

ABSTRACT
Muraymycins are antibacterial natural products from Streptomyces spp. that inhibit translocase I (MraY), which is involved in cell wall biosynthesis. Structurally, muraymycins consist of a 5′-C-glycyluridine (GlyU) appended to a 5″-amino-5″-deoxyribose (ADR), forming a disaccharide core that is found in several peptidyl nucleoside inhibitors of MraY. For muraymycins, the GlyU-ADR disaccharide is further modified with an aminopropyl-linked peptide to generate the simplest structures, annotated as the muraymycin D series. Two enzymes encoded in the muraymycin biosynthetic gene cluster, Mur29 and Mur28, were functionally assigned in vitro as a Mg·ATP-dependent nucleotidyltransferase and a Mg·ATP-dependent phosphotransferase, respectively, both modifying the 3″-OH of the disaccharide. Biochemical characterization revealed that both enzymes can utilize several nucleotide donors as cosubstrates and the acceptor substrate muraymycin also behaves as an inhibitor. Single-substrate kinetic analyses revealed that Mur28 preferentially phosphorylates a synthetic GlyU-ADR disaccharide, a hypothetical biosynthetic precursor of muraymycins, while Mur29 preferentially adenylates the D series of muraymycins. The adenylated or phosphorylated products have significantly reduced (170-fold and 51-fold, respectively) MraY inhibitory activities and reduced antibacterial activities, compared with the respective unmodified muraymycins. The results are consistent with Mur29-catalyzed adenylation and Mur28-catalyzed phosphorylation serving as complementary self-resistance mechanisms, with a distinct temporal order during muraymycin biosynthesis.
KEYWORDS: biosynthesis, antibiotic, nucleoside, translocase I, resistance, MraY inhibitor
INTRODUCTION
Muraymycins, produced as a mixture of nearly 20 congeners by Streptomyces sp. NRRL 30471 (also known as Streptomyces sp. LL-AA896), are peptidyl nucleoside antibiotics that were discovered using a screen to identify inhibitors of the lipid cycle of peptidoglycan cell wall biosynthesis (1, 2) (G. Carter, J. Lotvin, and L. A. McDonald, international patent application WO 2002085310 A2 20021031). Detailed investigations into their mechanisms of action subsequently revealed muraymycins to be potent and selective inhibitors of translocase I (MraY), which catalyzes the initiating step of the lipid cycle (3, 4). As a consequence of MraY inhibition, many muraymycin congeners are endowed with antibacterial activity. Notably, muraymycin A1, one of the more active congeners, displays a relatively broad antibacterial spectrum with modest activity against Gram-positive bacteria, including Staphylococcus aureus, and Gram-negative bacteria, including strains of Escherichia coli and Pseudomonas aeruginosa (1, 2). Additionally, muraymycin D1 was recently shown to have good activity against Mycobacterium tuberculosis (4).
Structurally, muraymycins belong to a larger family of peptidyl nucleoside MraY inhibitors that share a core consisting of a 5′-C-glycyluridine (GlyU) that is appended with a 5″-amino-5″-deoxyribose (ADR) moiety via a standard O-glycosidic bond (ADR-GlyU disaccharide) (Fig. 1). FR-900493 is structurally the simplest of this family known to have antibacterial activity and consists of the ADR-GlyU disaccharide core 6′-N-alkylated with an aminopropyl group (K. Ochi, M. Ezaki, M. Iwami, T. Komori, and M. Kohsaka, European patent application EP 333177 A2 19890920). A-90289 compounds, caprazamycins, and liposidomycins (herein collectively termed A-90289-type compounds) (5–7), sphaerimicins (8), and muraymycins also contain a 6′-N-alkyl substituent that is further modified, leading to structural distinctions among these three types (Fig. 1). Notably, muraymycins are the sole members of this family to have a linear peptide attached to the 6′-N-alkyl substituent, and variations within this peptide lead to the different series of muraymycin congeners that were initially reported (1). The D series of muraymycins contain l-Leu as part of the peptide, while the C series contain a β-hydroxylated l-Leu that can be acylated with an aliphatic, branched-chain, fatty acid (B series) or an acyl group containing a terminal guanidinium group (A series). In addition to the standard 2″-OH version of the ADR moiety that is exemplified in muraymycin D2, congeners with a 2″-methoxy ADR (for example, muraymycin D1) or a 2″-deoxy ADR (for example, muraymycin D3) have been isolated. Although they tend to be the least potent, the D series muraymycins are nonetheless strong inhibitors of MraY, with 50% inhibitory concentration (IC50) values near 10 nM, depending on the source of MraY (2, 3). Hydroxylation and acylation, which lead to the A to C series, significantly improve the IC50 values to the low picomolar range (9).
FIG 1.

Structures of representative peptidyl nucleoside antibiotics with an ADR-GlyU disaccharide core.
Recently, a new muraymycin (C6) acetylated at the 5″-amine of the ADR moiety, with modestly reduced (≥6-fold) IC50 values against recombinant MraYs from different sources, was isolated (10). In a separate report, a 3″-O-phosphorylated caprazamycin precursor called caprazol-3″-phosphate was unexpectedly isolated following inactivation of cpz23, encoding a putative SGNH hydrolase, although in that case the inhibitory or antibacterial activity of the phosphorylated variant was not tested (11). Covalent modifications of these types (phosphorylation and acetylation), along with nucleotidylation, are often found as mechanisms of resistance to antibiotics, most notably aminoglycosides (12–15). The same types of covalent modifications appear to be utilized by aminoglycoside-producing strains, inasmuch as homologous genes coding for such resistance enzymes are often clustered with the biosynthetic genes (16–18). However, the reported muraymycin gene cluster lacks any candidate gene coding for an acetyltransferase (19). Instead, the gene cluster codes for a pair of proteins, Mur29 and Mur28, that potentially catalyze the remaining two types of covalent modifications that are commonplace for aminoglycoside resistance, namely, nucleotidylation and phosphorylation, respectively. Of the known biosynthetic gene clusters for peptidyl nucleoside MraY inhibitors, Mur28 homologs are encoded in all of the biosynthetic gene clusters with two homologs encoded within the gene cluster for A-90289-type MraY inhibitors (20–23); the only exception to this is the sphaerimicin gene cluster, which lacks a Mur28 homolog. Conversely, a Mur29 homolog is encoded only within the sphaerimicin gene cluster (8).
As part of our goal to elucidate potential mechanisms of resistance to the peptidyl nucleoside antibiotics, we now provide in vitro data to support the functional assignment of Mur29 and Mur28 as a Mg·ATP-dependent nucleotidyltransferase and a Mg·ATP-dependent phosphotransferase, respectively, which covalently modify the same site, the 3″-OH of the ADR component of muraymycins. Examination of the substrate specificity and single-substrate kinetic analysis using muraymycins D1 to D3 and a hypothetical biosynthetic intermediate prepared through synthesis, however, suggest a distinct temporal order for Mur29 and Mur28 catalysis during biosynthesis. The antimicrobial and MraY inhibitory activities of the adenylated or phosphorylated products, compared with the respective unmodified versions, are reported, enabling us to propose distinct and complementary roles for Mur29 and Mur28 in the resistance and biosynthesis of muraymycin. Finally, a preliminary attempt to functionally characterize TmrB from Bacillus subtilis is reported, and the unexpected results are provided herein.
RESULTS
Functional insight through bioinformatic analysis.
