

Abstract
Clerocidin, a diterpenoid with antibacterial and antitumor activity, stimulates in vitro DNA cleavage mediated by mammalian and bacterial topoisomerase (topo) II. Different from the classical topoisomerase poisons, clerocidin-stimulated breaks at guanines immediately preceding the sites of DNA cleavage are not resealed upon heat or salt treatment. To understand the mechanism of irreversible trapping of the topo II-cleavable complex, we have investigated the reactivity of clerocidin per se towards DNA. We show here that the drug is able to nick negatively supercoiled plasmids. DNA cleavage by clerocidin in enzyme-free medium is due to the ability of the drug to form covalent adducts with guanines. Indeed, clerocidin was able to specifically react with short oligonucleotides when the guanines were unpaired and exposed as in bulges or in the single-strand form. The clerocidin epoxy group attacks the nitrogen at position 7 of guanines, leading to strand scission at the modified site. Our findings also demonstrate that trapping of topoisomerases by clerocidin is specific for type II enzymes. The guanine-alkylating ability of clerocidin suggests an unprecedented mechanism of topo II poisoning, according to which the enzyme renders the drug reactive toward DNA by distorting the double-helical structure of the nucleic acid at the cleavage site.
INTRODUCTION
Clerocidin (CL) is a microbial terpenoid with potent antibacterial and antitumor activity (1). Recent studies have shown that the molecular targets for this compound are, respectively, DNA gyrase and eukaryotic DNA topoisomerase (topo) II (2–4). In fact, CL induces enzyme-mediated DNA damage, producing a distinct cleavage pattern (2). Its molecular mechanism of action is different from other known topoisomerase poisons since the drug, as well as its derivatives, can induce the formation of a cleavage complex which is partially stable to heat and salt treatment (2). In this regard, we have recently established the structural determinants for drug activity, by investigating the sequence specificity of CL-stimulated topo II-mediated DNA damage (5). In agreement with the above cited results, most drug-stimulated cleavage sites did not revert upon salt addition, whereas some sites showed complete reversion. Each set of sites was characterized by different drug-preferred nucleotides at the cleavage site. In particular, the base immediately preceding the site of enzyme attack, i.e. position –1, was in both cases distinctively biased: guanine (G) residues were always preferred in the case of the irreversible cleavage sites, whereas cytosine (C) residues were normally found at the reversible sites. Furthermore, the DNA bands at the irreversible cleavage sites showed an abnormal electrophoretic mobility in sequencing gels, suggesting a chemical alteration caused by the drug (5).
Similar findings were obtained in vivo: pulsed field gel electrophoresis and genomic primer extension experiments showed that some CL cleavage sites did not undergo spontaneous reversion, indicating that this agent can stimulate irreversible DNA damage in chromatin. Again, these sites had a G–1 preference (6). These data, besides confirming the biological significance of in vitro studies, prompted further investigations aimed at understanding the molecular basis of the unusual behavior of CL as a topo II poison.
A close inspection of the chemical structure of CL, reported in Figure 1, shows the presence of several electrophilic groups, namely an α-ketoaldehyde function in equilibrium with the hemi-acetalic form, a strained epoxy ring (region C12–C15) and a second aldehyde at position 4 of the diterpenoid structure. This latter aldehyde, however, is not related to the poisoning activity of CL, since congeners such as terpenticin and UTC 4B, lacking this group, are still able to inhibit the eukaryotic enzyme (2,3,7). The C12–C15 region, on the other hand, can undergo chemical modification after prolonged storage in ethanol and this modification parallels an impaired ability of the compound to generate irreversible topo II-mediated DNA cleavage (5). It is therefore conceivable that at least one of these electrophilic moieties is capable of reacting with appropriate nucleophiles in DNA and/or the enzyme component (5,8). Several dicarbonyl compounds and aldehydes can in fact react with DNA at specific sites (9,10) and epoxides can induce DNA damage with a mechanism similar to nitrogen mustards (11–14).
Figure 1.

Structure of CL. The dicarbonylic form of the drug (left) is shown in equilibrium with the closed hemi-acetalic ring (right). The numbered atoms outline electrophilic carbons possibly involved in the reaction with nucleophilic moieties in the DNA.
On this line of thinking, in this paper we will address the issue of CL reactivity towards DNA per se. We show that the compound can nick supercoiled DNA and can react with guanines in appropriate oligodeoxynucleotide (oligo) structures. The reaction involves addition of the epoxy group to N7 of G residues and leads to phosphodiester backbone scission. On the other hand, linear double-stranded (ds)DNA is a poor substrate for CL since access to the nucleic acid is hindered by the bulky terpenoid substituent. Different from other known DNA-reactive agents, alkylation by CL is dictated by the availability of accessible structures in a double helix, thus supporting the idea that topo II creates such structures at the site of cleavage. As a consequence, DNA becomes alkylated at G nucleotides in the cleavable complex, eventually resulting in irreversible topo II poisoning.
