σI | 8.5 (1.0) | 10.0 (1.8) | 4.7 (1.2) | 10.5 (1.8) | 6.1 (1.0) | 9.7 (1.9) |
| Completeness (%) | 100.0 (99.8) | 100.0 (100.0) | 100.0 (99.8) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) |
| Multiplicity | 3.8 (3.6) | 4.2 (4.2) | 3.7 (3.5) | 4.3 (4.3) | 3.8 (3.7) | 4.3 (4.3) |
WT, wild type; CC1/2, half-dataset correlation coefficient.
TABLE 2.
Structural statistics for models of APH(2″)-Ia with aminoglycosides and of APH(2″)-Ia S376N
| Parameter | Value or description |
|||||
|---|---|---|---|---|---|---|
| WT APH-GMPPNP-ribostamycin (alternate) | WT APH-GMPPNP- lividomycin | WT APH-GMPPNP- dibekacin | WT APH-GMPPNP- amikacin | WT APH-GMPPNP- arbekacin | APH-S376N- GMPPNP | |
| Resolution | 2.42 | 2.40 | 2.60 | 2.20 | 2.40 | 2.30 |
| No. of unique reflections | 41,701 | 63,273 | 28,436 | 77,748 | 62,642 | 75,134 |
| Rwork/Rfree | 0.1803/0.2272 | 0.1714/0.2160 | 0.1918/0.2440 | 0.1719/0.2089 | 0.1780/0.2233 | 0.1701/0.2097 |
| No. of atoms | ||||||
| Protein | 9,600 | 9,588 | 9,486 | 9,812 | 9,627 | 9,632 |
| Ligands | 232 | 220 | 259 | 237 | 245 | 134 |
| Water | 513 | 541 | 503 | 634 | 716 | 579 |
| Mean B factors | ||||||
| Protein | 73.9 | 61.5 | 63.8 | 51.7 | 59.5 | 53.4 |
| Ligands | 81.4 | 63.5 | 57.8 | 50.7 | 57.0 | 56.4 |
| Water | 69.1 | 59.8 | 55.2 | 50.9 | 57.3 | 53.3 |
| RMSDa | ||||||
| Bond lengths (Å) | 0.0179 | 0.0208 | 0.0171 | 0.0156 | 0.0183 | 0.0211 |
| Bond angles (°) | 1.846 | 2.0378 | 1.8362 | 1.6584 | 1.910 | 2.0143 |
| Ramachandran values | ||||||
| % favored | 97.98 | 97.13 | 98.05 | 97.01 | 97.57 | 97.23 |
| % allowed | 1.67 | 2.44 | 1.24 | 2.13 | 1.83 | 2.25 |
| % outlier | 0.35 | 0.44 | 0.71 | 0.85 | 0.61 | 0.52 |
Root mean square deviation.
Figures were generated using the PyMOL molecular graphics system, version 1.3 (Schrödinger, LLC), and Inkscape (https://inkscape.org/).
Accession number(s).
The structures described in this report have been deposited in the Protein Data Bank under accession codes 6C5U (APH-GMPPNP-ribostamycin-2), 6CEY (APH-GMPPNP-lividomycin A), 6CGD (APH-GMPPNP-amikacin), 6CGG (APH-GMPPNP-arbekacin), 6CAV (APH-GMPPNP-dibekacin), and 6CH4 (APH S376N-GMPPNP).
ACKNOWLEDGMENTS
The arbekacin and dibekacin used in this study were generously supplied by Meiji Seika Pharma Co., Ltd. We acknowledge the excellent assistance provided by the staff of the CMCF facility at the Canadian Light Source in Saskatoon, Canada, in collecting the diffraction data for this study.
This research was funded by CIHR operating grant MOP-13107. S.J.C. was a recipient of a CIHR Banting and Best graduate fellowship and was previously also supported by the STIHR training program in chemical biology and the NSERC CGS-M program.
We declare that we have no conflicts of interest.
S.J.C. designed and conducted experiments, processed data, and wrote the manuscript. A.M.B. supervised the project and secured funding for this research.