Primary sequence analysis by BLAST revealed that Mur29 consists of two conserved domains, an N-terminal nucleotidyltransferase domain of DNA polymerase subunit β and a C-terminal DUF4111 superfamily/PRK13746 domain. PRK13746-domain-containing proteins are provisionally annotated as aminoglycoside resistance proteins. Secondary structure-based prediction using HHPred suggested structural similarity to Salmonella enteric aminoglycoside O-adenylyltransferase (annotated as AadA; PDB accession no. 4CS6), which catalyzes Mg·ATP-dependent adenylation of spectinomycin and streptomycin (24), and Staphylococcus aureus kanamycin nucleotidyltransferase [annotated as ANT(4′); PDB accession no. 1KNY], which adenylates several distinct aminoglycosides at the 4′- or 4″-hydroxyl substituent, depending on the identity of the aminoglycoside (25). Pairwise alignment, however, revealed only 17% sequence identity with respect to those proteins. Sequence alignment of Mur29 and the homologous gene product SphT encoded in the sphaerimicin gene cluster revealed 48% sequence identity.
Bioinformatic analysis revealed that Mur28 belongs to the nucleoside triphosphate hydrolases containing the P-loop conserved domain, which include several types of nucleoside/nucleotide kinases. Proteins with the greatest sequence identity (∼47%), notably present in both Gram-positive and Gram-negative bacteria, are annotated as tunicamycin resistance proteins (TmrB). Pairwise alignment of Mur28 with Bacillus subtilis TmrB, which contains the same conserved domain and confers resistance to tunicamycin upon heterologous expression of the gene, had lower sequence identity, which was nonetheless significant at 34% (see Table S1 in the supplemental material) (26–28). Sets of homologous genes encoding proteins with similarity to Mur28 are located in each of the A-90289-type gene clusters, represented by LipX (36% sequence identity) and LipI (23% sequence identity) for A-90289 (Table S1).
In vitro functional assignment and characterization of recombinant Mur29.
The three known congeners of the muraymycin D series, which differ only at the 2″ substituent of the ADR-GlyU disaccharide core (Fig. 1), were isolated from Streptomyces sp. NRRL 30475, a previously reported mutant strain of Streptomyces sp. NRRL 30471 (Carter et al., international patent application WO2002085310 A2 20021031). Mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy data were consistent with their identities as muraymycin D1 (Fig. S1 to S6), muraymycin D2 (Fig. S7 to S11), and muraymycin D3 (Fig. S12 and S13). The ADR-GlyU disaccharide, the likely biosynthetic precursor of muraymycin D1 and D2, as well as the other members of the peptidyl nucleoside MraY inhibitors (Fig. 1), was prepared based on the synthetic methodology developed by Ichikawa and coworkers, with slight modifications (29, 30). MS and NMR spectroscopy data were comparable to those in the prior report and similarly consistent with the identity of the disaccharide (Fig. S14 to S18).
Mur29 was produced in E. coli BL21(DE3) as a maltose-binding protein (MBP) fusion, to obtain soluble protein for in vitro assays. Using high-performance liquid chromatography (HPLC) analysis to monitor the reaction, incubation of recombinant Mur29 with muraymycin D1 and ATP revealed a new peak eluting with a longer retention time, relative to muraymycin D1 (Fig. 2A). High-resolution (HR) MS analysis of the purified product revealed an (M+H)+ ion at m/z 1,259.5045, consistent with the molecular formula of C48H75N16O22P, corresponding to monoadenylated muraymycin D1 (expected m/z 1,259.5052) (Fig. S19). 1H NMR spectroscopy data (Fig. S20 and S21) were also consistent with the adenylation of muraymycin D1 (Fig. 2B), and comparison of the substrate and product spectra revealed a clear downfield shift of the 3″ proton (Fig. 2C). Thus, the results are consistent with Mur29 catalyzing regioselective adenylation to yield the product 3″-AMP-muraymycin D1 (D1-AMP).
FIG 2.

Functional assignment of Mur29. (A) HPLC analysis after 12 h of incubation without Mur29 (i) and 12 h of Mur29-catalyzed reaction (ii). Data were collected using GP1, as described in the supplemental material. The arrowhead denotes the peak corresponding to D1-AMP. A260, absorbance at 260 nm. (B) Structure of 3″-monoadenylated muramycin D1. (C) Comparison of the 1H NMR spectra for muraymycin D1 and the Mur29 product.
To provide insight into the specificity and timing of adenylation during muraymycin biosynthesis, the activity of Mur29 was tested with the remaining two D series congeners and the ADR-GlyU disaccharide. In contrast to the disaccharide, which was not a substrate for Mur29, both muraymycin D2 and muraymycin D3 yielded products, with (M+H)+ ions at m/z 1,246.2 and 1,229.2, respectively, consistent with monoadenylated products (expected m/z 1,245.5 for D2-AMP and 1,229.5 for D3-AMP) (Fig. S22 and S23). The lack of activity with the ADR-GlyU disaccharide is consistent with the Mur29 reaction occurring after the addition of the aminopropyl linker. Single-substrate kinetic analysis with different muraymycin D1, D2, and D3 concentrations revealed non-Michaelis-Menten kinetics, with modest substrate inhibition, for all three substrates (Fig. 3A to C). Comparison of the second-order rate constants from the extracted kinetic parameters did not reveal a clear substrate preference (Table 1).
FIG 3.
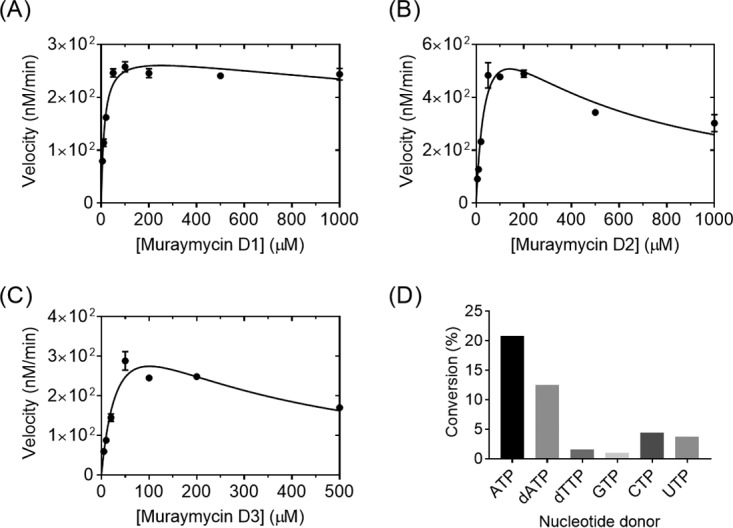
Biochemical characterization of Mur29. (A to C) Single-substrate kinetic analyses using a near-saturating ATP concentration and various concentrations of muraymycin D1 (A), muraymycin D2 (B), and muraymycin D3 (C). (D) Relative activity of Mur29 with muraymycin D1 and different nucleotide donors.
TABLE 1.
Kinetic parameters for Mur29 and Mur28
| Enzyme and substrate | Km (μM)a | Ki (μM) | kcat (min−1) | kcat/Km (M−1 s−1) | Relative kcat/Km value (%) |
|---|---|---|---|---|---|
| Mur29 | |||||
| Muraymycin D1 | 14 ± 2 | (4.6 ± 1.4) × 103 | 1.4 ± 0.1 | 1.7 × 103 | 100 |
| Muraymycin D2 | 41 ± 8 | (4.9 ± 1.1) × 102 | 4.0 ± 0.4 | 1.6 × 103 | 94 |
| Muraymycin D3 | 39 ± 10 | (2.6 ± 0.8) × 102 | 2.4 ± 0.4 | 1.0 × 103 | 59 |
| ADR-GlyU | NA | NA | NA | NA | NA |
| Mur28 | |||||
| Muraymycin D1 | NA | NA | NA | NA | NA |
| Muraymycin D2 | (1.7 ± 0.4) × 102 | (5.6 ± 1.0) × 102 | 5.2 ± 0.7 | 5.1 × 102 | 1.5 |
| Muraymycin D3 | ND | ND | ND | ND | ND |
| ADR-GlyU | 38 ± 4 | NA | 75 ± 2 | 3.3 × 104 | 100 |
NA, not applicable; ND, not determined.