MATERIALS AND METHODS
Enzymes, oligos and chemicals
CL was a kind gift of Leo Pharmaceutical Products (Ballerup, Denmark), while camptothecin was purchased from Sigma. CL was dissolved in absolute ethanol and the concentration determined by measuring absorbance in ethanol at 230 nm on a UV/VIS Spectrometer Lambda 12 (Perkin Elmer), using the experimentally determined molar extinction coefficient of 11 818 M–1 cm–1. Working CL solutions were obtained by diluting fresh or aged stocks, where indicated, in the appropriate buffer. Buffer components were all from Sigma, while electrophoretic reagents were from Amersham Pharmacia Biotech or Bio-Rad. Human topo I was purchased from TopoGEN (Columbus, OH). SV40 plasmid, restriction enzymes and T4 polynucleotide kinase were purchased from Gibco BRL. [γ-32P]ATP and [α-32P]dATP were from NEN Life Sciences, while all oligos were from Eurogentec Bel SA (Belgium).
Clerocidin nicking of supercoiled SV40
The supercoiled form of SV40 was reacted at 37°C with CL at the indicated concentrations in 50 mM phosphate buffer, pH 7.4, in a total volume of 20 µl. The final ethanol concentration was 1.25% (v/v) in all sample and control reactions. After the indicated times each reaction was stopped in ice and type II gel loading buffer added. The treated samples were then loaded on a 1% agarose gel in 44.5 mM Tris-borate, 1 mM EDTA, pH 8.3, run and stained with ethidium bromide. The nicked standard was obtained as reported (15), while the linearized standard was obtained by BamHI digestion. Both the nicked and the linearized SV40 were purified by phenol/chloroform extraction followed by ethanol precipitation.
Reaction with oligos
The ability of CL to damage DNA in the absence of enzyme was tested at the sequencing level using short oligos whose sequences span a strong irreversible site for CL cleavage in the presence of murine topo II, namely position 2640 in the SV40 genome (5). The oligo sequences are, respectively, CTATTGCTTTATTTGTAACCATTATAAGCT (CL2640-up), in which the irreversibly cleaved G is underlined, and its complementary oligo AGCTTATAATGGTTACAAATAAAGCAATAG (CL2640-down). The other oligos used to extrude 3 and 4 nt bulges (Bulge3 and Bulge4) upon annealing to 5′-end-labeled CL2640-up or 5′-end-labeled CL2640-down are, respectively, AGCTTATAATGGTTAATAAAGCAATAG (CL2640-B-down) and (CL2640-B-up) CTATTGCTTTATTTGTATTATAAGCT. The G underlined in oligo CL2640-up is substituted by 7-deaza-G in oligo CL2640-d7G.
Oligos were obtained desalted and lyophilized from Eurogentec and were gel purified before use. They were then labeled with [γ-32P]ATP by T4 polynucleotide kinase and ethanol precipitated. If oligos were to be annealed, equimolar amounts were mixed in sterile water and the annealing reaction was performed by heating at 90°C and letting the system cool down to room temperature. The annealed oligos were further purified in native polyacrylamide gels and electroeluted using the Biotrap method (Schleicher & Schuell).
CL reaction with the labeled oligos was performed at 37°C in 50 mM phosphate buffer, pH 7.4, containing 80 mM KCl and 10 mM MgCl2. Tris was avoided since its primary amine could react forming Schiff bases with the aldehyde groups of CL. After the indicated times, samples were ethanol precipitated to eliminate non-reacted drug, then resuspended and split into two aliquots, one of which was kept on ice while the other was treated at 90°C for 30 min with 1 M piperidine. Samples were finally lyophilized twice, resuspended in formamide gel loading buffer, heated at 90°C for 5 min and cleavage products resolved in 20% denaturing polyacrylamide gels alongside a G+A and/or G ladder obtained by the Maxam and Gilbert method (16).
Topo I cleavage assay
SV40 was cut at the BamHI site and labeled by fill-in with the large (Klenow) fragment of DNA polymerase I and [α-32P]dATP. Aliquots of 0.020 µg DNA were reacted with the indicated concentration of drug and 3.6 U human topo I (TopoGEN Inc., Columbus, OH) as described (17), using 50 mM HEPES or 20 mM phosphate buffer instead of Tris. Following 30 min incubation at 37°C, samples were stopped by incubation with 0.5 mg/ml proteinase K and 1% SDS at 37°C for 5 h and then made alkaline before loading onto a 1% agarose gel (0.09 M Tris-borate, pH 8.3, 2.5 mM EDTA). The gel was run for 15 h at 40 V, dried and autoradiographed.
NMR
All 1H NMR spectra were recorded using a 300 MHz Bruker AMX spectrometer. In the present study, CL was dissolved in deuterated ethanol and NMR determination was performed immediately after dissolution of the sample. After 6 weeks the inactive stock of clerocidin was lyophilized, dissolved in CDCl3 and 1H NMR spectra were recollected. The NMR spectrum of freshly ethanol dissolved CL is consistent with the peak assignments already published by Binaschi et al. (5).