REFERENCES
- 1.Wright GD, Berghuis AM, Mobashery S. 1998. Aminoglycoside antibiotics. Structures, functions, and resistance. Adv Exp Med Biol 456:27–69. doi: 10.1007/978-1-4615-4897-3_4. [DOI] [PubMed] [Google Scholar]
- 2.Wright GD, Berghuis AM. 2007. Structural aspects of aminoglycoside-modifying enzymes, p 21–33. In Bonomo RA, Tolmasky M (ed), Enzyme-mediated resistance to antibiotics. ASM Press, Washington, DC. [Google Scholar]
- 3.Andac CA, Stringfellow TC, Hornemann U, Noyanalpan N. 2011. NMR and amber analysis of the neamine pharmacophore for the design of novel aminoglycoside antibiotics. Bioorg Chem 39:28–41. doi: 10.1016/j.bioorg.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 4.François B, Russell RJM, Murray JB, Aboul-ela F, Masquida B, Vicens Q, Westhof E. 2005. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res 33:5677–5690. doi: 10.1093/nar/gki862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kondo S, Hotta K. 1999. Semisynthetic aminoglycoside antibiotics: development and enzymatic modifications. J Infect Chemother 5:1–9. doi: 10.1007/s101560050001. [DOI] [PubMed] [Google Scholar]
- 6.Kawaguchi H, Naito T, Nakagawa S, Fujisawa K. 1972. BB-K8, a new semisynthetic aminoglycoside antibiotic. J Antibiot (Tokyo) 25:695–708. doi: 10.7164/antibiotics.25.695. [DOI] [PubMed] [Google Scholar]
- 7.Kondo S, Iinuma K, Yamamoto H, Maeda K, Umezawa H. 1973. Syntheses of 1-n-(S)-4-amino-2-hydroxybutyryl)-kanamycin B and -3′,4′-dideoxykanamycin B active against kanamycin-resistant bacteria. J Antibiot (Tokyo) 26:412–415. doi: 10.7164/antibiotics.26.412. [DOI] [PubMed] [Google Scholar]
- 8.Hotta K, Kondo S. 2018. Kanamycin and its derivative, arbekacin: significance and impact. J Antibiot (Tokyo) 71:417–424. doi: 10.1038/s41429-017-0017-8. [DOI] [PubMed] [Google Scholar]
- 9.Ramirez MS, Tolmasky ME. 2017. Amikacin: uses, resistance, and prospects for inhibition. Molecules 22:E2267. doi: 10.3390/molecules22122267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kondo J, François B, Russell RJM, Murray JB, Westhof E. 2006. Crystal structure of the bacterial ribosomal decoding site complexed with amikacin containing the gamma-amino-alpha-hydroxybutyryl (haba) group. Biochimie 88:1027–1031. doi: 10.1016/j.biochi.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 11.Vetting MW, Park CH, Hegde SS, Jacoby GA, Hooper DC, Blanchard JS. 2008. Mechanistic and structural analysis of aminoglycoside N-acetyltransferase AAC(6′)-Ib and its bifunctional, fluoroquinolone-active AAC(6′)-Ib-cr variant. Biochemistry 47:9825–9835. doi: 10.1021/bi800664x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fong DH, Berghuis AM. 2002. Substrate promiscuity of an aminoglycoside antibiotic resistance enzyme via target mimicry. EMBO J 21:2323–2331. doi: 10.1093/emboj/21.10.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vetting MW, Hegde SS, Javid-Majd F, Blanchard JS, Roderick SL. 2002. Aminoglycoside 2′-N-acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat Struct Biol 9:653–658. doi: 10.1038/nsb830. [DOI] [PubMed] [Google Scholar]