Specificity toward the nucleotide triphosphate cosubstrate was also examined using muraymycin D1 as an acceptor. HPLC and liquid chromatography (LC)-MS analysis revealed that the other canonical ribonucleotide triphosphates could substitute for ATP to generate the corresponding mononucleotide muraymycin D1 (Fig. S24 to S26). Nucleotides dATP and dTTP were also used by Mur29 as group donors (Fig. S27 and S28). The highest specific activity, however, was observed with ATP (Fig. 3D). Finally, the addition of 20 mM EDTA abolished the reaction, consistent with the requirement for magnesium for activity. Thus, the data are consistent with the functional assignment of Mur29 as a Mg·ATP:muraymycin 3″-adenylyltransferase.
In vitro functional assignment and characterization of Mur28.
His6-Mur28 was initially produced in Streptomyces lividans TK24, to obtain soluble protein for in vitro assays. In contrast to Mur29, incubation of recombinant Mur28 with muraymycin D1 and ATP did not reveal any new peaks. However, HPLC analysis of an overnight incubation of Mur28 with ATP and muraymycin D2, which is the likely direct biosynthetic precursor of muraymycin D1, revealed the appearance of a new peak eluting after muraymycin D2 (Fig. 4A). The new peak was collected by semipreparative HPLC, and HR MS analysis of the purified product revealed an (M+H)+ ion at m/z 996.4050, consistent with the molecular formula of C37H62N11O19P, corresponding to monophosphorylated muraymycin D2 (expected m/z 996.4034) (Fig. S29). 1H NMR spectroscopy data (Fig. S30 and S31) were also consistent with phosphorylation of muraymycin D2 (Fig. 4B) and, similarly to the Mur29-catalyzed adenylation of muraymycin D1, comparison of the substrate and product spectra revealed a clear downfield shift of the 3″ proton (Fig. 4C). To provide further evidence for classic kinase activity, the Mur28-catalyzed reaction was performed with [γ-P18O4]ATP. LC-MS analysis of the monophosphorylated product revealed a shift of +6.0 amu, consistent with the incorporation of the γ-phosphate of ATP into muraymycin D2 (Fig. S32). Mur28 was also produced from E. coli BL21(DE3) as a MBP fusion protein, and comparable results were obtained. Thus, the data are consistent with Mur28 catalyzing regioselective phosphorylation to yield the product 3″-phospho-muraymycin D2 (D2-PHOS).
FIG 4.

Functional assignment of Mur28. (A) HPLC analysis after 12 h of incubation without Mur28 (i) and 12 h of Mur28-catalyzed reaction (ii). Data were collected using GP1. The arrowhead denotes the peak corresponding to D2-PHOS, and the asterisk denotes an unidentified peak. A260, absorbance at 260 nm. (B) Structure of 3″-phospho-muraymycin D2. (C) Comparison of the 1H NMR spectra for muraymycin D2 and the Mur28 product.
The specificity and timing of phosphorylation during muraymycin biosynthesis were subsequently investigated. LC-MS analysis of Mur28 reactions with the hypothetical substrates muraymycin D3 and ADR-GlyU disaccharide yielded new peaks with (M+H)+ ions at m/z 980.1 and 527.0, respectively, consistent with the formation of a monophosphorylated product (expected m/z 980.4 for D3-PHOS and 527.1 for ADR-GlyU-PHOS) (Fig. S33 and S34). Single-substrate kinetic analysis with different muraymycin D2 and D3 concentrations revealed non-Michaelis-Menten kinetics with modest substrate inhibition, while analysis with the ADR-GlyU disaccharide revealed typical Michaelis-Menten kinetics (Fig. 5). Second-order rate constants from the extracted kinetic parameters revealed >60-fold higher catalytic efficiency with ADR-GlyU, relative to muraymycin D2, consistent with phosphorylation occurring prior to the attachment of the aminopropyl-linked peptide.
FIG 5.
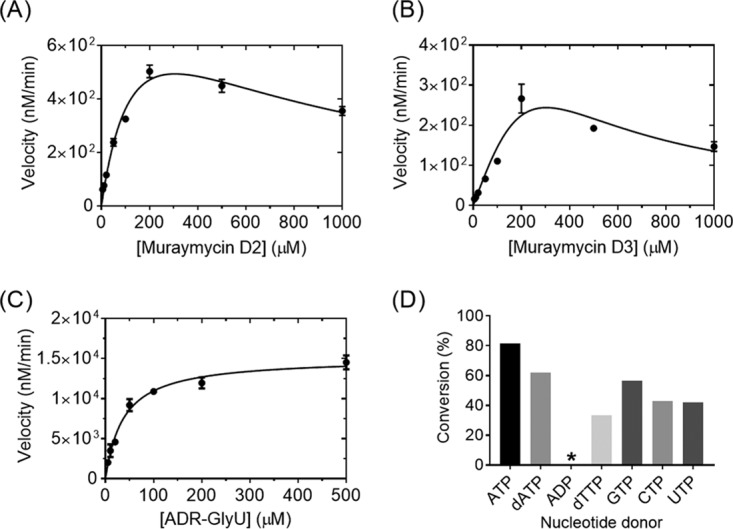
Biochemical characterization of Mur28. (A to C) Single-substrate kinetic analyses using a near-saturating ATP concentration and various concentrations of muraymycin D2 (A), muraymycin D3 (B), and ADR-GlyU (C). (D) Relative activity of Mur28 with muraymycin D2 and different nucleotide donors. *, 0.74%.
LC-MS analysis of reactions using muraymycin D2 as the acceptor revealed that Mur28 uses alternative phosphate donors, similar to results for Mur29 (Fig. S35). The highest activity was achieved with ATP, although good conversion was observed with dATP, dTTP, and the remaining ribonucleotide triphosphates (Fig. 5D). Unexpectedly, ADP, the commercial source of which had no detectable ATP (Fig. S36), was also utilized as a cosubstrate, although D2-PHOS was detected in only trace amounts, suggesting that this activity is insignificant in vivo. Finally, similar to results for Mur29, the inclusion of magnesium was essential for activity. In total, the data are consistent with the functional assignment of Mur28 as a Mg·ATP:ADR-GlyU 3″-phosphotransferase.
Insight into the function of the Mur28 homolog B. subtilis TmrB.
B. subtilis TmrB, which has never been functionally assigned in vitro except for its ability to bind ATP (24, 26) and tunicamycin (25), was initially targeted to serve as a control, due to its modest sequence similarity to Mur28. Soluble His6-TmrB and MBP-TmrB fusion protein were obtained using E. coli BL21(DE3) as an expression host. With the use of HPLC to monitor the reaction, recombinant TmrB did not have any phosphotransferase activity when incubated with ATP and commercial tunicamycin. Unexpectedly, however, LC-MS analysis revealed that TmrB was able to phosphorylate muraymycin D2 and ADR-GlyU using ATP as the phosphate donor (Fig. S37). The conversion of both was comparable to the Mur28-catalyzed reactions. Similarly, a trace amount of D2-PHOS was detected with ADP as the phosphate donor. In contrast to Mur28, however, no other nucleotide triphosphates were substrates for B. subtilis TmrB.
Potential roles of Mur29 and Mur28 in self-resistance.