Computational methodologies
Calculations were performed on a Silicon Graphics O2 R10000 workstation. A decanucleotide duplex of sequences d(5′-CGCGC-3′)2 in B-form was built using the DNA Builder module of Molecular Operating Environment (MOE 1999.05). The decanucleotide was minimized using the Amber94 all-atom force field (18) MOE modeling package until the r.m.s. value of the Truncated Newton method was <0.001 kcal mol–1 Å–1. The dielectric constant was assumed to be distance-independent with a magnitude of 4.
The CL model was constructed using the Molecule Builder module of Molecular Operating Environment (MOE 1999.05) (19). This structure was minimized using the MMFF94 force field (20–24) until the r.m.s. value of the Truncated Newton method was <0.001 kcal mol–1 Å–1. The optimized geometry of CL was fully minimized using the RHF/AM1//RHF/3–21G(*) ab initio level of Gaussian 98 (25). Atomic charges were calculated by fitting to electrostatic potential maps (CHELPG method) (26).
CL was docked into both the major and minor grooves of DNA using the flexible MOE-Dock methodology. The purpose of MOE-Dock is to search for favorable binding configurations between a small, flexible ligand and a rigid macromolecular target. Searching is conducted within a user-specified 3-dimensional docking box, using simulated annealing and a molecular mechanics force field. MOE-Dock performs a user-specified number of independent docking runs and writes the resulting conformations and their energies to a database file.
The resulting DNA–ligand complexes were subjected to Amber94 all-atom energy minimization until the r.m.s. of the conjugate gradient was <0.1 kcal mol–1 Å–1. Charges for the ligands were imported from the Gaussian output files. The interaction energy values were calculated as the energy of the complex minus the energy of the ligand, minus the energy of DNA: ΔEinteraction = Ecomplex – (Eligand + EDNA).
RESULTS
CL cleaves supercoiled DNA
To check CL reactivity towards DNA in the absence of topoisomerase we incubated the drug with a negatively supercoiled plasmid. DNA strand scission can be easily identified by differential migration of the nicked form in an agarose gel. As shown in Figure 2A, CL at 37°C interconverts the supercoiled form of the plasmid (CCC) into the nicked (OC) form, and the effect is both time and concentration dependent (Fig. 2B). CL can therefore cause direct damage to supercoiled DNA in the absence of gyrase or topo II.
Figure 2.

CL can nick negatively supercoiled DNA. The reactions between CL and the plasmid in the absence of topoisomerases were carried out as described in Materials and Methods and visualized by EtBr staining. CCC refers to the negatively supercoiled form of the plasmid SV40, while OC refers to the open circular form. The standards are the supercoiled (S), nicked (N) and linear (L) forms of the plasmid. Nicking of DNA by CL is confirmed by running the same samples in a gel containing EtBr (not shown). (A) Time course of CL nicking. The SV40 plasmid (2 µg/ml) was treated with buffer or with 200 µM CL. Aliquots of the reaction or the no drug control were taken at the indicated times (h) and loaded on the gel. (B) CL nicking of DNA is time and concentration dependent. SV40 plasmid (2 µg/ml) was treated with buffer (lanes 1 and 6) or with CL at 50 (lanes 2 and 7), 100 (lanes 3 and 8), 200 (lanes 4 and 9) and 400 (lanes 5 and 10) µM. Aliquots of the reactions were taken at 24 (lanes 1–5) or 48 (lanes 6–10) h and loaded on the gel alongside controls.
CL stocks stored for more than 7 days in EtOH solutions gradually lost their ability to irreversibly poison topo II and this loss was correlated with structural modification, as inferred by NMR spectra (5). To test if aging of CL stocks also impairs drug reactivity towards DNA, we tested our active CL stock in the nicking assay after 6 weeks. The results are shown in Figure 3. Freshly dissolved CL, as expected, caused DNA strand scission (lanes 4, 5, 9 and 10), whereas the stock kept frozen in EtOH for 6 weeks was totally inactive towards the nucleic acid (lanes 2, 3, 7 and 8) and no evidence of nicking can be seen, even after 48 h. To check which chemical modification of the structure of CL is linked to the loss of reactivity, 1H NMR spectra of the aged inactive stock of CL were taken and compared to those of fresh stocks (5). Again, the spectrum of the aged stock revealed major modifications, the most notable being disappearance of the signals of the epoxide protons at 3.35 and 2.85 p.p.m. (not shown).
Figure 3.

Aged CL stocks do not cleave DNA. Aged CL stocks were diluted to 200 (lanes 2 and 7) and 100 (lanes 3 and 8) µM and reacted with SV40 plasmid, along with freshly dissolved CL diluted to 200 (lanes 4 and 9) and 100 (lanes 5 and 10) µM for the time indicated. No-drug controls are shown in lanes 1 and 6.