- 14.Caldwell SJ, Berghuis AM. 2012. Small-angle X-ray scattering analysis of the bifunctional antibiotic resistance enzyme aminoglycoside (6′) acetyltransferase-Ie/aminoglycoside (2″) phosphotransferase-Ia reveals a rigid solution structure. Antimicrob Agents Chemother 56:1899–1906. doi: 10.1128/AAC.06378-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith CA, Toth M, Weiss TM, Frase H, Vakulenko SB. 2014. Structure of the bifunctional aminoglycoside-resistance enzyme AAC(6′)-Ie-APH(2″)-Ia revealed by crystallographic and small-angle X-ray scattering analysis. Acta Crystallogr D Biol Crystallogr 70:2754–2764. doi: 10.1107/S1399004714017635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frase H, Toth M, Vakulenko SB. 2012. Revisiting the nucleotide and aminoglycoside substrate specificity of the bifunctional aminoglycoside acetyltransferase(6′)-Ie/aminoglycoside phosphotransferase(2″)-Ia enzyme. J Biol Chem 287:43262–43269. doi: 10.1074/jbc.M112.416453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kondo S, Tamura A, Gomi S, Ikeda Y, Takeuchi T, Mitsuhashi S. 1993. Structures of enzymatically modified products of arbekacin by methicillin-resistant Staphylococcus aureus. J Antibiot (Tokyo) 46:310–315. doi: 10.7164/antibiotics.46.310. [DOI] [PubMed] [Google Scholar]
- 18.Daigle DM, Hughes DW, Wright GD. 1999. Prodigious substrate specificity of AAC(6′)-APH(2″), an aminoglycoside antibiotic resistance determinant in enterococci and staphylococci. Chem Biol 6:99–110. doi: 10.1016/S1074-5521(99)80006-4. [DOI] [PubMed] [Google Scholar]
- 19.Caldwell SJ, Huang Y, Berghuis AM. 2016. Antibiotic binding drives catalytic activation of aminoglycoside kinase APH(2″)-Ia. Structure 24:935–945. doi: 10.1016/j.str.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 20.Ishino K, Ishikawa J, Ikeda Y, Hotta K. 2004. Characterization of a bifunctional aminoglycoside-modifying enzyme with novel substrate specificity and its gene from a clinical isolate of methicillin-resistant Staphylococcus aureus with high arbekacin resistance. J Antibiot (Tokyo) 57:679–686. doi: 10.7164/antibiotics.57.679. [DOI] [PubMed] [Google Scholar]
- 21.Machiyama N. 1971. Mechanism of action of lividomycin A, a new aminoglycosidic antibiotic. J Antibiot (Tokyo) 24:706–707. doi: 10.7164/antibiotics.24.706. [DOI] [PubMed] [Google Scholar]
- 22.Daigle DM, McKay GA, Thompson PR, Wright GD. 1999. Aminoglycoside antibiotic phosphotransferases are also serine protein kinases. Chem Biol 6:11–18. doi: 10.1016/S1074-5521(99)80016-7. [DOI] [PubMed] [Google Scholar]
- 23.Thompson PR, Hughes DW, Wright GD. 1996. Regiospecificity of aminoglycoside phosphotransferase from enterococci and staphylococci (APH(3′)-IIIa). Biochemistry 35:8686–8695. doi: 10.1021/bi960389w. [DOI] [PubMed] [Google Scholar]
- 24.Matesanz R, Diaz JF, Corzana F, Santana AG, Bastida A, Asensio JL. 2012. Multiple keys for a single lock: the unusual structural plasticity of the nucleotidyltransferase (4′)/kanamycin complex. Chemistry 18:2875–2889. doi: 10.1002/chem.201101888. [DOI] [PubMed] [Google Scholar]
- 25.Courvalin P, Davies J. 1977. Plasmid-mediated aminoglycoside phosphotransferase of broad substrate range that phosphorylates amikacin. Antimicrob Agents Chemother 11:619–624. doi: 10.1128/AAC.11.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carlier C, Courvalin P. 1990. Emergence of 4′,4″-aminoglycoside nucleotidyltransferase in enterococci. Antimicrob Agents Chemother 34:1565–1569. doi: 10.1128/AAC.34.8.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martel A, Moreau N, Capmau ML, Soussy CJ, Duval J. 1977. 2″-O-phosphorylation of gentamicin components by a Staphylococcus aureus strain carrying a plasmid. Antimicrob Agents Chemother 12:26–30. doi: 10.1128/AAC.12.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dowding JE. 1977. Mechanisms of gentamicin resistance in Staphylococcus aureus. Antimicrob Agents Chemother 11:47–50. doi: 10.1128/AAC.11.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kayser FH, Homberger F, Devaud M. 1981. Aminocyclitol-modifying enzymes specified by chromosomal genes in Staphylococcus aureus. Antimicrob Agents Chemother 19:766–772. doi: 10.1128/AAC.19.5.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bacot-Davis VR, Bassenden AV, Berghuis AM. 2016. Drug-target networks in aminoglycoside resistance: hierarchy of priority in structural drug design. Med Chem Commun 7:103–113. doi: 10.1039/C5MD00384A. [DOI] [Google Scholar]