The possibility that Mur29 or Mur28 plays a role in self-resistance to muraymycins was explored using three complementary approaches. We first examined the antibacterial activity of muraymycin D1 and D2 against E. coli BL21(DE3) harboring the mur29, mur28, or tmrB expression plasmids (Table 2). E. coli BL21(DE3) harboring an expression plasmid for mur20, encoding a putative aminotransferase likely involved in ADR biosynthesis, was used as a control. Both muraymycin D1 and D2 have low antibacterial activity against plasmid-free E. coli BL21(DE3) or strains expressing mur20, with MICs determined in the range of 16 to 32 μg/ml. A similar MIC for muraymycin D1 (32 μg/ml) was obtained for E. coli BL21(DE3) expressing mur28 or tmrB. However, E. coli BL21(DE3) expressing mur29 was resistant to muraymycin D1 (MIC of >256 μg/ml). E. coli BL21(DE3) cells expressing mur29, mur28, or tmrB were all resistant to muraymycin D2 (MIC of >256 μg/ml). These results were consistent with the substrate selectivity of each enzyme that was revealed through in vitro assays.
TABLE 2.
Bacterial MraY in vitro assays and antimicrobial activity
| Compounda | MIC (μg/ml) |
IC50 (nM), S. aureus MraY | ||||||
|---|---|---|---|---|---|---|---|---|
|
E. coli BL21(DE3) |
E. coli ΔtolC, no plasmid | M. smegmatis | ||||||
| No plasmid | pET30-mur20 | pET30-mur29 | pET30-mur28 | pET30-tmrB | ||||
| Muraymycin D1 | 32 | 32 | >256 | 32 | 32 | 1 | 4 | 0.48 ± 0.13 |
| Muraymycin D2 | 32 | 16 | >256 | >256 | >256 | 1 | 4 | 0.39 ± 0.11 |
| D1-AMP | >256 | ND | ND | ND | ND | 16 | >256 | 82 ± 32 |
| D2-PHOS | >256 | ND | ND | ND | ND | 16 | >256 | 20 ± 5 |
D1-AMP, monoadenylated muraymycin D1; D2-PHOS, monophosphorylated muraymycin D2; ND, not determined.
The purified Mur29 and Mur28 products (D1-AMP and D2-PHOS, respectively) were next screened against Mycobacterium smegmatis and E. coli DH5α/ΔtolC (Table 2). Both strains are more sensitive to the unmodified muraymycins than is E. coli BL21(DE3), yielding MICs of 4 μg/ml for muraymycin D1 and D2 against M. smegmatis and 1 μg/ml for muraymycin D1 and D2 against E. coli DH5α/ΔtolC. In contrast, D1-AMP and D2-PHOS were inactive against M. smegmatis (MICs of >256 μg/ml). Somewhat unexpectedly, both modified muraymycins retained some activity, albeit significantly reduced, against efflux-deficient E. coli DH5α/ΔtolC (MICs of 16 μg/ml). Consequently, we explored the possibility that D1-AMP was hydrolyzed in the presence of E. coli DH5α/ΔtolC or upon treatment with the growth conditions, leading to some muraymycin D1 that could effectively kill the strain. However, only D1-AMP was recovered after overnight incubations in LB medium with or without E. coli ΔtolC (Fig. S38), suggesting that D1-AMP and likely D1-PHOS are stable under these conditions. Nevertheless, the significant decrease in antibacterial activity upon covalent modification of the muraymycins is consistent with the conferred resistance that was observed upon heterologous expression of mur29 and mur28 in E. coli BL21(DE3).
The third and final approach to examine a potential role in self-resistance was to screen D1-AMP and D2-PHOS for inhibitory activity against recombinant Staphylococcus aureus MraY in vitro. In contrast to muraymycin D1 and D2, which were determined to be subnanomolar inhibitors of S. aureus MraY, with IC50 values of 0.48 ± 0.13 nM and 0.39 ± 0.11 nM, respectively, D1-AMP and D2-PHOS were significantly less potent, with IC50 values of 82 ± 32 nM and 20 ± 5 nM, respectively. Therefore, direct inhibition of the translocase I target is positively correlated with the whole-cell antibacterial activity.
DISCUSSION
The results support the functional assignment of Mur29 as a nucleotidyltransferase and Mur28 as a phosphotransferase, both of which were predicted from bioinformatic analyses. Unexpected, however, was the discovery that the two enzymes covalently modify the same site (the 3″-hydroxyl substituent of the ADR-GlyU disaccharide core), as clearly evident from the 1H NMR spectra, which revealed a downfield shift of the C-3″ proton of the enzymatic products relative to the respective substrates. The magnitude of the chemical shift is consistent with findings for other enzymes that adenylate or phosphorylate sugars, including the numerous aminoglycoside-modifying enzymes that have been investigated because of their roles in resistance (31–34). Covalent modification at the same site by Mur29 and Mur28 raised the question of whether these modifications occur contemporaneously or at different stages during biosynthesis. Thus, an in-depth investigation into the biochemical properties of Mur29 and Mur28 was pursued, with the goal of establishing the substrate specificity while providing additional evidence to support the functional role of each enzyme.
We first characterized the nucleotidyltransferase Mur29 with the structurally simplest muraymycins, i.e., muraymycins D1, D2, and D3. The D series was utilized as representatives of all muraymycins in part due to (i) the higher fermentation titers, relative to the other congeners, and (ii) the realization that, in our hands, the D series was the only group for which we could readily isolate all three muraymycin congeners with 2″ variations in the ADR. Furthermore, although they are generally the congeners with the weakest antibacterial activity, the D series muraymycins are still very potent MraY inhibitors; therefore, the potential role in self-resistance upon modification could be examined. Biochemical investigation of Mur29 revealed properties that were quite comparable to those observed for aminoglycoside nucleotidyltransferases, such as the aforementioned AadA and S. aureus ANT(4′), as well as the kinetically well-characterized aminoglycoside adenylyltransferase ANT(2″)-Ia (35–40). The comparable properties include the ability to catalyze nucleotidyl transfer with several nucleotide donors and the observed substrate inhibition with respect to the nucleotide acceptor. Furthermore, the extracted Km and Ki values for the D series muraymycins with Mur29 are close in magnitude to those observed for ANT(2″)-Ia with gentamicin B1 and C1, two of the many aminoglycoside antibiotics that are turned over by ANT(2″)-Ia (38). It is noteworthy, however, that the catalytic rate constants for Mur29 with each of the D series muraymycins (kcat values of 1 to 4 min−1) are significantly reduced, compared to those for ANT(2″)-Ia with the aforementioned substrates (kcat values of 75 and 466 min−1, respectively). Given that the D series muraymycins have been shown to be at least 175-fold less potent MraY inhibitors than the corresponding congeners of the A to C series (9), it is tempting to speculate that turnover of Mur29, assuming that it truly provides a self-resistance mechanism, would be significantly increased with the more potent muraymycin congeners. Unfortunately, the production levels for the muraymycins are limiting; therefore, this potential correlation will need to be evaluated following the resolution of this bottleneck.
Mur28 was next functionally assigned as a Mg·ATP-dependent 3″-phosphotransferase that, in contrast to Mur29, modifies the potential biosynthetic intermediate ADR-GlyU disaccharide. Although Mur28 can recognize and modify muraymycin D2, the catalytic efficiency is significantly reduced (>60-fold), compared to that for ADR-GlyU disaccharide. Furthermore, the extracted kinetic constants (Km and kcat) for Mur28 with ADR-GlyU are comparable to those reported for several aminoglycoside phosphotransferases, such as the E. coli aminoglycoside 3′-phosphotransferase APH(3′)-IIIa (Km and kcat values in the range of 10 to 35 μM and 60 to 240 min−1, respectively) (31). Thus, the data are consistent with Mur28 catalysis occurring prior to the attachment of the aminopropyl-linked peptide. These conclusions are supported by the discovery of phosphorylated caprazol from a caprazamycin-producing strain (11), suggesting that phosphorylation occurs prior to complete fabrication of A90289-type antibiotics.