CL reacts with unpaired bases in DNA
To identify the site(s) of covalent attack by CL, the drug was reacted with short oligos and the cleavage bands obtained were resolved and mapped on a sequencing gel. The sequence chosen corresponds to one CL hot-spot for irreversible topo II-mediated cleavage in linear SV40 DNA (5). To precisely localize the site of cleavage, an aliquot of the samples and relative controls were further reacted with hot piperidine as in the Maxam and Gilbert reaction (16).
Since the torsional stress in negatively supercoiled DNA stabilizes locally distorted structures (27), we analyzed the reactivity of CL towards G residues either base paired to their complementary cytosines, as in all-annealed oligos, or non-base paired, as in locally distorted structures. These bulges were obtained by annealing 5′-end-labeled oligos to a partially complementary sequence which locates the hot-spot G in a 3 (Bulge3) or 4 nt bulge (Bulge4). Besides the extruded G residues, both bulged oligos contained paired G residues as internal controls. The results of CL treatment of annealed and bulged oligos are shown in Figure 4A. The extent of CL-induced DNA cleavage at the hot-spot G is not significantly changed relative to the control lanes in the annealed oligo, whereas it is known that CL reacts with dsDNA in the presence of topo II (5). Hence, the nicking observed in the supercoiled form of the plasmid cannot be ascribed to CL attack on paired bases in B-form DNA. In contrast, CL per se strongly stimulates DNA strand scission at G residues exposed in the loops (see bands marked G in Fig. 4A), while, again, no enhancement of cleavage with respect to the controls was observed for any paired G (bands marked g). DNA cleavage at the non-base paired G residues is evident as a set of bands in the samples not treated with piperidine (lanes 3 and 5), which, after CL reaction and precipitation, were kept on ice. Of these bands, a weak one (G band) co-migrated with the fragment corresponding to G obtained by the Maxam and Gilbert reaction, while the most intense bands were retarded in the sequencing gel. These slower migrating bands could be generated by strand scission 3′ of a CL-induced apurinic (AP) site, similarly to that observed for other alkylators (28,29). As expected, all these bands were transformed into a single band by hot piperidine incubation (see lanes 4 and 6 for each set of bulged oligos), which completes chemical degradation at the AP site, leading to DNA cleavage 5′ of the modified base (16).
Figure 4.

CL cleaves DNA at guanines. (A) Guanines in bulges are the substrate for CL attack. The 30 nt oligos depicted in the figure are CL2640-up, marked by an asterisk on the left, and its complementary sequence CL2640-down, marked with an asterisk on the right. Their sequences, obtained by the Maxam and Gilbert reaction for purines (G+A) and for guanines (G), are shown at each side of the gel. The three different structures, schematized at the top of the gel, are, from left to right, Bulge3 (TGT), Bulge4 (TGGT) and the all-annealed (double-stranded) form. To obtain them, each 5′-labeled oligo (strand marked by an asterisk) was annealed to an equimolar amount of its complementary sequence as detailed in Materials and Methods. G in the gel marks the guanines which are extruded in the bulges, while g refers to the annealed guanines. In the double-stranded form on the right side of the gel all guanines are annealed. Lanes are numbered 0 to 6 for each structure. Lanes 0 refer to untreated oligos, while all other samples (lanes 1–6) were reacted at 37°C for 24 h. Samples in lanes 2, 4 and 6 were further subjected to hot piperidine. Oligos in lanes 1 and 2 were treated with buffer only, those in lanes 3 and 4 with 100 µM CL and those in lanes 5 and 6 with 200 µM CL. (B) CL damages other unpaired bases besides G. The single-stranded form of CL2640-up, labeled at its 5′-end, was reacted at 37°C with buffer only (lanes 1–4) or 200 µM CL (lanes 5–8). Samples were ethanol precipitated after 24 (lanes 1, 3, 5 and 7) or 48 (lanes 2, 4, 6 and 8) h. Samples in lanes 3, 4, 7 and 8 were further treated with hot piperidine.
An increase in cleavage is observed at the T immediately preceding the reactive G, which is unpaired and exposed in the bulge. Since the same phenomenon is observed in the oligo subjected to dimethyl sulfate reaction (G ladder), we believe this is due to a similar lability of the phosphodiester backbone adjacent to the AP site rather than to a direct attack on thymidine, which lacks nucleophilic sites. On the other hand, we cannot exclude a reaction of CL with other unpaired bases besides G. CL was therefore reacted with the oligo CL2640-up in its single-stranded form, and the results are reported in Figure 4B. Oligo cleavage and band retardation at G residues is again evident before hot alkaline treatment (lanes 5 and 6), while DNA damage at C residues is observed after heat and piperidine (lanes 7 and 8). The level of damage at A residues (lanes 7 and 8) is only slightly higher than the control lanes (lanes 3 and 4). Cleavage at T is again observed only when the pyrimidine precedes the reactive G.