- 31.Holbrook SYL, Garneau-Tsodikova S. 2016. Expanding aminoglycoside resistance enzyme regiospecificity by mutation and truncation. Biochemistry 55:5726–5737. doi: 10.1021/acs.biochem.6b00770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bergthorsson U, Andersson DI, Roth JR. 2007. Ohno's dilemma: evolution of new genes under continuous selection. Proc Natl Acad Sci U S A 104:17004–17009. doi: 10.1073/pnas.0707158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuo H, Kobayashi M, Kumagai T, Kuwabara M, Sugiyama M. 2003. Molecular mechanism for the enhancement of arbekacin resistance in a methicillin-resistant Staphylococcus aureus. FEBS Lett 546:401–406. doi: 10.1016/S0014-5793(03)00644-6. [DOI] [PubMed] [Google Scholar]
- 34.Copley SD. 2015. An evolutionary biochemist's perspective on promiscuity. Trends Biochem Sci 40:72–78. doi: 10.1016/j.tibs.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lambert T, Ploy MC, Courvalin P. 1994. A spontaneous point mutation in the aac(6′)-Ib′ gene results in altered substrate specificity of aminoglycoside 6′-N-acetyltransferase of a Pseudomonas fluorescens strain. FEMS Microbiol Lett 115:297–304. [DOI] [PubMed] [Google Scholar]
- 36.Robicsek A, Strahilevitz J, Jacoby GA, Macielag M, Abbanat D, Hye Park C, Bush K, Hooper DC. 2006. Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat Med 12:83–88. doi: 10.1038/nm1347. [DOI] [PubMed] [Google Scholar]
- 37.Lee HK, Vakulenko SB, Clewell DB, Lerner SA, Chow JW. 2002. Mutations in the aph(2″)-Ic gene are responsible for increased levels of aminoglycoside resistance. Antimicrob Agents Chemother 46:3253–3256. doi: 10.1128/AAC.46.10.3253-3256.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toth M, Frase H, Chow JW, Smith C, Vakulenko SB. 2010. Mutant APH(2″)-IIa enzymes with increased activity against amikacin and isepamicin. Antimicrob Agents Chemother 54:1590–1595. doi: 10.1128/AAC.01444-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Studier FW. 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 40.Nørholm MHH. 2010. A mutant Pfu DNA polymerase designed for advanced uracil-excision DNA engineering. BMC Biotechnol 10:21. doi: 10.1186/1472-6750-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McPherson A, Gavira JA. 2013. Introduction to protein crystallization. Acta Crystallogr Sect F Struct Biol Cryst Commun 70:2–20. doi: 10.1107/S2053230X13033141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. 2011. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.Sunada A, Nakajima M, Ikeda Y, Kondo S, Hotta K. 1999. Enzymatic 1-N-acetylation of paromomycin by an actinomycete strain #8 with multiple aminoglycoside resistance and paromomycin sensitivity. J Antibiot 52:809–814. [DOI] [PubMed] [Google Scholar]
- 46.Houghton JL, Biswas T, Chen W, Tsodikov OV, Garneau-Tsodikova S. 2013. Chemical and structural insights into the regioversatility of the aminoglycoside acetyltransferase Eis. Chembiochem 14:2127–2135. doi: 10.1002/cbic.201300359. [DOI] [PMC free article] [PubMed] [Google Scholar]