The functional assignment of Mur28 was guided by the annotations of similar proteins, such as the tunicamycin resistance protein TmrB. The modest sequence identity of Mur28 with respect to bona fide B. subtilis TmrB suggested that these two proteins might have similar if not identical functions. Indeed, B. subtilis TmrB was shown to phosphorylate muraymycin D2, with enzymatic activity comparable to that of Mur28. Rather perplexing, however, was the lack of phosphotransferase activity with tunicamycin, although this is consistent with past efforts to characterize the function of B. subtilis TmrB, in which phosphorylation of tunicamycin was never detected (26–28). As a consequence of these negative data, the mechanism of how B. subtilis TmrB confers resistance to tunicamycin remains an enigma. Nevertheless, the results do demonstrate a phosphotransferase activity associated with B. subtilis TmrB, which has long been predicted based on bioinformatic analyses but has never been demonstrated in vivo or in vitro.
The cocrystal structure of Aquifex aeolicus MraY with muraymycin D2 has recently been solved (41, 42), and the ADR-GlyU component plays a critical role in muraymycin binding. Notably, the ADR component fits into a compact pocket, with the 2″-OH forming a hydrogen bond with the side chain of T75 and the 3″-OH forming a hydrogen bond with the nitrogen of the peptide backbone between M263 and G264. Interestingly, however, the muraymycin ADR variants D1, D2, and D3 have comparable A. aeolicus MraY inhibition and antibacterial activities, suggesting that the 2″-OH hydrogen bond is not essential. In contrast, the results provided here suggest that modification of the 3″-OH matters, as adenylation or phosphorylation significantly decreases the inhibitory activity (170-fold and 51-fold, respectively) with S. aureus MraY, which has a high level (∼50%) of sequence similarity to A. aeolicus MraY, including strict conservation of residues involved in inhibitor binding and catalysis. The reduction in IC50 values, notably upon adenylation, is comparable in magnitude to that observed upon phosphorylation of capuramycin, a structurally distinct nucleoside antibiotic that also inhibits MraY (34).
In total, the data support covalent modifications by phosphorylation and adenylation playing a role in self-resistance. The finding of two distinct types of covalent modifications as self-resistance mechanisms is perhaps not surprising, but the realization that the two modifications occur at the same site is quite unusual and unexpected. From a chemical and biosynthetic perspective, muraymycin is likely produced using convergent assembly from two distinct entities, the alkylated ADR-GlyU disaccharide core and the peptide component. In support of this, as noted previously, the former component is found in several nucleoside antibiotics, including FR-900493 from Bacillus cereus, which, unlike the other nucleoside antibiotics, is not modified with any additional functional groups. It is tempting to speculate that the muraymycin-producing strain acquired the genetic information, including the Mur28-encoding gene, to produce the “simple” antibiotic unit (FR-900493 or a similar alkylated disaccharide core) via a horizontal gene transfer event. As a consequence, the phosphorylation mechanism of self-resistance was in place to protect against the production of this simple antibiotic unit, which is consistent with the biochemical data provided here for Mur28, as well as the aforementioned discovery of phosphorylated caprazol. The biosynthesis of the remaining peptide component, which not only is structurally unique to the muraymycins but also imparts substantially improved biological activity to the compounds, is directed by a distinct set of genes. It is likely that the producing strain evolved the ability to assembly the peptide component and to attach it to the alkylated disaccharide core, essentially generating a hybrid molecule. The merging of the two genetic units to generate such a hybrid, a general process that is colloquially described as “changes to the gene roster” and is observed for several other natural products (43), was concurrently accompanied by the installment of a nucleotidylation resistance mechanism, given the inefficiency of Mur28-catalyzed phosphorylation following peptidation. Despite the acquisition of self-resistance through Mur29-catalyzed nucleotidylation, phosphorylation is likely retained due to the substrate selectivity of the N-alkylated enzyme and/or other enzymes or transport proteins functioning downstream of Mur28. This hypothetical scenario is now under investigation.
In conclusion, we have provided evidence to support the functional assignment of Mur29 as a Mg·ATP-dependent muraymycin 3″-adenylyltransferase and Mur28 as a Mg·ATP-dependent ADR-GlyU 3″-phosphotransferase. The biochemical data, along with consideration of MraY inhibition, lead us to conclude that ADR-GlyU is the authentic in vivo substrate of Mur28, as the kinetic parameters are in line with those of other phosphotransferases and attachment of the aminopropyl group leads to the first active antibiotic scaffold, FR-900493. Thus, we propose that transient phosphorylation occurs inside the cell and protects the producing strain from MraY self-inhibition during antibiotic maturation. Following phosphorylation, muraymycin biosynthesis is proposed to be completed by downstream, currently uncharacterized enzymes, with muraymycin then being transported out of the cell and dephosphorylated by an unknown enzyme. In contrast to Mur28, the biochemical data indicate that Mur29 functions after peptidation. Therefore, as opposed to a preventive resistance mechanism that is the likely consequence of Mur28-catalyzed phosphorylation, we hypothesize that Mur29-catalyzed adenylation provides a protective resistance mechanism in the case of intracellular accumulation of muraymycins. In terms of clinical implications, the functional assignment of both Mur29 and Mur28 will now enable sequence-based searches to identify potential related catalysts encoded in the genomes of pathogenic bacteria. Minimally, the results suggest that the 3″-OH of muraymycins might be labile to covalent modifications in the clinic, thus presenting a potential Achilles heel for muraymycin therapeutic applications, assuming the status quo.
MATERIALS AND METHODS
Chemicals, reagents, and instrumentation.
Nucleotides, buffers, salts, and medium components were purchased from Sigma-Aldrich (St. Louis, MO) or Alfa-Aesar (Ward Hill, MA). Synthetic oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA) and are listed in Table S2 in the supplemental material. Genomic DNA from Streptomyces sp. NRRL30473 was extracted using the UltraClean microbial DNA isolation kit (MoBio Laboratories), following the manufacturer's protocol. DNA quality and concentrations were confirmed using a Nanodrop 2000c spectrophotometer (Thermo Scientific) and gel electrophoresis. The tmrB gene from Bacillus subtilis (GenBank accession number WP_042977443) was synthesized by GenScript to give pET28-tmrB.
Analytical HPLC was performed with an Agilent 1200 series (Agilent Technologies, Santa Clara, CA) or Dionex Ultimate 3000 (Dionex, Sunnyvale, CA) HPLC system equipped with a diode array detector and an analytical Apollo C18 column (250 by 4.6 mm, 5 μm) purchased from Grace (Deerfield, IL). Semipreparative HPLC was performed with a Waters 600 controller and pump (Waters, Milford, MA) equipped with a 996 diode array detector, a 717plus autosampler, and an Apollo C18 column (250 by 10 mm, 5 μm). LC-MS was conducted with an Agilent 6120 quadrupole MSD mass spectrometer (Agilent) equipped with an Agilent 1200 series quaternary LC system and an Eclipse XDB-C18 column (150 by 4.6 mm, 5 μm). HR electrospray ionization-MS spectra were acquired with an AB Sciex Triple TOF 5600 system (AB Sciex, Framingham, MA). All solvents used were minimally of ACS grade and were purchased from Pharmco-AAPER (Brookfield, CT) or Fisher Scientific (Pittsburgh, PA). NMR data were recorded at 400 MHz for 1H and 100 MHz for 13C with a Varian Inova NMR spectrometer (Agilent) or at 600 MHz for 1H and 150 MHz for 13C with an Agilent DD2 NMR spectrometer (Agilent).