Species of slightly lower mobility than the oligo itself appear in the presence of the drug before alkaline treatment (lanes 5 and 6). These are likely the result of DNA retardation caused by non-cleaved and non-cleavable CL adduct formation at bases less reactive than G in DNA.
CL alkylates N7 of guanine
We have demonstrated that CL can attack specific accessible sites of bases and, in the case of G, the adduct formed easily depurinates, leading to single strand scission. Since G is the base at which irreversible poisoning of topo II takes place, we further investigated the mechanism of reaction of CL with guanines. The lability of the adduct formed suggests that the site of reaction is the nitrogen at position 7, which is the strongest nucleophile of G. Indeed, N7-alkylated G residues easily depurinate, causing spontaneous strand scission in DNA (28). To test this hypothesis, we incubated CL (100 and 200 µM) with an oligo in which one G was substituted with 7-deaza-2′-deoxyguanosine (7dG in Fig. 5), while the other G is wild-type and represents an internal control (wild-type versus 7-deaza-G). The reaction was performed directly with the oligo in its single-stranded form, under the same conditions as shown in Figure 4B.
Figure 5.
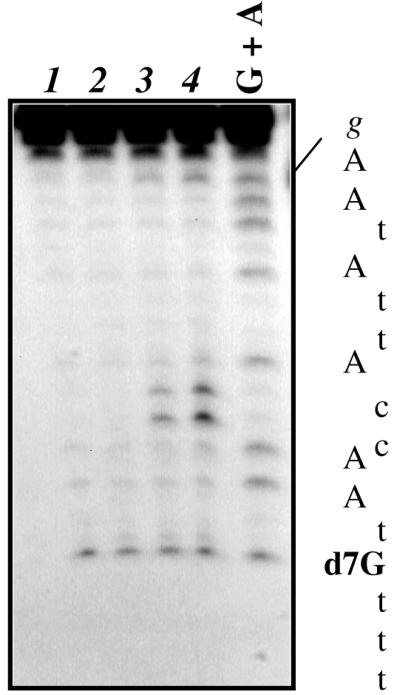
CL attacks N7 of guanines. The single-stranded form of CL2640-d7G, in which the central G is substituted by 7-deaza-G (d7G), was reacted at 37°C with buffer (lanes 1–2) or 100 (lane 3) or 200 µM (lane 4) CL for 24 h (lanes 2–4). After EtOH precipitation samples were further treated with hot piperidine. G+A refers to the purine ladder obtained by treating the wild-type G oligo.
The results reported in Figure 5 show how, upon CL incubation followed by hot piperidine treatment, the relevant wild-type bases react with CL as expected, but 7-deaza-G shows no increase in cleavage with respect to the control. The same result is obtained when all G residues are substituted by 7-deaza-2′-deoxyguanosine (not shown). The analog 7-deaza-G is therefore not affected by CL. We can hence conclude that N7 of G is the nucleophile targeted by the reactive group(s) of CL, this adduct being in turn responsible for strand scission at accessible positions.
CL accessibility to the major groove of DNA is sterically hindered
Our molecular modeling simulations show that CL, which does not intercalate into DNA (2), is a bulky molecule which fits poorly into either groove in B-DNA. In fact, the CL–DNA interaction energies after the docking/minimization protocol are 12 kcal mol–1 for the major groove complex and 39 kcal mol–1 for the minor groove complex, respectively. In particular, a strong electrostatic repulsion is expected to occur between the negatively charged phosphates of the minor groove and the CL moiety. According to the molecular modeling studies, CL can approach its target solely from the major groove and react only when the DNA helix is substantially distorted.
CL does not attack DNA in the presence of topo I
CL can attack and cleave DNA, but its reactivity depends on the accessibility of the reactive groups of CL to nitrogen 7 of G. Since both prokaryotic and eukaryotic topo II allow linear dsDNA to become a substrate for CL, we checked if the structure of the cleavable complex of topo I could be similarly accessible to CL. The drug was incubated with 3′-end-labeled SV40 and the human enzyme, using buffers excluding Tris, and several enzyme:DNA ratios. The result of a typical experiment is shown in Figure 6. As expected, CL alone does not cleave linear double-stranded plasmid after 30 min incubation (lanes 2–4). Also, it cannot trap the cleavable complex of linear DNA and human topo I (lanes 8–10). CL, therefore, can access reactive bases of DNA in the topo II but not topo I cleavable complex.
Figure 6.
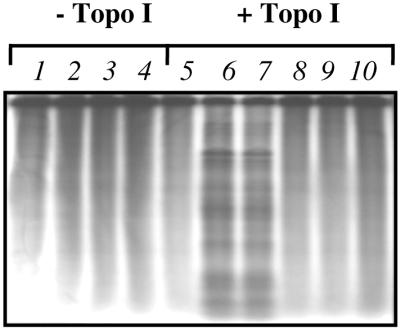
CL is not a topo I poison. Labeled SV40 DNA (0.020 µg) was reacted with CL alone (lanes 2–4) or with drug in the presence of human topo I (lanes 8–10) at 37°C in 20 mM phosphate buffer, pH 7.4, 10 mM MgCl2, 60 mM KCl, 0.5 mM DTT, 30 µg/ml BSA. The concentrations of CL were 25 (lanes 2 and 8), 50 (lanes 3 and 9) and 100 (lanes 4 and 10) µM, respectively, while camptothecin at 1 and 10 µM is present in lanes 6 and 7. The control lacking enzyme and drug is in lane 1, while DNA in lane 5 was incubated with enzyme but no drugs.