Cloning and heterologous expression of mur28, mur29, and tmrB.
The genes encoding Mur28 and Mur29 were amplified by PCR, using Phusion DNA polymerase, from genomic DNA extracted from Streptomyces sp. NRRL 30473. The gene encoding TmrB was amplified from pET28a-tmrB. The gel-purified PCR product was inserted into pET-30 Xa/LIC using ligation-independent cloning, as described in the provided protocol, to yield pET30-mur28 and pET30-mur29 or was digested with NdeI-BamHI and ligated into the identical sites of pXY200 or pDB.His.MBP to yield pXY200-mur28, pDB.His.MBP-mur28, pDB.His.MBP-mur29, and pDB.His.MBP-tmrB. The identity of the cloned genes was confirmed by DNA sequencing. The pDB.His.MBP-mur28, pDB.His.MBP-mur29, pET28-tmrB, and pDB.His.MBP-tmrB plasmids were expressed in E. coli BL21(DE3) cells, and the pXY200-mur28 plasmid was expressed in S. lividans TK24 cells, using a standard procedure provided in the supplemental material.
Production of muraymycin D congeners.
Streptomyces sp. NRRL 30475 was cultivated for 72 h at 30°C in 250-ml Erlenmeyer flasks containing 50 ml of tryptic soy broth (Difco) supplemented with 20 g/liter glucose, on a rotary shaker (250 rpm). The seed culture was used to inoculate flasks (250 ml) containing 50 ml of PM-1 medium. PM-1 was composed of 2% glucose, 1% soluble starch, 0.9% pressed yeast, 0.5% peptone (Bacto), 0.5% meat extract (Fluka), 0.5% NaCl, 0.3% CaCO3, and 0.01% CB-442 (NOF Co.) (pH 7.4, before sterilization). The fermentation was continued for 7 days at 23°C on a rotary shaker (210 rpm).
The contents of all culture flasks were combined and centrifuged at 5,000 rpm for 15 min, to separate the mycelium and the water phase. The mycelial cake portion was extracted with methanol by sonication, and the organic phase was evaporated to yield a crude brown extract. Amberlite XAD-16 resin (4%) was added to the water phase and stirred for 12 h. The resin was washed with water until the effluent became colorless, and then methanol elution was performed. The methanol extract was concentrated under reduced pressure to yield a crude extract. Components of the mycelium crude extract and the water-phase crude extract were subjected to separation on a Sephadex LH-20 column (25 to 100 μm; GE Healthcare), and methanol was used to elute compounds at a flow rate at 2 ml/min. The major fraction was further purified by using semipreparative HPLC. A series of linear gradients from 0.025% trifluoroacetic acid in water (solvent A) to 0.025% trifluoroacetic acid in acetonitrile (solvent B) was developed in the following manner: 0 min, 10% solvent B; 0 to 25 min, 22% solvent B; 25 to 26 min, 100% solvent B; 26 to 31 min, 100% solvent B; 31 to 32 min, 10% solvent B (beginning and ending times, with linear changes in solvent B). The flow rate was kept constant at 3.5 ml/min.
Synthesis of the ADR-GlyU disaccharide.
The ADR-GlyU disaccharide was synthesized mainly through a route reported by Ichikawa and coworkers (29, 30) but with slight modifications, including a new deprotection protocol to yield the desired unprotected disaccharide. The synthetic scheme employed and experimental details are provided in the supplemental material.
Enzyme reactions.
Reaction mixtures consisted of 50 mM Tris (pH 7.5), 10 mM MgCl2, 1 mM ATP, dATP, dTTP, GTP, CTP, or UTP, 1 mM muraymycin D1, D2, or D3 or 0.5 mM ADR-GlyU disaccharide, and 1 μM Mur29, Mur28, or TmrB, at 30°C. The reactions were terminated by the addition of 2 volumes of methanol, followed by centrifugation (14,000 rpm for 30 min) to remove the precipitated proteins. After centrifugation, the reaction mixtures were analyzed by HPLC or LC-MS, using one of four gradient programs (GPs) provided in the supplemental material.
Single-substrate kinetics.
For Mur29, assay mixtures consisted of 50 mM Tris (pH 7.5), 10 mM MgCl2, a near-saturating ATP concentration (1 mM), and various concentrations of muraymycin D1 (1 μM to 1,000 μM), muraymycin D2 (1 μM to 1,000 μM), or muraymycin D3 (5 μM to 500 μM). Reactions were performed at 30°C for 4 h with muraymycin D1, D2, or D3 and 200 nM Mur29. For Mur28, assay mixtures consisted of 50 mM Tris (pH 7.5), 10 mM MgCl2, a near-saturating ATP concentration (1 mM), and various concentrations of muraymycin D2 (1 μM to 1,000 μM), muraymycin D3 (5 μM to 500 μM), or ADR-GlyU disaccharide (5 μM to 500 μM). Reactions were performed at 30°C for 4 h with muraymycin D2 or D3 and for 15 min with ADR-GlyU disaccharide, with 200 nM Mur28. Product formation was determined using HPLC with GP2. All reactions were analyzed under near-initial-velocity conditions (≤10% product). Each data point represents a minimum of three replicate endpoint assays; kinetic constants were obtained by nonlinear regression analysis using GraphPad Prism (GraphPad Software, La Jolla, CA), by fitting the data to the following equations, depending on the presence of substrate inhibition: v = Vmax[S]/(Km + [S]) or v = Vmax[S]/[(Km + [S] + [S]2)/Ki].
Antimicrobial activity.
The protocol used for the determination of MICs was as described previously, with minor modifications (44). The bacterial strains Mycobacterium smegmatis ATCC 14468, E. coli DH5α/ΔtolC, and E. coli BL21(DE3), with introduced pET30-mur28, pET30-mur29, pET28a-tmrB, or pET30-mur20 plasmids, were used as model strains for antimicrobial susceptibility assays. All strains were grown in appropriate liquid medium or on agar plates, using Middlebrook 7H9 medium with albumin-dextrose-catalase (ADC) enrichment for M. smegmatis or LB medium supplemented with 50 μg/ml kanamycin for E. coli. Individual strains were grown for 16 h at 37°C in 5 ml of medium, with shaking (250 rpm), and then were diluted 1,000-fold into 4.5 ml of medium and incubated until the optical density at 600 nm (OD600) reached 0.4. Aliquots of the suspensions were diluted 1,000-fold. Aliquots (90 μl) of each diluted culture were transferred into the individual wells of a 96-well plate supplied with 10 μl of the test compound. The maximal final concentration of 256 μg/ml with serial dilutions was maintained to determine the antimicrobial activities, and values were compared to the negative-control (water) values. The culture plates were incubated for 16 h at 37°C, with shaking (160 rpm) for E. coli and without shaking for M. smegmatis. The OD600 of each well was measured using a BioTek Synergy 2 multimode microplate reader. The acquired OD600 values were normalized to those for the negative-control wells (100% viability). Resazurin solution (5 μl) was also added to each well, and the plates were shaken for 10 s and incubated at 37°C for another 3 h. The minimal concentration of the test compound that caused growth inhibition was recorded as the MIC.
Stability of D1-AMP.