DISCUSSION
We have shown in this paper how CL, a topo II poison able to stabilize irreversible cleavage at G at the position immediately preceding the cleavage site (–1), can react with DNA per se. Direct damage to supercoiled DNA caused by CL in the absence of enzyme has not been reported before (2,4). Our data show that the drug can react with unpaired bases and attack the nitrogen on position 7 of G. Indeed, this position is known to represent the most reactive site for DNA alkylation by drugs like psorospermins and nitrogen mustards (30–33). As expected, the adduct formed at N7 of G is rather labile and can depurinate, eventually leading to phosphodiester cleavage, which explains the nicking we observe in supercoiled DNA. The reaction responsible for DNA attack and strand scission involves the epoxide at position 13: a similar reactivity has also been described for the pluramycin family of antibiotics and for the active metabolite of aflatoxin B1, DNA intercalators which, upon binding to DNA, alkylate N7 of guanines through their epoxy group (12,14,33). However, unlike these and other small alkylating agents which can easily diffuse into the major groove and react with dsDNA (30–32), CL is unique since it cannot alkylate dsDNA in its canonical B-form, either from the major or from the minor groove. Reactivity at G residues clearly depends on the presence of locally distorted and unpaired structures in the nucleic acid, consistent with the molecular modeling findings that CL does not access the bases of a regular double helix.
The reactivity of CL towards DNA can explain the irreversible cleavage at G residues in the presence of topo II. In fact, irreversible cleavage stimulation is substantially reduced when using aged CL samples (5), which also have impaired direct reactivity towards the nucleic acid: our data support the hypothesis that, in the context of the cleavable complex, the drug can directly alkylate N7 of G residues at –1, leading to the formation of an adduct which, in turn, traps the complex irreversibly. The enzyme hence catalyzes formation of the adduct, since cleavage at G residues in plasmid DNA or oligos requires higher doses and longer reaction times than those necessary in the presence of topo II (5). Irreversible poisoning of topo II by alkylation of specific nucleotides has only been reported, to the best of our knowledge, for specifically designed alkylating anthraquinones (34) and for azidoamsacrine, a photoactivable amsacrine which, following intercalation, can react covalently with T residues at the cleavage site (35). These compounds, however, are all DNA intercalators which interact efficiently with dsDNA and therefore, differently from CL, can produce random, topoisomerase-independent alkylation along the genome.
Alkylation at specific positions of G and A residues has recently been reported to stimulate DNA cleavage by topo II (36); similarly to what was observed for psorospermin (13), stimulation of cleavage was maximal when the alkylated base was at position +4 or inside the stagger, whereas alkylation of the G at position –1 was shown to strongly inhibit topo II-mediated DNA cleavage (36). Similarly, AP lesions at this site result in inhibition, rather than stimulation, of enzyme-mediated cleavage (37). Based on these results, we can safely conclude that alkylation and depurination at –1 by CL prior to formation of the cleavable complex is unlikely to happen, but rather, as we propose, enzyme intervention is necessary to allow CL to attack DNA at G residues. Depurination following the formation of topo II-induced CL adducts cannot be excluded and could similarly result in further irreversible damage by the terpenoid antibiotic.
In conclusion, according to our results CL accesses dsDNA in the presence of topo II, likely producing irreversible cleavage when the drug epoxy group faces N7 of a G immediately preceding the cutting site in the cleavable complex. To confirm the peculiarity of this process, reaction with the nucleic acid in the presence of topo I is not allowed.
These results, besides giving further insight into the mechanism of action of CL, represent a very useful tool to dissect the structural features of the DNA–topoisomerase complex. In this connection, it would be very interesting to check CL poisoning activity toward type IA enzymes, whose mechanism of breakage and rejoining of DNA is surprisingly similar to type IIA enzymes (38).
Acknowledgments
ACKNOWLEDGEMENT
G.C. acknowledges grant support from the Associazione Italiana per la Ricerca sul Cancro, Milan, Italy.
References
- 1.Andersen N.R., Lorck,H.O. and Rasmussen,P.R. (1983) Fermentation, isolation and characterization of antibiotic PR-1350. J. Antibiot. (Tokyo), 36, 753––760.. [DOI] [PubMed] [Google Scholar]
- 2.Kawada S., Yamashita,Y., Fujii,N. and Nakano,H. (1991) Induction of a heat-stable topoisomerase II-DNA cleavable complex by nonintercalative terpenoides, terpentecin and clerocidin. Cancer Res., 51, 2922––2925.. [PubMed] [Google Scholar]
- 3.Kawada S., Yamashita,Y., Ochiai,K., Ando,K., Iwasaki,T., Takiguchi,T. and Nakano,H. (1995) Terpentecin and ECT4B, new family of topoisomerase II targeting antitumor antibiotics produced by Streptomyces: producing organism, fermentation and large scale purification. J. Antibiot. (Tokyo), 48, 211––216.. [DOI] [PubMed] [Google Scholar]
- 4.McCullough J.E., Muller,M.T., Howells,A.J., Maxwell,A., O’Sullivan,J., Summerill,R.S., Parker,W.L., Wells,J.S., Bonner,D.P. and Fernandes,P.B. (1993) Clerocidin, a terpenoid antibiotic, inhibits bacterial DNA gyrase. J. Antibiot. (Tokyo), 46, 526––530.. [DOI] [PubMed] [Google Scholar]
- 5.Binaschi M., Zagotto,G., Palumbo,M., Zunino,F., Farinosi,R. and Capranico,G. (1997) Irreversible and reversible topoisomerase II DNA cleavage stimulated by clerocidin: sequence specificity and structural drug determinants. Cancer Res., 57, 1710––1716.. [PubMed] [Google Scholar]
- 6.Borgnetto M.E., Tinelli,S., Carminati,L. and Capranico,G. (1999) Genomic sites of topoisomerase II activity determined by comparing DNA breakage enhanced by three distinct poisons. J. Mol. Biol., 285, 545––554.. [DOI] [PubMed] [Google Scholar]
- 7.Kawada S.Z., Yamashita,Y., Uosaki,Y., Gomi,K., Iwasaki,T., Takiguchi,T. and Nakano,H. (1992) UCT4B, a new antitumor antibiotic with topoisomerase II mediated DNA cleavage activity, from Streptomyces sp. J. Antibiot. (Tokyo), 45, 1182––1184.. [DOI] [PubMed] [Google Scholar]
- 8.Sehested M. and Jensen,P.B. (1996) Mapping of DNA topoisomerase II poisons (etoposide, clerocidin) and catalytic inhibitors (aclarubicin, ICRF-187) to four distinct steps in the topoisomerase II catalytic cycle. Biochem. Pharmacol., 51, 879––886.. [DOI] [PubMed] [Google Scholar]
- 9.Dorado L., Ruis Montoya,M.R. and Rodriguez Mellado,J.M. (1992) A contribution to the study of the structure-mutagenicity relationship for alpha-dicarbonyl compounds using the Ames test. Mutat. Res., 269, 301––306.. [DOI] [PubMed] [Google Scholar]
- 10.Kasai H., Iwamoto Tanaka,N. and Fukada,S. (1998) DNA modifications by the mutagen glyoxal: adduction to G and C, deamination of C and GC and GA cross-linking. Carcinogenesis, 19, 1459––1465.. [DOI] [PubMed] [Google Scholar]
- 11.Millard J. and White,M. (1993) Diepoxybutane cross-links DNA at 5′-GNC sequences. Biochemistry, 32, 2120––2123.. [DOI] [PubMed] [Google Scholar]
- 12.Hansen M., Lee,S.-J., Cassady,J. and Hurley,L. (1996) Molecular details of the structure of a psorospermin-DNA covalent/intercalation complex and associated DNA sequence selectivity. J. Am. Chem. Soc., 118, 5553––5561.. [Google Scholar]
- 13.Kwok Y., Zeng,Q. and Hurley,L. (1998) Topoisomerase II-mediated site-directed alkylation of DNA by psorospermin and its use in mapping other topoisomerase II poison binding sites. Proc. Natl Acad. Sci. USA, 95, 13531––13536.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gopalakrishnan S., Harris,T.M. and Stone,M.P. (1990) Intercalation of aflatoxin B1 in two oligodeoxynucleotide adducts: comparative 1H NMR analysis of d(ATCAFBGAT).d(ATCGAT) and d(ATAFBGCAT)2. Biochemistry, 29, 10438––10448.. [DOI] [PubMed] [Google Scholar]
- 15.Wang J.C. (1974) Interactions between twisted DNAs and enzymes. The effects of superhelical turns. J. Mol. Biol., 87, 797––816.. [DOI] [PubMed] [Google Scholar]
- 16.Maxam A.M. and Gilbert,W. (1980) Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol., 65, 499––560.. [DOI] [PubMed] [Google Scholar]
- 17.Gatto B., Sanders,M.M., Yu,C., Wu,H.Y., Makhey,D., Lavoie,E.J. and Liu,L.F. (1996) Identification of topoisomerase I as the cytotoxic target of the protoberberine alkaloid coralyne. Cancer Res., 56, 2795––2800.. [PubMed] [Google Scholar]
- 18.Weiner S.J., Kollman,P.A., Nguyen,D.T. and Case,D.A. (1986) An all-atom force field for simulation of protein and nucleic acids. J. Comput. Chem., 7, 230––254.. [DOI] [PubMed] [Google Scholar]
- 19. Chemical Computing Group Inc. (1999) Molecular Operating Environment 1999.05. Chemical Computing Group, Montreal, Canada.