M. smegmatis ATCC 14468 and E. coli DH5α/ΔtolC were used as model strains to test the stability of D1-AMP. Strains were grown in Middlebrook 7H9 medium with ADC enrichment or in LB medium supplemented with 50 μg/ml kanamycin, respectively. Liquid cultures (5 ml) were grown for 16 h at 37°C with shaking (250 rpm), diluted 1,000-fold into 4.5 ml of fresh medium, and incubated until the OD600 reached 0.4. Aliquots of the suspensions were diluted 1,000-fold. Aliquots (45 μl) of each diluted culture or a filtered culture sample (0.22-μm membrane filter) were transferred to a 96-well plate supplied with 5 μl of 2,560-μg/ml D1-AMP. The culture plates were incubated for 16 h at 37°C, with shaking (160 rpm) for E. coli and without shaking for M. smegmatis. Cells and insoluble material were removed by centrifugation at 4,000 rpm for 10 min. The supernatant was analyzed by HPLC using GP2.
MraY inhibition.
Fluorescence-based in vitro inhibition assays against S. aureus MraY were performed as reported previously (9, 45).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the National Institutes of Health (grant AI087849), the National Center for Advancing Translational Sciences (grant UL1TR000117), the Fonds der Chemischen Industrie (Germany) (Sachkostenzuschuss to C.D.), and the German federal state of Lower Saxony (Lichtenberg doctoral fellowship [CaSuS program] to A.L.).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00193-18.
REFERENCES
- 1.McDonald LA, Barbieri LR, Carter GT, Lenoy E, Lotvin J, Petersen PJ, Siegel MM, Singh G, Williamson RT. 2002. Structures of the muraymycins, novel peptidoglycan biosynthesis inhibitors. J Am Chem Soc 124:10260–10261. doi: 10.1021/ja017748h. [DOI] [PubMed] [Google Scholar]
- 2.Wiegmann D, Koppermann S, Wirth M, Niro G, Leyerer K, Ducho C. 2016. Muraymycin nucleoside-peptide antibiotics: uridine-derived natural products as lead structures for the development of novel antibacterial agents. Beilstein J Org Chem 12:769–795. doi: 10.3762/bjoc.12.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanino T, Al-Dabbagh B, Mengin-Lecreulx D, Bouhss A, Oyama H, Ichikawa S. 2011. Mechanistic analysis of muraymycin analogues: a guide to the design of MraY inhibitors. J Med Chem 54:8421–8439. doi: 10.1021/jm200906r. [DOI] [PubMed] [Google Scholar]
- 4.Mitachi K, Aleiwi BA, Schneider CM, Siricilla S, Kurosu M. 2016. Stereocontrolled total synthesis of muraymycin D1 having a dual mode of action against Mycobacterium tuberculosis. J Am Chem Soc 39:12975–12980. doi: 10.1021/jacs.6b07395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujita Y, Kizuka M, Funabashi M, Ogawa Y, Ishikawa T, Nonaka K, Takatsu T. 2011. A-90289 A and B, new inhibitors of bacterial translocase I, produced by Streptomyces sp. SANK 60405. J Antibiot (Tokyo) 64:495–501. doi: 10.1038/ja.2011.38. [DOI] [PubMed] [Google Scholar]
- 6.Igarashi M, Takahashi Y, Shitara T, Nakamura H, Naganawa H, Miyake T, Akamatsu Y. 2005. Caprazamycins, novel lipo-nucleoside antibiotics, from Streptomyces sp. II. Structure elucidation of caprazamycins. J Antibiot (Tokyo) 58:327–337. doi: 10.1038/ja.2005.41. [DOI] [PubMed] [Google Scholar]
- 7.Ubukata M, Isono K, Kimura K, Nelson CC, McCloskey JA. 1988. The structure of liposidomycin B, an inhibitor of bacterial peptidoglycan synthesis. J Am Chem Soc 110:4416–4417. doi: 10.1021/ja00221a052. [DOI] [Google Scholar]
- 8.Funabashi M, Baba S, Takatsu T, Kizuka M, Ohata Y, Tanaka M, Nonaka K, Spork AP, Ducho C, Chen WCL, Van Lanen SG. 2013. Structure-based gene targeting discovery of sphaerimicin, a bacterial translocase I inhibitor. Angew Chem Int Ed Engl 52:11607–11611. doi: 10.1002/anie.201305546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koppermann S, Cui Z, Fischer PD, Wang X, Ludwig J, Thorson JS, Van Lanen SG, Ducho C. 2018. Insights into the target interaction of naturally occurring muraymycin nucleoside antibiotics. ChemMedChem 13:779–784. doi: 10.1002/cmdc.201700793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui Z, Wang X, Koppermann S, Thorson JS, Ducho C, Van Lanen SG. 2018. Antibacterial muraymycins from mutant strains of Streptomyces sp. NRRL 30471. J Nat Prod 81:942–948. doi: 10.1021/acs.jnatprod.7b01054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiraishi T, Hiro N, Igarashi M, Nishiyama M, Kuzuyama T. 2016. Biosynthesis of the antituberculous agent caprazamycin: identification of caprazol-3″-phosphate, an unprecedented caprazamycin-related metabolite. J Gen Appl Microbiol 62:164–166. doi: 10.2323/jgam.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 12.Wright GD. 2008. Mechanisms of aminoglycoside antibiotic resistance, p 71–101. In Wax RG. (ed), Bacterial resistance to antimicrobials, 2nd ed CRC Press, Boca Raton, FL. [Google Scholar]
- 13.Smith CA, Baker EN. 2002. Aminoglycoside antibiotic resistance by enzymatic deactivation. Curr Drug Targets Infect Disord 2:143–160. doi: 10.2174/1568005023342533. [DOI] [PubMed] [Google Scholar]
- 14.Magnet S, Blanchard JS. 2005. Molecular insights into aminoglycoside action and resistance. Chem Rev 105:477–498. doi: 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]
- 15.Vakulenko SB, Mobashery S. 2003. Versatility of aminoglycosides and prospects for their future. Clin Microbiol Rev 16:430–450. doi: 10.1128/CMR.16.3.430-450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wehmeier UF, Piepersberg W. 2009. Enzymology of aminoglycoside biosynthesis: deduction from gene clusters. Methods Enzymol 459:459–491. doi: 10.1016/S0076-6879(09)04619-9. [DOI] [PubMed] [Google Scholar]
- 17.Kudo F, Eguchi T. 2009. Biosynthetic enzymes for the aminoglycosides butirosin and neomycin. Methods Enzymol 459:493–519. doi: 10.1016/S0076-6879(09)04620-5. [DOI] [PubMed] [Google Scholar]
- 18.Barka EA, Vatsa P, Sanchez L, Gaveau-Vaillant N, Jacquard C, Klenk H-P, Clément C, Ouhdouch Y, van Wezel GP. 2016. Taxonomy, physiology, and natural products of Actinobacteria. Microbiol Mol Biol Rev 80:1–43. doi: 10.1128/MMBR.00019-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng L, Chen W, Zhai L, Xu D, Huang T, Lin S, Zhou X, Deng Z. 2011. Identification of the gene cluster involved in muraymycin biosynthesis from Streptomyces sp. NRRL 30471. Mol Biosyst 7:920–927. doi: 10.1039/C0MB00237B. [DOI] [PubMed] [Google Scholar]
- 20.Funabashi M, Baba S, Nonaka K, Hosobuchi M, Fujita Y, Sibata T, Van Lanen SG. 2010. The biosynthesis of liposidomycin-like A-90289 antibiotics featuring a new type of sulfotransferase. ChemBioChem 11:184–190. doi: 10.1002/cbic.200900665. [DOI] [PubMed] [Google Scholar]
- 21.Kaysser L, Siebenberg S, Kammerer B, Gust B. 2010. Analysis of the liposidomycin gene cluster leads to the identification of new caprazamycin derivatives. ChemBioChem 11:191–196. doi: 10.1002/cbic.200900637. [DOI] [PubMed] [Google Scholar]