- 20.Halgren T. (1996) Merck Molecular Force Field. I. Form, scope, parameterization and performance of MMFF94. J. Comput. Chem., 17, 490––519.. [Google Scholar]
- 21.Halgren T. (1996) Merck Molecular Force Field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interaction. J. Comput. Chem., 17, 520––552.. [Google Scholar]
- 22.Halgren T. (1996) Merck Molecular Force Field. III. Molecular geometrics and vibrational frequencies for MMFF94. J. Comput. Chem., 17, 553––586.. [Google Scholar]
- 23.Halgren T. (1996) Merck Molecular Force Field. IV. Conformational energies and geometries for MMFF94. J. Comput. Chem., 17, 587––615.. [DOI] [PubMed] [Google Scholar]
- 24.Halgren T. and Nachbar,R. (1996) Merck Molecular Force Field. V. Extension of MMFF94 using experimental data, additional computational data and empirical rules. J. Comput. Chem., 17, 616––632.. [Google Scholar]
- 25.Frisch M., Trucks,G.W., Schlegel,H., Scuseria,G., Robb,M., Cheeseman,J., Zakrzewski,V., Montgomery,J., Stratmann,R.E., Burant,J.C. et al. (1998) Gaussian 98. Gaussian Inc., Pittsburgh, PA.
- 26.Chirlian L.E. and Francl,M.M. (1987) Atomic charges derived from electrostatic potentials: a detailed study. J. Comput. Chem., 8, 894––905.. [Google Scholar]
- 27.Palecek E. (1991) Local supercoil-stabilized DNA structures. Crit. Rev. Biochem. Mol. Biol., 26, 151––226.. [DOI] [PubMed] [Google Scholar]
- 28.Hara M., Yoshida,M. and Nakamo,H. (1990) Covalent modification and single-strand scission of DNA by a new antitumor antibiotic Kapurimycin A3. Biochemistry, 29, 10449––10455.. [DOI] [PubMed] [Google Scholar]
- 29.Sun D., Hansen,M., Clement,J. and Hurley,L. (1993) Structure of the altromycin B (N7-guanine)-DNA adduct. A proposed prototypic DNA adduct structure for the pluramycin antitumor antibiotic. Biochemistry, 32, 8068––8074.. [DOI] [PubMed] [Google Scholar]
- 30.Kohn K.W., Hartley,J.A. and Mattes,W.B. (1987) Mechanisms of DNA sequence selective alkylation of guanine-N7 positions by nitrogen mustards. Nucleic Acids Res., 15, 10531––10549.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartley J.A., Gibson,N.W., Kohn,K.W. and Mattes,W.B. (1986) DNA sequence selectivity of guanine-N7 alkylation by three antitumor chloroethylating agents. Cancer Res., 46, 1943––1947.. [PubMed] [Google Scholar]
- 32.Mattes W.B., Hartley,J.A. and Kohn,K.W. (1986) DNA sequence selectivity of guanine-N7 alkylation by nitrogen mustards. Nucleic Acids Res., 14, 2971––2987.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hansen M. and Hurley,L. (1995) Altromycin B threads the DNA helix interacting with both the major and the minor grooves to position itself for site-directed alkylation of guanine N7. J. Am. Chem. Soc., 117, 2421––2429.. [Google Scholar]
- 34.Kong X.B., Rubin,L., Chen,L.I., Ciszewska,G., Watanabe,K.A., Tong,W.P., Sirotnak,F.M. and Chou,T.C. (1992) Topoisomerase II-mediated DNA cleavage activity and irreversibility of cleavable complex formation induced by DNA intercalator with alkylating capability. Mol. Pharmacol., 41, 237––244.. [PubMed] [Google Scholar]
- 35.Freudenreich C.H. and Kreuzer,K.N. (1994) Localization of an aminoacridine antitumor agent in a type II topoisomerase-DNA complex. Proc. Natl Acad. Sci. USA, 91, 11007––11011.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sabourin M. and Osheroff,N. (2000) Sensitivity of human type II topoisomerases to DNA damage: stimulation of enzyme-mediated DNA cleavage by abasic, oxidized and alkylated lesions. Nucleic Acids Res., 28, 1947––1954.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kingma P.S. and Osheroff,N. (1997) Apurinic sites are position-specific topoisomerase II poisons. J. Biol. Chem., 272, 1148––1155.. [DOI] [PubMed] [Google Scholar]
- 38.Liu Q. and Wang,J. (1999) Similarity in the catalysis of DNA breakage and rejoining by type IA and IIA DNA topoisomerases. Proc. Natl Acad. Sci. USA, 96, 881––886.. [DOI] [PMC free article] [PubMed] [Google Scholar]