- 22.Kaysser L, Lutsch L, Siebenberg S, Wemakor E, Kammerer B, Gust B. 2009. Identification and manipulation of the caprazamycin gene cluster lead to new simplified liponucleoside antibiotics and give insights into the biosynthetic pathway. J Biol Chem 284:14987–14996. doi: 10.1074/jbc.M901258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chi X, Baba S, Tibrewal N, Funabashi M, Nonaka K, Van Lanen SG. 2013. The muraminomicin biosynthetic gene cluster and enzymatic formation of the 2-deoxyaminoribosyl appendage. MedChemComm 4:239–243. doi: 10.1039/C2MD20245J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, Nasvall J, Wu S, Andersson DI, Selmer M. 2015. Structure of AadA from Salmonella enterica: a monomeric aminoglycoside (3″)(9) adenyltransferase. Acta Crystallogr Sect D Struct Biol 71:2267–2277. doi: 10.1107/S1399004715016429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pedersen LC, Benning MM, Holden HM. 1995. Structural investigation of the antibiotic and ATP-binding sites in kanamycin nucleotidyltransferase. Biochemistry 34:13305–13311. doi: 10.1021/bi00041a005. [DOI] [PubMed] [Google Scholar]
- 26.Noda Y, Yoda K, Takatsuki A, Yamasaki M. 1992. TmrB protein, responsible for tunicamycin resistance of Bacillus subtilis, is a novel ATP-binding membrane protein. J Bacteriol 174:4302–4307. doi: 10.1128/jb.174.13.4302-4307.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noda Y, Takatsuki A, Yoda K, Yamasaki M. 1995. TmrB protein, which confers resistance to tunicamycin on Bacillus subtilis, binds tunicamycin. Biosci Biotechnol Biochem 59:321–322. doi: 10.1271/bbb.59.321. [DOI] [PubMed] [Google Scholar]
- 28.Kapp U, Macedo S, Hall DR, Leiro I, McSweeney SM, Mitchell E. 2008. Structure of tunicamycin-resistance protein (TmrD), a phosphotransferase. Acta Crystallogr Sect F Struct Biol Cryst Commun 64:479–486. doi: 10.1107/S1744309108011822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanino T, Ichikawa S, Shiro M, Matsuda A. 2010. Total synthesis of (−)-muraymycin D2 and its epimer. J Org Chem 75:1366–1377. doi: 10.1021/jo9027193. [DOI] [PubMed] [Google Scholar]
- 30.Hirano S, Ichikawa S, Matsuda A. 2007. Development of a highly β-selective ribosylation reaction without using neighboring group participation: total synthesis of (+)-caprazol, a core structure of caprazamycins. J Org Chem 72:9936–9946. doi: 10.1021/jo701699h. [DOI] [PubMed] [Google Scholar]
- 31.McKay GA, Thompson PR, Wright GD. 1994. Broad spectrum aminoglycoside phosphotransferase type III from Enterococcus: overexpression, purification, and substrate specificity. Biochemistry 33:6936–6944. doi: 10.1021/bi00188a024. [DOI] [PubMed] [Google Scholar]
- 32.Barrasa MI, Tercero JA, Jiménez A. 1997. The aminonucleoside antibiotic A201A is inactivated by a phosphotransferase activity from Streptomyces capreolus NRRL 3817, the producing organism: isolation and molecular characterization of the relevant gene and its DNA flanking regions. Eur J Biochem 245:54–63. doi: 10.1111/j.1432-1033.1997.00054.x. [DOI] [PubMed] [Google Scholar]
- 33.Naganawa H, Kondo S, Maeda K, Umezawa H. 1971. Structure determinations of enzymatically phosphorylated products of aminoglycosidic antibiotics by proton magnetic resonance. J Antibiot (Tokyo) 24:823–829. doi: 10.7164/antibiotics.24.823. [DOI] [PubMed] [Google Scholar]
- 34.Yang Z, Funabashi M, Nonaka K, Hosobuchi M, Shibata T, Pahari P, Van Lanen SG. 2010. Functional and kinetic analysis of the phosphotransferase CapP conferring selective self-resistance to capuramycin antibiotics. J Biol Chem 285:12899–12905. doi: 10.1074/jbc.M110.104141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Goffic F, Martel A, Capmau ML, Baca B, Goebel P, Chardon H, Soussy CJ, Duval J, Bouanchaud DH. 1976. New plasmid-mediated nucleotidylation of aminoglycoside antibiotics in Staphylococcus aureus. Antimicrob Agents Chemother 10:258–264. doi: 10.1128/AAC.10.2.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith AL, Smith DH. 1974. Gentamicin: adenine mononucleotide transferase: partial purification, characterization, and use in the clinical quantitation of gentamicin. J Infect Dis 129:391–401. doi: 10.1093/infdis/129.4.391. [DOI] [PubMed] [Google Scholar]
- 37.Jana S, Deb JK. 2005. Kinetic mechanism of streptomycin adenylyltransferase from a recombinant Escherichia coli. Biotechnol Lett 27:519–524. doi: 10.1007/s10529-005-2544-9. [DOI] [PubMed] [Google Scholar]
- 38.Gates CA, Northrop DB. 1988. Substrate specificities and structure-activity relationships for the nucleotidylation of antibiotics catalyzed by aminoglycoside nucleotidyltransferase 2″-I. Biochemistry 27:3820–3825. doi: 10.1021/bi00410a045. [DOI] [PubMed] [Google Scholar]
- 39.Gates CA, Northrop DB. 1988. Alternative substrate and inhibition kinetics of aminoglycoside nucleotidyltransferase 2″-I in support of a Theorell-Chance kinetic mechanism. Biochemistry 27:3826–3833. doi: 10.1021/bi00410a046. [DOI] [PubMed] [Google Scholar]
- 40.Van Pelt JE, Northrop DB. 1984. Purification and properties of gentamicin nucleotidyltransferase from Escherichia coli: nucleotide specificity, pH optimum, and the separation of two electrophoretic variants. Arch Biochem Biophys 230:250–263. doi: 10.1016/0003-9861(84)90106-1. [DOI] [PubMed] [Google Scholar]
- 41.Chung BC, Mashalidis EH, Tanino T, Kim M, Matsuda A, Hong J, Ichikawa S, Lee SY. 2016. Structural insights into inhibition of lipid I production in bacterial cell wall synthesis. Nature 533:557–560. doi: 10.1038/nature17636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chung BC, Zhao J, Gillespie RA, Kwon DY, Guan Z, Hong J, Zhou P, Lee SY. 2013. Crystal structure of MraY, an essential membrane enzyme for bacterial cell wall synthesis. Science 341:1012–1016. doi: 10.1126/science.1236501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fischbach MA, Walsh CT, Clardy J. 2008. The evolution of gene collectives: how natural selection drives chemical innovation. Proc Natl Acad Sci U S A 105:4601–4608. doi: 10.1073/pnas.0709132105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cai W, Wang X, Elshahawi SI, Ponomareva LV, Liu X, McErlean MR, Cui Z, Arlinghaus AL, Thorson JS, Van Lanen SG. 2016. Antibacterial and cytotoxic actinomycins Y6-Y9 and Zp from Streptomyces sp. strain Gö-GS12. J Nat Prod 79:2731–2739. doi: 10.1021/acs.jnatprod.6b00742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wohnig S, Spork AP, Koppermann S, Mieskes G, Gisch N, Jahn R, Ducho C. 2016. Total synthesis of dansylated Park's nucleotide for high-throughput MraY assays. Chem Eur J 22:17813–17819. doi: 10.1002/chem.201604279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.